

Background: A high sugar diet leads to obesity and insulin resistance in Drosophila.
Results: The metabolic fate of dietary glucose is reprogrammed in high sugar-fed and lean animals.
Conclusion: Obesity is protective against the deleterious effects of a high sugar diet.
Significance: An emerging perspective that obesity is protective against sequelae of human metabolic disease is conserved in the fly.
Keywords: Diabetes, Drosophila Metabolism, Lipogenesis, Metabolic Tracers, Obesity, ChREBP, Caloric Excess, High Sugar Diet, Stearoyl-CoA Desaturase
Abstract
The Drosophila fat body is a liver- and adipose-like tissue that stores fat and serves as a detoxifying and immune responsive organ. We have previously shown that a high sugar diet leads to elevated hemolymph glucose and systemic insulin resistance in developing larvae and adults. Here, we used stable isotope tracer feeding to demonstrate that rearing larvae on high sugar diets impaired the synthesis of esterified fatty acids from dietary glucose. Fat body lipid profiling revealed changes in both carbon chain length and degree of unsaturation of fatty acid substituents, particularly in stored triglycerides. We tested the role of the fat body in larval tolerance of caloric excess. Our experiments demonstrated that lipogenesis was necessary for animals to tolerate high sugar feeding as tissue-specific loss of orthologs of carbohydrate response element-binding protein or stearoyl-CoA desaturase 1 resulted in lethality on high sugar diets. By contrast, increasing the fat content of the fat body by knockdown of king-tubby was associated with reduced hyperglycemia and improved growth and tolerance of high sugar diets. Our work supports a critical role for the fat body and the Drosophila carbohydrate response element-binding protein ortholog in metabolic homeostasis in Drosophila.
Introduction
Caloric restriction has been the subject of much study in model organisms. More recently, caloric excess has been shown to elicit many of the same negative consequences in model organisms as have long been observed in humans (1–7). Work from our laboratory and that of others has shown that feeding Drosophila a high sugar diet (HSD)2 results in a model of type 2 diabetes (T2D) that includes hyperglycemia, obesity, insulin resistance, cardiac arrhythmias, and reduced lifespan (4, 8, 73).
Drosophila larvae eat continually and store fat for metamorphosis as they develop, making them a convenient model system for studying relationships between nutrition and obesity. The Drosophila fat body (FB) is an organ that serves the roles of adipose tissue, liver, and the immune system. The FB is the animal's primary fat storage depot as well as the source of antimicrobial peptides, endocrine mediators of neuronal function, and cytochrome p450 enzymes that catalyze detoxification and biosynthetic reactions (9–12). FB insulin and adipokinetic hormone (glucagon ortholog) signaling can control lipid storage in a manner similar to that in human liver (13, 14). Therefore, we studied the role of the Drosophila FB in the response to an HSD.
High calorie diets are associated with non-alcoholic fatty liver disease, obesity, and insulin resistance in rodents and humans (2, 15–18), but the role of lipid accumulation remains controversial. An emerging concept is that obesity is protective against T2D (19, 20). It has been postulated that obesity itself is not harmful, but when the capacity to store triglyceride (TAG) is exceeded, accumulation of saturated free fatty acids or their derivatives (called lipotoxicity) acts to damage tissues and contribute to the complications of T2D (21, 22).
We used biochemical and genetic approaches to understand lipid biology in the setting of caloric excess. We demonstrated that the Drosophila FB plays an essential role in systemic metabolic homeostasis in the face of dietary excess. The rates of synthesis from labeled glucose into fatty acids and lipid storage pools were reduced upon HSD rearing. Lipids were stored in larger droplets and contained TAG species with fatty acid substituents that had shorter carbon chains and a greater degree of unsaturation in the FBs of HSD-fed larvae compared with those in larvae fed control diets. TAG storage in the FB ameliorated the adverse metabolic effects of HSD feeding because hyperglycemia was exacerbated in lean animals and reduced when FB TAG content was increased in tissue-specific loss of function experiments. Our data support a model where TAG storage is protective when consuming an HSD. When the capacity to store TAG is exceeded during this form of caloric excess, we propose that dysregulated lipogenesis results in adverse consequences, which may include an increase in free fatty acids.
EXPERIMENTAL PROCEDURES
Fly Lines and Husbandry
Canton-S and cgGAL4 lines were from Bloomington Drosophila Stock Center. The control w1118, UAS-Dcr2, UAS-Mioi, UAS-desat1i, and UAS-King-Tubbyi lines were from the Vienna Drosophila RNAi Center. Knockdown was confirmed by semiquantitative RT-PCR of RNA from transgene-expressing flies. Diets were a modification of Bloomington semi-defined food and contained 0.15 or 0.7 m sucrose as the sugar source as described previously (4). Larvae were reared from egg lay until wandering third instar and harvested from the vial wall for all experiments except tracer studies.
Stable Isotope Tracer Experiments
[U-13C6]Glucose was used as 10% of dietary sugar in either 5 (control) or 34% (high) glucose Bloomington semi-defined food diets. Third instar Canton-S larvae were fed unlabeled diets until early third instar and transferred to tracer diets (Fig. 1A). Twenty-five larvae were homogenized in water, and aqueous and hexane phases were obtained as described (23). Lipids from the organic phase were derivatized to fatty acid methyl esters using either iodomethane (nonesterified fatty acids (NEFAs)) or 10% acetyl chloride in methanol (total free + esterified fatty acids (TFAs)) and analyzed by GC/MS as described previously (23). TFA and NEFA concentrations were determined by quantitative GC using a flame ionization detector relative to a known amount of C17:0 internal standard fatty acid added to the sample prior to processing (23). We calculated the lipid derived from dietary sugar by multiplying the FA 13C percent enrichment (Fig. 1, C and D) and the FA pool size (Table 1) for each condition. This product was then divided by the 13C atom percent enrichment in the tracer food (Fig. 1E) as extrapolated from the trehalose labeling. The rates of synthesis of NEFA and TFA were then derived from the slopes of the plotted values. The distribution of mass isotopomers of TFA or NEFA methyl ester species was determined by GC/MS as described previously (24, 25) on an Agilent 5973 MSD instrument using a 30-m × 0.25-mm DB-5MS column. Isotopomer distributions of labeled glucose and trehalose were measured by GC/MS on trimethylsilyl derivatives. All GC/MS isotopic enrichments were corrected for the natural abundance of 13C in larvae prior to the consumption of tracer diets. The total atom percent excess 13C content within TFA and NEFA was estimated from the measured fatty acid distribution profile and the total atom percent excess 13C for each fatty acid species calculated from the distribution of labeled isotopomers.
FIGURE 1.

Stable isotope tracer studies of glucose metabolism. A, larvae were reared on control or HSD foods and then transferred as early L3 to diets containing 10% [U-13C6]glucose. Larvae were fed tracer diets as indicated, and lipid concentrations and 13C (*) enrichment were measured in non-esterified (NEFA) and total fatty acids (TEFA) by GC/MS. Labeled glucose can be metabolized into acetyl-CoA (which can be either singly or doubly labeled with 13C and incorporated into de novo synthesized non-esterified fatty acids (1). These labeled fatty acids can be esterified as TAG (2). B, larval weights at 24 h after transfer to tracer diets. C, tracer incorporation from dietary [U-13C6]glucose into NEFAs. D, tracer incorporation from dietary [U-13C6]glucose into TFAs. E, tracer incorporation from dietary [U-13C6]glucose into trehalose. F, normalized NEFA synthesis rates from dietary carbohydrate. G, normalized TFA synthesis rates from dietary carbohydrate. Shown is the weighted average of 14-, 16-, and 18-carbon fatty acids. Error bars are ±S.E. An unpaired, two-tailed t test was used to derive p values. ctl, control; carbs, carbohydrates.
TABLE 1.
Lipid pool sizes in control and HSD-reared larvae
TFA and NEFA contents of larvae reared on unlabeled diets until early L3 were measured by GC analysis at the indicated time after transfer of the larvae to tracer diets. Pool sizes were calculated as mean nmol/larva ±S.D. ctl, control.
| 2 h | 6 h | 24 h | |
|---|---|---|---|
| nmol/larva | nmol/larva | nmol/larva | |
| TFA | |||
| ctl → 13C ctl | 95.6 ± 21.7 | 117.8 ± 4.0 | 251.7 ± 28.6 |
| ctl → 13C HS | 110.2 ± 20.1 | 116.6 ± 12.7 | 287.9 ± 45.9 |
| HS → 13C ctl | 149.1 ± 25.9 | 127.9 ± 3.3 | 199.8 ± 21.2 |
| HS → 13C HS | 143.7 ± 5.5a | 161.3 ± 20.0a | 299.1 ± 14.8 |
| NEFA | |||
| ctl → 13C ctl | 5.34 ± 0.87 | 8.43 ± 1.38 | 13.85 ± 2.62 |
| ctl → 13C HS | 6.11 ± 0.56 | 6.99 ± 0.71 | 16.94 ± 0.45 |
| HS → 13C ctl | 3.74 ± 0.80 | 4.87 ± 0.52a | 12.43 ± 1.51 |
| HS → 13C HS | 4.81 ± 0.17a | 4.79 ± 0.90a | 11.06 ± 1.00a |
a A significant difference from larvae fed the same tracer diet but reared on control food at the same time point, p < 0.05 using a two-tailed, unpaired Student's t test.
Electrospray Ionization Mass Spectrometric Analyses of Glycerolipids
Larvae were homogenized in PBS and extracted by the method of Bligh and Dyer (26). The extract was concentrated to dryness under nitrogen and reconstituted in chloroform/methanol (1:1) to which LiCl was added (final [Li+], 2 mm). Phosphatidylcholine (PC) (27) and TAG (28) species were analyzed as Li+ adducts by positive ion ESI-MS/MS, and phosphatidylethanolamine and phosphatidylinositol (PI) species were analyzed as [M − H]− ions by negative ion ESI-MS/MS (29) on a Finnigan (San Jose, CA) TSQ-7000 triple stage quadrupole mass spectrometer with an ESI source controlled by Finnigan ICIS software. Lipid extracts were infused (1 μl/min) into the source with a Harvard syringe pump and analyzed under previously described instrumental conditions (27). For tandem MS, precursor ions selected in the first quadrupole were accelerated (32–36-eV collision energy) into a chamber containing argon (2.3–2.5 millitorrs) to induce collisionally activated dissociation, and product ions were analyzed in the final quadrupole. Identities of lipid molecular species in the total ion current profiles were determined from their tandem spectra as described.
Lipid Droplet Staining
For Nile Red staining, larvae were inverted and fixed in 4% paraformaldehyde for 20 min and then treated with 0.001% Nile Red in PBS for 30 min. Animals were washed, and then fat bodies were dissected out and mounted in Vectashield. Slides were imaged at 60× magnification on a Nikon confocal microscope.
