

Background: The structures of neutral and acidic amino acid transporters of the SLC1 family are very similar, yet they function quite differently.
Results: Three residues involved in determining substrate selectivity have been identified.
Conclusion: A threonine residue modulates the behavior of substrate and proton binding residues.
Significance: We have identified how SLC1 members distinguish between substrates.
Keywords: Amino Acid Transport, Aspartate, Chloride Channels, Electrophysiology, Membrane Proteins, Mutagenesis Site Specific, Neurotransmitter Transport, ASCT, EAAT, GltPh
Abstract
The ASCTs (alanine, serine, and cysteine transporters) belong to the solute carrier family 1 (SLC1), which also includes the human glutamate transporters (excitatory amino acid transporters, EAATs) and the prokaryotic aspartate transporter GltPh. Despite the high degree of amino acid sequence identity between family members, ASCTs function quite differently from the EAATs and GltPh. The aim of this study was to mutate ASCT1 to generate a transporter with functional properties of the EAATs and GltPh, to further our understanding of the structural basis for the different transport mechanisms of the SLC1 family. We have identified three key residues involved in determining differences between ASCT1, the EAATs and GltPh. ASCT1 transporters containing the mutations A382T, T459R, and Q386E were expressed in Xenopus laevis oocytes, and their transport and anion channel functions were investigated. A382T and T459R altered the substrate selectivity of ASCT1 to allow the transport of acidic amino acids, particularly l-aspartate. The combination of A382T and T459R within ASCT1 generates a transporter with a similar profile to that of GltPh, with preference for l-aspartate over l-glutamate. Interestingly, the amplitude of the anion conductance activated by the acidic amino acids does not correlate with rates of transport, highlighting the distinction between these two processes. Q386E impaired the ability of ASCT1 to bind acidic amino acids at pH 5.5; however, this was reversed by the additional mutation A382T. We propose that these residues differences in TM7 and TM8 combine to determine differences in substrate selectivity between members of the SLC1 family.
Introduction
The cellular uptake of small neutral amino acids is regulated by various transport systems. One class of transporters found in a number of tissues is the alanine-serine-cysteine transport system (ASCT)4 (1, 2). Two isoforms have been identified, ASCT1 and ASCT2. ASCT1 is found in the brain, skeletal muscle, and pancreas (3, 4) and is largely selective for alanine, serine, and cysteine but can also transport threonine, asparagine, and to a lesser extent, proline. ASCT2 exhibits a broader substrate selectivity than ASCT1 and is located in the kidney, lung, large intestine, skeletal muscle, testes, and the brain (5, 6). In the brain, ASCT1 is preferentially expressed by glial cells and has been suggested to be responsible for the release of l-serine from intracellular stores, in exchange for other extracellular substrates (7). This exchange of small neutral amino acids is regulated to accommodate the metabolic needs of the surrounding neurons. ASCT2 also plays a role in the brain, where it may contribute to glutamine homeostasis of neurons and astrocytes (6). ASCT2 has been shown to be significantly up-regulated in cancerous tissues and is believed to play a role in the growth and survival of cancer cells (8). Both ASCT1 and ASCT2 exhibit pH-dependent substrate selectivity. Acidic amino acids become substrates at pH 5.5 (5, 9), which may be physiologically important in the acidic environment of the digestive tract where ASCT1 and ASCT2 are involved in nutrient uptake (10).
The ASCTs belong to the solute carrier family 1 (SLC1) (3–5), which also includes a range of human and prokaryotic glutamate and aspartate transporters such as the human glutamate transporters (otherwise known as excitatory amino acid transporters or EAATs) and GltPh (3, 11–13). GltPh is an aspartate transporter from Pyrococcus horikoshii that was crystallized in 2004 by Yernool et al. (13), revealing the complex structure of this transporter family. GltPh exists as a homotrimer with each protomer containing eight transmembrane domains (TM1–8) and two hairpin loops (HP1,2) (see Fig. 1A). GltPh shares ∼23% amino acid sequence identity with ASCT1 and 36% amino acid sequence identity with the EAATs, with many of the residues implicated in substrate and ion binding/transport conserved throughout the family (see Fig. 1C) (13). Thus, GltPh provides a good structural model for investigating the transport mechanism of ASCTs and EAATs.
