

Background: Better treatments are needed for combating influenza.
Results: LSTc-sialoside-bearing decoy liposomes competitively bind to influenza A virus, as assessed by hemagglutination inhibition, flow cytometry, and growth inhibition studies. Decoy liposomes co-localize with influenza virus, as assessed by confocal imaging.
Conclusion: LSTc-sialoside-bearing decoy liposomes are highly effective in capturing influenza virus.
Significance: Decoy liposomes may serve as an effective platform for presenting anti-pathogen receptors.
Keywords: Cell Culture, Confocal Microscopy, Influenza Virus, Liposomes, Sialic Acid
Abstract
Influenza is a severe disease in humans and animals with few effective therapies available. All strains of influenza virus are prone to developing drug resistance due to the high mutation rate in the viral genome. A therapeutic agent that targets a highly conserved region of the virus could bypass resistance and also be effective against multiple strains of influenza. Influenza uses many individually weak ligand binding interactions for a high avidity multivalent attachment to sialic acid-bearing cells. Polymerized sialic acid analogs can form multivalent interactions with influenza but are not ideal therapeutics due to solubility and toxicity issues. We used liposomes as a novel means for delivery of the glycan sialylneolacto-N-tetraose c (LSTc). LSTc-bearing decoy liposomes form multivalent, polymer-like interactions with influenza virus. Decoy liposomes competitively bind influenza virus in hemagglutination inhibition assays and inhibit infection of target cells in a dose-dependent manner. Inhibition is specific for influenza virus, as inhibition of Sendai virus and respiratory syncytial virus is not observed. In contrast, monovalent LSTc does not bind influenza virus or inhibit infectivity. LSTc decoy liposomes prevent the spread of influenza virus during multiple rounds of replication in vitro and extend survival of mice challenged with a lethal dose of virus. LSTc decoy liposomes co-localize with fluorescently tagged influenza virus, whereas control liposomes do not. Considering the conservation of the hemagglutinin binding pocket and the ability of decoy liposomes to form high avidity interactions with influenza hemagglutinin, our decoy liposomes have potential as a new therapeutic agent against emerging influenza strains.
Introduction
Influenza A virus (IAV),3 a member of the orthomyxovirus family, causes upper and lower respiratory tract infections in humans that range from mild, non-life threatening illness to lethality (1). The current therapeutic options for influenza are limited, with vaccination being the most effective tool against the disease. Vaccines for influenza require a large lead time from production to delivery, and if the circulating strain drifts, the vaccine would offer little protection (2). Furthermore, in pandemic outbreaks, vaccines cannot be generated and delivered at the required speed or level (3). Treatments for influenza include small-molecule inhibitors such as amantadine, and neuraminidase inhibitors, including oseltamivir and zanamivir. Resistance against these inhibitors in the circulating influenza strains restricts their utility (2). In the past decade, resistance to amantadine, which inhibits the viral M2 ion channel, emerged in clinical isolates of H1N1 influenza and quickly reached nearly 100% for circulating H3N2 influenza (4, 5). Resistance to the neuraminidase inhibitor oseltamivir has rapidly emerged also; only 12.3% of influenza A (H1N1) viruses tested were oseltamivir-resistant in the 2007–2008 season but escalated to 98.5% in the 2008–2009 season (6). Alternative treatment options are needed, as resistance becomes an increasing threat.
One therapeutic strategy is to target the attachment and fusion of influenza virus particles to host cells. Influenza hemagglutinin (HA) binds to specific carbohydrate structures on surface proteins and lipids. Human-adapted influenza virus binds to terminal sialic acid (SA) in α2–6 linkage (7, 8). Vaccines work by primarily eliciting neutralizing antibodies that target the head region of HA and sterically inhibiting the HA-glycan interaction (9, 10). However, the virus can easily mutate to escape from neutralizing antibodies while still binding to the host-glycan receptors (9, 11).
Targeting the glycan-receptor binding site using a decoy offers a promising alternate therapeutic strategy. Although SA is the key monosaccharide on the host glycan that interacts with HA, the monovalent HA-SA interaction is weak with a 50% inhibitory concentration (IC50) of influenza virus attachment ranging from the millimolar (12, 13) to the micromolar range (14). Oligomerization of sialosides greatly increases the apparent affinity, making them strong inhibitors (15). However, the polymers needed to create a backbone for these multivalent sialosides are often cytotoxic, insoluble, or immunogenic, and therefore oligomerized sialosides are not ideal as therapeutic agents (16). Several groups have created polymer-like sialosides by creating SA analog glycolipids and incorporating them into liposomes. SA functionalized liposomes are capable of binding influenza to similar extents as SA polymers. Kingery-Wood et al. (17) and Guo et al. (18) created neutrally charged liposomes functionalized with SA analogs that bound tightly to influenza, with IC50 values at the micromolar to nanomolar range. They did not, however, attempt to block virus infectivity, and interestingly, Guo et al. (18) failed to inhibit PR/8 influenza. Spevak et al. (19) used a series of liposomes with different amounts of SA on the surface to inhibit hemagglutination and infection, but their results did not correlate between the two assays.
These early experiments demonstrate the validity of using SA functionalized liposomes to inhibit influenza. The liposomal approach can further be improved upon by adding a net negative charge to decrease interactions with host cells (20) while increasing the interaction with HA (21). Furthermore, the SA analog and its attachment chemistry incorporated into the liposomes can be optimized for stronger binding to HA. IAV HA interacts with the host-glycan motif beyond the terminal SA monosaccharide. Monosaccharides extending beyond the SA critically interact with HA and govern the HA-glycan interaction. A common feature of previous pandemic strains is their high affinity binding to α2–6-sialylated glycans that are tetrasaccharides or longer (22, 23).
Here we describe a novel approach to constructing a SA receptor-based therapeutic agent capable of multivalent presentation of SA residues to influenza. Sialylneolacto-N-tetraose c (LSTc) is a well studied sialooligosaccharide that is a high affinity binder to HA (24–26). A liposome-based delivery platform allows spherical display and lateral diffusion of SA in the plane of the membrane. This display leads to a proper presentation of SA, as distribution and configuration affects the avidity of SA analogs for influenza (27). We demonstrate that LSTc-sialoside-bearing liposomes can present SA in a multivalent fashion to enable high avidity binding to influenza HA. Our data show that decoy liposomes bind strongly to influenza viral particles and can inhibit IAV infectivity in vitro as well as extend survival of mice challenged with a lethal dose of influenza A virus in vivo.
EXPERIMENTAL PROCEDURES
Purification of LSTc
LSTc was either obtained from a commercial source (Dextra, Reading, UK) or purified from milk. Frozen bovine milk was thawed and centrifuged at 4000 × g for 10 min at 4 °C. The upper fatty layer was discarded, and the lower aqueous layer was mixed with 2 volumes of ethanol and kept at 4 °C overnight. Precipitate was removed by centrifugation at 12,000 × g for 10 min at 4 °C; the supernatant, consisting primarily of oligosaccharides and lactose, was dried under nitrogen at room temperature and then reconstituted with 0.2 volumes of fresh 20% methanol and stored at −20 °C until use. To separate LSTc from other oligosaccharides, the reconstituted solution was subjected to dual stage purification using size exclusion as the first step to remove high molecular weight material and to exchange the sample into a suitable buffer. As the second step, weak anion exchange purification was performed using ammonium formate as the eluting agent. Fractions were monitored by mass spectrometry. LSTc fractions were pooled and lyophilized. Purity of sample was assessed using capillary electrophoresis.
Glycolipid Synthesis
1,2-Dioleoyl-sn-glycero-3-phosphoethanolamine (DOPE) (Avanti Polar Lipids, Alabaster, AL) and N-(FMoc-13-amino-4,7,10-trioxa-tridecyl)succinamic acid (FMOC) (Polypeptide Laboratories, San Diego, CA) were purchased from commercial sources and used without further purification. Ethyl-3-[3-dimethylaminopropyl] carbodiimide hydrochloride (EDC) was purchased from Sigma. Thin layer chromatography (TLC) was performed on silica-coated glass plates. Column chromatography was performed using silica gel 60 Å. Mass spectrometry was performed using 4800 MALDI-MS and MALDI-TOF instruments (Voyager DE-STR, Applied Biosystems, Carlsbad, CA). Solvent evaporation was performed on a rotary evaporator under reduced pressure at 30–35 °C. All other synthetic lipids were purchased as solutions in chloroform (Avanti Polar Lipids, Alabaster, AL) including: DOPC (1,2-dioleoyl-sn-glycero-3-phosphocholine), DOPG (1,2-dioleoyl-sn-glycero-3-[phospho-rac-(1-glycerol)] (sodium salt)), and DOPE-NBD (DOPE-N-(7-nitro-2–1,3-benzoxadiazol-4-yl) (ammonium salt)). Cholesterol (purity ≥99%, Mr 386.65) was purchased dry (Sigma) and dissolved in chloroform. Chloroform (Acros Organics, Morris Plains, NJ) was >99.8% pure and stabilized with 200 proof ethanol (Goldshield Chemical Co., Hayward, CA), hydrochloric acid was certified ACS plus (Fisher), and all water used was ultra-purified (MilliQ A10, Millipore, Billerica, MA).
