

Background: 8-Oxo-7,8-dihydroguanine induces base mispairing, thereby altering genetic information.
Results: 8-Oxoguanine-containing ribonucleotides are eliminated from the RNA precursor pool by the concerted actions of guanylate kinase and MutT protein with distinct substrate specificities.
Conclusion: Formation of RNA carrying 8-oxoguanine is efficiently prevented.
Significance: This study reveals a novel process by which bacterial cells protect themselves against oxidative damage to RNA.
Keywords: Enzyme Mechanisms, Mutagenesis, Nucleoside Nucleotide Biosynthesis, Nucleoside Nucleotide Metabolism, Nucleotide, RNA Synthesis, Ribonucleotide Pool, oxidative stress
Abstract
Reactive oxygen species are produced as side products of oxygen utilization and can lead to the oxidation of nucleic acids and their precursor nucleotides. Among the various oxidized bases, 8-oxo-7,8-dihydroguanine seems to be the most critical during the transfer of genetic information because it can pair with both cytosine and adenine. During the de novo synthesis of guanine nucleotides, GMP is formed first, and it is converted to GDP by guanylate kinase. This enzyme hardly acts on an oxidized form of GMP (8-oxo-GMP) formed by the oxidation of GMP or by the cleavage of 8-oxo-GDP and 8-oxo-GTP by MutT protein. Although the formation of 8-oxo-GDP from 8-oxo-GMP is thus prevented, 8-oxo-GDP itself may be produced by the oxidation of GDP by reactive oxygen species. The 8-oxo-GDP thus formed can be converted to 8-oxo-GTP because nucleoside-diphosphate kinase and adenylate kinase, both of which catalyze the conversion of GDP to GTP, do not discriminate 8-oxo-GDP from normal GDP. The 8-oxo-GTP produced in this way and by the oxidation of GTP can be used for RNA synthesis. This misincorporation is prevented by MutT protein, which has the potential to cleave 8-oxo-GTP as well as 8-oxo-GDP to 8-oxo-GMP. When 14C-labeled 8-oxo-GTP was applied to CaCl2-permeabilized cells of a mutT− mutant strain, it could be incorporated into RNA at 4% of the rate for GTP. Escherichia coli cells appear to possess mechanisms to prevent misincorporation of 8-oxo-7,8-dihydroguanine into RNA.
Introduction
Reactive oxygen species (ROS),2 such as superoxide and hydroxyl radicals, are produced during normal cellular metabolism, and the formation of such radicals is further enhanced by exposure of cells to ionizing radiation and certain chemicals (1–3). Although most of these radicals are eliminated by the actions of cellular antioxidant systems, some of the radicals remain and can attack various cellular constituents, including nucleic acids, proteins, and lipids. Among the many types of oxidized purine and pyrimidine bases thus produced, 8-oxo-7,8-dihydroguanine (8-oxo-Gua) is the most abundant and seems to be important with respect to the maintenance and transfer of genetic information (4–7). The 8-oxo-Gua can pair with adenine as well as cytosine during nucleic acid synthesis and thus can cause errors in DNA replication and gene expression.
Organisms are equipped with elaborate mechanisms for counteracting such deleterious effects of 8-oxo-Gua. In Escherichia coli, three proteins, MutT, MutM, and MutY, function to exclude 8-oxo-Gua from the DNA (8–12). The MutT protein hydrolyzes 8-oxo-Gua-containing nucleoside di- and triphosphates (8-oxo-dGDP and 8-oxo-dGTP) to the monophosphate, thereby preventing the misincorporation of 8-oxo-Gua into DNA. When 8-oxo-Gua is present in DNA, the MutM protein removes the 8-oxo-Gua paired with cytosine, and the MutY protein excises adenine paired with 8-oxo-Gua. By the concerted actions of these enzymes, the spontaneous mutation frequency of E. coli is kept very low. Similar mechanisms appear to function in mammalian cells, although the systems are more complex than those found in bacteria. Human cells contain three types of MutT-related proteins, MTH1 (NUDT1), MTH2 (NUDT15), and MTH3 (NUDT18), each of which exhibits distinct substrate preferences (13). MTH1 cleaves 8-oxo-dGTP but not 8-oxo-dGDP, whereas MTH2 can degrade both 8-oxo-dGTP and 8-oxo-dGDP, although the intrinsic enzyme activity of MTH2 is considerably lower than that of MTH1. On the other hand, MTH3 is specifically active against 8-oxo-dGDP and hardly cleaves 8-oxo-dGTP.
The oxidation of guanine also occurs in the ribonucleotide pool of cells, and the 8-oxo-GTP thus produced can be misincorporated into RNA (14). The 8-oxo-Gua present in messenger RNA would cause errors during codon-anticodon pairing in the translation process, and thus, the persistence of 8-oxo-Gua in RNA may lead to erroneous protein synthesis. In addition to its action on the oxidized DNA precursors, MutT can degrade 8-oxo-Gua-containing RNA precursor nucleotides 8-oxo-GDP and 8-oxo-GTP (15). Because the RNA and the DNA precursor pools are separated by a distinct nucleoside-diphosphate reductase system, which prevents the formation of 8-oxo-dGDP from 8-oxo-GDP (16), MutT appears to function independently for the two pools.
