

Background: Mechanisms of sodium-coupled substrate binding/release are largely unknown in the family of glutamate transporters.
Results: Opening of helical hairpin HP2 leads to spontaneous substrate release from an aspartate transporter in the inward-facing state.
Conclusion: Substrate dissociation is preceded by the release of a sodium ion.
Significance: Exploring the dynamics of substrate gating is crucial for understanding the mechanisms of the full transport cycle.
Keywords: Aspartate, Membrane Transport, Neurotransmitter Transport, Sodium Transport, Transporters, Inward-facing, Sodium-coupled, Substrate Release
Abstract
Sodium-coupled neurotransmitter transporters play a key role in neuronal signaling by clearing excess transmitter from the synapse. Structural data on a trimeric archaeal aspartate transporter, GltPh, have provided valuable insights into structural features of human excitatory amino acid transporters. However, the time-resolved mechanisms of substrate binding and release, as well as that of coupling to sodium co-transport, remain largely unknown for this important family. We present here the results of the most extensive simulations performed to date for GltPh in both outward-facing and inward-facing states by taking advantage of significant advances made in recent years in molecular simulation technology. The generated multiple microsecond trajectories consistently show that the helical hairpin HP2, not HP1, serves as an intracellular gate (in addition to its extracellular gating role). In contrast to previous proposals, HP1 can neither initiate nor accommodate neurotransmitter release without prior opening of HP2 by at least 4.0 Å. Aspartate release invariably follows that of a sodium ion located near the HP2 gate entrance. Asp-394 on TM8 and Arg-276 on HP1 emerge as key residues that promote the reorientation and diffusion of substrate toward the cell interior. These findings underscore the significance of examining structural dynamics, as opposed to static structure(s), to make inferences on the mechanisms of transport and key interactions.
Introduction
Neurotransmitter transporters harness the electrochemical potential gradient of ions, namely sodium, across the cell membrane to import neurotransmitters against their electrochemical gradient into neuronal or glial cells. Effective translocation from extracellular (EC)2 space regulates neuronal signaling by keeping the levels of neurotransmitters sufficiently low at the synapse. Glutamate is the main excitatory neurotransmitter in the central nervous system. Its high concentrations at the synaptic cleft have been linked to neurological diseases such as epilepsy, stroke, ischemia, and Huntington disease (1–4). Therefore, glutamate transporters play a key role in preventing excitotoxic effects.
Glutamate transporters belong to the solute carrier 1 (SLC1) family composed of eukaryotic and prokaryotic members that transport acidic or neutral amino acids. The only available high resolution structure for a member of the SLC1 is currently that of the archaeal aspartate transporter from Pyrococcus horikoshii, GltPh. GltPh is a homotrimer. Each monomer is composed of eight transmembrane (TM) helices, TM1–8, and two helical hairpins, HP1 and HP2 (see Fig. 1, A–C), organized in two structural regions: a transport “core” that binds and transports the substrate and sodium ions and a “scaffold” that provides support for the transport core and forms the intersubunit interface. The scaffold is composed of TM1–6, with the trimerization domain formed by TM2, -4, and -5, and the transport core contains the binding pocket that is composed of TM7, TM8, HP1, and HP2. The hairpins reach from opposite sides of the membrane with their tips coming into very close proximity as has been experimentally shown (see Fig. 1D) (5, 6).
FIGURE 1.

Structure of the aspartate transporter GltPh. A displays the trimeric structure in the outward-facing state. B displays one of the subunits with the transport core shown in colored ribbon representation. The transport core is magnified in C and D to highlight the substrate (Asp)-binding pocket composed of broken helices TM7 and -8 and helical hairpins HP1 and -2 (helices/coils) and two bound sodium ions, designated as Na2 and Na1 (purple and cyan, respectively). Labeled residues that make close (less than 2.5 Å) interatomic contacts with Asp, Na2, and Na1 are colored according to the respective helix or hairpin loop they are located on. E and F compare the location of the binding pocket in the outward- (E) and inward- (F) facing structures. TM2 and -5 are omitted for clarity.