Hemolymph Glucose
Hemolymph was collected and assayed as described previously (4).
Triglyceride Assays
Total triglycerides were assayed as described previously (4).
Lipase Assays
Measurement of lipase activity in larval homogenates was adapted from Ref. 30 as follows. Ten larvae were homogenized in 200 μl of citrate buffer, pH 5. 40 μl of homogenate was incubated with 150 μl of excess glycerol tributyrate substrate (170 mm; sonicated to mix into citrate buffer) for 4 h at 37 °C. After 4 h, samples were spun to pellet debris, and 20 μl of supernatant was added to 100 μl of Free Glycerol Reagent (Sigma F6428), incubated at 37 °C for 30 min, and read according to the manufacturer's instructions. Free glycerol levels in larvae measured immediately after homogenization were negligible (<5%).
Ketone Assays
Six larvae were homogenized in PBS on ice and analyzed using the Wako Total Ketone Body assay kit (415-73301 and 411-73401) according to the manufacturer's instructions.
Coenzyme A (CoA) Determinations
Eighty larvae were homogenized in PBS on ice, treated with 25 mm DTT, and frozen at −80 °C. After defrosting, larval pellets and debris were removed by centrifugation, and protein was removed by TCA precipitation. The supernatant was washed with water-saturated ether, and the aqueous phase was dried and resuspended in buffer A. Free CoA was quantified using reversed-phase HPLC with buffers and methods as described (31) using a CoA standard curve with a range of 0–20 nmol/sample for quantification. CoA eluted as a single peak around 28 min. Recovery of a CoA standard using this method was ∼30%.
Gene Expression
RNA was isolated from 75–150 wandering L3 FBs using TRIzol, DNase-treated, and purified using RNeasy columns (Qiagen). Poly(A) RNA was used to generate a cDNA library as described (32). An Illumina sequencer was used to generate 3–10 million reads per sample, and the reads were aligned to the Drosophila genome sequence using TopHat (33). Reads that aligned were considered for gene expression quantification with Cufflinks (34) using the MB8 genome annotation (which the Brent laboratory produced in its role as the informatics group for the Fly Transcriptome Group of the National Human Genome Research Institute modENCODE project). Gene expression was normalized using quantile normalization, and statistically significant differences (p < 0.05) were identified using an unpaired, two-tailed Student's t test. Complete expression data can be viewed at the Gene Expression Omnibus (GEO) under accession number GSE43734.
RESULTS
Metabolic Channeling of Glucose Is Reduced by HSD Feeding
We have shown previously that consumption of an HSD by Drosophila larvae results in phenotypes that model type 2 diabetes, including elevated hemolymph sugars, obesity, and insulin resistance. When fed excess calories in the form of an HSD, larvae routed energy from the diet into lipid storage in the fat body (4). We hypothesized that in the face of chronic exposure to HSD this storage capacity is limiting, leading to altered processing of dietary carbon and the model T2D phenotypes. To determine how HSD feeding alters carbon flux, we undertook a novel approach using stable isotope tracer methodology with uniformly 13C-labeled glucose to study the partitioning of carbons from dietary glucose ([U-13C6]glucose). Flies were fed diets in which [U-13C6]glucose made up 10% of the total added glucose content. The fate of dietary [U-13C6]glucose was tracked into NEFA and TFA. These two pools were of interest because free, non-esterified fatty acids are thought to be toxic, whereas TAG-esterified fatty acids are likely to be inert in human adipose and liver (35, 36). Wild-type animals were reared on unlabeled control or HSD foods until the early third larval instar and then transferred to labeled foods (Fig. 1A). Transfer experiments allowed us to evaluate the chronic effects of HSD rearing and simplified the comparison between labeled foods. HSDs reduced larval weight in these experiments, similar to published data from our laboratory (Fig. 1B and Ref. 4).
After consuming labeled food for up to 24 h, larvae were collected and homogenized, and lipids were extracted for determination of free and total fatty acid 13C content by GC/MS. For both labeling conditions, we observed significant 13C labeling of individual fatty acids (C14:0, C16:1, C16:0, C18:1, and C18:0) in the NEFA and TFA pools (data not shown). The relative percentage of 13C labeling increased over the 24-h labeling period and was similar for each free fatty acid except for C18:0 in both NEFA and TFA, which consistently exhibited lower 13C enrichment at each time point (data not shown). NEFA species exhibited higher 13C enrichment after consumption of the labeled HSD compared with labeled control food and was not affected by the rearing diet (Fig. 1C). When reared on control diets, TFA 13C enrichment was similar to that observed for NEFA (Fig. 1, C and D). In contrast to control rearing, larvae reared on HS diets synthesized TFA that incorporated a lower fraction of 13C-labeled carbons than did NEFA (Fig. 1, C and D). The lower TFA 13C enrichment in HSD-reared larvae persisted for at least 24 h after transfer to 13C-labeled HS or control food (3.6 ± 0.06 (S.E.) versus 5.2 ± 0.06 atom % and 1.1 ± 0.02 versus 2.0 ± 0.1 atom %, respectively; Fig. 1D). These results support a model that larvae fed an HSD increased the incorporation of dietary sugar into lipids, but chronic exposure to HSD resulted in a diminished ability to store dietary sugar as lipid.
From a technical standpoint, the higher level of 13C enrichment observed in larvae fed an HS tracer diet might have been due in part to less dilution of the tracer by other metabolically active sugars and carbohydrates. To estimate the atom percent content of metabolically available 13C in each diet, we measured the 13C enrichment into trehalose. The incorporation of tracer into trehalose occurred more rapidly than for NEFA or TFA (Fig. 1, C, D, and E). Based on the monoexponential rise to plateau in trehalose 13C content, we calculated that the bioavailable 13C was 8.1% in control (0.3 m glucose) and 9.2% in HSD (2 m glucose) foods, respectively (Fig. 1E). These values were used to determine the synthesis rates of NEFA and TFA derived from dietary carbohydrates (see below). The rate of 13C enrichment observed in the fatty acids is a function of both the rate of synthesis from the bioavailable 13C and the size of the pre-existing pool of that fatty acid. The pre-existing lipid pool size for TFA was larger in HSD-reared larvae (Table 1), contributing to the initial decrease in the observed 13C enrichment in TFA.
The NEFA and TFA synthesized from dietary bioavailable carbohydrate carbons after transfer to the tracer diets are shown in Fig. 1, F and G. NEFA synthesis on a 13C control diet was independent of the rearing diet (94.4 ± 3.3 (S.E.) pmol of FA/larva/h for control-reared and 95.7 ± 4.0 pmol of FA/larva/h for HS-reared). By contrast, on the HS 13C tracer diet, larvae reared on control food had a higher rate of synthesis of NEFA from dietary carbohydrates compared with HS-reared larvae (359.9 ± 9.5 versus 212.4 ± 6.7 pmol of FA/larva/h; Fig. 1F). When compared with NEFAs, the rates of synthesis of TFAs were 20–30 times higher yet also demonstrated effects of the rearing diets. HSD-reared larvae exhibit a much reduced rate of TFA synthesis from dietary carbohydrates compared with those reared on control food regardless of the tracer diet: 5.06 ± 0.25 versus 6.92 ± 0.30 nmol of FA/larva/h for HS 13C labeling and 1.19 ± 0.05 versus 2.56 ± 0.11 nmol of FA/larva/h for control labeling conditions (Fig. 1G). These differences between NEFA and TFA synthesis rates supported the hypothesis that prolonged consumption of an HSD diminished the rates of synthesis of lipids from dietary sugar and led us to more closely examine the processing of dietary glucose into NEFA and TFA.
Incorporation of glucose carbons into fatty acids occurs through a two-carbon acetyl-CoA intermediate that can be either singly (m + 1) or doubly (m + 2) labeled. An (m + 2)-labeled acetyl-CoA molecule arises directly from [U-13C6]glucose through glycolysis-derived pyruvate by way of citrate and is exported for FA synthesis, whereas singly labeled acetyl-CoA molecules have undergone one or more rounds of the tricarboxylic acid cycle and/or been diverted to other paths, such as conversion to phosphoenolpyruvate for gluconeogenesis, before export from the mitochondrion as citrate for lipogenesis (Fig. 2A). Therefore, the ratio of (m + 2)/(m + 1) isotopically labeled fatty acids (isotopomers) reflects the metabolic route of carbon atoms derived from dietary glucose. Larvae fed the labeled HSD exhibited higher m + 2 and m + 1 content in NEFA compared with larvae fed labeled control food that was due in part to the higher percentage of 13C labeling of sugars in the food as well as a general increase in lipogenesis induced by the HSD. Interestingly, neither the diet on which the larvae were reared nor the tracer diet had much effect on the NEFA isotopomer distribution 6 h after transfer to 13C diets (Fig. 2B). By contrast, the isotopomer labeling pattern in TFA clearly reflected the sugar content of the diet on which the larvae were reared. HSD-reared larvae transferred to either labeled HSD or control food exhibited marked reductions of the (m + 2)/(m + 1) ratio in TFA compared with larvae reared on control food and then switched to a 13C-labeled diet (Fig. 2C). The reduced ability of larvae to generate TFA from m + 2 FAs persisted for 24 h, although transfer of the larvae from HS to control diets did partially restore the ability to use m + 2 acetyl-CoA (Fig. 2D), correlating with an increase in growth (Fig. 1B). The strikingly different isotopomer labeling patterns of NEFA compared with those of TFA indicate that NEFA and TFA were derived from different pools of acetyl-CoA and that TFA synthesis was more greatly affected by HSD rearing. Taken together, these results support a model in which processing of dietary carbohydrate into esterified fatty acids stored in the FB is fundamentally altered in chronically HSD-fed larvae (Fig. 2A).
FIGURE 2.

A shift in metabolic channeling of dietary glucose occurs upon HSD feeding. A, proposed route of dietary glucose into lipid in larvae reared on control or chronic HS diets. Dietary [U-13C6]glucose undergoes glycolysis and then enters the tricarboxylic acid cycle (TCA) to follow a metabolic route either directly (m + 2; solid line) or indirectly (m + 1; dotted line) into fatty acids. OAA, oxaloacetate; PEP, phosphoenolpyruvate; pyr, pyruvate; G-6-P, glucose 6-phosphate. This diagram is simplified, and some steps have been omitted. B–D, isotopomer composition (m + 2 or m + 1) of NEFA at 6 h (B) or TFA at 6 h (C) or TFA at 24 h (D) after transfer to labeled diets. Shown is the weighted average of 14-, 16-, and 18-carbon fatty acids. Error bars are ±S.E. The ratio of (m + 2)/(m + 1) for each condition is shown beneath the graphs. An unpaired, two-tailed t test was used to derive p values. ctl, control.