FIGURE 1.
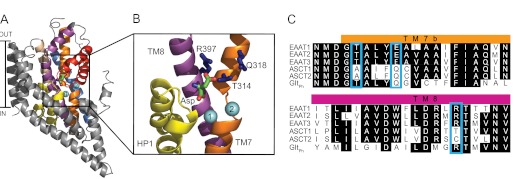
The structure of a GltPh protomer and sequence alignments of the EAATs, ASCT1, and GltPh. A, GltPh protomer (Protein Data Bank code 2NWX) shown in the plane of the membrane, with the trimerization domain (TM1, -2, -4, and -5) in gray and the transport domain: TM3 (light brown), TM6 (blue), TM7 (orange), TM8 (purple), HP1 (yellow), and HP2 (red). l-Aspartate is shown in stick representation, and two bound Na+ ions are shown as light blue spheres. B, close view of the substrate binding site with the residues targeted for mutation in this study shown in stick representation (dark blue). Arg-397 is equivalent to Thr-459 in ASCT1, Gln-318 is equivalent to Gln-386 and Thr-314 is equivalent to Ala-382. TM1–6 and HP2 have been omitted, and the protein was tilted toward the viewer for ease of visualization. Images were made using PyMOL (33). C, sequence alignment of TM7b and part of TM8 in EAAT1–3, ASCT1, ASCT2, and GltPh, where conserved residues are highlighted in black, and mutated residues are highlighted by blue boxes.
Despite the high degree of sequence identity, ASCTs function quite differently from the EAATs. The EAATs transport the acidic amino acids glutamate and aspartate against a concentration gradient generating a net movement of substrate and charge across the membrane. In contrast, ASCTs exchange small, neutral amino acids in an electroneutral fashion without generating net movement of substrate. ASCTs also exhibit pH-dependent substrate selectivity, in that at low pH (< pH 6) small acidic amino acids are substrates. For example, l-cysteate is a substrate of ASCT1 at pH 5.5 (9) and l-glutamate is a substrate of ASCT2 at pH 5.5 (5). The EAATs do not exhibit pH-dependent changes in substrate selectivity; however acidic amino acid transport by the EAATs is coupled to H+. Glutamate transport by the EAATs is coupled to the co-transport of three Na+ ions and one H+ ion, and the counter transport of one K+ ion (14, 15). It is thought that only one Na+ is co-transported with each substrate molecule in ASCT1, and no K+ or H+ transport occurs (15). Similar to the EAATs, ASCTs possess a thermodynamically uncoupled Cl− conductance, which is responsible for the current observed during transport (16, 17).
Numerous mutational studies have been performed in the EAATs that have successfully knocked out substrate binding and/or ion coupling thereby identifying key residues involved in the transport process. Three residues in the EAATs, which are not conserved in the ASCTs, stand out as potentially playing a role in determining differences between the transporters. First, mutations of Arg-479 in EAAT1 abolish glutamate transport (18), indicating a role of this residue in substrate binding. Furthermore, replacing this arginine with cysteine, glutamate, glycine, or serine not only impaired glutamate transport but also introduced neutral amino acid exchange (19). Interestingly, the equivalent residue in ASCT1 is a threonine. Second, the TM7 motif NMDGT includes residues involved in substrate (20) and Na+ (21–23) binding and is conserved throughout the EAATs and GltPh. The threonine in this motif is replaced with an alanine in ASCT1. Third, a H+/K+ binding site in the EAATs has been proposed to be formed by Glu-404 in TM7 of EAAT2 (24, 25). Mutation of this glutamate to glutamine abolishes both H+ and K+ coupling in EAAT2. Glu-404 is highly conserved throughout the EAAT family (25), but it is a glutamine at the corresponding site in ASCTs and also GltPh. ASCT- and GltPh-mediated transport are not coupled to H+ or K+ (26); however, the substrate selectivity of ASCTs is pH-dependent (5, 9).