GltPh has been resolved in several functional states, including outward-facing (OF) (5, 6) and inward-facing (IF) (7) states, and an intermediate state (8). The most prominent difference between the OF and IF states is the almost rigid body translation of the transport core, together with TM3 and -6, by ∼15 Å into the cytoplasm, accompanied by rigid body rotation of ∼30° in each subunit (see Fig. 1, E and F). This difference is clearly observed upon structural alignment of the trimerization domains in the OF and IF subunits (7). We recently proposed that EAAT1 and/or GltPh would undergo a sequential transition between these two endpoints during the transport cycle, visiting two intermediates composed of 1 (or 2) OF and 2 (or 1) IF subunits (9). Notably, the recently resolved intermediate structure confirmed this prediction. The latter, composed of two IF subunits and one intermediate OF, with the transport domain shifted by ∼3.5 Å and ∼15° toward the IF position (8), overall shows a root mean square deviation of 1.3 Å only from the predicted (9) 2IF-1OF intermediate.
In all structures resolved in the presence of substrate, the bound aspartate is coordinated by residues on TM7 and -8 and on HP1 and HP2 loops. Additionally, two Na+-binding sites located ∼7 Å from the substrate and from each other have been identified; the first (Na1) is more buried and lies between TM7 and -8, whereas the second (Na2) is located between HP2 and TM7 (5–7) (see Fig. 1, C and D). These structures are in accord with the topology and function of mammalian and prokaryotic glutamate transporters (10–15).
Despite this significant progress, much remains to be elucidated with regard to the time-resolved events and inter-residue interactions that mediate sodium-coupled substrate binding or release. For instance, the role of HP2 as an EC gate that controls the binding (or unbinding) of substrate and cations in the OF state has been suggested both by the crystallization of the transporter with an antagonist (5) and by molecular dynamics simulations (16, 17). This role seems plausible because of its high exposure to the EC environment and ability to move therein unobstructed by the rest of the transporter. On the other hand, the IC gating mechanism is far less clear. Crystallographic (7) and substrate uptake studies performed by introducing cysteines in a cysteine-less version of EAAT1 (18) suggested that HP1 might be involved in IC gating, whereas support from time-resolved examinations at an atomic level has been lacking. Here we conducted a series of molecular dynamics runs, using the high performance computing system, Anton, which permits us to examine processes on the microsecond to millisecond timescale (19) depending on the system size. We were able to view for the first time multiple incidences of IC gate opening and substrate and ion release so as to deduce reproducible patterns and extract statistically reliable information on gating mechanism. The picture that emerged differs from that indirectly inferred from static crystal structures; HP2 (and not HP1) opening is the major event enabling the release of neurotransmitter to the cell interior. HP2 therefore serves as IC gate in the IF state, in addition to its established EC gate role. Our study further highlights the sequence of events that enable the release of substrate, including prior release of Na2 to weaken local interactions and promote substrate dissociation.
MATERIALS AND METHODS
Systems
A summary of the simulation systems and conditions is provided in Table 1. The OF or IF structures of GltPh (Protein Data Bank (PDB) identifiers 2NWX and 3KBC, respectively) were used to construct the initial conformation embedded in a hydrated 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphoethanolamine bilayer, with aspartate and Na+ ions bound at their crystallographically observed/modeled binding sites (runs 1–3) or removed (apoIF, run 4). Water molecules were modeled explicitly, whereas the united atom model was used for lipid tail groups, resulting in simulation systems of 121,000 and 137,000 atoms for the IF and OF states, respectively. More details are presented in supplemental Table S1. A summary of observed release times of aspartate, Na1, and Na2 in runs 1–3 is presented in Table 1.
TABLE 1.
Summary of MD runs and observed aspartate/sodium release times
| Run | State | Substrate + ions/medium | Duration | Subunit | r.m.s.d.a | Observed time of release forb |
|
|---|---|---|---|---|---|---|---|
| Asp | Na2 | ||||||
| μs | Å | μs | |||||
| 1 | OF | Asp− + 2Na+/NaCl | 5 | A | 1.6 ± 0.2 | 3.61 | 3.54 |
| 1 | OF | Asp− + 2Na+/NaCl | 5 | B | 2.5 ± 0.4 | 0.92 | 0.85 |
| 1 | OF | Asp− + 2Na+/NaCl | 5 | C | 2.2 ± 0.3 | 0.23 | 0.38 |
| 2 | IF | Asp− + 2Na+/NaCl | 6 | A | 2.1 ± 0.3 | 2.34 | 1.94 |
| 2 | IF | Asp− + 2Na+/NaCl | 6 | B | 3.1 ± 0.5 | 0.58 | 0.07 |
| 2 | IF | Asp− + 2Na+/NaCl | 6 | C | 2.0 ± 0.4 | 1.34 | 0.08 |
| 3 | IF | Asp− + 2Na+/NaCl | 6 | A | 1.7 ± 0.3 | 2.49 | |
| 3 | IF | Asp− + 2Na+/NaCl | 6 | B | 2.2 ± 0.2 | 1.51 | 0.23 |
| 3 | IF | Asp− + 2Na+/NaCl | 6 | C | 3.1 ± 0.5 | 0.39 | 0.22 |
| 4 | IF | None/KCl | 6 | A | 3.6 ± 1.0 | ||
| 4 | IF | None/KCl | 6 | B | 3.2 ± 0.3 | ||
| 4 | IF | None/KCl | 6 | C | 2.8 ± 0.3 | ||
a r.m.s.d., root mean square deviation. r.m.s.d. values refer to the backbone of each subunit, excluding the EC loop between TM3 and -4, and are averages of the entire trajectories.