1,2-Diamino-4,5-methylene Dioxybenzene-HPLC Quantification
LSTc incorporation into glycolipid was quantified by HPLC as described by Klein et al. (28); briefly, glycolipid or standard was treated with 2 n glacial acetic acid at 80 °C for 5 h then dried by centrifugal evaporation. Samples were treated with 30 μl of 1,2-diamino-4,5-methylene dioxybenzene labeling mixture (1.6 mg of 1,2-diamino-4,5-methylenedioxybenzene dihydrochloride, 3.1 mg of sodium hydrosulfite, 58 μl of β-mercaptoethanol, 82 μl of glacial acetic acid per ml) at 50 °C for 2.5 h in the dark. Samples were diluted with 20 μl of double distilled H2O and analyzed by reverse phase HPLC using a TSKgel ODS-120T column (Tosoh Corp., South San Francisco, CA) running 7% methanol in water. Fluorescence of the 1,2-diamino-4,5-methylene dioxybenzene-SA complex is detected at excitation 373/emission 448.
Liposome Preparation
Gas-tight syringes (Hamilton Co., Reno, NV) and 4 ml borosilicate glass vials with Teflon-lined caps (National Scientific, Rockwood, TN) were thoroughly cleaned before use. Syringes were rinsed 10× with 100% ethanol and then 10× with chloroform. Vials were soaked in 300 mm HCl for 1.5 h and then rinsed thoroughly with water, 3× with ethanol, and 3× with chloroform. Residual solvent was evaporated under a filtered stream of dry nitrogen gas.
Lipids were mixed and deposited in clean vials using clean syringes. Solvent was evaporated under a filtered stream of dry nitrogen gas while manually rotating the vial until only a thin layer of lipid remained on the inner walls. Residual solvent was removed by placing uncapped vials in a desiccator (Dry Seal; Wheaton, Millville, NJ) followed by application of reduced pressure for 24 h using an oil-free diaphragm vacuum pump (Gast, Benton Harbor, MI).
Aqueous lipid solutions were made by hydrating lipid films in 150 mm phosphate-buffered saline (PBS) (140 mm NaCl, 8.5 mm NaH2PO4, 1.5 mm Na2HPO4, pH 7.4) and vortexing for 2 min in 30 s intervals. Lipid solutions were then subjected to 10 rapid cycles of freeze-thawing by submersion in liquid nitrogen and 70 °C water, respectively, to break apart multi-lamellar structures and then extruded through 200 nm pores. Extrusion consisted of either 10 passes through an aluminum oxide membrane using a Lipex Thermobarrel Extruder (Northern Lipids, Burnaby, BC, Canada) or 21 passes through a polycarbonate membrane using a LiposoFast-Basic Extruder (Avestin; Ottowa, ON, Canada). Extruders were thoroughly cleaned and primed with buffer before use. After the final pass, samples were collected in a clean vial, sealed with a Teflon-lined cap, and stored at 4 °C until use.
Lipid concentration post-extrusion relative to pre-extrusion was determined by fluorimetry. Typical recoveries were ∼50% with the Lipex Thermobarrel Extruder and ∼80% with the LiposoFast-Basic Extruder. The concentration of lipid in a final solution made from a stock mixture of 3 mol% DOPE-NBD and 97 mol% DOPC was determined colorimetrically as described (29) and served as a reference for fluorimetric measurements on solutions made from the same lipid stock. Results were consistent with concentrations estimated based on mass lipid deposited and volume hydrated. Samples containing DOPE-NBD or DOPE-rhodamine were hydrated to an estimated concentration of 7.7 mm total lipid.
Liposome Characterization
Diameter and polydispersity of liposomes were determined by dynamic light scattering (Zetasizer Nano; Malvern Instruments, Worcestershire, UK) specifying a lipid refractive index of 1.480 and a dispersant (150 mm PBS) refractive index of 1.332. Measurements were taken using 40 μl of disposable cuvettes at room temperature (20 °C) and a backscattering angle of 173 degrees. Data are reported as the average of 5 measurements separated by 15 s. Decoy liposomes were stored at 4 °C for 12 months and were stable over this period, retaining their anti-influenza properties.
Viral Strains
Influenza A/Puerto Rico/8/34 virus (PR/8, H1N1) was generously provided by Susan Swain (University of Massachusetts, Worcester, MA). Influenza A/Philippines X-79 (H3N2) was provided by Richard Dutton (University of Massachusetts, Worcester, MA). Influenza A/Aichi/68 (X-31, H3N2) and Sendai virus (Cantell Strain) were purchased from Charles River Laboratories (North Franklin, CT). Influenza A/Beijing/262/95 (H1N1) was purchased from Meridian Life Science (Saco, ME). Influenza viruses and Sendai virus were originally grown in the allantoic cavity of embryonated chicken eggs. Influenza viruses were stored at −80 °C before use and titered on Madin-Darby canine kidney (MDCK) cells. Respiratory syncytial virus (RSV) strain A2 was grown in Vero cells in 5% fetal bovine serum (FBS), and cell debris was frozen at −80 °C and subsequently titered on Vero cells. Recombinant influenza A/WSN/33 with AlexaFluor 647 covalently attached to the HA protein via sortase (WSN HA-647) was prepared as described (30) and stored at 4 °C before use.
Cell Lines
MDCK cells were obtained from the American Type Culture Collection (ATCC, Manassas, VA) and were cultured in Eagle's minimal essential medium with 10% FBS, 2 mm l-glutamine, 2 mm penicillin/streptomycin, 0.1 mm nonessential amino acids, and 1 mm sodium pyruvate. A549 cells were obtained from ATCC and cultured in Dulbecco's modified Eagle's medium (DMEM) with 10% FBS and 2 mm penicillin/streptomycin. Vero cells were obtained from ATCC and cultured in DMEM with 10% FBS and 2 mm penicillin/streptomycin.
Mouse Infection Studies
Female C57BL/6 wild-type mice were purchased from The Jackson Laboratory (Bar Harbor, ME). Mice were 8–10 weeks of age at the time of infection. IAV (1000 plaque forming units (PFU)) was combined with LSTc decoy liposomes (170 μm SA) or the equivalent amount of control liposomes in a final volume of 30 μl and incubated at 37 °C for 30 min. Samples were stored on ice until use. Mice were infected intratracheally with 30 μl of sample and monitored daily. Mice were scored as deceased when found dead or were clearly imminently moribund, in which case they were euthanized. The University of Massachusetts Medical School Institutional Animal Care and Use Committee approved all experimental protocols.
Hemagglutination Inhibition Assay
Red blood cells (RBCs) were isolated from normal human peripheral blood, blood type O. Whole blood was washed in sterile PBS 3 times to remove serum. Packed RBCs were diluted 1:30 in sterile PBS and stored at 4 °C before use. All procedures involving human subjects were approved by the University of Massachusetts Medical School Committee for the Protection of Human Subjects in Research and in accordance with the Declaration of Helsinki.
HA titers for each virus were determined on RBCs before inhibition studies per standard protocol (31). For hemagglutination inhibition (HAI) assays, liposome samples were diluted 2-fold in PBS. Four HA units of virus in 25 μl of PBS were added to all dilutions. Samples were incubated for 30 min at room temperature. 50 μl of diluted RBCs were added to the wells and incubated for an additional hour at room temperature to allow agglutination. The HAI titer is the reciprocal of the last dilution of liposomes that results in non-agglutinated RBCs.
Plaque Assay with Immunostaining
MDCK cells were seeded into 12-well plates and incubated at 37 °C for 24 h to form monolayers. Liposome samples were diluted to the desired concentration in sterile PBS, 1% bovine serum albumin (BSA, Sigma) in a final volume of 225 μl. IAV was diluted to 300 PFU/ml and mixed 1:1 (v/v) with liposome samples and incubated at 37 °C for 30 min. MDCK cells were washed twice with PBS, 1% BSA, and samples were added to wells in duplicate (200 μl per well). Samples were incubated at 37 °C for 1 h on MDCK cells. Cells were washed with PBS, 1% BSA and overlaid with freshly prepared 0.5% agar in DMEM-F-12 and incubated at 37 °C for 48 h. Cells were fixed and stained with anti-hemagglutinin antibody MAB8261 (Millipore). Plaques were visualized with anti-mouse horseradish peroxidase-conjugated secondary antibody (BD Biosciences) and developed with peroxidase substrate kit (Vector Laboratories, Burlingame, CA). Viral plaques in the MDCK monolayer were counted, and the PFU/ml was determined.
For some experiments, RSV was used in a similar fashion, except as noted. Vero cells were grown to confluency in 24-well plates. Liposome samples were diluted to the desired concentration in serum-free DMEM in a final volume of 130 μl. RSV was diluted to 1000 PFU/ml and mixed 1:1 (v/v) with liposome samples and incubated at 37 °C for 30 min. Vero cells were prewashed in serum-free DMEM before the addition of 100 μl of sample in duplicate. After 1 h of infection, cells were washed with DMEM, 10% FBS and incubated in DMEM, 10% FBS and 2 mm penicillin/streptomycin for 3 days at 37 °C. Cells were fixed with 80/20 (v/v) acetone/PBS and stained with anti-F and anti-G glycoprotein antibodies (MAB8599 and MAB858, respectively; 1:1000 dilution, Millipore). RSV plaques were visualized and quantified as for IAV.