These experiments have been useful in identifying residues involved in the transport process, but very little is understood about how these residues work together to establish the substrate-ion coupling mechanism. An alternative to knocking out a function is to introduce a function, which provides a deeper understanding of the mechanism of substrate-ion coupling. In this study, we have mutated residues that are predicted to be involved in substrate and ion binding in ASCT1 to the EAAT counterpart (A382T, T459R, Q386E) (Fig. 1). The T459R difference in TM8 and the Q386E difference in TM7 play key roles in determining substrate selectivity and pH dependence of substrate selectivity, but it is interesting to note that the additional A382T difference in TM7 appears to modulate the properties of the other two residues to facilitate these functional differences.
EXPERIMENTAL PROCEDURES
Site-directed Mutagenesis
ASCT1 was subcloned into the plasmid oocyte transcription vector (pOTV). Site-directed mutagenesis was performed using the QuikChange site-directed mutagenesis kit (Stratagene) using Velocity DNA polymerase (Bioline, Sydney, Australia). Primers were designed and synthesized by Sigma Genosys (Sydney, Australia), and the DNA sequences of all mutations were confirmed by the Australian Genome Research Facility (Sydney, Australia). DNA was prepared using the Qiagen Miniprep kit (Qiagen Pty, Ltd.). All transporter cDNAs were linearized with Spe1 (Promega) and mRNA transcribed with T7 polymerase using the mMESSAGE mMACHINE kit (Ambion).
Electrophysiology
All chemicals were obtained from Sigma unless otherwise stated. Stage V oocytes were harvested from Xenopus laevis as described previously (27), and all surgical procedures followed a protocol approved under the Australian Code of Practice for the Care and Use of Animals for Scientific Purposes. 20 ng of cRNA was injected into oocytes and incubated in Cl− containing buffer (96 mm NaCl, 2 mm KCl, 1 mm MgCl2, 1.8 mm CaCl2, 5 mm HEPES, pH 7.5) supplemented with 50 μg/ml gentamycin, 2.5 mm sodium pyruvate, 0.5 mm theophylline at 16–18 °C.
Two to 4 days after microinjection, current recordings were made using the two electrode voltage clamp technique with a Geneclamp 500 amplifier (Axon Instruments, Foster City, CA) interfaced with a MacLab 2e chart recorder (ADI Instruments, Sydney, Australia) using the chart software, and a Digidata 1322A (Axon Instruments) controlled by an IBM-compatible computer using the pClamp software (version 10, Molecular Devices, Union City, CA). The current-voltage relationships for substrate elicited conductances were obtained by subjecting cells to 200-ms voltage pulses between −100 mV and +60 mV in 10-mV steps. Current voltage relationships were calculated by subtracting steady state current measurements in the absence of substrate from corresponding current measurements in the presence of substrate.
Recording solution for all experiments (except where otherwise stated) was normal frog Ringer's solution (Cl− containing buffer) with complete NO3− substitution for Cl−. The pH of recording solutions was adjusted using HNO3 and NaOH. Recordings were made with the bath grounded via a 3 m KCl/agar bridge connected to a 3 m KCl reservoir to minimize offset potentials. Cells were washed with Cl− containing buffer between substrate applications to ensure that NO3− loading of the cell was not significant.
Current (I) as a function of substrate concentration was fitted by least-squares analysis to a derivation of the Michaelis-Menten equation,
where Imax is the maximum current generated and EC50 is the substrate concentration which generates a half-maximal response.