b Only a single event of Na1 release took place and was observed in subunit B in run 3 at ∼1.5 μs.
Protocol
All titratable residues were left in the dominant protonation state at pH 7.0. Each system was neutralized, and NaCl (runs 1–3) or KCl (run 4) was added to reach a final concentration of 150 mm. All systems were first equilibrated for 10 ns with the protein backbone and bound substrate molecules constrained followed by another 10 ns of free simulation with Desmond on the 64-node Anton machine before starting the productive simulations on the 512-node machine. All runs were performed under constant temperature and pressure using Berendsen barostat/thermostat, at 310 K and 1 bar. CHARMM27 force field parameters with CMAP terms were used for the protein and water molecules, and CHARMM36 parameters were used for lipid molecules (20, 21). For all productive runs, a RESPA integrator with a time step of 2 fs was used (with all bond lengths to hydrogen atoms fixed). A cutoff distance of 11.5 Å was used for short range electrostatic and van der Waals interactions, whereas long range electrostatic interactions were computed every 6 fs with the particle mesh Ewald method using a 64 × 64 × 64 grid with σ = 2.85 Å. The structural integrity of GltPh was maintained throughout the entire duration of all runs (5 μs in run 1, 6 μs in runs 2–4, each) (supplemental Fig. S1).
RESULTS
Substrate and Na+ Release in the Outward-facing State Correlates with Opening of HP2 but Not That of HP1
As illustrated in Fig. 1D, the sodium ion at the Na2 site is coordinated in the OF state by the backbone carbonyl group of Thr-308 on TM7 and those of Ser-349, Gly-351, and Thr-352 on HP2a, and that at the Na1 site is coordinated by the backbone carbonyls of Gly-306 and Asn-310 on TM7 and Asn-401 on TM8 and by the carboxyl group of Asp-405 on TM8. Fig. 2 (upper panels) displays the change in the position of the substrate (mass center) and sodium ions, as a function of time, recorded in run 1 for each subunit. Aspartate and Na2 dissociate almost simultaneously in all three subunits, indicating a strong correlation between their unbinding events. In contrast, Na1 remains bound in all three subunits, although it undergoes fluctuations in its position up to 4 Å away from its original binding site. Strikingly, in all three subunits, substrate/Na2 dissociation takes place shortly after, or together with, the opening of HP2, and not before that (Fig. 2, lower panels), demonstrating that HP2 serves as an EC gate. This nicely complements earlier observations regarding the opening of HP2 for substrate uptake in the OF state (16, 17). In contrast, little or no movement is observed in HP1. The largest displacement observable at the tip of HP1 is about ∼4 Å in subunit B, which takes place only after the release of the substrate, whereas HP2 loop moves by more than ∼12 Å away in all subunits.
FIGURE 2.

Coupling of substrate/cation unbinding to HP2 opening in the OF substrate-bound GltPh. The upper three panels display the change in positions of aspartate mass center (black), Na2 (purple), and Na1 (blue), for subunits A–C, during the course of 5-μs simulations (run 1). Aspartate and Na1 unbind almost simultaneously in all three subunits, whereas Na1 remains bound. Their unbinding strongly correlates with prior HP2 opening (red, lower panels), whereas HP1 (black) shows no detectable correlation. HP1 and HP2 motions are based on the displacements of Ser-277 and Val-355 Cα atoms, respectively, relative to the crystal structure positions. These two residues lie at the tip of the respective hairpin loops.
Weak Interactions between Na2 and Its Coordinating Residues on HP2 and TM7 Prompt the Release of Na2 to the Cytoplasm with Minimal Reconfigurations of HP1 and HP2
Examination of the trajectories of aspartate and Na+ ions in the IF state (Fig. 3 and supplemental Fig. S2 for the respective runs 2 and 3) showed that Na1 was again highly stable; it remained bound in five out of six observations (two runs, three subunits per run). The single event of Na1 dissociation took place in run 3, subunit B (supplemental Fig. S2), around t = 0.5 μs where Na1 left after 4 μs into the IC region (long after Na2 and aspartate were released). In contrast, Na2 left the transporter in all cases, and aspartate left in five out of six cases.