The FB Lipidome Is Affected by Diet
To determine whether HSD consumption affects FB content of individual glycerolipid molecular species, ESI-MS/MS analyses were performed on FB lipids extracted from larvae raised on control food or a 0.7 m sucrose HSD (Fig. 3, A and B). Wandering third instar larvae were studied because this postfeeding stage exhibits many diet-dependent phenotypes, including obesity and insulin resistance (4). The relative abundances of various glycerolipid classes in FB were found to be similar to those reported previously for whole flies. The major lipid classes were TAG and phospholipids, and less than 10% of total lipid comprised diglycerides, ceramides, and free fatty acids (data not shown and Ref. 37). We therefore examined the molecular species of TAG and phospholipids further. Tandem mass spectra indicated that the predominant fatty acid substituents in larval FB TAG had carbon chain lengths of C16 or C18 with some C14 and even less with C12 (Fig. 3C).
FIGURE 3.

An HSD induced changes in TAG fatty acid length and saturation. Wandering third instar FBs were isolated from larvae reared on control (ctl) or 0.7 m sucrose HSD and subjected to ESI-MS analyses. A and B represent total ion current tracings that display the distribution of TAG molecular species as [M + Li]+ ions. C, collisionally activated dissociation tandem mass spectrum analysis of fatty acid composition of m/z 811 (D–F) TAG composition of FBs. Error bars are ±S.E. *, p < 0.05; **, p < 0.01.
When TAG species were represented by the sum of the carbon chain lengths of and the total number of double bonds in their FA substituents, HSD consumption was associated with a reduction in mean total chain length compared with a control diet (C47.053 ± 0.042 versus C47.300 ± 0.017, p = 0.0054). This is illustrated by the lower relative abundance of longer chain (C50 and C52) TAG species and a higher relative abundance of shorter chain (C44, C46, and C48) TAG species in FB of HSD-fed larvae compared with those fed a control diet (Fig. 3, D and E). HSD-fed larvae also exhibited an increase in the mean total number of double bonds (Δ1.549 ± 0.002 versus Δ1.390 ± 0.006, p = 1.31 × 10−5) in their FB TAG FA substituents as reflected by the higher relative abundance of doubly and triply unsaturated species and a lower relative abundance of monounsaturated and saturated species in HSD-fed larval FB compared with FB from larvae fed control food (Fig. 3F).
PC was the most abundant phospholipids in larval FBs with phosphatidylethanolamine and PI making up most of the remainder. Only one species of phosphatidylglycerol was observed in larval FB, albeit at levels just slightly above background (data not shown). Tandem mass spectra indicated that the most abundant PC species contained C16:1 and C18:1 FA substituents (data not shown). When FB PC species were represented by the sum of the carbon chain lengths of and the total number of double bonds in their FA substituents (Fig. 4A), HSD consumption led to a reduction in mean total chain length (C32.259 ± 0.016 versus C32.429 ± 0.027, p = 0.00543) compared with a control diet as reflected by a lower relative abundance of longer chain (C34 and C36) PC species and a higher relative abundance of shorter chain (C32 and C28) PC species in HSD-fed larvae compared with those fed control food (Fig. 4B). HSD feeding had little effect on the double bond content of FB PC fatty acid substituents (Fig. 4C).
FIGURE 4.

An HSD induced changes in phospholipid fatty acid length and saturation. Wandering third instar FBs were isolated from larvae reared on control or 0.7 m sucrose HSD and subjected to ESI-MS analyses. A–C, phosphatidylcholine composition. D–F, phosphatidylethanolamine composition. G–I, phosphatidylinositol composition. An unpaired, two-tailed t test was used to derive p values. *, p < 0.05; **, p < 0.01. Error bars are ±S.E.
Phosphatidylethanolamine in FBs from larvae reared on an HSD exhibited trends in chain length and unsaturation that were similar to those for TAG but of smaller magnitude (Fig. 4, D–F). The composition of larval FB PI (Fig. 4G) was also affected by HSD in a manner similar to that for TAG with a decrease in mean total carbon chain length (C33.180 ± 0.015 versus C33.322 ± 0.033, p = 0.017) as reflected by a lower relative abundance of longer chain (C34 and C36) PI species in FB and a higher relative abundance of shorter chain (C32) PI species for HSD larvae compared with those fed control food (Fig. 4H). An increase in mean double bond content (Δ1.438 ± 0.002 versus Δ1.292 ± 0.009, p = 2.28 × 10−4) of FA substituents in FB PI was also observed for HSD-fed larvae as reflected by a higher relative abundance of doubly unsaturated species and a lower relative abundance of monounsaturated species for HSD-fed larvae compared with those fed control food (Fig. 4I). These findings indicate that caloric excess affected the carbon chain lengths and degree of unsaturation of fatty acid substituents of glycerolipids in larval FBs.
HSDs Increase the Steady-state Levels of NEFA and Ketones
Our tracer studies demonstrated differences in the ability of larvae to channel dietary glucose carbons into stored fat versus NEFA. Given the proposed role of NEFA in pathophysiology of insulin resistance, we measured NEFAs in larvae and found them to be elevated on an HSD (Fig. 5A). One potential source of NEFA could be lipolysis; however, lipase activity was markedly reduced in larvae fed HSD (Fig. 5B), consistent with an increase in NEFA synthesis by an HSD and an attempt by the animal to retain these FAs in a stored form. Expression profiling studies also suggested that lipogenesis, lipid storage, and β-oxidation were increased on an HSD (4). β-Oxidation could lead to increased ketones, which are another potential source for use in generating NEFA. Ketone concentrations increased in larvae reared on an HSD compared with those reared on control food (Fig. 5C). Finally, CoA is required for NEFA to undergo β-oxidation or esterification into TAG and might be expected to be limiting in a setting of excess NEFA. CoA is also required for ketone production and many other cellular processes. Free CoA was measured and was decreased on an HSD (Fig. 5D), although the difference was not significant (p = 0.18).
FIGURE 5.
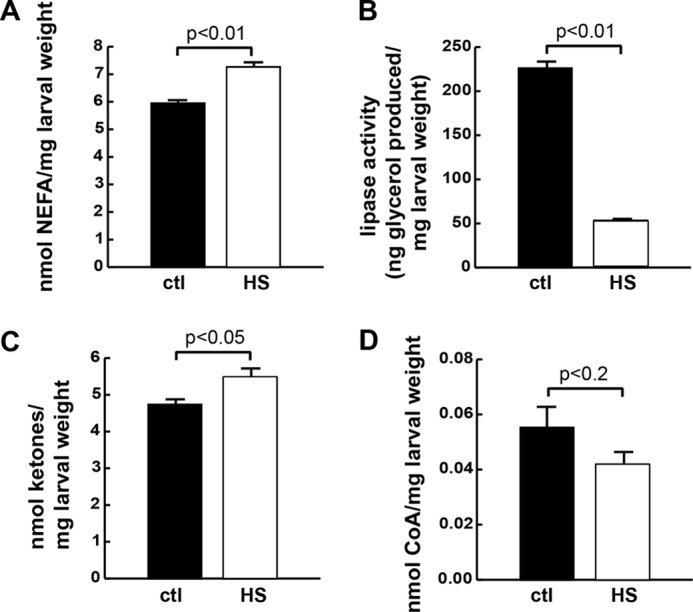
Steady state levels of lipid metabolic substrates are affected by an HSD. Wandering wild-type (cg-GAL4; Vw1118) third instar larvae were reared on control (ctl) (0.15 m sucrose) or HSD (0.7 m), homogenized, and analyzed. A, NEFA concentrations. B, lipase activity measured over 4 h. C, total ketone body concentrations. D, free CoA concentrations. Error bars are ±S.E. An unpaired, two-tailed t test was used to derive p values.
Lean Larvae Are Intolerant of an HSD
In response to excessive nutrient consumption, both mammals and flies increase fatty acid synthesis and esterification into TAG storage pools (2, 4, 38, 39). We previously observed that HSD-fed wandering third instar larvae exhibited gene expression changes that reflect increased lipogenesis and decreased lipolysis (4). These results are consistent with TAG accumulation in the FB, the major lipid storage organ, in HSD-fed larvae (4). To determine whether the HSD-induced rise in FB TAG content is protective or maladaptive, we used fat body-specific RNA interference (indicated by superscript i) to manipulate lipid storage. The expression of two putative lipogenic genes, Mio (CG18362) and desat1 (CG5887), was increased by feeding an HSD to larvae (4), so these genes became candidates of interest.
To generate lean FBs, we targeted Mio, which encodes a protein homologous to mammalian ChREBP, a carbohydrate-responsive transcription factor that up-regulates glycolytic and lipogenic gene expression in response to increased intracellular sugar levels (40). We also targeted stearoyl-CoA desaturase 1 (Desat1), which is a transcriptional target of vertebrate ChREBP. Vertebrate Desat1 is an enzyme that catalyzes conversion of saturated long chain fatty acids into monounsaturated fatty acids and participates in TAG synthesis (41). Interestingly, larvae with FB-specific loss of either Mio or Desat1 were unable to tolerate an HSD of 1 m sucrose (data not shown). A high sucrose diet is likewise lethal to lean mouse ChREBP mutants (42), implying that up-regulation of these genes in the FB upon chronic HSD feeding protects against caloric excess (Table 2). We therefore used an HSD of 0.7 m sucrose to study these mutants. Both cg-GAL4; UAS-Mioi and cg-GAL4; UAS-desat1i larvae were smaller than wild-type larvae reared on the 0.7 m HSD (Fig. 6A). HSD-reared cg-GAL4; UAS-Mioi and cg-GAL4; UAS-desat1i larvae contained less TAG than control larvae (Fig. 6B). Imaging the lipid droplets in the FB revealed an apparent reduction of lipid storage in cg-GAL4; UAS-Mioi and cg-GAL4; UAS-desat1i larval fat bodies compared with controls (Fig. 6, C–F). Upon consumption of the 0.7 m HSD, both of these lean animals exhibited increased hemolymph glucose concentrations compared with controls (Fig. 6, G and H). This suggests that fat storage in the FB protects against deleterious effects of an HSD.
TABLE 2.