Inhibition of Viral Growth
MDCK cells were seeded into 24-well plates and allowed to grow to confluency overnight at 37 °C. MDCK monolayers were washed with PBS, 1% BSA and treated with decoy liposomes ranging in concentration from 1 to 1000 nm LSTc or control liposomes at lipid concentrations equivalent to the 1000 nm LSTc decoy liposome treatment for 30 min at 37 °C. Treated MDCK cells were then infected at a multiplicity of infection of 0.01 or 0.001 of PR/8 diluted in PBS, 1% BSA without removal of liposome solution. After an infection period of 1 h at 37 °C, the inocula were removed, and MDCK cells were washed with PBS, 1% BSA. Viral growth medium (0.5 ml) with identical liposome treatment as during the infection step was added. After 24 h, supernatants were taken, and virus growth was measured by plaque assays.
Flow Cytometry
Adherent A549 cells were detached with 0.25% trypsin, 2.21 mm EDTA. Cells were washed twice with PBS, enumerated using a hemocytometer, and transferred to 96-well round-bottom plate (Costar, Washington, D. C.) at 50,000 cells per well. WSN HA-647 and decoy liposomes were co-incubated for 30 min. WSN HA-647 was tested at 1.3, 6.5, and 13 HA units with 1000 nm, 7.5 mol% LSTc decoy liposomes, diluted in PBS, 1% BSA in a final volume of 50 μl. Control liposomes without LSTc were diluted to the same lipid concentration as decoy liposomes. Cells were treated with virus/decoy mixtures for 15 min at 37 °C before being fixed with formalin (final concentration 1%). Cells were analyzed using a BD Biosciences LSR II flow cytometer and FlowJo Version 9.4.11 (TreeStar software).
Confocal Microscopy
30 HA units of WSN HA-647 were absorbed onto glass coverslips overnight, then treated with either 7.5 mol% LSTc decoy liposomes or control liposomes, both containing NBD-labeled lipids. Decoy liposomes were diluted to 1000 nm LSTc in a final volume of 50 μl; control liposomes were diluted to the same lipid concentration as the decoy liposomes. Decoy or control liposomes were incubated on the WSN HA-647 coverslips for 30 min at 4 °C and washed 3 times with PBS, 1% BSA before imaging. Images were taken on a Leica SP2 AOBS confocal laser-scanning microscope with a 63× objective using the Leica Confocal Software (Version 2.6.1). Multichannel images were obtained by sequential scanning with only one laser active for each scan to avoid cross-excitation. Overall brightness and contrast of images were optimized using Image J (32).
Statistics
Statistical analysis was performed using the unpaired, two-tailed Student's t test. Values of p ≤ 0.05 were considered significant. Error bars are ± S.D. or ± S.E. as listed in each figure legend. For survival analysis, the Mantel-Cox Log-rank test was performed. Statistics were calculated using Prism Version 6.0 (GraphPad Software).
RESULTS
Generation of Decoy Liposomes Containing the Sialic Acid Moiety, LSTc
We chose a lipid backbone for our decoys to promote LSTc flexibility and permit multivalent binding to influenza HA trimers. We formulated decoy liposomes from a mixture of phospholipids with two 18:1 fatty acyl chains such as DOPC, a principal component of mammalian membranes (Fig. 1A). DOPC has a low gel transition temperature, which allows it to form relatively disordered membranes that remain fluid. Into this background we incorporated LSTc-DOPE. DOPE carries a primary amine that is easily linked to LSTc and that allows controlled addition of SA moieties with distinct linker chemistries and of defined stoichiometry. This allows optimal presentation of the decoys to influenza HA. The DOPE primary amine also enables ready modification with the fluorescent dye NBD or rhodamine, a marker to assist with tracking and quantification. We added DOPG in some formulations to compensate for the net negative charge imparted by LSTc-DOPE.
FIGURE 1.

Summary of decoy liposome construction. A, the different components of decoy liposomes are described. B, the chemical synthesis of LSTc-DOPE is outlined in the diagram. Additional details are described in the accompanying text. Rh, rhodamine; EDC, ethyl-3-[3-dimethylaminopropyl] carbodiimide hydrochloride; NaCNBH3, sodium cyanoborohydride; DMSO, dimethyl sulfoxide.
Previous studies using glycan-specific lectins in histological analysis of tissue sections of the human respiratory tract demonstrated the upper respiratory tract is found to predominately contain glycans with α2–6-linked SA, whereas alveolar cells of the deep lung are dominated by glycans bearing α2–3-linked SA. Furthermore, solid phase glycan array analysis of human-adapted influenza viruses from H1N1, H2N2, and H3N2 subtypes has revealed a high affinity binding to long (tetrasaccharide or longer) α2–6-linked sialylated glycans as a key common feature (22, 33). Therefore, a readily available “long” α2–6-sialylated glycan LSTc (Neu5Aca2–6Galb1–4GlcNAcb1–3Galb1–4Glc) was selected for use in decoy liposomes for human-adapted influenza viruses. LSTc is also an ideal sialylated glycan due to its ability to bind to multiple HA subtypes, a feature not true for all sialylated glycans (25, 26, 33).
Glycolipid synthesis is schematically represented in Fig. 1B. We attached DOPE, a commercially available amine-linked unsaturated phospholipid (Fig. 1B, compound 1) to an Fmoc-protected linker acid (compound 2) using carbodiimide coupling. We obtained compound 3 by purification of the crude product by silica gel column chromatography. We used piperidine to deprotect the FMOC-modified lipid-linker conjugate (compound 3) and monitored the reaction by TLC. The crude amine (compound 4) was purified by column chromatography and eluted in organic solvent. The free amine of compound 4 is available for coupling to the sugar aldehyde (reducing end). Conjugation with the free non-reducing sugar aldehyde of LSTc was achieved via reductive β-elimination. LSTc (compound 5) and compound 4 were conjugated at 60 °C in minimal acid to minimize hydrolysis (desialylation) of LSTc. The final product (compound 6) was partially purified using flash chromatography and further purified by HPLC to obtain LSTc-containing glycolipids. The final LSTc-DOPE product was assessed by 1,2-diamino-4,5-methylene dioxybenzene-HPLC to confirm the presence of SA in the glycolipid. We characterized the purified product by MALDI-MS and found the final glycolipid had m/z = 2029–2031 Da, expected m/z = 2032 Da (data not shown).
We generated decoy liposomes with variable amounts of LSTc to determine the optimal amount of SA required for efficacy. The LSTc glycan is negatively charged, and all decoys containing less than 7.5 mol% LSTc were made with DOPG as needed to generate decoy liposomes with the same net negative charge (Fig. 1A). SA concentrations for each preparation were estimated based on the percent of input LSTc-DOPE and by the nanomolar concentration of total lipid. Liposomes with 0 mol% LSTc (and 7.5 mol% DOPG) were used as control liposomes. We used extrusion through 200-nm pores to create the liposomes and measured their average size and polydispersity index using dynamic light scattering.
Decoy Liposomes Containing LSTc Bind to Different Strains of IAV
We used a standard hemagglutination inhibition (HAI) assay to assess binding of decoy liposomes to influenza A/Puerto Rico/8/34 (PR/8, H1N1). The indicated percentage of sialoside at the liposome surface represents the mole percentage of lipid monomers used in liposome synthesis reaction. IAV binds to SA on the surface of RBCs, causing hemagglutination. LSTc decoy liposomes provide an alternative SA binding option for IAV and will inhibit hemagglutination when present in sufficient quantity. The ability of liposomes to inhibit hemagglutination can be expressed either as the HAI titer (i.e. the reciprocal of the last dilution of liposomes required to inhibit hemagglutination) or as the concentration (molarity of SA) that results in 90% inhibition (IC90). For PR/8, decoy liposomes with 7.5 mol% LSTc had HAI titers of 256 and an IC90 of 0.041 μm SA, and decoy liposomes with 5 mol% LSTc had HAI titers of 128 and an IC90 of 0.04 μm SA (Table 1). Decoy liposomes containing 1 mol% LSTc did not inhibit hemagglutination at the highest concentration tested, 0.52 μm SA. Control liposomes, used at similar lipid concentrations as the LSTc decoy liposomes, did not inhibit hemagglutination. Monovalent LSTc, used at 5 × 105 μm, did not inhibit hemagglutination (Table 1). We also tested the activity of decoy liposomes containing either 10, 20, or 30 mol% LSTc by HAI against various strains of IAV but did not see any significant increase in HAI titers compared with liposomes containing 7.5 mol% LSTc (data not shown).
TABLE 1.