FIGURE 3.

Intracellular gating dynamics in the inward-facing substrate-bound conformation. Unbinding of aspartate (black) or Na2 (purple) out of the binding pocket into the IC solvent, and corresponding HP1 and HP2 displacements observed in run 2, are shown here as a function of time. The upper and middle panels are in the same format as in Fig. 2. Unbinding of Na2 precedes that of aspartate in all subunits. The time evolution of the space between HP2 and TM8 (violet, lower panels) indicates an opening of at least 10 Å for aspartate to be released to the cytoplasm. The HP2-TM8 distance is based on the instantaneous coordinates of the Cα atoms of Pro-356 (HP2) and Asp-394 (TM8). See also supplemental Fig. S2 for similar results from run 3.
An important observation was the ability of Na2 to wiggle by up to 3 Å within the confines of its original binding site while being coordinated by the carbonyl and hydroxyl groups of Thr-308 on the unwound part of TM7 and by the carbonyls of Ser-349 to Thr-352 on HP2 (Fig. 4 and supplemental Fig. S3), before leaving the transporter within a maximal duration of 2.5 μs. The unbinding of Na2 preceded that of aspartate by hundreds of nanoseconds in most cases, which suggests that Na2 release induced a local relaxation in the network of interactions near its (original) binding site between the TM7 and HP2 loop, which, in turn, facilitated the translocation of the aspartate. A careful look at the mechanism of substrate unbinding indeed reveals interesting patterns as described next.
FIGURE 4.

Pathway of aspartate release into the intracellular medium. Snapshots shown here, at the indicated times (∼500-ns period before substrate release), are of subunit A in run 2. Each snapshot, viewed from two angles (60° difference, left and right panels), shows the substrate-binding site with TM7 and -8 and HP1 and -2 colored as in Fig. 1. Residues distinguished by their close interaction with the aspartate or the sodium at the Na2 site are shown in stick representation and labeled (labels are colored by the structural elements to which they belong). Note the increase in the distance between HP2 (red) and TM8 (violet) before substrate release (right panels). See also supplemental Fig. S3 for similar results from run 3.
Subtle Structural Rearrangements Succeeding Na2 Release Induce the Reorientation and Translation of Aspartate toward Arg-276 on HP1
The mobility of sodium near its Na2-binding site indicates that the interactions that stabilize this ion in the crystallized structure are relatively weak under physiological conditions. Following the dissociation of Na2, a loosening in the packing of structural elements in its vicinity takes place as can be discerned upon comparing the respective panels at 1.828 and 1.935 μs (before and after the release of Na2) in Fig. 4. Notably, HP2 moves away from the transport core, its distance from TM8 increases, and the aspartate itself undergoes a significant rearrangement (a rotation of about 90°) between these two snapshots, presumably enabled by the relaxation of the neighboring network of interactions. A similar mechanism can be observed upon comparison of the top panels in supplemental Fig. S3, where a local relaxation succeeding the departure of Na2 allows for an overall reorientation of the neurotransmitter. Notably, in both runs, Asp-394 plays a role in stabilizing the intermediate pose of the aspartate before release. Also, in both runs, the electrostatic attraction between the guanidinium group of Arg-276 and the aspartate β-carboxylate is instrumental in promoting the substrate orientation and translocation toward the IC region.
Our runs have produced five events of full aspartate departure from the IF transporter (Fig. 3 and supplemental Fig. S2). The above described mechanism, depicted in Fig. 5, appears to control substrate release in all cases. The process is initiated by the release of Na2 and movement of HP2 away from the transport core, including an increase in the distance between TM8 and HP2, that enables the reorientation and sliding of the bound aspartate across the space between HP1, HP2, and TM8 toward Arg-276. Along this path, we distinguish the interactions of the substrate with Pro-356 and Gly-357 (HP2) and Asp-394 (TM8). Amino acids that exhibit close interactions with the substrate at successive stages of this translocation process are shown in Fig. 4 and supplemental Fig. S3. Supplemental Movies S1, A and B provide a clear view of the recurrent mechanism of substrate release.
FIGURE 5.