In the de novo pathway of purine nucleotide biosynthesis, GMP is formed from xanthylate by the action of GMP synthase (17). GMP is then phosphorylated to GDP by guanylate kinase (GMK) (18, 19). The phosphorylation of GDP to GTP is carried out by nucleoside-diphosphate kinase (NDK) (20–23) and by adenylate kinase (ADK), which is capable of phosphorylating nucleoside diphosphates in addition to its originally defined activity (24–26). The GTP thus formed is used for RNA synthesis together with other ribonucleoside triphosphates. During these processes, guanine nucleotides may be subjected to the action of ROS, yielding 8-oxo-Gua-containing nucleoside mono-, di-, and triphosphates.
To elucidate the metabolic fates of 8-oxo-Gua-containing nucleotides in the RNA precursor pool, we investigated how the GMK, NDK, ADK, and MutT proteins act on normal and oxidized guanine nucleotides. We then asked whether 8-oxo-GTP, the final product of these enzymatic processes, can be utilized for RNA synthesis. By using permeabilized cells, we found that 8-oxo-Gua-containing nucleotides are incorporated into RNA but that its degree of incorporation is lower than that found for normal guanine nucleotides. We herein present the results of these studies.
EXPERIMENTAL PROCEDURES
Bacterial Strains
E. coli strain CC101 and its mutT− deficient derivative, CC101T, were used in these studies (13, 15). Strain DH5α (27) was used for subcloning E. coli genes and producing their product proteins.
Preparation of 8-Oxo-Gua-containing Nucleotides
Oxidized forms of guanine nucleotides were prepared as described (13, 28) with slight modifications. For preparation of 8-oxo-GTP, the reaction was performed in a mixture containing 100 mm sodium phosphate (pH 6.8), 6 mm GTP, 30 mm ascorbic acid, and 100 mm H2O2 at 37 °C for 5 h in the dark. One hundred microliters of the reaction mixture were applied to an ion exchange column (Mono Q HR 5/5 5 × 50 mm, GE Healthcare), and nucleotides were separated with a linear gradient (5–100%) of 1 m triethylammonium hydrogen carbonate (pH 7.0) at a flow rate of 1 ml/min using HPLC (Model LC-10AD, Shimadzu Co., Kyoto, Japan). Fractions containing 8-oxo-GTP were combined and applied again to the Mono Q column to remove unoxidized nucleotides. The fractions containing 8-oxo-GTP were lyophilized, dissolved in deionized-distilled water, and stored at −20 °C. Radioactive 8-oxo-GTP was prepared as described above with the use of 6 mm GTP and 10 μCi (51.8 mCi/mmol) of [8-14C]guanosine 5′-triphosphate tetraammonium salt (catalogue number MC-194, Moravek, Brea, CA). To prepare 8-oxo-GDP and 8-oxo-GMP, 6 mm GDP and 6 mm GMP, respectively, were oxidized and purified by Mono Q column to homogeneity as described above for the preparation for 8-oxo-GTP.
Purification of GMK
Cloning of the E. coli GMK gene (gmk/spoR) was performed by polymerase chain reaction (PCR) to amplify the DNA fragments from E. coli strain CC101 with a GMK primer set (forward primer, 5′-GGGGATCCATGGCTCAAGGCACGCTT-3′; reverse primer, 5′-GGCTCGAGTCAGTCTGCCAACAATTTG-3′) using PrimeSTAR HS DNA polymerase (Takara Shuzo, Kyoto, Japan). The amplified DNA fragment was subcloned into the pCRblunt vector (Invitrogen), and its nucleotide sequence was determined. pCRblunt-GMK DNA was digested by the BamHI and XhoI restriction enzymes. The BamHI-XhoI fragment with GMK was inserted into the BamHI and XhoI sites of pGEX-KG. A total of 5 ml of overnight culture of DH5α cells harboring pGEX-KG-GMK was diluted with 100 ml of LB broth and grown to an A540 of 0.2 at 37 °C. Then the cells were incubated with 0.5 mm isopropyl β-d-thiogalactoside at 37 °C for 2 h to induce the expression of GST-GMK. Cells were dispersed in lysis solution (10 mm Tris-HCl (pH 7.5), 150 mm NaCl, and 1 mm PMSF) at a ratio of 1:30 (cell volume:lysis solution) and sonicated on ice two times for 2 min each with a microtip. After two rounds of centrifugation at 2300 × g for 10 min at 4 °C, the supernatant (3 ml) was filtered through a 0.45-μm filter (Sartorius, Germany) to remove bacterial debris, mixed with 0.5 ml of a 50% (v/v) slurry of glutathione-Sepharose 4B beads (GE Healthcare), and rotated for 30 min at 4 °C. The beads were washed four times with the lysis buffer and resuspended in 0.5 ml of stock solution (50% (v/v) glycerol, 50 mm Hepes-KOH (pH 7.4), 50 mm KCl, and 1 mm DTT). Purified proteins were resolved by SDS-polyacrylamide gel (5–20%) electrophoresis followed by Coomassie Brilliant Blue staining (see Fig. 1).
FIGURE 1.