Assessment of different decoy liposome formulations by hemagglutination inhibition assay
LSTc-containing decoy liposomes competitively bind to A/PR/8 (H1N1). Several different liposome formulations were assessed. Decoy liposomes containing 0, 1, 5, or 7.5 mol% LSTc were effective in inhibiting hemagglutination of both strains of influenza virus. Monovalent LSTc did not inhibit PR/8. “No inhibition” indicates that no inhibition was observed at the highest concentration tested. IC90 values are shown as molarity of sialic acid.
| Mol% LSTc on liposomes | HAI titer | IC90 |
|---|---|---|
| μm SA | ||
| 0 | No inhibition | N/A |
| 1 | No inhibition | >0.52 |
| 5 | 128 | 0.04 |
| 7.5 | 256 | 0.041 |
| Monovalent LSTc | No inhibition | >500,000 |
To test the specificity of our decoy liposomes, we performed the HAI assay with several additional strains of IAV as well as the related RNA virus, Sendai virus (SeV). SeV is a Paramyxoviridae family virus that binds specifically to α2–3-linked SA (34), which is not present in our LSTc decoy liposomes. Decoy liposomes containing 7.5 mol% LSTc inhibited all strains of IAV tested: A/Philippines/2/82/X-79 (Philippines, H3N2) with an HAI titer of 16 (IC90 = 0.98 μm SA), X-31 (A/Aichi/68, H3N2) with an HAI titer of 32 (IC90 = 0.26 μm SA), Beijing (H1N1) with an HAI titer of 64 (IC90 = 0.11 μm SA) and PR/8 (H1N1) with an HAI titer of 256 (IC90 = 0.041 μm SA) (Table 2). However, 7.5 mol% LSTc decoy liposomes did not inhibit SeV agglutination (Table 2), demonstrating that the observed binding is specific for α2–6-linked SA binding rather than a nonspecific binding event.
TABLE 2.
Assessment of 7.5 mol% LSTc decoy liposomes by hemagglutination inhibition assay of multiple strains of influenza A virus and Sendai virus
Decoy liposomes competitively bind to several strains of influenza virus. Decoy liposomes containing 7.5 mol% LSTc are effective at inhibiting hemagglutination of Philippines, X-31, Beijing, and PR/8 influenza strains. IC90 values are shown as molarity of sialic acid. Hemagglutination of Sendai virus is not inhibited by 7.5 mol% decoy liposomes at the highest concentration tested.
| Virus | HAI titer | IC90 |
|---|---|---|
| μm SA | ||
| Philippines H3N2 | 16 | 0.98 |
| X-31 H3N2 | 32 | 0.26 |
| Beijing H1N1 | 64 | 0.11 |
| PR/8 H1N1 | 256 | 0.041 |
| Sendai | No inhibition | >84 |
Decoy Liposomes Containing LSTc Prevent IAV Infection of MDCK Cells
To investigate the impact of decoy liposomes on IAV infectivity, we co-incubated either PR/8 with LSTc-containing decoy liposomes or control liposomes before infection of MDCK cell monolayers. In this experiment viral particles are expected to bind to native SA on MDCK cells only when not competitively bound to decoy liposomes. Increasing concentrations of LSTc decoy liposomes inhibited infectivity of PR/8 (Fig. 2A) in these cells, whereas control liposomes did not inhibit infection. The molarity of SA in the solution was calculated by multiplying the mole percent input of LSTc-DOPE glycolipids by the total lipid concentration. At 1 nm SA, all decoy liposomes displayed weak inhibition. As the total concentration of SA increased, each series with different mole percentages of LSTc on the liposome surface inhibited more PR/8. Decoy liposomes with 1 mol% LSTc inhibited weakly, blocking only 22 ± 5.6% of PR/8 at 515 nm SA. LSTc decoy liposomes at 5 mol% inhibited weakly at low concentrations of LSTc, but increasing the concentration of these decoys had a more pronounced inhibitory effect, inhibiting 73 ± 10% of PR/8 at 1000 nm SA. LSTc decoy liposomes at 7.5 mol% blocked PR/8 infection to the largest extent, inhibiting PR/8 almost completely at 1000 nm SA, 93.8 ± 1.3% compared with control liposomes (Fig. 2A).
FIGURE 2.
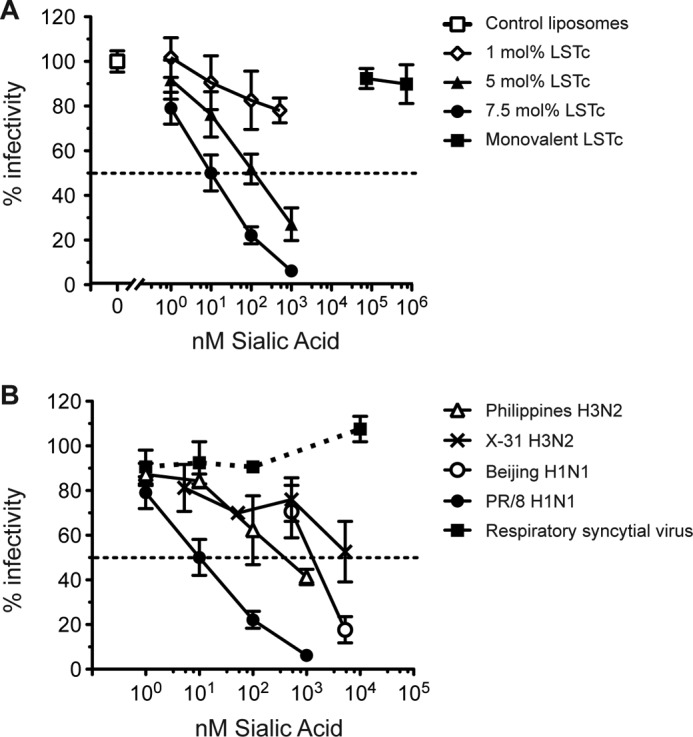
LSTc decoy liposomes specifically inhibit influenza infection of MDCK cells. A, PR/8 (50 PFU/well) was incubated with control liposomes, LSTc-containing decoy liposomes, or monovalent LSTc before the addition to MDCK cells. Plaques per well of virus treated with control liposomes (0 mol% LSTc) are defined as 100% infectivity (open square), and virus treated with different liposome formulations are expressed as the percent reduction from its respective control. Decoy liposomes with 7.5 mol% LSTc (circles) inhibited PR/8 to the greatest extent. Decoy liposomes with 5 mol% LSTc (triangles) also significantly inhibited infection; however, decoy liposomes with 1 mol% LSTc (open diamonds) poorly inhibited infection. B, decoy liposomes containing 7.5 mol% LSTc inhibit multiple strains of influenza. Decoy liposomes inhibited Philippines (open triangles, 50 PFU/well), X-31 (crosses, 50 PFU/well), Beijing (open circles, 15 PFU/well), and PR/8 (filled circles, 50 PFU/well) strains of influenza. LSTc decoy liposomes did not inhibit RSV (filled squares, 50 PFU/well). The data represent the average ± S.E. of three experiments for PR/8 and two experiments for Philippines, X-31, Beijing, and RSV.
These data show not only a dose-dependent response of LSTc in the medium on influenza inhibition but also that the density of LSTc displayed on the surface of each liposome at a given concentration of LSTc affects the extent of viral inhibition. For example, when each series of LSTc decoy liposomes was diluted to 100 nm concentrations of total SA and challenged with PR/8, 7.5 mol% LSTc decoy liposomes inhibit to a greater degree than decoy liposomes with either 5 or 1 mol% LSTc (Fig. 2A). This inhibition occurs despite the fact that decoy preparations with a higher density of LSTc per liposome have fewer liposomes in solution at equimolar concentrations of LSTc. Decoy liposomes with denser LSTc are, therefore, more efficient at inhibiting influenza. We also tested the infectivity inhibition of decoy liposomes containing more than 7.5 mol% LSTc against several strains of IAV. We saw no significant increase of inhibition from liposomes with up to 30 mol% LSTc as compared with 7.5 mol% LSTc (data not shown). These results were similar to the finding that decoy liposomes with more than 7.5 mol% did not increase HAI titers of IAV.
Next, we tested our decoy liposomes at 7.5 mol% LSTc on several additional IAV strains. Decoy liposomes at 7.5 mol% inhibited all IAV strains in a dose-dependent manner. Decoy liposomes inhibited Philippines up to 58.3 ± 3.4% at 1000 nm SA. At 5250 nm SA, decoy liposomes at 7.5 mol% inhibited infectivity of X31 by 47.3 ± 13.5% and infectivity of Beijing by 82.3 ± 5.9%. Control liposomes lacking LSTc did not inhibit any of these strains. Decoy liposomes displayed high avidity for influenza in both the HAI and infectivity assays. We also compared LSTc-containing decoy liposomes with monovalent LSTc at SA concentrations well in excess of the estimated SA concentrations for LSTc-containing liposomes. However, 74,000 nm monovalent LSTc did not inhibit the infectivity of either PR/8 (Fig. 2) or Philippines (data not shown). At this high concentration, monovalent LSTc would be expected to bind to both PR/8 and Philippines HA during the preincubation period. Given that infection was not inhibited, monovalent LSTc cannot make multivalent, high avidity interactions with IAV, whereas LSTc complexed in liposomes can make multivalent, high avidity interactions with IAV.
Decoy Liposomes Containing LSTc Do Not Prevent Infection of RSV in Vero Cells
To test the specificity of LSTc-containing decoy liposomes, we assessed whether decoy liposomes would affect RSV infection of Vero cells. RSV interacts with cellular heparan sulfate for attachment and infectivity (35) and has not been reported to interact with SA receptors. We co-incubated 7.5 mol% LSTc decoy liposomes or control liposomes with RSV before infection of Vero cells. LSTc-containing liposomes did not affect RSV infectivity (Fig. 2B) even at a SA concentration of 10,000 nm, an amount that inhibits nearly 100% of PR/8 influenza. This lack of inhibition along with the lack of hemagglutination inhibition of SeV demonstrates that our LSTc-decoy liposomes specifically inhibit via α2–6-linked SA.