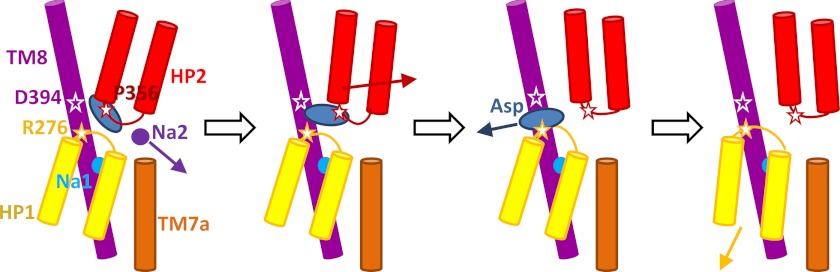
Schematic description of the steps of aspartate and sodium release in the inward-facing structure of GltPh. The depiction shows that a sodium ion at the Na2 site (dark purple circle) can leave the symporter with little or no structural changes followed by displacement of the HP2 domain (red) away from the TM8 domain (violet) that allows the bound aspartate (blue ellipse) to leave into the IC solvent, assisted by the mediating action of Asp-394 on TM8 followed by attraction by Arg-276 on HP1 loop prior to complete release. For clarity, only the intracellular part of TM7 (TM7a; orange) is shown here. HP1 motions do not seem to be essential for aspartate or Na2 release and become more pronounced only after the release of aspartate.
Roles of HP1 and HP2 in Intracellular Gating Indicate That HP2 Displacement Is a Prerequisite for Substrate Release
Our trajectories clearly show that to enable aspartate translocation, the distance between Asp-394 (TM8) and Pro-356 (HP2) Cα atoms needs to increase from ∼7 Å, as in the crystal structure, to ∼10 Å (Fig. 3 and supplemental Fig. S2, lower panels), and this is essentially accomplished by HP2 movement away from the core (Fig. 4 and supplemental Fig. S3). However, the coupling between aspartate/Na+ release and HP1/HP2 motions appears to be more complex than that in the OF state where large displacements in HP2 were clearly seen to correlate with the release of Na2 and aspartate (Fig. 2). First, the release of the substrate and Na2 is not synchronous in the IF state; there is usually a time delay of 0.5 μs or longer before the release of aspartate, after that of Na2 (except for subunit C in run 3 where the delay is shorter (supplemental Fig. S2)). However, the release of aspartate always follows that of Na2, whether the time elapsed in between is of the order of tenths of microseconds or microseconds. This is in contrast to the OF state behavior where aspartate release could even precede that of Na2 (subunit C in Fig. 2). Second, although the amplitudes of HP2 movements were significantly larger than those of HP1 in the OF state, this is not the case in the IF state. The histograms in supplemental Fig. S4 display the contrast between the two states. Panel A compares the occurrence of HP1 and HP2 displacements in the OF state, exposing the significantly higher mobility of HP2 in this state (the data from the three subunits have been combined in each case for viewing the overall picture averaged over all three subunits). Panel B shows the counterpart for the IF state (where the data from all subunits in both run 2 and run 3 are compiled), clearly demonstrating that HP2 is more restrained in the IF state.
Timing, Rather than Size of Motions, Controls Substrate Release
Although HP1 enjoys larger movements in the IF form when compared with HP2 (as shown in supplemental Fig. S4), this does not mean that it serves as an IC gate. Based on the relative sizes of motions, one might infer that HP1 is the critical structural element that is mediating substrate release (as previously inferred from structural examinations (6)) in the IF state. However, critical examination of the sequence of events that lead to substrate release in the IF state, confirmed in both run 2 and run 3, shows that the release of Na2 is always the first event that is followed by (increased) opening of HP2 and the subsequent release of aspartate. The movement of HP1 follows that of HP2, as a consequence of the weakening of local interactions near the substrate-binding region, as discussed above. Although at “later” stages of simulations (succeeding the release of substrate) HP1 exhibits larger excursions, these motions are manifested only after the HP2 tip has moved at least 4 Å away from its original position. In fact, a displacement of HP2 away from TM8 invariably appears to be a prerequisite for substrate release that may be facilitated by the consequent movement of HP1. The only case where substrate dissociation did not take place is in subunit A of run 3 (supplemental Fig. S2) where HP2 displacement remained lower than around 4 Å, and TM8 and HP2 remained closely associated throughout the entire duration of a 6-μs run. Notably, the lack of HP2 movement away from TM8 also restricted the mobility of the substrate, and that of HP1, and prevented the release.