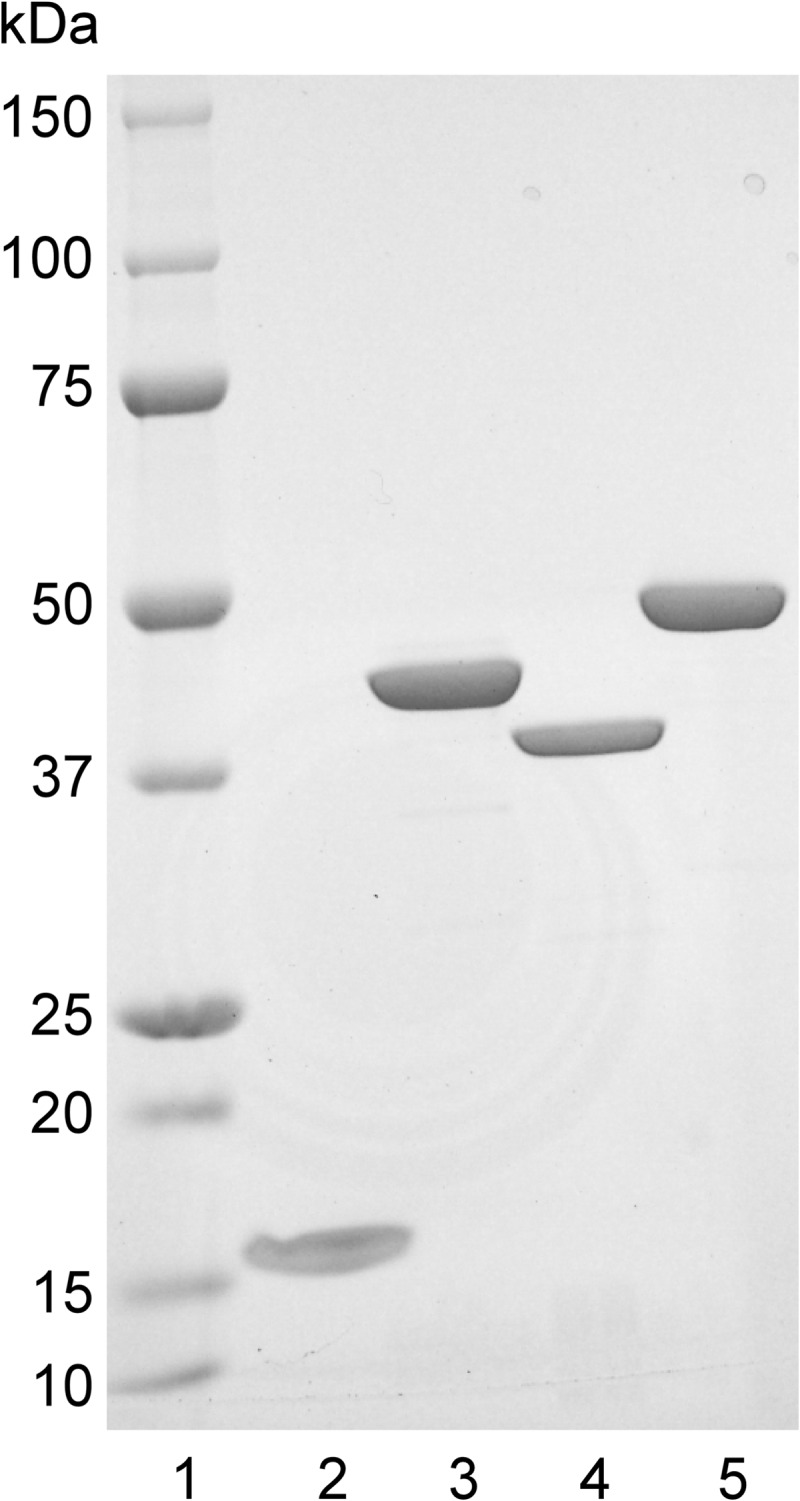
SDS-PAGE of purified preparations of E. coli enzymes. Purified proteins (1 μg each) were subjected to 5–20% SDS-PAGE. Lane 1, molecular mass markers; lane 2, N-terminally His-tagged MutT; lane 3, GST-guanylate kinase fusion protein; lane 4, GST-nucleoside-diphosphate kinase fusion protein; lane 5, GST-adenylate kinase fusion protein.
Purification of NDK
Cloning of the E. coli NDK gene (ndk) was performed by PCR to amplify DNA fragments from E. coli CC101 using a primer set for NDK (forward primer, 5′-GGGGATCCATGGCTATTGAACGTACT-3′; reverse primer, 5′-GGCTCGAGTTAACGGGTGCGCGGGCAC-3′). The amplified DNA fragment was subcloned into the pCRblunt vector, and its nucleotide sequence was determined. The BamHI-XhoI fragment for NDK was subcloned into the BamHI and XhoI sites of pGEX-KG. A total of 5 ml of an overnight culture of DH5α cells harboring pGEX-KG-NDK was mixed with 100 ml of LB broth and grown to an A540 of 0.2 at 37 °C. Then the cells were treated with 0.5 mm isopropyl β-d-thiogalactoside at 37 °C for 2 h to induce the expression of the GST-NDK protein, which was purified as described above.
Purification of ADK
Cloning of the E. coli ADK gene (adk/plsA/dnaW) was performed by PCR to amplify DNA fragments from E. coli strain CC101 using a primer set for ADK (forward primer, 5′-GGGGATCCATGCGTATCATTCTGCTT-3′; reverse primer, 5′-GGCTCGAGTTAGCCGAGGATTTTTTCC-3′) as described above. The amplified DNA fragment was subcloned into the pCRblunt vector, and its nucleotide sequence was determined. The BamHI-XhoI fragment of ADK was subcloned into the BamHI and XhoI sites of pGEX-KG. A total of 5 ml of overnight culture of DH5α cells harboring pGEX-KG-ADK was mixed with 100 ml of LB broth and grown to an A540 of 0.2 at 37 °C. Then the cells were treated with 0.5 mm isopropyl β-d-thiogalactoside at 37 °C for 2 h to induce the expression of the GST-ADK protein, which was purified as described above.