Decoy Liposomes Containing LSTc Block Viral Growth of IAV in MDCK Cells
To investigate the decoy ability to inhibit influenza during multiple rounds of infection, we infected MDCK cells with influenza virus in the presence of increasing concentrations of decoy or control liposomes and allowed continued viral replication and spread in the presence of either decoy or control liposomes. Control liposomes or decoy liposomes with 7.5 mol% LSTc at concentrations of 1–1000 nm SA (10-fold increments) were added to MDCK cells 30 min before the addition of PR/8 influenza. All inoculating virus was removed, and wells were treated with the same concentration of LSTc decoy or control liposomes as during the infection in serum-free media to allow virus replication in host cells. Twenty-four hours after infection, supernatants were sampled and tested for PR/8 plaque-forming units. The amount of virus recovered from wells treated with highly concentrated LSTc decoy liposomes was significantly reduced compared with wells with control liposomes or with PR/8 and assay medium alone (Fig. 3). Infected MDCK cells treated with 100 or 1000 nm LSTc decoy liposomes had viral titers below the limit of detection, >100-fold less than control liposome-treated cells (decoy liposomes 2.7 ± 0 versus control liposomes 4.8 ± 0.07; p < 0.001, based on log10-transformed PFU/ml data) (Fig. 3). 10 nm LSTc decoy liposomes also significantly inhibited PR/8 replication (2.8 ± 0.2 versus control liposomes 4.8 ± 0.07; p < 0.01) (Fig. 3). Decoy liposomes diluted to 1 nm LSTc or less did not prevent PR/8 replication (4.6 ± 0.04 versus control liposomes 4.8 ± 0.07; p = 0.069). These results suggest that adhesion of viral particles to LSTc decoy liposomes depletes the amount of free virus capable of infecting in successive rounds of replication and that this interaction is long-lasting because the bound virus is not cleared in this in vitro system.
FIGURE 3.
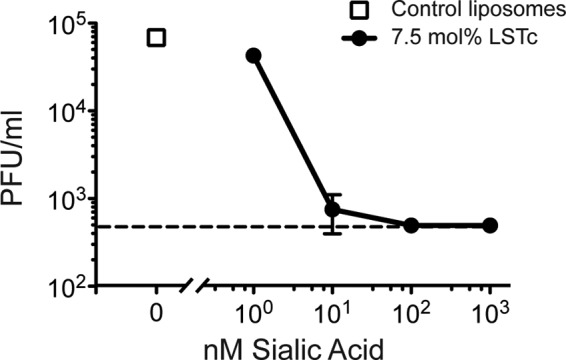
Inhibition of influenza replication by LSTc decoy liposomes. Control or LSTc-containing decoy liposomes were added onto confluent monolayers of MDCK cells 30 min before infection with PR/8 (multiplicity of infection 0.01). After infection, cells were incubated in the presence of liposomes for 48 h. Viral growth was measured by a plaque forming unit assay and plotted as PFU/ml versus total LSTc concentration present in the solution. Control liposomes did not inhibit influenza growth (open squares); however, increasing concentrations of 7.5 mol% LSTc decoy liposomes inhibited PR/8 infectivity (filled circles). The dotted line denotes the limit of detection of the assay. Data shown are representative of two experiments, each with similar results.
Decoy Liposomes Containing LSTc Extend Survival of IAV-infected Mice
To investigate the decoy ability to inhibit influenza in a physiological setting, we evaluated the effects of LSTc-containing decoy liposomes on mice infected with a lethal dose of IAV. LSTc decoy liposomes or control liposomes were co-incubated with 1000 PFU of PR/8 at 37 °C for 30 min. This dose of PR/8 typically causes 90% lethality (LD90) in C57BL/6 mice after intratracheal delivery. We predicted that any IAV not initially associating with LSTc decoy liposomes could subsequently infect susceptible cells within the lungs. Thus, mice administered virus with LSTc decoy liposomes might be expected to succumb to IAV infection less quickly than mice administered virus in combination with control liposomes or mice receiving virus alone.
Mice that received control liposomes and 1000 PFU of PR/8 died at the same rate as mice that received 1000 PFU of PR/8 alone (Fig. 4, median survival of 8 days for both groups, Log Rank test, χ2 = 0.01, p = 0.9). However, mice that received LSTc decoy liposomes and 1000 PFU of PR/8 had significantly extended survival, with a median survival of 12 days compared with 8 days for mice that received control liposomes and 1000 PFU of PR/8 (Log Rank test, χ2 = 13.6, p ≤ 0.01), resulting in a 33.3% mean increase in lifespan post-infection (Fig. 4). The median survival and survival curve slope was reproducible over multiple experiments (the data in Fig. 4 represents the sum of three independent experiments with a combined n ≥ 19 mice for each group). Thus, these data demonstrate that LSTc decoy liposomes significantly extend survival of mice challenged with a LD90 of IAV and that the inhibition observed in vitro extends to a physiological infection setting in vivo. Additionally, LSTc decoy liposomes may form long-lasting interactions with viral particles, even in the presence of the mucociliary system, lung surfactants, and resident professional phagocytes. No apparent toxicity was observed in uninfected mice that were administered LSTc decoy liposomes and monitored for 3 weeks (data not shown).
FIGURE 4.
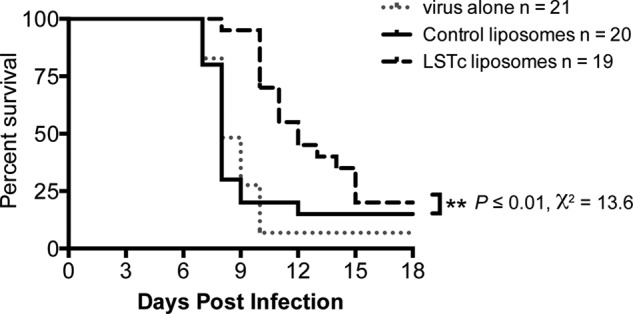
LSTc decoy liposomes extend survival after lethal IAV challenge. 1000 PFU of PR/8 was incubated with control liposomes or LSTc-containing decoy liposomes at 37 °C for 30 min, then used for intratracheal infection of C57BL/6 mice. Mice were monitored daily for survival. Mice that received LSTc decoy liposomes and PR/8 (dashed black line) had a 33% increase of mean survival time post infection as compared with mice that received control liposomes and PR/8 (solid black line) or PR/8 alone (gray dotted line). The LSTc decoy liposome and PR/8-treated mice survival advantage was statistically significant (χ2 = 13.6, p ≤ 0.01; n ≥ 19 for each strain). Data shown are combined from three independent experiments, each having similar results.
Decoy Liposomes Containing LSTc Co-localize with IAV and Inhibit Binding of IAV at the Surface of A549 Human Lung Epithelial Cells
We surmised that inhibition of infection results from the direct binding of SA on LSTc decoy liposomes to HA. To directly observe contact between decoy and influenza virus, we took advantage of a method for enzymatically attaching a fluorophore onto viral particles for single virion tracking (30). WSN HA-647 was absorbed onto glass coverslips, then treated with either decoy or control liposomes containing NBD-labeled lipids. WSN HA-647, decoy, and control liposomes were each readily visible by confocal microscopy when fixed to glass coverslips (data not shown). We observed significant co-localization when WSN HA-647 was treated with 1,000 nm SA decoy liposomes with 7.5 mol% LSTc (Fig. 5, A and C, Pearson's correlation coefficient: r = 0.423, p < 0.01). Control liposomes did not co-localize with adhered virus (Fig. 5, B and D, Pearson's correlation coefficient r = 0.023, p = 0.87). We used cross-correlation analysis to validate the association of virus and decoy liposomes. The cross-correlation function (CCF) establishes whether there is a relationship between two channels of a complex three-dimensional stacked image (36). Fig. 5E shows the CCF of WSN HA-647 treated with decoy or control liposomes. The maximum CCF for LSTc decoy liposomes is at 0 x-voxel displacement and follows the standard decay curve for x shift in both positive and negative directions. The distribution of LSTc decoy liposomes and virus have significantly more overlap than control liposomes and virus, where the CCF does not peak at 0 x-voxel displacement but peaks at 20×-voxel shift with an r = 0.023 and has a flat distribution of CCF throughout the x-voxel shift.
FIGURE 5.

LSTc decoy liposomes bind directly to influenza A virus. Immunofluorescent Z-stack images were obtained of WSN HA-647 and fluorescent liposomes, viewed from above (A and B) or rotated 37 degrees (C and D). A and C, LSTc decoy liposomes, which contain NBD (green) bound specifically to WSN HA-647 (red) and can be seen binding to the apical portion of virus foci when the field of view is rotated 37 degrees. B and D, control liposomes do not show significant co-localization with WSN HA-647. Scale bar, 1 μm. E, Van Steensel's CCF coefficient analysis of images A and B is shown. The CCF is the Pearson coefficient of WSN HA-647 and liposomes at each pixel shift in the x dimension; this calculation is based on a representative field of 45 by 45 μm. Decoy liposomes (open circles) significantly correlate to WSN HA-647 (p < 0.001), whereas control liposomes (black squares) do not (p = 0.21).