Supplemental Fig. S4C displays the size distribution of HP1 and HP2 movements in the apo structure (run 4) upon replacement of sodium in the aqueous environment by potassium to avoid interference in the gating mechanism caused by Na+ binding to the transporter. We notice a shift toward larger movements in both hairpins. Notably, this time, the large movements of HP1 do not require prior relaxation of the interactions that stabilize HP2 and may be manifested even before HP2 opening (see subunit B in supplemental Fig. S5), consistent with the weaker local interactions in the apo state.
DISCUSSION
Despite the huge amount of data revealed by high resolution crystal structures, the dynamic nature of proteins necessitates the examination of molecular properties beyond those provided by single, static images, such as time-resolved events at the atomic scale. Molecular simulations, empowered in recent years with important advances in computing hardware and software technology such as the Anton supercomputer, serve as a valuable tool to derive detailed information on mechanisms of function, provided that structural data are available (22, 23). Using the structures resolved for GltPh, a prototype for learning about the behavior of EAATs, we were able to delineate the mechanism of substrate unbinding out of the transport core under physiological conditions not necessarily present under the crystallization conditions. The emerging behavior, schematically shown in Fig. 5, is different from that inferred from static structures; a displacement by ∼4 Å at the tip of HP2 loop away from the transport core, or more precisely, an increase by at least 3 Å in the distance between HP2 and TM8 around the space between Pro-356 and Asp-394 is required to initiate the release of neurotransmitter in the IF state, suggesting that HP2 serves as the IC gate. Although the EC gating role of HP2 has been established by several studies, our extensive simulations provide for the first time a concrete visualization of its role as an IC gate in the release of the substrate to the cytoplasm.
It is important to note that the motion of HP2 is required, but may not be sufficient alone to prompt the release of aspartate in the IF state; the increased reorientation and translation ability of the substrate in the transport core succeeding the release of Na2, allowed by an increased separation between HP2 and TM8, and the electrostatic attraction by Arg-276 (neighboring the Ser3 (Ser-277–279) motif at the HP1 tip) all play important roles in driving and completing substrate release. We also note the assisting role of Asp-394 on TM8, which tends to move away from TM7 and HP2 and mediate the interactions of the substrate with Arg-276. The equivalent position of this aspartate in EAAT1 was shown to be essential for substrate interaction with the transporter (24).
It should be stressed, however, that although the crystal structures of GltPh were obtained with an aspartate and two thallium (6) or sodium ions in the transport core (7, 8), the transport stoichiometry in GltPh (25) and mammalian transporters (26–28) is three Na+ per substrate. In addition, in mammalian transporters, but not GltPh (29), a proton is co-transported with the substrate while a potassium ion is counter-transported (28, 30, 31). The crystallized Na2 site and the aspartate pose thus may not necessarily represent the correct or optimal conformation that supports transport. In support of this is the previous experimental observation that in the homologous EAAC1, glutamate in the cytoplasmic binding site dissociates before the three sodium ions (32). The binding pocket shows a tendency to open and release aspartate and sodium at Na2, as shown in this study, rather than remain closed. The closure of the binding pocket and tight binding of the substrate and co-transported ions are prerequisites for the translation of the binding pocket between the external and internal sides of the membrane as the transporter undergoes a global transition from the OF to the IF state. Whether the transition back to an OF state requires the closure of the binding pocket and how anion (chloride) channeling and potassium counter-transport play out in the complete transport cycle are issues that remain to be clarified. In addition, although there is experimental evidence supporting the role of the mammalian equivalent of Asp-405 (in GltPh) at Na1 in cation binding (33), no support for the Na2 site has been reported, mostly due to the proposed coordination of a cation at Na2 by backbone carbonyls, rather than side chain groups that can be mutated to investigate their role in binding or transport.
Therefore, further studies that can ultimately lead to the elucidation of a stable binding pocket conformation that contains an aspartate/glutamate, a proton, and three sodium ions in mammalian transporters, and to the visualization of the other steps of transport cycle including anion transport, are required for fully understanding the mechanism of excitatory amino acid transport from the synapse to the cell interior.
Acknowledgments
The Anton machine has been provided generously by David E. Shaw Research (19). Anton computer time was provided by the Pittsburgh Supercomputing Center through Grant RC2GM09337. We thank M. Dittrich, N. Simakov, and other staff at the Pittsburgh Supercomputing Center for their support.
This work was supported, in whole or in part, by National Institutes of Health Grants R01GM086238 from the NIGMS (to I. B.) and R01 LM007994-08 (to I. B.) and the National Institutes of Health-funded Biomedical Technology Research Center (Grant P41 GM103712) (to I. B.).