MutT Protein
The MutT protein used in this study was prepared as described previously (15). The reaction with purified MutT protein was performed in a mixture containing 20 mm Tris-HCl (pH 8.0), 0.08 μg/μl BSA, 8 mm MgCl2, 40 mm NaCl, 5 mm DTT, 2% glycerol, and a combination of oxidized and normal guanine-containing nucleotides. The reaction was carried out at 30 °C, and 10-μl aliquots of the mixture were withdrawn at the times indicated and mixed with 40 μl of 0.1% SDS to terminate the reaction. The products were analyzed by HPLC using a Mono Q column. One-milliliter fractions were collected, and the radioactivity of each sample was measured by a liquid scintillation counter (LSC-6000, Aloka, Mitaka, Japan).
Incorporation of Labeled Nucleotides into RNA
[14C]GTP or 8-oxo-[14C]GTP was added to the Ca2+-treated permeabilized cell suspension. The cell suspensions with labeled nucleotides were placed on ice for 30 min, kept at 42 °C for 2 min, and then placed on ice for 2 min. To each cell suspension, LB broth was added, and the mixture was incubated at 37 °C. After incubation, samples were mixed with trichloroacetic acid to give a final concentration of 5% (w/v). After being washed with 1 ml of 5% (w/v) trichloroacetic acid twice, the cells were suspended in 0.6 ml of 1 n NaOH and incubated at 80 °C for 15 min. Next 0.15 ml of 1 n HCl and 0.1 ml of 50% trichloroacetic acid were added, and the mixture was centrifuged. The supernatant obtained was the RNA fraction. The radioactivity was measured using a liquid scintillation counter.
RESULTS
Action of Guanylate Kinase on Oxidized GMP
The 8-oxo-Gua-containing ribonucleoside monophosphate 8-oxo-GMP can be formed by the direct oxidation of GMP by ROS and by the cleavage of 8-oxo-GDP and 8-oxo-GTP by the MutT protein. Because GMP is converted to GDP by GMK, we first examined whether this enzyme can act on 8-oxo-GMP. The E. coli guanylate kinase was produced in cells harboring multicopy plasmids expressing the gmk gene, and a nearly homogeneous enzyme preparation was obtained (see Fig. 1). Using the purified GMK, we compared its actions on GMP and 8-oxo-GMP. The reactions were carried out at pH 7.4 in the presence of ATP and Mg2+, and the reaction products were analyzed by HPLC. When the reaction was performed with a small amount of GMK for 8 min, GMP was converted to GDP upon consumption of an equimolar amount of ATP (Fig. 2A). On the other hand, in the case of 8-oxo-GMP, no appreciable amount of the diphosphate form was produced even when 50 times larger amounts of the enzyme were used and the reaction time was extended to 30 min (Fig. 2B). Fig. 2C shows the time courses of the reactions for the two types of nucleotides, confirming that GMK hardly phosphorylates 8-oxo-GMP.
FIGURE 2.

Actions of guanylate kinase on nucleoside monophosphates containing normal and oxidized guanine bases. A, the action of GMK on GMP. The reaction mixture (20 μl) contained 0.2 mm GMP, 0.2 mm ATP, 50 mm Hepes (pH 7.4), 4 mm MgCl2, 40 mm (NH4)2SO4, and 20 ng of a purified preparation of GMK. The reaction was carried out at 37 °C for 8 min, and then samples were applied to HPLC using a TSKgel DEAE-2 SW column (Tosoh, Tokyo, Japan). B, the action of GMK on 8-oxo-GMP. The reaction was performed as described above except that 8-oxo-GMP was treated with 1 μg of the GMK protein for 30 min. C, the time courses of reactions for GMP (●) and 8-oxo-GMP (○) with GMK. D, the time courses of reactions for dGMP (●) and 8-oxo-dGMP (○) with GMK. For C and D, the reactions were carried out with 20 ng of GMK preparation and 0.2 mm substrate nucleotides in 20 μl of a reaction mixture containing 50 mm Hepes (pH 7.4), 0.2 mm ATP, 4 mm MgCl2, and 40 mm (NH4)2SO4.
Deoxyribonucleotides, which are used for the synthesis of DNA, are produced by the reduction of ribonucleoside diphosphates (29). The enzyme responsible, ribonucleoside-diphosphate reductase, catalyzes the reduction of four types of ribonucleotides, ADP, GDP, UDP, and CDP, but has minimal effects on 8-oxo-GDP (16). This implies that 8-oxo-Gua-containing deoxyribonucleotides, if present at all, are formed by the oxidation of dGDP and dGTP in the DNA precursor pool. Because the 8-oxo-dGDP and 8-oxo-dGTP thus formed may be subjected to the action of MutT, 8-oxo-dGMP would be produced as the cleavage product. Fig. 2D indicates that GMK phosphorylates dGMP, but not 8-oxo-dGMP, as is the case with ribonucleotides. Thus, for both RNA and DNA precursor pools, GMK acts as a gatekeeper to prevent oxidized guanine nucleotides from being used for nucleic acid synthesis.