Expression changes induced by an HSD in wild-type larval fat body
Wild-type Vw1118; UAS-Dcr2; cgGAL4 larvae were reared on a 0.15 m (control (ctl)) or 0.7 m HSD, and FBs were isolated for RNA extraction. A two-tailed Student's t test was used to derive p values. For simplicity, only a selection of genes is shown here; for full data, please see GEO accession number GSE43734. TCA, tricarboxylic acid; PEPCK, phosphoenolpyruvate carboxykinase.
| CG number/category | Name | Fold change HSD/ctl | p vs. ctl |
|---|---|---|---|
| Trehalose | |||
| CG4104 | Tps1 | 2.23 | 0.040 |
| CG30035 | Tret-1 | 1.72 | 0.018 |
| CG8234 | Tret-2 | 1.89 | 0.026 |
| Glycolysis | |||
| CG8251 | Pgi | 2.10 | 0.003 |
| CG6058 | Aldolase | 1.72 | 0.005 |
| CG1721 | Pglym78 | 1.84 | 0.013 |
| CG17654 | Eno | 1.95 | 0.007 |
| CG7070 | PyK | 1.76 | 0.008 |
| CG9042 | Gpdh | 5.72 | 0.000 |
| Lipogenesis | |||
| CG11198 | ACC | 0.54 | 0.197 |
| CG3523 | Fatty-acid synthase | 1.88 | 0.012 |
| CG17374 | Fatty-acid synthase | 0.51 | 0.152 |
| CG6178 | Fatty-acid-CoA ligase | 3.64 | 0.023 |
| CG3209 | GPAT | 1.32 | 0.028 |
| CG4729 | GPAT | 1.41 | 0.021 |
| CG17608 | GPAT | 1.92 | 0.047 |
| CG31991 | mdy (DGAT) | 1.72 | 0.034 |
| CG8522 | SREBP | 0.88 | 0.078 |
| CG18362 | Mio/dChREBP | 1.71 | 0.000 |
| CG5887 | desat1 | 2.90 | 0.001 |
| Lipolysis | |||
| CG11325 | AKHR | 2.64 | 0.001 |
| CG5560 | Doppelgänger of brummer | 0.63 | 0.001 |
| β-Oxidation | |||
| CG12891 | CPT1 | 1.50 | 0.018 |
| CG2107 | CPT2 | 1.25 | 0.091 |
| CG4630 | Carnitine acyl-carnitine translocase | 2.63 | 0.008 |
| CG6178 | Acyl-CoA synthase | 3.64 | 0.023 |
| CG9527 | Acyl-CoA dehydrogenase | 1.88 | 0.002 |
| CG17544 | Acyl-CoA dehydrogenase | 0.24 | 0.040 |
| CG9547 | Acyl-CoA dehydrogenase | 0.61 | 0.018 |
| CG9709 | Acyl-CoA oxidase 57D-d | 3.09 | 0.073 |
| CG9707 | Acyl-CoA oxidase 57D-p | 0.73 | 0.031 |
| CG8778 | Enoyl-CoA hydratase | 0.77 | 0.040 |
| CG5044 | 3-Hydroxyisobutyryl-CoA hydrolase | 0.76 | 0.002 |
| CG14630 | Carnitine biosynthesis; γ-butyrobetaine dioxygenase activity | 0.76 | 0.027 |
| TCA cycle | |||
| CG9244 | Aconitase | 1.51 | 0.001 |
| CG7998 | Malate dehydrogenase | 1.47 | 0.027 |
| CG7430 | α-Ketoglutarate dehydrogenase | 1.57 | 0.006 |
| Ketone and CoA metabolism | |||
| CG1140 | SCOT (acetoacyl-CoA synthase) | 1.30 | 0.557 |
| CG5725 | fbl (pantothenate kinase) | 1.82 | 0.036 |
| CG10932 | Acetoacyl-CoA thiolase | 1.11 | 0.533 |
| CG9149 | Acetoacyl-CoA thiolase | 1.04 | 0.898 |
| CG10399 | HMG-CoA lyase | 1.79 | 0.456 |
| CG4311 | HMG-CoA synthase | 1.39 | 0.062 |
| CG8322 | ATP-citrate lyase | 1.11 | 0.570 |
| CG1774 | Acyl-CoA thioester hydrolase | 10.59 | 0.000 |
| Gluconeogenesis | |||
| CG17725 | PEPCK | 2.32 | 0.027 |
| CG10924 | PEPCK | 1.75 | 0.024 |
| CG31692 | fbp | 1.56 | 0.060 |
| CG1516 | Pyruvate carboxylase | 2.63 | 0.019 |
| Glycogenolysis | |||
| CG11325 | AKHR | 2.64 | 0.001 |
| CG7254 | GlyP | 1.81 | 0.003 |
FIGURE 6.

TAG accumulation is necessary to tolerate an HSD. Wandering third instar larvae were reared on a 0.7 m HSD for these studies. A, larval size in cg-GAL4; UAS-Mioi and cg-GAL4; UAS-desat1i larvae compared with controls. Larval TAG in cg-GAL4; UAS-Mioi (B) and cg-GAL4; UAS-desat1i larvae (E and F) are compared with controls. C, FB lipid storage droplets in wild-type larvae reared on a control (0.15 m sucrose) diet. D, FB lipid storage in wild-type larvae reared on an HSD. E, FB lipid storage in cg-GAL4; UAS-Mioi larvae reared on an HSD. F, FB lipid storage in cg-GAL4; UAS-desat1i larvae reared on an HSD. G, hemolymph glucose concentrations in cg-GAL4; UAS-Mioi larvae compared with controls. H, hemolymph glucose concentrations in cg-GAL4; UAS-desat1i larvae compared with controls. I, larval size in cg-GAL4; UAS-King-Tubbyi larvae compared with controls. J, larval TAG in cg-GAL4; UAS-King-Tubbyi larvae compared with controls. K, hemolymph glucose concentrations in cg-GAL4; UAS-King-Tubbyi larvae compared with controls. An unpaired, two-tailed t test was used to derive p values. Error bars are ±S.E.
Lipogenesis Is Protective against Hyperglycemia in HSD-fed Drosophila Larvae
To examine further the functional role of FB lipid storage in the face of an HSD, we increased larval fat content by generating an FB-specific loss of function mutant of king-tubby (CG9398), which is the fly ortholog of the mammalian Tubby gene. Loss of Tubby is correlated with obesity in mice, worms, and humans (43–47). Wandering third instar cg-GAL4; UAS-king-tubbyi larvae were larger than stage-matched controls when reared on the HSD (Fig. 6I). Consistent with the role of Tubby in other organisms, cg-GAL4; UAS-king-tubbyi larvae exhibited increased TAG accumulation compared with control animals fed an HSD (Fig. 6J). Obese cg-GAL4; UAS-king-tubbyi larvae had significantly lower hemolymph glucose levels (Fig. 6K), consistent with a protective role of fat storage against the development of diet-induced diabetes in Drosophila.
An HSD Affects Gene Expression and Lipid Content in a Mio-dependent Fashion in the FB
To study how HSD consumption causes changes in FB physiology, we profiled gene expression in the FB of control and HSD-reared wandering third instar larvae. A total of 456 down-regulated genes and 528 up-regulated genes was determined to be differentially expressed in FBs of HSD-fed larvae compared with controls using a paired t test with a cutoff of p < 0.05 and no -fold change requirements (supplemental Table S1). As expected, genes encoding enzymes involved in glycolysis, lipogenesis, and trehalose synthesis and transport were up-regulated in HSD-fed larvae (Table 2). Surprisingly, chronic HSD feeding resulted in increased expression of enzymes involved in glycogenolysis, gluconeogenesis, and fatty acid β-oxidation (Table 2). GOstat (48) was used to group expression changes into categories that are expected to influence cellular pathway activity. A relatively large number of cytochrome P450 oxidoreductases were significantly down-regulated by chronic HSD feeding (Table 3) as was the expression of many stress response proteins (Table 3). Note, however, that cytochrome P450 4e3 is the fourth most highly up-regulated gene (18-fold, p < 0.01) in HSD-fed larvae (supplemental Table S1). Genes encoding five glutathione S-transferases were down-regulated in HSD-fed larvae (GEO accession number GSE43734). Other genes that GOstat identified as up-regulated in response to HSD-feeding encoded several proteins localized to endoplasmic reticulum and Golgi and enzymes involved in tRNA aminoacylation (Table 3).
TABLE 3.
Gene ontology (GO) categories of mRNAs significantly up- or down-regulated in the fat body by an HSD
COPI, coatomer protein 1.
| GO category | p value | Genes | Up- or down-regulated |
|---|---|---|---|
| Endoplasmic reticulum | 3.39 × 10−10 | axs, ca-p60a, cg6512, cg10908, cg14476, cnx99a, cpr, crc, gtp-bp, hsc70-3, l(1)g0320, l(2)not, oststt3, p115, pek, pdi, sec31, sec61α, spase18–21, spase25, spp, surf4, syx5, tango14, ter94, tram | Up |
| Golgi apparatus | 3.26 × 10−6 | αcop,β cop,β ′cop,δ cop,ε cop,β 4galt7, fws, garz, grasp65, lva, nucb1, p115, pgant5, pgant6, sec31, syx5, tango1, tango12 | Up |
| Aminoacyl-tRNA ligase activity | 5.84 × 10−8 | aats-ala, aats-asn, aats-asp, aats-cys, aats-glupro, aats-gly, aats-ile, aats-thr, aats-trp, aats-val, cg16912, cg17259, cg31133, cg33123 | Up |
| COPI vesicle coat | 1.80 × 10−4 | αcop,β cop,β ′cop,δ cop,ε cop | Up |
| Oxidoreductase | 4.94 × 10−8 | acox57d-p, cat, cg1319, cg3699, cg5493, cg5840, cg6870, cg7280, cg8193, cg9512, cg9547, cg10962, cg14630, cg14946, cg17544, cg31169, cg33099, cyp6a8, cyp6a9, cyp6a18, cyp6a21, cyp6d4, cyp6d5, cyp6v1, cyp12a4, cyp12a5, cyp12d1-d, cyp12d1-p, cyp28d1, eo, gdh, ho, ifc, nmdmc, phgpx, ppox, pxn, sodh-1, sodh-2, uro | Down |
| Response to stress | 0.00966 | ark, cat, cdc42, cdk9, hml, hsp23, hsp70ba, hsp70bb, hsp70bbb, hsp70bc, ip3k1, mei-41, mthl10, pain, phgpx, spel1 | Down |
| Actin cytoskeleton organization and biogenesis | 0.00966 | arc-p20, arpc3b, capt, cdc42, ced-12, chic, hem, kel, mei-41, msp-300, rac1, rhogef2, spir, src42a | Down |
| UDP-N-acetylglucosamine-peptide N-acetylglucosaminyltransferase complex | 0.045 | cg4050, cg5038 | Down |
To understand how the transcription factor Mio might lead to reduced viability and a systemic disruption of lipid homeostasis in response to HSD feeding, we profiled gene expression in cg-GAL4; UAS-Mioi larval FBs on a 0.7 m sucrose HSD. A total of 1683 down-regulated genes and 1869 up-regulated genes was found to be differentially expressed in FBs of control and Mio mutant HSD-fed larvae using a paired t test with a cutoff of p < 0.05 without -fold change requirements. Consistent with the hypothesis that ChREBP is largely a transcriptional activator of glucose catabolism, we observed a decrease in expression of genes involved in glycolysis, pyruvate metabolism, and lipogenesis in Mio mutant fat bodies, many of which were induced by the HSD (Tables 2 and 4 and supplemental Table S1). One of the most highly up-regulated categories, however, was lipases, consistent with the leanness observed in these mutants. Expression of enzymes controlling the levels of ketone and CoA substrates for fatty acid synthesis were also misregulated in cg-GAL4; UAS-Mioi FBs (Table 4). Transcription of genes encoding many members of the insulin signaling pathway was significantly reduced in mutants, which may account for part of the hyperglycemia observed in cg-GAL4; UAS-Mioi HSD-fed larvae (Table 4). Proteases and mitochondrial and ribosomal proteins in cg-GAL4; UAS-Mioi larval FBs were GO categories of interest because they may be repressed by ChREBP (Table 5). GO analysis also suggested that growth and the immune response are reduced at the transcriptional level in Mio mutant fat bodies (Table 5).