To further test the competiveness of LSTc decoy liposomes for IAV, we challenged human alveolar basal epithelial cells (A549 cells) with three different doses of WSN HA-647 combined with either LSTc-bearing decoy liposomes or control liposomes. Mixtures of virus and liposome were added to A549 cells where free virus would bind to cells. Binding of virus at the single cell level was assessed by detection of WSN HA-647 by flow cytometry. WSN HA-647 in combination with control liposomes allowed a high degree of binding to A549 cells (Fig. 6A, gray lines). No significant shifts were noted for A549 cells challenged with control liposomes mixed with 1.3, 6.5, or 13 HA units of WSN-HA-647 (Fig. 6A, left, middle, and right panels). However, decoy liposomes at 7.5 mol% LSTc reduced viral binding when challenged with WSN HA-647 (Fig. 6A, black lines). Decoy liposomes decreased 75, 79, and 54% of binding when challenged with 1.3, 6.5, or 13 HA units of WSN HA-647 (Fig. 6A, left, middle, and right panels, respectively). At the 1.3 HA unit dose of WSN HA-647, the mean fluorescence intensities ± S.E. for control liposomes and decoy liposomes were 369 (± 158) and 89 (± 88) (p = 0.11) (Fig. 6B). The mean fluorescence intensities for the 6.5 HA unit dose of WSN HA-647 were 1071 (±145) and 223 (±86) (p < 0.001) and for the 13 HA unit dose of WSN HA-647, 1142 (±113) and 522 (±89) (p < 0.001) (Fig. 6B). Together with our infectivity data above, these results show that our decoy liposomes competitively bind to influenza, block its adhesion to SA on uninfected cells, and thus block infection.
FIGURE 6.

LSTc-containing decoy liposomes inhibit binding of influenza A virus to A549 cells. A, shown are representative flow cytometry plots of A549 cells treated with control liposomes (gray lines) combined with WSN HA-647 (1.3 HA units (left panel), 6.5 HA units (middle panel), and 13 HA units (right panel)), LSTc-containing decoy liposomes combined WSN HA-647 (black lines), or A549 cells without virus (gray-shaded). LSTc-decoy liposomes contained 7.5 mol% LSTc. B, shown is mean fluorescence intensity (MFI) quantification of data in A; data are presented as the mean ± S.E. ***, p < 0.001 decoy liposomes versus control liposomes.
DISCUSSION
Decoy receptors have the potential to attenuate infections by diverting the pathogen away from susceptible tissues (37). Pathogen receptors are especially suited for use as decoys because the target virus is unlikely to develop resistance through mutation. Viruses require binding specificity and avidity to replicate efficiently and transmit between hosts. Our approach utilized a liposome platform to create a series of synthetic decoy receptors that can bind and neutralize multiple strains of influenza virus (Fig. 7).
FIGURE 7.

Summary: LSTc-containing decoy liposomes inhibit influenza A virus binding and infection. A, influenza infects host cells by first attaching to α2–6 terminally linked SA. Control liposomes that do not contain LSTc do not inhibit influenza adhesion or infection. B, decoy liposomes with limited amounts of LSTc on their surfaces do not form high multivalent interactions with influenza and only partially inhibit influenza binding to host cells. C, decoy liposomes with 5 mol% or more LSTc on the surface are capable of competitively binding multiple strains of influenza, thereby preventing binding and infection of host cells.
SA-bearing receptor molecules can bind and inhibit IAV strains (38). Indeed, many different sialosides containing a single SA residue can inhibit influenza virus (12, 14, 38–40). Because influenza uses a polyvalent interaction between HA and the host cell, these monosialosides have relatively weak inhibitory properties, the strongest having an IC50 of 3.7 μm (14). To take advantage of the multivalent nature of HA binding, polymer-based sialosides are more potent inhibitors of RBC hemagglutination (13, 15, 27, 41, 42). The evidence that polymerized sialosides are capable of inhibiting infectivity is not particularly compelling (19, 43, 44). Gamian et al. (45) were unable to block H3N2 influenza infection of embryonated chicken eggs with polyvalent SA glycoconjugates or their monovalent parental building blocks. Mochalova et al. (46) and Tuzikov et al. (47) were able to prevent some influenza strains from infecting MDCK cells. However, the doses required to block infection were significantly higher than those required to block RBC hemagglutination. In contrast, our decoy liposomes inhibited infectivity at similar concentrations as they inhibited hemagglutination. One important distinction between decoy liposomes and polymer-based sialosides is the issue of potential toxicity when used in vivo. The liposomes are constructed with lipids found in the human body, such as DOPC, and the entire liposome structure is non-toxic, whereas many polymer-based sialosides can cause cell death at high doses.
Increasing the valence and flexibility of SA analogs improves virus binding and potentially the capacity to inhibit infection. Therefore, we used SA-functionalized liposomes to allow sialoside movement and multivalent complex formation as a means of increasing the avidity for IAV. We incorporated LSTc sialosides into liposomes (Fig. 1). These LSTc-containing decoy liposomes competitively bound to both H1N1 and H3N2 subtypes of influenza A (Table 1 and Table 2), whereas control liposomes lacking LSTc did not. Increasing the percentage of LSTc displayed on the surface of our decoy liposomes increased the HAI titer against IAV. We also tested monovalent LSTc, the building block used to create our decoy liposomes. Monovalent LSTc did not inhibit hemagglutination even when tested at concentrations well in excess of the estimated molarity of LSTc incorporated into decoy liposomes. Monovalent SA analogs have dissociation constants for HA of ∼2 mm SA (12, 13, 38). We tested monovalent LSTc at 5 mm, yet saw no inhibition of IAV. Furthermore, the interaction was virus-specific, as high concentrations of LSTc decoy liposomes did not competitively bind to SeV (Table 2) but were clearly capable of inhibiting IAV.
The ability of LSTc decoy liposomes to bind to influenza, as observed in the HAI assay, also protected cells from infection with both H1N1 and H3N2 influenza A (Fig. 2). The degree of binding and inhibition of infectivity was also dependent on the percentage of functionalized LSTc lipids incorporated into decoy liposomes. Decoy liposomes with only 1 mol% of their surfaces functionalized with LSTc did not block hemagglutination and blocked only a small fraction of infectious virus. By increasing from 5 to 7.5 mol% LSTc, HAI was increased, and infectivity was reduced. 7.5 mol% LSTc decoy liposomes were the most efficient at inhibiting infectivity; they were able to significantly decrease infectivity at the lowest concentrations of LSTc, inhibiting PR/8 at an effective concentration of 10 nm SA. All other series of LSTc decoy liposomes did not significantly inhibit IAV at this concentration. In contrast, the monovalent form of LSTc did not inhibit either PR/8 or Philippines when tested at equimolar (data not shown) or at far higher concentrations of LSTc incorporated into decoy liposomes (Fig. 2). The inability of monovalent LSTc to inhibit IAV agrees with published data that show other monovalent SA analogs to be incapable of inhibiting IAV infectivity (45–47). LSTc incorporated into our decoy liposomes can make polymer-like, multivalent interactions with influenza virus due to the increased efficacy of the liposomes compared with monovalent LSTc.
Our decoy liposomes are specific for influenza. We tested decoy liposomes containing 7.5 mol% LSTc against multiple strains of influenza and show that they are capable of binding and inhibiting not just the PR/8 and Beijing H1N1 strains, but also the Philippines and X-31 H3N2 strains (Table 2 and Fig. 2B). They do not, however, inhibit either SeV hemagglutination (Table 2) or RSV infectivity (Fig. 2B). Inhibition of influenza is thus specific for the binding of HA to α2–6-linked SA and is not a nonspecific adhesion mediated by some other interaction.
Although each of the IAV strains tested was inhibited by LSTc decoy liposomes, the degree of inhibition for each IAV strain varied considerably. Each of the four strains tested is a conventional research strain propagated in chicken eggs. PR/8, which exhibited the greatest degree of inhibition (see Table 2), is a commonly used H1N1 strain that is adapted to growth in mice. X31 and Philippines are each mouse-adapted reassortant strains. Beijing 262/95 is a relatively recent human clinical isolate that resembles modern H1N1 variants that emerged after 1994 (48) and has undergone relatively little selection in the laboratory. One might expect that the Beijing human isolate would be most greatly inhibited by LSTc decoy liposomes given its adaptation to the human airway, which has a high prevalence of α2–6-linked SA (49). However, the Beijing strain, like the other viruses used in this study, was expanded in chicken eggs, which may have allowed for the selection of virions that favor binding to α2–3-linked SA. Human RBC and MDCK cells, which were used in binding and infectivity assays, respectively, display a combination of α2–6 and α2–3-linked SAs. The affinity between virus and host either in the presence or absence of decoy liposomes is influenced by virion preferential binding to host α2–3-linked SA. Of note, the antigenic subtype did not appear to predispose viruses to greater or lesser susceptibility to LSTc decoy liposomes.