Formation of 8-Oxo-GTP by NDK
E. coli NDK catalyzes the ATP-dependent synthesis of nucleoside triphosphates from diphosphates. The enzyme is able to act on all types of RNA- and DNA-related nucleotides (22). To determine whether this enzyme also acts on 8-oxo-Gua-containing nucleotides, a homogeneous preparation of NDK was obtained with the aid of the cloned gene (see Fig. 1). When GDP was incubated with NDK in the presence of ATP, GTP was generated with concomitant conversion of ATP to ADP (Fig. 3A). The 8-oxo-GDP was converted to 8-oxo-GTP in a similar manner (Fig. 3B). The time courses of the reactions for GDP and 8-oxo-GDP revealed that NDK acts on the two types of nucleotides with almost the same efficiency (Fig. 3C). The experiment was extended to deoxyribonucleotides, and almost the same result was obtained with ribo- and deoxyribonucleotides (Fig. 3D). Because NDK acts on both 8-oxo-GDP and 8-oxo-dGDP, oxidized forms of guanine nucleoside triphosphates, which may be used for nucleic acid synthesis, can be produced.
FIGURE 3.

Actions of nucleoside diphosphate kinase on nucleoside diphosphates containing normal and oxidized guanine bases. A, the actions of NDK on GDP. The reaction mixture (10 μl) contained 0.5 mm GDP, 0.5 mm ATP, 50 mm Hepes (pH 7.4), 4 mm MgCl2, 40 mm (NH4)2SO4, and 100 ng of a purified preparation of NDK. The reaction was carried out at 37 °C for 4 min, and the reaction products were analyzed by HPLC. B, the actions of NDK on 8-oxo-GDP. The reaction was performed as described above. C, the time courses of reactions for GDP (●) and 8-oxo-GDP (○) with NDK. D, the time course of reactions for dGDP (●) and 8-oxo-dGDP (○) with NDK. For C and D, the reactions were carried out with 100 ng of NDK preparation and 0.5 mm substrate nucleotides in 10 μl of a reaction mixture containing 50 mm Hepes (pH 7.4), 0.5 mm ATP, 4 mm MgCl2, and 40 mm (NH4)2SO4.
Formation of 8-Oxo-GTP by Adenylate Kinase
Because disruption of ndk, the gene encoding NDK, does not affect the viability of cells (21), it was supposed that there is (an)other enzyme(s) that also catalyzes this reaction. ADK, which catalyzes the conversion of AMP to ADP (24), has been implicated to have such an activity (25). To examine whether ADK is capable of acting on 8-oxo-Gua-containing nucleotides, we cloned adk, the gene encoding ADK, from E. coli and overexpressed the enzyme. Using a homogeneous preparation of ADK (see Fig. 1), we examined whether it acts on 8-oxo-GDP. As shown in Fig. 4, 8-oxo-GTP was produced from 8-oxo-GDP by the action of ADK, although the rate of formation of 8-oxo-GTP was approximately one-fifth of that for GTP. Therefore, ADK can substitute for NDK to generate 8-oxo-GTP.
FIGURE 4.

Time courses of reactions for GDP and 8-oxo-GDP with adenylate kinase. The reaction was carried out with 800 ng of ADK preparation and 0.2 mm GDP (●) or 8-oxo-GDP (○) in a reaction mixture (10 μl) containing 50 mm Hepes (pH 7.4), 0.2 mm ADP, 4 mm MgCl2, and 40 mm (NH4)2SO4.
Specific Cleavage of 8-Oxo-GTP by the MutT Protein
The Km values of MutT for the hydrolysis of 8-oxo-GTP and 8-oxo-GDP are about 4000 times lower than those for GTP and GDP (15), providing an enzymatic basis for the high fidelity of RNA synthesis under oxidative stress. Because the measurements of these parameters were made under conditions where only MutT and the substrate were present, we performed the MutT reaction under more physiologically relevant conditions. A small amount of 8-oxo-[14C]GTP was incubated with MutT in the presence of large amounts of ATP, GTP, CTP, and UTP, the levels of which were adjusted to those found in E. coli cells (17). Under these conditions, radioactive 8-oxo-GTP was efficiently converted to the monophosphate form, whereas no appreciable change in the distribution of other nucleoside triphosphates was observed (Fig. 5). Thus, even in the presence of large amounts of GTP and other ribonucleoside triphosphates, 8-oxo-GTP can be selectively and efficiently converted to the monophosphate by MutT.
FIGURE 5.

Selective degradation of 8-oxoGTP by MutT in the presence of a large amount of ATP, GTP, CTP, and UTP. The reaction mixture (25 μl) contained 1 μm 8-oxo-[14C]GTP, 0.9 mm GTP, 3 mm ATP, 0.52 mm CTP, 0.89 mm UTP, 1 ng of MutT, 20 mm Tris-HCl (pH 8), 8 mm MgCl2, 40 mm NaCl, 5 mm DTT, 80 μg/ml BSA, and 2% (v/v) glycerol. The reaction was performed at 30 °C and terminated at 0 or 30 min by adding SDS (final concentration, 0.1%). The samples were resolved by HPLC with a Mono Q column. Nucleotides were separated with a linear gradient (5–100%) of 1 m triethylammonium hydrogen carbonate (pH 7.0) at a flow rate of 1 ml/min using HPLC.