TABLE 4.
Mio-dependent gene expression in the wandering third instar fat body
Vw1118; UAS-Dcr2; cgGAL4 and UAS-Mioi; UAS-Dcr2; cgGAL4 larvae were reared on a 0.7 m HSD, and FBs were isolated for RNA extraction. A two-tailed Student's t test was used to derive p values. For simplicity, only a selection of genes is shown here; for full data, please see GEO accession number GSE43734. ctl, control.
| CG number/category | Name | Fold change Mioi/ctl | p vs. ctl |
|---|---|---|---|
| Glycolysis | |||
| CG8251 | Pgi | 0.38 | 0.024 |
| CG6058 | Aldolase | 0.27 | 0.012 |
| CG3127 | Pgk | 1.79 | 0.003 |
| Pyruvate metabolism | |||
| CG8808 | Pdk | 0.21 | 0.001 |
| CG7010 | PDH | 0.38 | 0.002 |
| CG5261 | PDH | 0.39 | 0.000 |
| Lipogenesis | |||
| CG3523 | Fas | 0.24 | 0.010 |
| CG3209 | GPAT | 1.60 | 0.007 |
| CG4729 | GPAT | 0.43 | 0.010 |
| CG17608 | GPAT | 2.22 | 0.006 |
| CG18362 | Mio/dChREBP | 0.09 | 0.000 |
| Lipolysis | |||
| CG1882 | Putative CGI-58 homology, ATGL activator | 1.84 | 0.032 |
| CG5295 | Brummer TAG lipase | 0.26 | 0.002 |
| CG5932 | TAG lipase | 6.38 | 0.005 |
| CG6277 | TAG lipase | 23.01 | 0.000 |
| CG6283 | TAG lipase | 18.92 | <0.0001 |
| CG6295 | TAG lipase | 28.01 | 0.000 |
| CG8093 | TAG lipase | >300 | 0.002 |
| CG8823 | Lip3 | 4.02 | 0.041 |
| CG10374 | Lsd-1 | 0.34 | 0.002 |
| CG9057 | Lsd-2 | 0.23 | 0.004 |
| β-Oxidation | |||
| CG2107 | CPT2 | 0.54 | 0.035 |
| CG4335 | Carnitine biosynthesis; γ-butyrobetaine dioxygenase activity | 0.27 | 0.026 |
| CG5321 | Carnitine biosynthesis; γ-butyrobetaine dioxygenase activity | 2.63 | 0.001 |
| CG11637 | Acyl-CoA dehydrogenase | 2.12 | 0.039 |
| CG5009 | Acyl-CoA dehydrogenase | 1.68 | 0.001 |
| CG9527 | Acyl-CoA dehydrogenase | 0.44 | 0.025 |
| CG9547 | Acyl-CoA dehydrogenase | 1.75 | 0.021 |
| CG6984 | Enoyl-CoA hydratase | 0.24 | 0.010 |
| CG8778 | Enoyl-CoA hydratase | 3.91 | 0.000 |
| CG4598 | Dodecenoyl-CoA Δ-isomerase | 1.99 | 0.003 |
| CG5844 | Dodecenoyl-CoA Δ-isomerase | 1.88 | 0.035 |
| Glycogenolysis | |||
| CG7254 | GlyP | 0.25 | 0.009 |
| Ketone and CoA metabolism | |||
| CG1140 | SCOT (acetoacyl-CoA synthase) | 0.95 | 0.933 |
| CG5725 | fbl (pantothenate kinase) | 0.54 | 0.198 |
| CG10932 | Acetoacyl-CoA thiolase | 2.67 | 0.005 |
| CG9149 | Acetoacyl-CoA thiolase | 3.32 | 0.000 |
| CG10399 | HMG-CoA lyase | 1.66 | 0.603 |
| CG4311 | HMG-CoA synthase | 8.03 | 0.000 |
| CG8322 | ATP-citrate lyase | 0.63 | 0.138 |
| Insulin signaling | |||
| CG5686 | Chico | 0.12 | 0.005 |
| CG3727 | Dock; dreadlocks | 0.18 | 0.016 |
| CG4141 | PI3K; Dp110 | 0.15 | 0.000 |
| CG1210 | PDK1 | 0.40 | 0.003 |
| CG5092 | TOR | 0.22 | 0.015 |
| CG6147 | TSC1 | 0.48 | 0.024 |
| CG10539 | S6K | 0.05 | 0.004 |
| CG8846 | Thor | 0.12 | 0.011 |
| CG4006 | Akt | 0.32 | 0.030 |
| CG14049 | Dilp6 | 0.12 | 0.004 |
TABLE 5.
GO categories of mRNAs significantly up- or down-regulated by loss of Mio/dChREBP in the fat body
COPI, coatomer protein 1.
| GO category | p value | Genes | Up- or down-regulated |
|---|---|---|---|
| Ribosome | 1.18 × 10−45 | cg6764, mrpl10, mrpl17, mrpl21, mrpl23, mrpl24, mrpl27, mrpl30, mrpl36, mrpl39, mrpl40, mrpl41, mrpl46, mrpl51, mrpl52, mrps5, mrps14, mrps16, mrps17, mrps18c, mrps22, mrps25, mrps26, mrps28, qm, rpl7a, rpl8, rpl9, rpl11, rpl12, rpl13a, rpl19, rpl21, rpl23, rpl29, rpl30, rpl36, rpl38, rpl39, rplp1, rplp2, rps3, rps5a, rps6, rps9, rps10b, rps11, rps14a, rps14b, rps15aa, rps15ab, rps17, rps18, rps23, rps27a, rps29, tko | Up |
| Mitochondrion | 1.07 × 10−35 | aldh, arg, cchl, cg1824, cg1907, cg2543, cg2658, cg2789, cg3621, cg3683, cg3719, cg3803, cg4095, cg4225, cg4592, cg4598, cg4769, cg4995, cg5037, cg5254, cg5548, cg6020, cg6404, cg6412, cg6439, cg7430, cg7598, cg7943, cg8680, cg8728, cg9172, cg9547, cg10320, cg12400, cg14482, cg31075, cg32230, cg32250, cg32649, cova, cyp12a4, dnapol-γ35, eftum, ferrochelatase, gata, got2, hsp60, hsp68, idh, l(1)g0255, l(2)37cc, mrpl10, mrpl17, mrpl21, mrpl23, mrpl24, mrpl27, mrpl30, mrpl36, mrpl39, mrpl40, mrpl41, mrpl46, mrpl51, mrpl52, mrps5, mrps14, mrps16, mrps17, mrps18c, mrps22, mrps25 mrps26, mrps28, mtacp1, mt:coi, mt:coii, mt:coiii, mt:cyt-b, mt:nd1, mt:nd2, mt:nd3, mt:nd4l, mt:nd5, mt:nd6, nd75, nemy, oat, pink1, porin, scpx, scsα, scu, sucb, sun, surf1, t-cp1, tko, yip2 | Up |
| COPI vesicle coat | 2.24 × 10−6 | chop24, αcop,β cop, γcop,ε cop,δ cop, ζcop | Up |
| Electron transport | 1.30 × 10−10 | cg3560, cg3621, cg3683, cg4169, cg4769, cg5548, cg6020, cg6666, cg7181, cg8680, cg9172, cg10320, cg11015, cg11455, cg12079, cg12859, cg14482, cg12400, cg17280, cg32230, cg32649, cg41623, cova, cyt-b5, mtacp1, mt:coi, mt:coii, pdsw, rfesp, ox | Up |
| Proteolysis | 6.91 × 10−8 | aos1, ate1, cg2200, cg7142, cg8299, cg8952, cg9631, cg10472, cg10908, cg11911, cg11912, cg12000, cg13779, cg16749, cg16996, cg16997, cg17571, cg17684, cg18179, cg18180, cg30025, cg30031, cg31728, cg32147, ice, jon66ci, pros45, jon25biii, jon25bii, jon25bi, jon44e, jon65ai, jon65aii, jon65aiii, jon65aiv, jon66cii,jon74e, jon99ci, jon99cii, jon99ciii, jon99fii, kaz1-orfb, roc1a, αtry,β try,δ try,ε try, ηtry, ιtry, θtry, pros26.4, pros28.1, pros29, pros35, prosma5, psn, rpn7, rpn11, rpn12, rps27a, rpt3, sras, ter94, th, uch-l3, w, yip7 | Up |
| Growth regulation | 1.58 × 10−5 | akt1, babo, chico, dsx, eif-4e, foxo, lack, lk6, path, phl, pi3k92e, pk61c, s6k, samuel,β -spec, thor, tkv, tor, tsc1, tsg101, wts, yki | Down |
| Protein transport | 1.64 × 10−5 | arr, cact, cas, cg4289, cg6359, cg6842, cg9426, cg14804, cora, cos, dco, emb, fbp1, fs(2)ket, hkl, hop, kap-α1, kap-α3, lwr, mbo, nup214, olf186-f, sec6, slmb, srp54k, srprβ, tim8, tom40, tom70 | Down |
| Programmed cell death | 4.66 × 10−5 | aac11, ago2, akap200, akt1, ark, art4, bruce, cas, cbt, cg3829, cg5651, cg17019, decay, dream, ecr, egfr, egr, eif-4e, eip74ef, eip75b, hkl, hr78, ik2, l(1)g0148, l(2)01424, ofs, pall, pk61c, pnt, pp2a-b′, pr2, puc, raw, sktl, slmb, usp, wts, yki, zip | Down |
| Immune system process | 8.17 × 10−6 | adgf-a, ago2, brm, cact, cdc42, cg6509, dcr-2, dif, egfr, egr, eip75b, gef26, hel89b, hep, hop, kis, lola, lwr, mask, mbo, mp1, mys, nej, par-1, pgrp-lc, pgrp-le, phl, pnt, pvr, r, rac1, rel, rho1, srp, stat92e, su(var)2–10, tab2, tepii, thor, tl, tlk, tub, ush, ytr | Down |
| Hemocyte differentiation | 8.65 × 10−6 | adgf-a, dif, egfr, hep, hop, lwr, phl, pnt, pvr, rac1, srp, stat92e, tl, ush, ytr | Down |
| Histone modification | 7.51 × 10−6 | art4, ash1, atac1, bur, cg1716, cg10542, cg15835, e(z), jil-1, parg, pr-set7, rpd3, rtf1, sir2, su(var)4–20, su(z)12, tlk | Down |
Because our analyses implicated Mio in the control of lipid metabolism and Mio is essential for tolerance of HSDs, we characterized the lipid composition of Mio mutant FBs by ESI-MS (Fig. 7). Lipid levels of all types (comprising TAG, PI, PC, and phosphatidylethanolamine) analyzed were reduced in Mio mutant FBs (data not shown). Specific substituents were also affected in cg-GAL4; UAS-Mioi mutants. We observed an increased mean carbon chain length of the fatty acid substituents (C47.908 ± 0.042 versus C47.053 ± 0.042, p = 0.0018) in FB TAG from HSD-fed cg-GAL4; UAS-Mioi mutants compared with controls (Fig. 7, A and B). Fatty acid substituents seemed to be more saturated in Mio mutant FBs compared with controls, although these data were not highly significant (Fig. 7C). NEFA and ketones were measured in cg-GAL4; UAS-Mioi mutants. FB Mio knockdown marginally affected NEFA levels (12% reduction, p < 0.08), whereas ketones were reduced significantly compared with wild-type larvae on the 0.7 m HSD (Fig. 7, D and E). Interestingly, lipase activity was not increased in cg-GAL4; UAS-Mioi larvae despite a strong increase in expression of most lipases compared with wild-type larvae on a 0.7 m HSD (Fig. 7F). Together, these data suggest that Mio transcriptionally controls lipogenesis, lipid storage, and fatty acid substituent length and saturation in the fly.