To further test our decoy liposome ability to inhibit viral spread, we used a serum-free system to allow continued replication in the presence or absence of LSTc-containing liposomes. Decoy liposomes, but not control liposomes, inhibited viral replication (Fig. 3). The ability of LSTc decoy liposomes to inhibit viral replication in this system shows that they can block not only the initial infection event but will remain active for days at body temperature and block progeny virus from re-infecting new target cells. Liposomal lifetime is also extended by the addition of a net negative charge, reducing the likelihood of phagocytosis (20). The LSTc decoy liposomes reduced the infectious titers in this experiment by more than two logs, demonstrating significant efficacy against influenza virus.
The ability of our LSTc decoy liposomes to bind IAV and protect cells from infection also led to extended survival of mice challenged with a lethal dose of IAV (Fig. 4). Mice had significantly longer survival times when infected with a LD90 of IAV preincubated with LSTc decoy liposomes than mice infected with the LD90 preincubated with control liposomes. These data demonstrate that LSTc decoy liposomes are capable of maintaining long-lasting interactions with IAV while in the hostile environment of the respiratory tract. An overall negative charge and the presence of glycan enable our decoy liposomes to be retained for longer periods of time within the lung (20). Although co-incubating decoy liposomes with virus does not reflect a typical therapeutic delivery system, these mouse experiments demonstrate a proof of concept that LSTc decoy liposomes remain active and inhibitory in vivo. The extended survival of these mice suggests that the IAV binding and neutralization observed in vitro also occurs in vivo.
We confirmed that influenza virus directly binds to LSTc-containing decoy liposomes using fluorescently labeled decoy liposomes and influenza covalently modified with AlexaFluor 647. Only LSTc-containing decoy liposomes, but not control liposomes, bound to fluorescently labeled influenza virus (Fig. 5). WSN HA-647 can also be tracked as it binds to human lung epithelial cells. When WSN HA-647 was mixed with control liposomes, strong binding to A549 cells was observed, whereas less binding was observed when virus was mixed with LSTc-containing decoy liposomes (Fig. 6). MDCK cells are widely used in IAV studies because they are very permissive to IAV infection and propagation and allow for analysis of viral binding, infection and viral growth kinetics. MDCK cells express both α2–6- and α2–3-linked SA, which presents a higher bar for inhibition with our LSTc decoy liposomes, which only prevents IAV from binding α2–6-linked SA. We have further demonstrated that the inhibition by LSTc decoy liposomes seen in MDCK cells is also true in a human respiratory epithelial cell line (A549 cells) as well as in vivo in mice.
In this study we have shown that decoy liposomes containing LSTc have higher avidity for influenza than monovalent LSTc alone. Decoy liposomes bind directly to IAV virions, and bound IAV cannot bind to SA-bearing epithelial cells. Importantly, the decoy liposomes specifically block infection of influenza but not other respiratory viruses that do not bind to α2–6-linked SA. Subsequent rounds of viral infection can be inhibited with decoy liposomes. Our results show that the decoy receptor liposome platform is a novel method of combating not only influenza but also possibly other pathogens with defined host receptors.
Acknowledgments
We thank all members of the 7-Day Immunity ENVELOP team.
This work was supported by Defense Advanced Research Projects Agency, Defense Sciences Office W911NF-10-1-0268.
- IAV
- influenza A virus
- SA
- sialic acid
- LSTc
- sialylneolacto-N-tetraose c
- DOPE
- 1,2-dioleoyl-sn-glycero-3-phosphoethanolamine
- DOPE-NBD
- DOPE-N-(7-nitro-2–1,3-benzoxadiazol-4-yl
- Fmoc
- N-(9-fluorenyl)methoxycarbonyl
- DOPC
- 1,2-dioleoyl-sn-glycero-3-phosphocholine
- DOPG
- 1,2-dioleoyl-sn-glycero-3-[phospho-rac-(1-glycerol)
- MDCK
- Madin-Darby canine kidney
- RSV
- respiratory syncytial virus
- PFU
- plaque forming units
- HAI
- HA inhibition
- SeV
- Sendai virus
- LD90
- 90% lethality
- CCF
- cross-correlation function.
REFERENCES
- 1. Sullivan K. M. (1996) Health impact of influenza in the United States. Pharmacoeconomics 9, 26–33 [DOI] [PubMed] [Google Scholar]
- 2. Potter C. W. (2001) A history of influenza. J. Appl. Microbiol. 91, 572–579 [DOI] [PubMed] [Google Scholar]
- 3. Moscona A. (2005) Neuraminidase inhibitors for influenza. N. Engl. J. Med. 353, 1363–1373 [DOI] [PubMed] [Google Scholar]
- 4. Hayden F. (2009) Developing new antiviral agents for influenza treatment. What does the future hold? Clin. Infect. Dis. 48, S3–S13 [DOI] [PubMed] [Google Scholar]
- 5. Deyde V. M., Xu X., Bright R. A., Shaw M., Smith C. B., Zhang Y., Shu Y., Gubareva L. V., Cox N. J., Klimov A. I. (2007) Surveillance of resistance to adamantanes among influenza A(H3N2) and A(H1N1) viruses isolated worldwide. J. Infect. Dis. 196, 249–257 [DOI] [PubMed] [Google Scholar]
- 6. Dharan N. J., Gubareva L. V., Meyer J. J., Okomo-Adhiambo M., McClinton R. C., Marshall S. A., St George K., Epperson S., Brammer L., Klimov A. I., Bresee J. S., Fry A. M. (2009) Infections with oseltamivir-resistant influenza A(H1N1) virus in the United States. JAMA 301, 1034–1041 [DOI] [PubMed] [Google Scholar]
- 7. Skehel J. J., Wiley D. C. (2000) Receptor binding and membrane fusion in virus entry. The influenza hemagglutinin. Annu. Rev. Biochem. 69, 531–569 [DOI] [PubMed] [Google Scholar]
- 8. Gambaryan A., Yamnikova S., Lvov D., Tuzikov A., Chinarev A., Pazynina G., Webster R., Matrosovich M., Bovin N. (2005) Receptor specificity of influenza viruses from birds and mammals. New data on involvement of the inner fragments of the carbohydrate chain. Virology 334, 276–283 [DOI] [PubMed] [Google Scholar]
- 9. Krause J. C., Tsibane T., Tumpey T. M., Huffman C. J., Albrecht R., Blum D. L., Ramos I., Fernandez-Sesma A., Edwards K. M., García-Sastre A., Basler C. F., Crowe J. E., Jr. (2012) Human monoclonal antibodies to pandemic 1957 H2N2 and pandemic 1968 H3N2 influenza viruses. J. Virol. 86, 6334–6340 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Ohkura T., Kikuchi Y., Kono N., Itamura S., Komase K., Momose F., Morikawa Y. (2012) Epitope mapping of neutralizing monoclonal antibody in avian influenza A H5N1 virus hemagglutinin. Biochem. Biophys. Res. Commun. 418, 38–43 [DOI] [PubMed] [Google Scholar]
- 11. Rudneva I., Ignatieva A., Timofeeva T., Shilov A., Kushch A., Masalova O., Klimova R., Bovin N., Mochalova L., Kaverin N. (2012) Escape mutants of pandemic influenza A/H1N1 2009 virus. Variations in antigenic specificity and receptor affinity of the hemagglutinin. Virus Res. 166, 61–67 [DOI] [PubMed] [Google Scholar]
- 12. Sauter N. K., Bednarski M. D., Wurzburg B. A., Hanson J. E., Whitesides G. M., Skehel J. J., Wiley D. C. (1989) Hemagglutinins from two influenza virus variants bind to sialic acid derivatives with millimolar dissociation constants. A 500-MHz proton nuclear magnetic resonance study. Biochemistry 28, 8388–8396 [DOI] [PubMed] [Google Scholar]
- 13. Glick G. D., Toogood P. L., Wiley D. C., Skehel J. J., Knowles J. R. (1991) Ligand recognition by influenza virus. The binding of bivalent sialosides. J. Biol. Chem. 266, 23660–23669 [PubMed] [Google Scholar]
- 14. Weinhold E. G., Knowles J. R. (1992) Design and evaluation of a tightly binding fluorescent ligand for influenza A hemagglutinin. J. Am. Chem. Soc. 114, 9270–9275 [Google Scholar]
- 15. Mammen M., Dahmann G., Whitesides G. M. (1995) Effective inhibitors of hemagglutination by influenza virus synthesized from polymers having active ester groups. Insight into mechanism of inhibition. J. Med. Chem. 38, 4179–4190 [DOI] [PubMed] [Google Scholar]
- 16. Gambaryan A. S., Tuzikov A. B., Chinarev A. A., Juneja L. R., Bovin N. V., Matrosovich M. N. (2002) Polymeric inhibitor of influenza virus attachment protects mice from experimental influenza infection. Antiviral Res. 55, 201–205 [DOI] [PubMed] [Google Scholar]
- 17. Kingery-Wood J. E., Williams K. W., Sigal G. B., Whitesides G. M. (1992) The agglutination of erythrocytes by influenza virus is strongly inhibited by liposomes incorporating an analog of sialyl gangliosides. J. Am. Chem. Soc. 114, 7303–7305 [Google Scholar]