Utilization of 8-Oxo-GTP for RNA Synthesis
By treating E. coli cells with 0.1 m CaCl2 at 4 °C, the cells become permeable to charged molecules, such as nucleotides (27). MutT-deficient cells were treated in this manner, and 14C-labeled GTP or 8-oxo-GTP was applied. After a 2-min pulse treatment at 42 °C, the cells were incubated in nutrient broth. At the times indicated, aliquots of the culture were withdrawn, and the radioactivity of the labeled nucleotides incorporated into RNA was determined. As shown in Table 1, [14C]GTP was actively incorporated into RNA, and the value at 30 min was slightly higher than that obtained at 15 min. In the case of 8-oxo-[14C]GTP, a small amount of radioactivity was incorporated into RNA at 15 min, and this value did not change at the later time point of the incubation. The ratio of the incorporation level of 8-oxo-GTP compared with that of GTP at 15 min was ∼4%. It is close to the value obtained in in vitro system in which E. coli RNA polymerase can incorporate 8-oxo-GTP into RNA at a rate 5–10% of that of GTP (14). It seems that 8-oxo-Gua-containing RNA may actually be formed, but its amount is lesser compared with normal RNA.
TABLE 1.
Incorporation of 14C-labeled GTP and 8-oxo-GTP into RNA in CaCl2-treated E. coli mutT− cells
E. coli CC101T cells (20-ml culture) were harvested during the exponential phase of growth at an A540 of 0.3, washed once with 10 ml of cold 0.1 m CaCl2, and then suspended in 4.6 ml of 0.1 m CaCl2. An aliquot of the suspension (0.4 ml) was mixed with 25 μl of [14C]GTP or 8-oxo-[14C]GTP ( 36 nmol). The suspension was kept on ice for 30 min and then at 42 °C for 2 min. Twenty-five volumes of LB broth were added, and the mixture was kept at 37 °C. Aliquots of the mixture were withdrawn at the times indicated, and the radioactivity of the nucleotides incorporated into RNA was determined. Three samples were analyzed at each time point and the standard deviations were calculated (±).
| Incubation time | Radioactivity |
8-Oxo-GTP/GTP | |
|---|---|---|---|
| GTP | 8-Oxo-GTP | ||
| dpm | % | ||
| 15 min | 1081 ± 33 | 45.5 ± 8.5 | 4.2 |
| 30 min | 1249 ± 145 | 43.7 ± 3.7 | 3.4 |
DISCUSSION
In the biosynthetic pathway of guanine-containing ribonucleotides, GMP is formed first and is phosphorylated progressively to GDP and then to GTP. These guanine-containing ribonucleotides may be subjected to the actions of ROS, yielding oxidized forms of all three types of guanine nucleotides, namely 8-oxo-GMP, 8-oxo-GDP, and 8-oxo-GTP. Once 8-oxo-GTP is formed, it can be used for RNA synthesis because RNA polymerase itself has the ability to use 8-oxo-GTP as a substrate. It has been shown that, in an in vitro system, E. coli RNA polymerase incorporates 8-oxo-Gua-containing nucleotides at rates of 5 and 10% of those of normal guanine nucleotides when DNA and poly(dA-dT) are used as templates, respectively (14). In the present study, we have shown that, in an in vivo system in which permeabilized E. coli cells were used, 8-oxo-GTP can be incorporated into RNA at a rate of 4% of that for GTP. Because the misincorporation of 8-oxo-Gua into RNA would cause errors in translation, it is important for cells to eliminate 8-oxo-Gua-containing nucleotides from the RNA precursor pool.
The first step of guanine nucleotide biosynthesis is the ATP-driven amination of xanthylate, which is catalyzed by a specific enzyme, GMP synthase (30). The GMP thus formed is phosphorylated to GDP by the action of GMK (19). It was unclear whether GMK can phosphorylate 8-oxo-GMP, which is produced by the action of ROS. In the present study, we have shown that GMK hardly phosphorylates 8-oxo-GMP. Even when exceedingly high concentrations of GMK were applied, no appreciable amount of 8-oxo-GDP was produced. This finding is important because the reutilization of 8-oxo-GMP, formed by the action of the MutT protein, is thus prevented. As shown in this and previous studies (9, 15), the MutT protein efficiently converts 8-oxo-GDP and 8-oxo-GTP to 8-oxo-GMP. In this regard, it is important to note that GMK functions to prevent the utilization of the 8-oxo-GMP formed by oxidation in situ and that derived from the MutT reaction for RNA synthesis.
Although the phosphorylation of 8-oxo-GMP is prevented in this way, 8-oxo-GDP may be formed by the direct oxidation of GDP. The question then arises whether any cellular enzyme(s) can phosphorylate 8-oxo-GDP to the triphosphate form. The most plausible candidate to carry out this reaction was NDK, which has the ability to phosphorylate a broad range of nucleoside diphosphates to triphosphates (23). By using a purified enzyme preparation obtained from E. coli, we showed that NDK can phosphorylate 8-oxo-GDP as efficiently as normal GDP.
In addition to NDK, ADK is also involved in this process. Lu and Inouye (25) reported that ADK promotes the formation of the nucleoside triphosphate from the diphosphate by transferring the γ-phosphoryl of ATP to nucleoside diphosphate. Subsequently, Willemoës and Kilstrup (26) revealed that the nucleoside triphosphate formation occurs by β-phosphoryl transfer from ADP. Because the former study was performed with the E. coli ADK enzyme, whereas the latter was carried out with a commercial ADK preparation from chicken muscle, we examined this problem using a homogeneous ADK preparation obtained from adk-overproducing E. coli cells. Our results clearly indicated that the GTP formation occurs in an ADP-dependent manner, confirming the findings of Willemoës and Kilstrup (26). We then investigated whether ADK can phosphorylate 8-oxo-GDP and found that the E. coli ADK enzyme can convert 8-oxo-GDP to 8-oxo-GTP, although the rate of reaction was about one-fifth of that for GDP. Thus, ADK can function as a substitute of NDK during the formation of 8-oxo-GTP as well as that of other ribonucleoside triphosphates.