This article contains supplemental Table S1, Figs. S1–S5, and supplemental Movie S1, A and B.
- EC
- extracellular
- IC
- extracellular
- EAAT
- excitatory amino acid transporter
- TM
- transmembrane
- HP
- helical hairpin
- OF
- outward-facing
- IF
- inward-facing.
REFERENCES
- 1. Tanaka K., Watase K., Manabe T., Yamada K., Watanabe M., Takahashi K., Iwama H., Nishikawa T., Ichihara N., Kikuchi T., Okuyama S., Kawashima N., Hori S., Takimoto M., Wada K. (1997) Epilepsy and exacerbation of brain injury in mice lacking the glutamate transporter GLT-1. Science 276, 1699–1702 [DOI] [PubMed] [Google Scholar]
- 2. Buckingham S. C., Campbell S. L., Haas B. R., Montana V., Robel S., Ogunrinu T., Sontheimer H. (2011) Glutamate release by primary brain tumors induces epileptic activity. Nat. Med. 17, 1269–1274 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Kanner B. I., Zomot E. (2008) Sodium-coupled neurotransmitter transporters. Chem. Rev. 108, 1654–1668 [DOI] [PubMed] [Google Scholar]
- 4. Storm-Mathisen J., Danbolt N. C., Rothe F., Torp R., Zhang N., Aas J. E., Kanner B. I., Langmoen I., Ottersen O. P. (1992) Ultrastructural immunocytochemical observations on the localization, metabolism and transport of glutamate in normal and ischemic brain tissue. Prog. Brain Res. 94, 225–241 [DOI] [PubMed] [Google Scholar]
- 5. Boudker O., Ryan R. M., Yernool D., Shimamoto K., Gouaux E. (2007) Coupling substrate and ion binding to extracellular gate of a sodium-dependent aspartate transporter. Nature 445, 387–393 [DOI] [PubMed] [Google Scholar]
- 6. Yernool D., Boudker O., Jin Y., Gouaux E. (2004) Structure of a glutamate transporter homologue from Pyrococcus horikoshii. Nature 431, 811–818 [DOI] [PubMed] [Google Scholar]
- 7. Reyes N., Ginter C., Boudker O. (2009) Transport mechanism of a bacterial homologue of glutamate transporters. Nature 462, 880–885 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Verdon G., Boudker O. (2012) Crystal structure of an asymmetric trimer of a bacterial glutamate transporter homolog. Nat. Struct. Mol. Biol. 19, 355–357 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Jiang J., Shrivastava I. H., Watts S. D., Bahar I., Amara S. G. (2011) Large collective motions regulate the functional properties of glutamate transporter trimers. Proc. Natl. Acad. Sci. U.S.A. 108, 15141–15146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Grunewald M., Bendahan A., Kanner B. I. (1998) Biotinylation of single cysteine mutants of the glutamate transporter GLT-1 from rat brain reveals its unusual topology. Neuron 21, 623–632 [DOI] [PubMed] [Google Scholar]
- 11. Slotboom D. J., Sobczak I., Konings W. N., Lolkema J. S. (1999) A conserved serine-rich stretch in the glutamate transporter family forms a substrate-sensitive reentrant loop. Proc. Natl. Acad. Sci. U.S.A. 96, 14282–14287 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Seal R. P., Leighton B. H., Amara S. G. (2000) A model for the topology of excitatory amino acid transporters determined by the extracellular accessibility of substituted cysteines. Neuron 25, 695–706 [DOI] [PubMed] [Google Scholar]
- 13. Seal R. P., Amara S. G. (1998) A reentrant loop domain in the glutamate carrier EAAT1 participates in substrate binding and translocation. Neuron 21, 1487–1498 [DOI] [PubMed] [Google Scholar]
- 14. Grunewald M., Kanner B. I. (2000) The accessibility of a novel reentrant loop of the glutamate transporter GLT-1 is restricted by its substrate. J. Biol. Chem. 275, 9684–9689 [DOI] [PubMed] [Google Scholar]
- 15. Groeneveld M., Slotboom D. J. (2007) Rigidity of the subunit interfaces of the trimeric glutamate transporter GltT during translocation. J. Mol. Biol. 372, 565–570 [DOI] [PubMed] [Google Scholar]