FIGURE 7.

Loss of Mio in the fat body affects lipid metabolism. Wandering third instar FBs were isolated from control or cg-GAL4; UAS-Mioi larvae reared on 0.7 m sucrose HSDs and subjected to ESI-MS analyses. A, composition of FB TAG. B, relative chain lengths of fatty acids esterified as TAG. C, saturation of fatty acids esterified as TAG. D, NEFA concentrations. E, total ketone body concentrations. F, lipase activity measured over 4 h. An unpaired, two-tailed t test was used to derive p values. *, p < 0.05; **, p < 0.01. Error bars are ±S.E.
DISCUSSION
Caloric excess is associated with several human pathologic states, including cancer, obesity, and cardiovascular and liver diseases, and aberrant tissue lipogenesis correlates with disease severity (49–54). Here, we have explored the effects of HSDs on lipid metabolism in the simple model organism Drosophila. We observed a switch in metabolism of dietary glucose in HSD-reared larvae that attenuated FA esterification. The fate of carbon atoms from dietary glucose was dependent on the rearing diet of the larvae: carbons were more directly incorporated into esterified lipids in larvae reared on the control diet compared with HS-reared animals. Lipogenesis was required in the fat body for tolerance of HSDs, suggesting that this pathway acts protectively against the deleterious consequences of HSD consumption, which may result from high free fatty acid or reduced CoA concentrations. Our results in the fruit fly support a model in which excess dietary sugar is stored as TAG; when the capacity of the FB to store fat is exceeded, negative consequences for growth and metabolism ensue.
A Metabolic Switch in Larvae Reared on an HSD
In healthy larvae, energy from dietary sugar is stored as FB TAG. When dietary sugar consumption is excessive, larvae must accommodate an increased flux of sugar-derived lipids into storage forms. Stable isotope labeling studies revealed that HSD feeding affected both the rate and route of dietary glucose metabolism (Figs. 1 and 2). Of note, some of the effects of HSD on growth and metabolic diversion of glucose carbons can be reversed after transfer to a control diet (Figs. 1B and 2, C and D). Animals reared on an HSD initiated a robust transcriptional response toward increasing TAG storage in the FB (Table 2), suggesting that larvae attempted to incorporate excess sugar into stored fat but that their ability to do so was limited. Rerouting of dietary carbons may be one mechanism by which the animal accommodates dietary excess. Both the tricarboxylic acid cycle and gluconeogenesis are increased at the transcriptional level by an HSD (Table 2). These could contribute to a metabolic detour for dietary carbon to produce more singly labeled acetate in HSD-reared larvae (Fig. 2A). Much of the decrease in TFA synthesized from dietary glucose can be accounted for by the reduction in doubly labeled, directly synthesized FAs, suggesting that this rerouting affects the rate of synthesis of TFA from dietary carbohydrate in HSD-reared larvae. Interestingly, HSD-induced rerouting of dietary glucose carbons did not occur in the NEFA pool, and this pool was affected differently by rearing diet than the TFA pool (Figs. 1, C, D, F, and G, and 2, B–D). Thus, the precursors for synthesis of free and esterified fatty acids reside in two distinguishable pools in whole larvae. These different fatty acid pools might reside in distinct anatomic compartments, such as FB and muscle or oenocyte, tissues known to be involved in fat metabolism (55). They could also represent lipogenesis in two subcellular locations, perhaps by one of three different fatty acid synthases (CG3523, CG3524, and CG17374) encoded in the fly genome.
Flux of Excess Dietary Carbon into Stored TAG Can Protect Drosophila from HSD-induced Hyperglycemia
Classical studies have identified several Drosophila mutants with perturbed glucose and lipid homeostasis (for a review, see Ref. 56). We used a tissue-specific approach to study the role of the FB in regulating glucose disposal and energy storage. Our results support a model in which FB lipogenesis protects against deleterious effects of HSD consumption. In general, we have found that lean animals consistently have decreased viability on an HSD, whereas fatter animals exhibit improved growth and hemolymph sugar homeostasis (Fig. 6 and data not shown). Our results indicate that Mio acts as the Drosophila ChREBP ortholog (dChREBP). Similarly, reducing king-tubby expression resulted in increased larval size and adiposity, resembling mammalian Tubby mutants (44, 45, 47). Interestingly, tubby mutant mice are normoglycemic despite severe adult onset obesity (44). These and other studies support an emerging, protective role for fat storage that appears to be evolutionarily conserved (see below).
Dysregulated Metabolite Pools May Contribute to Toxicity of HSD
Although it is tempting to speculate that an individual lipid or metabolite impairs growth and disrupts glucose and lipid metabolism in wild-type larvae fed an HSD, it is more likely to be dysregulation of multiple pathways that together result in insulin resistance. We found changes in NEFA, CoA, and ketone levels in HSD-fed larvae. NEFA excess might be expected to increase β-oxidation and lipid storage, and we observed an increase in expression of enzymes that direct these processes. The increased β-oxidation could deplete essential metabolites, such as CoA, which is required for more than 100 enzymatic reactions. HS diets may have reduced CoA modestly, but the difference was not statistically significant. It is difficult to know the physiologic relevance of a small decrease in CoA levels; however, there was a transcriptional response to increase CoA levels (pantothenate kinase and acyl-CoA thioester hydrolase were up-regulated 1.8- and 10-fold, respectively; Table 2). Finally, decreased CoA levels would also impair the ability to use ketones for lipogenesis and might explain the increased ketones observed in larvae fed HSD. Any or all of these metabolic disturbances could contribute to the toxicity of HSD feeding.
The transcription factor Mio/dChREBP clearly plays a central role in the regulation of the glucose and lipid metabolism of the animals exposed to caloric excess by HSD. On control diets, fat body-specific Mio/dChREBP mutants (cg-GAL4; UAS-Mioi) grow and develop normally and do not exhibit increased hemolymph glucose concentrations compared with wild-type larvae (data not shown), suggesting that Mio/dChREBP-dependent metabolism of dietary sugar is not essential under these conditions. The cg-GAL4; UAS-Mioi larvae are lean and do show reduced expression of lipogenic genes on a control diet (data not shown and supplemental Table S1). Therefore, an inability to store dietary glucose as TAG is deleterious only in the face of caloric excess.
On an HSD, lean, Mio/dChREBP mutant fat bodies displayed features of general metabolic imbalance. Despite transcriptional up-regulation of several lipases, ChREBP mutants had no increase in lipase activity and exhibited marginally reduced total NEFA. Mutants also increased expression of insulin signaling pathway components, yet growth was impaired. Ketones were dramatically reduced in Mio/dChREBP knockdown coincident with increased expression of enzymes involved in conversion of ketones into NEFA (Table 4). This might reflect that the Mio/dChREBP knockdown larvae consume ketones for the essential synthesis of lipids due to a diminished ability to use more direct paths from glucose to lipogenesis.
An Evolutionarily Conserved Maximum TAG Capacity Model for T2D
Previously published work demonstrates that despite resulting in caloric excess larval HSD consumption does not result in increased TAG per animal measured at the end of larval development when TAG storage reaches its maximum (4). HSD-reared larvae contain a larger pool of stored lipids midway through development than controls such that their capacity to further store lipids may be compromised (Table 1). Growing evidence in other models suggests that storage of excess NEFA in adipose TAG may be protective against T2D, an idea that challenges the current paradigm that obesity causes T2D (57, 58). Increasing fat storage in adipose tissue protects mice from T2D (59, 60), and animals that cannot store fat normally are insulin-resistant when fed normal diets (61, 62). A number of studies in humans are also consistent with a maximum TAG capacity model for T2D (for a review, see Ref. 63). Gastric bypass surgery ameliorates T2D (64), whereas liposuction does not (65), suggesting that the capacity to store fat is important. Peroxisome proliferator-activated receptor (PPAR)-γ agonists act as insulin sensitizers, and these drugs increase adiposity while decreasing hepatic and circulating lipid levels, effects that are also consistent with a protective function for fat storage in adipose tissue (66). In contrast, ectopic TAG accumulation in non-adipose tissues, such as muscle, liver, or blood, is associated with insulin resistance (for a review, see Ref. 67).
ESI-MS analyses of lipid molecular species revealed that fatty acid substituents of glycerolipids in HSD-reared larvae exhibit shorter carbon chains and increased unsaturation compared with those from larvae reared on a control diet (Figs. 3 and 4), and desaturase activity was protective in HSD-fed larvae (Fig. 6). Recent work describing the TAG signature of insulin resistance in humans associated increased risk with reduced carbon chain length and double bond content (68). Fatty acid carbon chain length has also been associated with insulin resistance in mice (69), and desaturation has been associated with insulin sensitivity in mice and cultured cells (70, 71). Esterified fatty acid substituents in Mio/dChREBP mutant FBs were longer compared with controls. Interestingly, expression of Elovl6, a mammalian ortholog of these elongases, is dependent on ChREBP (72), and knock-out of Elovl6 reduced fatty acid substituent length and decreased both lipogenic and oxidative gene expression in the face of a high fat/high sucrose diet (69). We hypothesize that saturated fatty acids are one source of cellular damage in HSD-fed Drosophila. The ability to store excess dietary carbon as TAG could be protective against free saturated long chain fatty acids or other lipotoxic molecules that accumulate when their safe storage capacity is exceeded.