It is evident, therefore, that 8-oxo-GTP can be formed by the phosphorylation of 8-oxo-GDP as well as by the oxidation of GTP itself. Regardless of its origin, 8-oxo-GTP is subjected to the action of the MutT protein, which converts it to 8-oxo-GMP, a form that is unusable for RNA synthesis. In the present study, we examined whether the MutT reaction occurs efficiently in the presence of large amounts of other nucleoside triphosphates, which are usually present in the cellular nucleotide pool. Even under these conditions, the selective breakdown of 8-oxo-GTP was achieved by the MutT protein. Thus, MutT plays a principal role in achieving precise RNA synthesis under aerobic conditions. Fig. 6 provides a scheme deduced from the results obtained from these experiments.
FIGURE 6.

Metabolism of guanine ribonucleotides leading to the synthesis of RNA under normal and oxidative conditions. RNP denotes RNA polymerase. O• indicates reactive oxygen species, and the closed dots to the upper left of chemicals represent 8-oxoguanine-containing molecules.
The conversion of ribonucleotides to deoxyribonucleotides occurs at the level of nucleoside diphosphate, and the enzyme responsible, ribonucleoside-diphosphate reductase, acts on four types of naturally occurring ribonucleoside diphosphates, ADP, GDP, UDP, and CDP (29). It was shown, however, that the enzyme is inactive for 8-oxo-GDP (16). Thus, it is likely that in the DNA precursor pool 8-oxo-Gua is formed by the oxidation of guanine-containing deoxyribonucleotides themselves. Because the present study revealed that NDK is capable of phosphorylating 8-oxo-dGDP to the triphosphate, the 8-oxo-dGTP thus formed can be used as a building material for DNA. It has been shown that DNA polymerase ΙΙΙ of E. coli can use 8-oxo-dGTP as efficiently as dGTP (9). To prevent the misincorporation of 8-oxo-Gua into DNA, which would cause accumulation of mutations in cells, MutT degrades 8-oxo-dGTP and 8-oxo-dGDP to form 8-oxo-dGMP, a form that is unusable for DNA synthesis (9). In the present study, we have shown that guanylate kinase, which can phosphorylate dGMP, hardly phosphorylates 8-oxo-dGMP. Thus, the scheme established for the RNA precursors can also be applied to the related process for DNA, although the metabolic significance of nucleoside monophosphates is different in the two cases; GMP is an essential precursor from which GDP and GTP are formed, whereas dGMP is merely a cleavage product.
Acknowledgments
We thank Hachiro Inokuchi for critical reading of the manuscript. We appreciate the technical support from the Research Support Center, Graduate School of Medical Sciences, Kyushu University.
This work was supported by grants from the Ministry of Education, Culture, Sports, Science and Technology (MEXT) of Japan, including the MEXT-supported Program for the Strategic Research Foundation at Private Universities and grants-in-aid for scientific research.
- ROS
- reactive oxygen species
- 8-oxo-Gua
- 8-oxo-7,8-dihydroguanine
- GMK
- guanylate kinase
- NDK
- nucleoside-diphosphate kinase
- ADK
- adenylate kinase.
REFERENCES
- 1. Bjelland S., Seeberg E. (2003) Mutagenicity, toxicity and repair of DNA base damage induced by oxidation. Mutat. Res. 531, 37–80 [DOI] [PubMed] [Google Scholar]
- 2. Imlay J. A. (2003) Pathways of oxidative damage. Annu. Rev. Microbiol. 57, 395–418 [DOI] [PubMed] [Google Scholar]
- 3. Nakatsu Y., Sekiguchi M. (2006) Oxidative Damage to Nucleotides: Consequences and Preventive Mechanisms. Oxidative Stress, Disease and Cancer, pp. 221–252, Imperial College Press, London [Google Scholar]
- 4. Grollman A. P., Moriya M. (1993) Mutagenesis by 8-oxoguanine: an enemy within. Trends Genet. 9, 246–249 [DOI] [PubMed] [Google Scholar]
- 5. Kuchino Y., Mori F., Kasai H., Inoue H., Iwai S., Miura K., Ohtsuka E., Nishimura S. (1987) Misreading of DNA templates containing 8-hydroxydeoxyguanosine at the modified base and at adjacent residues. Nature 327, 77–79 [DOI] [PubMed] [Google Scholar]
- 6. Sekiguchi M. (1996) MutT-related error avoidance mechanism for DNA synthesis. Genes Cells 1, 139–145 [DOI] [PubMed] [Google Scholar]
- 7. Sekiguchi M., Tsuzuki T. (2002) Oxidative nucleotide damage: consequences and prevention. Oncogene 21, 8895–8904 [DOI] [PubMed] [Google Scholar]
- 8. Fowler R. G., White S. J., Koyama C., Moore S. C., Dunn R. L., Schaaper R. M. (2003) Interactions among the Escherichia coli mutT, mutM, and mutY damage prevention pathways. DNA Repair 2, 159–173 [DOI] [PubMed] [Google Scholar]