- 16. Huang Z., Tajkhorshid E. (2008) Dynamics of the extracellular gate and ion-substrate coupling in the glutamate transporter. Biophys. J. 95, 2292–2300 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Shrivastava I. H., Jiang J., Amara S. G., Bahar I. (2008) Time-resolved mechanism of extracellular gate opening and substrate binding in a glutamate transporter. J. Biol. Chem. 283, 28680–28690 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Leighton B. H., Seal R. P., Watts S. D., Skyba M. O., Amara S. G. (2006) Structural rearrangements at the translocation pore of the human glutamate transporter, EAAT1. J. Biol. Chem. 281, 29788–29796 [DOI] [PubMed] [Google Scholar]
- 19. Shaw D. E., Deneroff M. M., Dror R. O., Kuskin J. S., Larson R. H., Salmon J. K, Young C., Batson B., Bowers K. J., Chao J. C., Eastwood M. P., Gagliardo J., Grossman J.P., Ho C. R., Ierardi D. J., István Kolossváry, Klepeis J. L., Layman T., McLeavey C., Moraes M. A., Mueller R., Priest E. C., Shan Y., Spengler J., Theobald M., Towles B., Wang S. C. (2007) Anton, a special-purpose machine for molecular dynamics simulation. Anton, a special-purpose machine for molecular dynamics simulation. Commun. ACM 51, 91–97 [Google Scholar]
- 20. Pastor R. W., Mackerell A. D., Jr. (2011) Development of the CHARMM force field for lipids. J. Phys. Chem. Lett. 2, 1526–1532 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Guvench O., MacKerell A. D., Jr. (2008) Comparison of protein force fields for molecular dynamics simulations. Methods Mol. Biol. 443, 63–88 [DOI] [PubMed] [Google Scholar]
- 22. Dror R. O., Dirks R. M., Grossman J. P., Xu H., Shaw D. E. (2012) Biomolecular simulation: a computational microscope for molecular biology. Annu. Rev. Biophys. 41, 429–452 [DOI] [PubMed] [Google Scholar]
- 23. Jensen M. Ø., Jogini V., Borhani D. W., Leffler A. E., Dror R. O., Shaw D. E. (2012) Mechanism of voltage gating in potassium channels. Science 336, 229–233 [DOI] [PubMed] [Google Scholar]
- 24. Teichman S., Kanner B. I. (2007) Aspartate-444 is essential for productive substrate interactions in a neuronal glutamate transporter. J. Gen. Physiol 129, 527–539 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Groeneveld M., Slotboom D. J. (2010) Na+:aspartate coupling stoichiometry in the glutamate transporter homologue GltPh. Biochemistry 49, 3511–3513 [DOI] [PubMed] [Google Scholar]
- 26. Owe S. G., Marcaggi P., Attwell D. (2006) The ionic stoichiometry of the GLAST glutamate transporter in salamander retinal glia. J. Physiol 577, 591–599 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Levy L. M., Warr O., Attwell D. (1998) Stoichiometry of the glial glutamate transporter GLT-1 expressed inducibly in a Chinese hamster ovary cell line selected for low endogenous Na+-dependent glutamate uptake. J. Neurosci. 18, 9620–9628 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Zerangue N., Kavanaugh M. P. (1996) Flux coupling in a neuronal glutamate transporter. Nature 383, 634–637 [DOI] [PubMed] [Google Scholar]
- 29. Ryan R. M., Compton E. L., Mindell J. A. (2009) Functional characterization of a Na+-dependent aspartate transporter from Pyrococcus horikoshii. J. Biol. Chem. 284, 17540–17548 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Danbolt N. C., Pines G., Kanner B. I. (1990) Purification and reconstitution of the sodium- and potassium-coupled glutamate transport glycoprotein from rat brain. Biochemistry 29, 6734–6740 [DOI] [PubMed] [Google Scholar]
- 31. Kavanaugh M. P., Bendahan A., Zerangue N., Zhang Y., Kanner B. I. (1997) Mutation of an amino acid residue influencing potassium coupling in the glutamate transporter GLT-1 induces obligate exchange. J. Biol. Chem. 272, 1703–1708 [DOI] [PubMed] [Google Scholar]
- 32. Zhang Z., Tao Z., Gameiro A., Barcelona S., Braams S., Rauen T., Grewer C. (2007) Transport direction determines the kinetics of substrate transport by the glutamate transporter EAAC1. Proc. Natl. Acad. Sci. U.S.A. 104, 18025–18030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Teichman S., Qu S., Kanner B. I. (2009) The equivalent of a thallium binding residue from an archeal homolog controls cation interactions in brain glutamate transporters. Proc. Natl. Acad. Sci. U.S.A. 106, 14297–14302 [DOI] [PMC free article] [PubMed] [Google Scholar]