Acknowledgments
We thank Alan Bohrer, Trey Coleman, Freida Custodio, Fong Fu-Hsu, Jennifer Shew, and Sarah Spencer for expert technical assistance and Peter Crawford for helpful discussions. We also thank the Bloomington Drosophila Stock Center and the Vienna Drosophila RNAi Center for fly strains.
This work was supported, in whole or in part, by National Institutes of Health Grants T32 GM007464 (to L. P. M.), 5K12HD001459-12 (to L. P. M.), P60 DK20579 through the Washington University Diabetes Research and Training Center (to J. T. and T. J. B.), P20 RR020643 (to T. J. B.), P30 DK56341 through the Nutrition Obesity Research Center (to A. L. O., B. W. P., and J. T.), P41-RR00954 through the Biomedical Mass Spectrometry Resource (to J. T.), and 1R41DK76338, a Small Business Technology Transfer grant (to Medros, Inc.). This work was also supported by Children's Discovery Institute Grant MD-II-2010-41 (to M. R. B. and T. J. B.) and Washington University (to T. J. B.). T. J. B. is a co-founder of Medros, Inc.

This article contains supplemental Table S1.
- HSD
- high sugar diet
- T2D
- type 2 diabetes
- FB
- fat body
- TAG
- triglyceride
- NEFA
- nonesterified fatty acid
- TFA
- total free + esterified fatty acid
- PE
- phosphatidylethanolamine
- PC
- phosphatidylcholine
- PI
- phosphatidylinositol
- ESI
- electrospray ionization
- HS
- high sugar
- FA
- fatty acid
- ChREBP
- carbohydrate response element-binding protein
- Desat1
- stearoyl-CoA desaturase 1
- GO
- gene ontology.
REFERENCES
- 1. Ceriello A. (2006) Effects of macronutrient excess and composition on oxidative stress: relevance to diabetes and cardiovascular disease. Curr. Atheroscler. Rep. 8, 472–476 [DOI] [PubMed] [Google Scholar]
- 2. Fukuchi S., Hamaguchi K., Seike M., Himeno K., Sakata T., Yoshimatsu H. (2004) Role of fatty acid composition in the development of metabolic disorders in sucrose-induced obese rats. Exp. Biol. Med. 229, 486–493 [DOI] [PubMed] [Google Scholar]
- 3. Gagliardi L., Wittert G. (2007) Management of obesity in patients with type 2 diabetes mellitus. Curr. Diabetes Rev. 3, 95–101 [DOI] [PubMed] [Google Scholar]
- 4. Musselman L. P., Fink J. L., Narzinski K., Ramachandran P. V., Hathiramani S. S., Cagan R. L., Baranski T. J. (2011) A high-sugar diet produces obesity and insulin resistance in wild-type Drosophila. Dis. Model. Mech. 4, 842–849 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Reed L. K., Williams S., Springston M., Brown J., Freeman K., DesRoches C. E., Sokolowski M. B., Gibson G. (2010) Genotype-by-diet interactions drive metabolic phenotype variation in Drosophila melanogaster. Genetics 185, 1009–1019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Skorupa D. A., Dervisefendic A., Zwiener J., Pletcher S. D. (2008) Dietary composition specifies consumption, obesity, and lifespan in Drosophila melanogaster. Aging Cell 7, 478–490 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Sullivan S. (2010) Implications of diet on nonalcoholic fatty liver disease. Curr. Opin. Gastroenterol. 26, 160–164 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Pasco M. Y., Léopold P. (2012) High sugar-induced insulin resistance in Drosophila relies on the lipocalin Neural Lazarillo. PLoS One 7, e36583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Chung H., Sztal T., Pasricha S., Sridhar M., Batterham P., Daborn P. J. (2009) Characterization of Drosophila melanogaster cytochrome P450 genes. Proc. Natl. Acad. Sci. U.S.A. 106, 5731–5736 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Géminard C., Rulifson E. J., Léopold P. (2009) Remote control of insulin secretion by fat cells in Drosophila. Cell Metab. 10, 199–207 [DOI] [PubMed] [Google Scholar]
- 11. Kühnlein R. P. (2011) The contribution of the Drosophila model to lipid droplet research. Prog. Lipid Res. 50, 348–356 [DOI] [PubMed] [Google Scholar]
- 12. Lemaitre B., Hoffmann J. (2007) The host defense of Drosophila melanogaster. Annu. Rev. Immunol. 25, 697–743 [DOI] [PubMed] [Google Scholar]
- 13. DiAngelo J. R., Birnbaum M. J. (2009) Regulation of fat cell mass by insulin in Drosophila melanogaster. Mol. Cell. Biol. 29, 6341–6352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Lee G., Park J. H. (2004) Hemolymph sugar homeostasis and starvation-induced hyperactivity affected by genetic manipulations of the adipokinetic hormone-encoding gene in Drosophila melanogaster. Genetics 167, 311–323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Clifton P. M. (2008) Dietary treatment for obesity. Nat. Clin. Pract. Gastroenterol. Hepatol. 5, 672–681 [DOI] [PubMed] [Google Scholar]
- 16. Hession M., Rolland C., Kulkarni U., Wise A., Broom J. (2009) Systematic review of randomized controlled trials of low-carbohydrate vs. low-fat/low-calorie diets in the management of obesity and its comorbidities. Obes. Rev. 10, 36–50 [DOI] [PubMed] [Google Scholar]
- 17. Leclercq I. A., Horsmans Y. (2008) Nonalcoholic fatty liver disease: the potential role of nutritional management. Curr. Opin. Clin. Nutr. Metab. Care 11, 766–773 [DOI] [PubMed] [Google Scholar]
- 18. Tappy L., Lê K. A., Tran C., Paquot N. (2010) Fructose and metabolic diseases: new findings, new questions. Nutrition 26, 1044–1049 [DOI] [PubMed] [Google Scholar]
- 19. Samuel V. T., Shulman G. I. (2012) Mechanisms for insulin resistance: common threads and missing links. Cell 148, 852–871 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Taubes G. (2009) Insulin resistance. Prosperity's plague. Science 325, 256–260 [DOI] [PubMed] [Google Scholar]
- 21. Brookheart R. T., Michel C. I., Schaffer J. E. (2009) As a matter of fat. Cell Metab. 10, 9–12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Postic C., Girard J. (2008) Contribution of de novo fatty acid synthesis to hepatic steatosis and insulin resistance: lessons from genetically engineered mice. J. Clin. Investig. 118, 829–838 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Patterson B. W., Zhao G., Elias N., Hachey D. L., Klein S. (1999) Validation of a new procedure to determine plasma fatty acid concentration and isotopic enrichment. J. Lipid Res. 40, 2118–2124 [PubMed] [Google Scholar]
- 24. Bohlin K., Patterson B. W., Spence K. L., Merchak A., Zozobrado J. C., Zimmermann L. J., Carnielli V. P., Hamvas A. (2005) Metabolic kinetics of pulmonary surfactant in newborn infants using endogenous stable isotope techniques. J. Lipid Res. 46, 1257–1265 [DOI] [PubMed] [Google Scholar]
- 25. Chinkes D. L., Aarsland A., Rosenblatt J., Wolfe R. R. (1996) Comparison of mass isotopomer dilution methods used to compute VLDL production in vivo. Am. J. Physiol. Endocrinol. Metab. 271, E373–E383 [DOI] [PubMed] [Google Scholar]
- 26. Bligh E. G., Dyer W. J. (1959) A rapid method of total lipid extraction and purification. Can. J. Biochem. Physiol. 37, 911–917 [DOI] [PubMed] [Google Scholar]
- 27. Hsu F. F., Bohrer A., Turk J. (1998) Formation of lithiated adducts of glycerophosphocholine lipids facilitates their identification by electrospray ionization tandem mass spectrometry. J. Am. Soc. Mass Spectrom. 9, 516–526 [DOI] [PubMed] [Google Scholar]
- 28. Hsu F. F., Turk J. (1999) Structural characterization of triacylglycerols as lithiated adducts by electrospray ionization mass spectrometry using low-energy collisionally activated dissociation on a triple stage quadrupole instrument. J. Am. Soc. Mass Spectrom. 10, 587–599 [DOI] [PubMed] [Google Scholar]
- 29. Hsu F. F., Turk J. (2000) Characterization of phosphatidylinositol, phosphatidylinositol-4-phosphate, and phosphatidylinositol-4,5-bisphosphate by electrospray ionization tandem mass spectrometry: a mechanistic study. J. Am. Soc. Mass Spectrom. 11, 986–999 [DOI] [PubMed] [Google Scholar]
- 30. Sieber M. H., Thummel C. S. (2009) The DHR96 nuclear receptor controls triacylglycerol homeostasis in Drosophila. Cell Metab. 10, 481–490 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Chakravarthy M. V., Pan Z., Zhu Y., Tordjman K., Schneider J. G., Coleman T., Turk J., Semenkovich C. F. (2005) “New” hepatic fat activates PPARα to maintain glucose, lipid, and cholesterol homeostasis. Cell Metab. 1, 309–322 [DOI] [PubMed] [Google Scholar]
- 32. Haynes B. C., Skowyra M. L., Spencer S. J., Gish S. R., Williams M., Held E. P., Brent M. R., Doering T. L. (2011) Toward an integrated model of capsule regulation in Cryptococcus neoformans. PLoS Pathog. 7, e1002411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Trapnell C., Pachter L., Salzberg S. L. (2009) TopHat: discovering splice junctions with RNA-Seq. Bioinformatics 25, 1105–1111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Trapnell C., Williams B. A., Pertea G., Mortazavi A., Kwan G., van Baren M. J., Salzberg S. L., Wold B. J., Pachter L. (2010) Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat. Biotechnol. 28, 511–515 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Neuschwander-Tetri B. A. (2010) Nontriglyceride hepatic lipotoxicity: the new paradigm for the pathogenesis of NASH. Curr. Gastroenterol. Rep. 12, 49–56 [DOI] [PubMed] [Google Scholar]
- 36. Sørensen T. I., Virtue S., Vidal-Puig A. (2010) Obesity as a clinical and public health problem: is there a need for a new definition based on lipotoxicity effects? Biochim. Biophys. Acta 1801, 400–404 [DOI] [PubMed] [Google Scholar]