- 9. Maki H., Sekiguchi M. (1992) MutT protein specifically hydrolyses a potent mutagenic substrate for DNA synthesis. Nature 355, 273–275 [DOI] [PubMed] [Google Scholar]
- 10. Michaels M. L., Cruz C., Grollman A. P., Miller J. H. (1992) Evidence that MutY and MutM combine to prevent mutations by an oxidatively damaged form of guanine in DNA. Proc. Natl. Acad. Sci. U.S.A. 89, 7022–7025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Tajiri T., Maki H., Sekiguchi M. (1995) Functional cooperation of MutT, MutM and MutY proteins in preventing mutations caused by spontaneous oxidation of guanine nucleotide in Escherichia coli. Mutat. Res. 336, 257–267 [DOI] [PubMed] [Google Scholar]
- 12. Setoyama D., Ito R., Takagi Y., Sekiguchi M. (2011) Molecular actions of Escherichia coli MutT for control of spontaneous mutagenesis. Mutat. Res. 707, 9–14 [DOI] [PubMed] [Google Scholar]
- 13. Takagi Y., Setoyama D., Ito R., Kamiya H., Yamagata Y., Sekiguchi M. (2012) Human MTH3 (NUDT18) protein hydrolyzes oxidized forms of guanosine and deoxyguanosine diphosphates: comparison with MTH1 and MTH2. J. Biol. Chem. 287, 21541–21549 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Taddei F., Hayakawa H., Bouton M., Cirinesi A., Matic I., Sekiguchi M., Radman M. (1997) Counteraction by MutT protein of transcriptional errors caused by oxidative damage. Science 278, 128–130 [DOI] [PubMed] [Google Scholar]
- 15. Ito R., Hayakawa H., Sekiguchi M., Ishibashi T. (2005) Multiple enzyme activities of Escherichia coli MutT protein for sanitization of DNA and RNA precursor pools. Biochemistry 44, 6670–6674 [DOI] [PubMed] [Google Scholar]
- 16. Hayakawa H., Hofer A., Thelander L., Kitajima S., Cai Y., Oshiro S., Yakushiji H., Nakabeppu Y., Kuwano M., Sekiguchi M. (1999) Metabolic fate of oxidized guanine ribonucleotides in mammalian cells. Biochemistry 38, 3610–3614 [DOI] [PubMed] [Google Scholar]
- 17. Kornberg A., Baker T. A. (1992) DNA Replication, pp. 55–61, W. H. Freeman & Co, New York [Google Scholar]
- 18. Gentry D., Bengra C., Ikehara K., Cashel M. (1993) Guanylate kinase of Escherichia coli K-12. J. Biol. Chem. 268, 14316–14321 [PubMed] [Google Scholar]
- 19. Oeschger M. P., Bessman M. J. (1966) Purification and properties of guanylate kinase from Escherichia coli. J. Biol. Chem. 241, 5452–5460 [PubMed] [Google Scholar]
- 20. Hama H., Almaula N., Lerner C. G., Inouye S., Inouye M. (1991) Nucleoside diphosphate kinase from Escherichia coli; its overproduction and sequence comparison with eukaryotic enzymes. Gene 105, 31–36 [DOI] [PubMed] [Google Scholar]
- 21. Lu Q., Zhang X., Almaula N., Mathews C. K., Inouye M. (1995) The gene for nucleoside diphosphate kinase functions as a mutator gene in Escherichia coli. J. Mol. Biol. 254, 337–341 [DOI] [PubMed] [Google Scholar]
- 22. Almaula N., Lu Q., Delgado J., Belkin S., Inouye M. (1995) Nucleoside diphosphate kinase from Escherichia coli. J. Bacteriol. 177, 2524–2529 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Parks R. E., Jr., Agarwal R. (1973) in The Enzymes (Boyer P. D., ed) pp. 307–333, Academic Press, New York [Google Scholar]
- 24. Noda L. (1973) in The Enzymes (Boyer P. D., ed) pp. 279–305, Academic Press, New York [Google Scholar]
- 25. Lu Q., Inouye M. (1996) Adenylate kinase complements nucleoside diphosphate kinase deficiency in nucleotide metabolism. Proc. Natl. Acad. Sci. U.S.A. 93, 5720–5725 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Willemoës M., Kilstrup M. (2005) Nucleoside triphosphate synthesis catalysed by adenylate kinase is ADP dependent. Arch. Biochem. Biophys. 444, 195–199 [DOI] [PubMed] [Google Scholar]
- 27. Sambrook J., Fritsch E. F., Maniatis T. (1989) in Molecular Cloning: A Laboratory Manual, 2nd Ed., pp. 116–118, Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY [Google Scholar]
- 28. Fujikawa K., Kamiya H., Yakushiji H., Nakabeppu Y., Kasai H. (2001) Human MTH1 protein hydrolyzes the oxidized ribonucleotide, 2-hydroxy-ATP. Nucleic Acids Res. 29, 449–454 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Elledge S. J., Zhou Z., Allen J. B. (1992) Ribonucleotide reductase: regulation, regulation, regulation. Trends Biochem. Sci. 17, 119–123 [DOI] [PubMed] [Google Scholar]
- 30. Lagerkvist U. (1958) Biosynthesis of guanosine 5′-phosphate. II. Amination of xanthosine 5′-phosphate by purified enzyme from pigeon liver. J. Biol. Chem. 233, 143–149 [PubMed] [Google Scholar]