- 18. Guo C. T., Sun X. L., Kanie O., Shortridge K. F., Suzuki T., Miyamoto D., Hidari K. I., Wong C. H., Suzuki Y. (2002) An O-glycoside of sialic acid derivative that inhibits both hemagglutinin and sialidase activities of influenza viruses. Glycobiology 12, 183–190 [DOI] [PubMed] [Google Scholar]
- 19. Spevak W., Nagy J. O., Charych D. H., Schaefer M. E., Gilbert J. H., Bednarski M. D. (1993) Polymerized liposomes containing C-glycosides of sialic acid. Potent inhibitors of influenza virus in vitro infectivity. J. Am. Chem. Soc. 115, 1146–1147 [Google Scholar]
- 20. Deol P., Khuller G. K. (1997) Lung specific stealth liposomes. Stability, biodistribution, and toxicity of liposomal antitubercular drugs in mice. Biochim. Biophys. Acta 1334, 161–172 [DOI] [PubMed] [Google Scholar]
- 21. van Meer G., Davoust J., Simons K. (1985) Parameters affecting low pH-mediated fusion of liposomes with the plasma membrane of cells infected with influenza virus. Biochemistry 24, 3593–3602 [DOI] [PubMed] [Google Scholar]
- 22. Chandrasekaran A., Srinivasan A., Raman R., Viswanathan K., Raguram S., Tumpey T. M., Sasisekharan V., Sasisekharan R. (2008) Glycan topology determines human adaptation of avian H5N1 virus hemagglutinin. Nat. Biotechnol. 26, 107–113 [DOI] [PubMed] [Google Scholar]
- 23. Srinivasan A., Viswanathan K., Raman R., Chandrasekaran A., Raguram S., Tumpey T. M., Sasisekharan V., Sasisekharan R. (2008) Quantitative biochemical rationale for differences in transmissibility of 1918 pandemic influenza A viruses. Proc. Natl. Acad. Sci. U. S. A. 105, 2800–2805 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Eisen M. B., Sabesan S., Skehel J. J., Wiley D. C. (1997) Binding of the influenza A virus to cell-surface receptors. Structures of five hemagglutinin-sialyloligosaccharide complexes determined by x-ray crystallography. Virology 232, 19–31 [DOI] [PubMed] [Google Scholar]
- 25. Ha Y., Stevens D. J., Skehel J. J., Wiley D. C. (2001) X-ray structures of H5 avian and H9 swine influenza virus hemagglutinins bound to avian and human receptor analogs. Proc. Natl. Acad. Sci. U. S. A. 98, 11181–11186 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Russell R. J., Stevens D. J., Haire L. F., Gamblin S. J., Skehel J. J. (2006) Avian and human receptor binding by hemagglutinins of influenza A viruses. Glycoconj. J. 23, 85–92 [DOI] [PubMed] [Google Scholar]
- 27. Lees W. J., Spaltenstein A., Kingery-Wood J. E., Whitesides G. M. (1994) Polyacrylamides bearing pendant α-sialoside groups strongly inhibit agglutination of erythrocytes by influenza A virus. Multivalency and steric stabilization of particulate biological systems. J. Med. Chem. 37, 3419–3433 [DOI] [PubMed] [Google Scholar]
- 28. Klein A., Diaz S., Ferreira I., Lamblin G., Roussel P., Manzi A. E. (1997) New sialic acids from biological sources identified by a comprehensive and sensitive approach. Liquid chromatography-electrospray ionization-mass spectrometry (LC-ESI-MS) of SIA quinoxalinones. Glycobiology 7, 421–432 [DOI] [PubMed] [Google Scholar]
- 29. Weirich K. L., Israelachvili J. N., Fygenson D. K. (2010) Bilayer edges catalyze supported lipid bilayer formation. Biophys. J. 98, 85–92 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Popp M. W., Karssemeijer R. A., Ploegh H. L. (2012) Chemoenzymatic site-specific labeling of influenza glycoproteins as a tool to observe virus budding in real time. PLoS Pathog. 8, e1002604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Choi S. K., Mammen M., Whitesides G. M. (1996) Monomeric inhibitors of influenza neuraminidase enhance the hemagglutination inhibition activities of polyacrylamides presenting multiple C-sialoside groups. Chem. Biol. 3, 97–104 [DOI] [PubMed] [Google Scholar]
- 32. Schneider C. A., Rasband W. S., Eliceiri K. W. (2012) NIH Image to ImageJ. 25 years of image analysis. Nat. Methods 9, 671–675 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Xu D., Newhouse E. I., Amaro R. E., Pao H. C., Cheng L. S., Markwick P. R., McCammon J. A., Li W. W., Arzberger P. W. (2009) Distinct glycan topology for avian and human sialopentasaccharide receptor analogues upon binding different hemagglutinins. A molecular dynamics perspective. J. Mol. Biol. 387, 465–491 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Markwell M. A., Paulson J. C. (1980) Sendai virus utilizes specific sialyloligosaccharides as host cell receptor determinants. Proc. Natl. Acad. Sci. U. S. A. 77, 5693–5697 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Feldman S. A., Audet S., Beeler J. A. (2000) The fusion glycoprotein of human respiratory syncytial virus facilitates virus attachment and infectivity via an interaction with cellular heparan sulfate. J. Virol. 74, 6442–6447 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. van Steensel B., van Binnendijk E. P., Hornsby C. D., van der Voort H. T., Krozowski Z. S., de Kloet E. R., van Driel R. (1996) Partial colocalization of glucocorticoid and mineralocorticoid receptors in discrete compartments in nuclei of rat hippocampus neurons. J. Cell Sci. 109, 787–792 [DOI] [PubMed] [Google Scholar]
- 37. Asher D. R., Cerny A. M., Finberg R. W. (2005) The erythrocyte viral trap. Transgenic expression of viral receptor on erythrocytes attenuates coxsackievirus B infection. Proc. Natl. Acad. Sci. U. S. A. 102, 12897–12902 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Pritchett T. J., Brossmer R., Rose U., Paulson J. C. (1987) Recognition of mono-valent sialosides by influenza virus H-3 hemagglutinin. Virology 160, 502–506 [DOI] [PubMed] [Google Scholar]
- 39. Sparks M. A., Williams K. W., Lukacs C., Schrell A., Priebe G., Spaltenstein A., Whitesides G. M. (1993) Synthesis of potential inhibitors of hemagglutination by influenza virus. Chemoenzymic preparation of N-5 analogs of N-acetylneuraminic acid. Tetrahedron 49, 1–12 [Google Scholar]
- 40. Toogood P. L., Galliker P. K., Glick G. D., Knowles J. R. (1991) Monovalent sialosides that bind tightly to influenza A virus. J. Med. Chem. 34, 3138–3140 [DOI] [PubMed] [Google Scholar]
- 41. Kiessling L. L., Pohl N. L. (1996) Strength in numbers. Non-natural polyvalent carbohydrate derivatives. Chem. Biol. 3, 71–77 [DOI] [PubMed] [Google Scholar]
- 42. Spaltenstein A., Whitesides G. M. (1991) Polyacrylamides bearing pendant α-sialoside groups strongly inhibit agglutination of erythrocytes by influenza virus. J. Am. Chem. Soc. 113, 686–687 [DOI] [PubMed] [Google Scholar]
- 43. Itoh M., Hetterich P., Isecke R., Brossmer R., Klenk H. D. (1995) Suppression of influenza virus infection by an N-thioacetylneuraminic acid acrylamide copolymer resistant to neuraminidase. Virology 212, 340–347 [DOI] [PubMed] [Google Scholar]
- 44. Reuter J. D., Myc A., Hayes M. M., Gan Z., Roy R., Qin D., Yin R., Piehler L. T., Esfand R., Tomalia D. A., Baker J. R., Jr. (1999) Inhibition of viral adhesion and infection by sialic acid-conjugated dendritic polymers. Bioconjug. Chem. 10, 271–278 [DOI] [PubMed] [Google Scholar]
- 45. Gamian A., Chomik M., Laferrière C. A., Roy R. (1991) Inhibition of influenza A virus hemagglutinin and induction of interferon by synthetic sialylated glycoconjugates. Can J. Microbiol. 37, 233–237 [DOI] [PubMed] [Google Scholar]
- 46. Mochalova L. V., Tuzikov A. B., Marinina V. P., Gambaryan A. S., Byramova N. E., Bovin N. V., Matrosovich M. N. (1994) Synthetic polymeric inhibitors of influenza virus receptor binding activity suppress virus replication. Antiviral. Res. 23, 179–190 [DOI] [PubMed] [Google Scholar]
- 47. Tuzikov A. B., Gambaryan A. S., Juneja L. R., Bovin N. V. (2000) Conversion of complex sialooligosaccharides into polymeric conjugates and their anti-influenza virus inhibitory potency. J. Carbohydr. Chem. 19, 1191–1200 [Google Scholar]
- 48. McDonald N. J., Smith C. B., Cox N. J. (2007) Antigenic drift in the evolution of H1N1 influenza A viruses resulting from deletion of a single amino acid in the haemagglutinin gene. J. Gen. Virol. 88, 3209–3213 [DOI] [PubMed] [Google Scholar]
- 49. Ibricevic A., Pekosz A., Walter M. J., Newby C., Battaile J. T., Brown E. G., Holtzman M. J., Brody S. L. (2006) Influenza virus receptor specificity and cell tropism in mouse and human airway epithelial cells. J. Virol. 80, 7469–7480 [DOI] [PMC free article] [PubMed] [Google Scholar]