- 37. Hammad L. A., Cooper B. S., Fisher N. P., Montooth K. L., Karty J. A. (2011) Profiling and quantification of Drosophila melanogaster lipids using liquid chromatography/mass spectrometry. Rapid Commun. Mass Spectrom. 25, 2959–2968 [DOI] [PubMed] [Google Scholar]
- 38. Magkos F., Yannakoulia M., Chan J. L., Mantzoros C. S. (2009) Management of the metabolic syndrome and type 2 diabetes through lifestyle modification. Annu. Rev. Nutr. 29, 223–256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Niswender K. (2010) Diabetes and obesity: therapeutic targeting and risk reduction—a complex interplay. Diabetes Obes. Metab. 12, 267–287 [DOI] [PubMed] [Google Scholar]
- 40. Postic C., Dentin R., Denechaud P. D., Girard J. (2007) ChREBP, a transcriptional regulator of glucose and lipid metabolism. Annu. Rev. Nutr. 27, 179–192 [DOI] [PubMed] [Google Scholar]
- 41. Sampath H., Ntambi J. M. (2011) The role of stearoyl-CoA desaturase in obesity, insulin resistance, and inflammation. Ann. N.Y. Acad. Sci. 1243, 47–53 [DOI] [PubMed] [Google Scholar]
- 42. Iizuka K., Bruick R. K., Liang G., Horton J. D., Uyeda K. (2004) Deficiency of carbohydrate response element-binding protein (ChREBP) reduces lipogenesis as well as glycolysis. Proc. Natl. Acad. Sci. U.S.A. 101, 7281–7286 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Ashrafi K., Chang F. Y., Watts J. L., Fraser A. G., Kamath R. S., Ahringer J., Ruvkun G. (2003) Genome-wide RNAi analysis of Caenorhabditis elegans fat regulatory genes. Nature 421, 268–272 [DOI] [PubMed] [Google Scholar]
- 44. Coleman D. L., Eicher E. M. (1990) Fat (fat) and tubby (tub): two autosomal recessive mutations causing obesity syndromes in the mouse. J. Hered. 81, 424–427 [DOI] [PubMed] [Google Scholar]
- 45. Shiri-Sverdlov R., Custers A., van Vliet-Ostaptchouk J. V., van Gorp P. J., Lindsey P. J., van Tilburg J. H., Zhernakova S., Feskens E. J., van der A D. L., Dollé M. E., van Haeften T. W., Koeleman B. P., Hofker M. H., Wijmenga C. (2006) Identification of TUB as a novel candidate gene influencing body weight in humans. Diabetes 55, 385–389 [DOI] [PubMed] [Google Scholar]
- 46. van Vliet-Ostaptchouk J. V., Onland-Moret N. C., Shiri-Sverdlov R., van Gorp P. J., Custers A., Peeters P. H., Wijmenga C., Hofker M. H., van der Schouw Y. T. (2008) Polymorphisms of the TUB gene are associated with body composition and eating behavior in middle-aged women. PLoS One 3, e1405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Wang Y., Seburn K., Bechtel L., Lee B. Y., Szatkiewicz J. P., Nishina P. M., Naggert J. K. (2006) Defective carbohydrate metabolism in mice homozygous for the tubby mutation. Physiol. Genomics 27, 131–140 [DOI] [PubMed] [Google Scholar]
- 48. Beissbarth T., Speed T. P. (2004) GOstat: find statistically overrepresented Gene Ontologies within a group of genes. Bioinformatics 20, 1464–1465 [DOI] [PubMed] [Google Scholar]
- 49. Fay J. R., Steele V., Crowell J. A. (2009) Energy homeostasis and cancer prevention: the AMP-activated protein kinase. Cancer Prev. Res. 2, 301–309 [DOI] [PubMed] [Google Scholar]
- 50. Knight J. A. (2011) Diseases and disorders associated with excess body weight. Ann. Clin. Lab. Sci. 41, 107–121 [PubMed] [Google Scholar]
- 51. Kritchevsky D. (1995) The effect of over- and undernutrition on cancer. Eur. J. Cancer Prev. 4, 445–451 [DOI] [PubMed] [Google Scholar]
- 52. Lim J. S., Mietus-Snyder M., Valente A., Schwarz J. M., Lustig R. H. (2010) The role of fructose in the pathogenesis of NAFLD and the metabolic syndrome. Nat. Rev. Gastroenterol. Hepatol. 7, 251–264 [DOI] [PubMed] [Google Scholar]
- 53. Mensink R. P., Plat J., Schrauwen P. (2008) Diet and nonalcoholic fatty liver disease. Curr. Opin. Lipidol. 19, 25–29 [DOI] [PubMed] [Google Scholar]
- 54. Reaven G., Abbasi F., McLaughlin T. (2004) Obesity, insulin resistance, and cardiovascular disease. Recent Prog. Horm. Res. 59, 207–223 [DOI] [PubMed] [Google Scholar]
- 55. Gutierrez E., Wiggins D., Fielding B., Gould A. P. (2007) Specialized hepatocyte-like cells regulate Drosophila lipid metabolism. Nature 445, 275–280 [DOI] [PubMed] [Google Scholar]
- 56. Baker K. D., Thummel C. S. (2007) Diabetic larvae and obese flies-emerging studies of metabolism in Drosophila. Cell Metab. 6, 257–266 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Tan C. Y., Vidal-Puig A. (2008) Adipose tissue expandability: the metabolic problems of obesity may arise from the inability to become more obese. Biochem. Soc. Trans. 36, 935–940 [DOI] [PubMed] [Google Scholar]
- 58. Unger R. H., Scherer P. E. (2010) Gluttony, sloth and the metabolic syndrome: a roadmap to lipotoxicity. Trends Endocrinol. Metab. 21, 345–352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Gavrilova O., Marcus-Samuels B., Graham D., Kim J. K., Shulman G. I., Castle A. L., Vinson C., Eckhaus M., Reitman M. L. (2000) Surgical implantation of adipose tissue reverses diabetes in lipoatrophic mice. J. Clin. Investig. 105, 271–278 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Kim J. Y., van de Wall E., Laplante M., Azzara A., Trujillo M. E., Hofmann S. M., Schraw T., Durand J. L., Li H., Li G., Jelicks L. A., Mehler M. F., Hui D. Y., Deshaies Y., Shulman G. I., Schwartz G. J., Scherer P. E. (2007) Obesity-associated improvements in metabolic profile through expansion of adipose tissue. J. Clin. Investig. 117, 2621–2637 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Aitman T. J., Glazier A. M., Wallace C. A., Cooper L. D., Norsworthy P. J., Wahid F. N., Al-Majali K. M., Trembling P. M., Mann C. J., Shoulders C. C., Graf D., St Lezin E., Kurtz T. W., Kren V., Pravenec M., Ibrahimi A., Abumrad N. A., Stanton L. W., Scott J. (1999) Identification of Cd36 (Fat) as an insulin-resistance gene causing defective fatty acid and glucose metabolism in hypertensive rats. Nat. Genet. 21, 76–83 [DOI] [PubMed] [Google Scholar]
- 62. Kim J. K., Gavrilova O., Chen Y., Reitman M. L., Shulman G. I. (2000) Mechanism of insulin resistance in A-ZIP/F-1 fatless mice. J. Biol. Chem. 275, 8456–8460 [DOI] [PubMed] [Google Scholar]
- 63. DeFronzo R. A. (2004) Dysfunctional fat cells, lipotoxicity and type 2 diabetes. Int. J. Clin. Pract. Suppl. 143, 9–21 [DOI] [PubMed] [Google Scholar]
- 64. Sjöström L., Lindroos A. K., Peltonen M., Torgerson J., Bouchard C., Carlsson B., Dahlgren S., Larsson B., Narbro K., Sjöström C. D., Sullivan M., Wedel H.; Swedish Obese Subjects Study Scientific Group (2004) Lifestyle, diabetes, and cardiovascular risk factors 10 years after bariatric surgery. N. Engl. J. Med. 351, 2683–2693 [DOI] [PubMed] [Google Scholar]
- 65. Klein S., Fontana L., Young V. L., Coggan A. R., Kilo C., Patterson B. W., Mohammed B. S. (2004) Absence of an effect of liposuction on insulin action and risk factors for coronary heart disease. N. Engl. J. Med. 350, 2549–2557 [DOI] [PubMed] [Google Scholar]
- 66. Mayerson A. B., Hundal R. S., Dufour S., Lebon V., Befroy D., Cline G. W., Enocksson S., Inzucchi S. E., Shulman G. I., Petersen K. F. (2002) The effects of rosiglitazone on insulin sensitivity, lipolysis, and hepatic and skeletal muscle triglyceride content in patients with type 2 diabetes. Diabetes 51, 797–802 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. van Herpen N. A., Schrauwen-Hinderling V. B. (2008) Lipid accumulation in non-adipose tissue and lipotoxicity. Physiol. Behav. 94, 231–241 [DOI] [PubMed] [Google Scholar]
- 68. Rhee E. P., Cheng S., Larson M. G., Walford G. A., Lewis G. D., McCabe E., Yang E., Farrell L., Fox C. S., O'Donnell C. J., Carr S. A., Vasan R. S., Florez J. C., Clish C. B., Wang T. J., Gerszten R. E. (2011) Lipid profiling identifies a triacylglycerol signature of insulin resistance and improves diabetes prediction in humans. J. Clin. Investig. 121, 1402–1411 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Matsuzaka T., Shimano H., Yahagi N., Kato T., Atsumi A., Yamamoto T., Inoue N., Ishikawa M., Okada S., Ishigaki N., Iwasaki H., Iwasaki Y., Karasawa T., Kumadaki S., Matsui T., Sekiya M., Ohashi K., Hasty A. H., Nakagawa Y., Takahashi A., Suzuki H., Yatoh S., Sone H., Toyoshima H., Osuga J., Yamada N. (2007) Crucial role of a long-chain fatty acid elongase, Elovl6, in obesity-induced insulin resistance. Nat. Med. 13, 1193–1202 [DOI] [PubMed] [Google Scholar]
- 70. Coll T., Eyre E., Rodríguez-Calvo R., Palomer X., Sánchez R. M., Merlos M., Laguna J. C., Vázquez-Carrera M. (2008) Oleate reverses palmitate-induced insulin resistance and inflammation in skeletal muscle cells. J. Biol. Chem. 283, 11107–11116 [DOI] [PubMed] [Google Scholar]
- 71. Miyazaki M., Flowers M. T., Sampath H., Chu K., Otzelberger C., Liu X., Ntambi J. M. (2007) Hepatic stearoyl-CoA desaturase-1 deficiency protects mice from carbohydrate-induced adiposity and hepatic steatosis. Cell Metab. 6, 484–496 [DOI] [PubMed] [Google Scholar]
- 72. Wang Y., Botolin D., Xu J., Christian B., Mitchell E., Jayaprakasam B., Nair M. G., Peters J. M., Busik J. V., Olson L. K., Jump D. B. (2006) Regulation of hepatic fatty acid elongase and desaturase expression in diabetes and obesity. J. Lipid Res. 47, 2028–2041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Na J., Musselman L. P., Pendse J., Baranski T. J., Bodmer R., Ocorr K., Cagan R. (2013) A Drosophila model of high sugar diet-induced cardiomyopathy. PLoS Genet. 9, e1003175. [DOI] [PMC free article] [PubMed] [Google Scholar]