Radiolabeled Uptake Experiments
Uptake of l-[3H]aspartate and l-[3H]serine (PerkinElmer Life Sciences) was measured in oocytes expressing wild type and mutant ASCT1 and uninjected oocytes. 4–6 oocytes were incubated in Cl− containing buffer with 10 μm l-[3H]aspartate or l-[3H]serine at room temperature. After 10 min, uptake was terminated by three rapid washes in ice-cold Cl− containing buffer followed by lysis in 50 mm NaOH and 50% SDS. [3H]Substrate was measured by scintillation counting using a Trilux beta counter (PerkinElmer Life Sciences).
RESULTS
The aim of this study was to determine the molecular basis for functional differences between ASCT1, the EAATs, and GltPh.
Neutral Amino Acid Transport at pH 7.5
In standard Cl−-based recording buffer, application of neutral amino acids to oocytes expressing wild type ASCT1 generates small currents, which reverse direction at approximately −20 mV (Fig. 2A). This is indicative of activation of the uncoupled anion conductance of ASCT1 (17). The uncoupled anion conductance is activated by the binding of substrate and Na+ and follows the permeability sequence of SCN− > NO3− > I− > Cl−. To enhance the amplitude and reliability of current measurements mediated by ASCT1, Cl− in the recording buffer was replaced with NO3−. Using this NO3− containing buffer, the application of l-serine to oocytes expressing ASCT1 at pH 7.5 generates large outward currents (Fig. 2A). The EC50 for l-serine activation of the anion current at +60 mV is 100 ± 16 μm (Fig. 2D, Table 1) and ASCT1 also supports significant levels of l-[3H]serine uptake (Fig. 2F). Previous studies indicate that amino acid transport by ASCT1 is not coupled to the counter-transport of K+ (17). We also confirmed this observation by measuring l-serine induced anion currents in the presence of elevated extracellular K+. In the EAATs, elevated extracellular K+ concentrations reduces the outward current observed at positive membrane potentials (data not shown), as a result of reducing the driving force for inward transport. In ASCT1 however, the anion conductance was not affected by elevated extracellular K+ (60 mm) when compared with low extracellular K+ (2 mm) (data not shown), confirming that ASCT1 is K+-independent.
FIGURE 2.
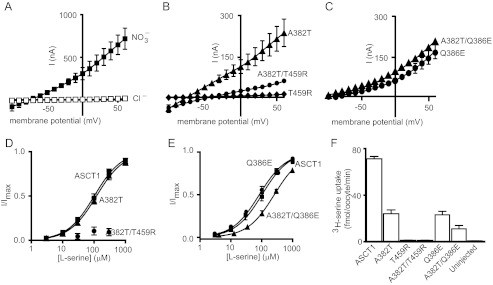
Neutral amino acid transport by wild type and mutant ASCT1 transporters. A, current voltage relationship elicited by 300 μm l-serine in Cl− containing buffer (open squares) and NO3− containing buffer (closed squares), at pH 7.5 in ASCT1. B, current voltage relationships elicited by 300 μm l-serine at pH 7.5 for A382T (triangles), A382T/T459R (circles), and T459R (diamonds). C, current voltage relationships elicited by 300 μm l-serine at pH 7.5 for A382T/Q386E (triangles) and Q386E (circles). D, l-serine concentration-response curves for ASCT1 (squares), A382T (triangles), and A382T/T459R (circles) at +60 mV. As an l-serine concentration response curve for A382T/T459R was not able to be established, the data were normalized to the predicted maximal current for l-cysteate of A382T/T459R. E, l-serine concentration-response curves for ASCT1 (squares), Q386E (circles), and Q386E/A382T (triangles) at +60 mV. F, l-[3H]serine uptake into oocytes expressing wild type and mutant ASCT1 transporters. Oocytes were incubated in Cl− containing buffer with 10 μm l-[3H]serine at room temperature at pH 7.5 for 10 min. Values presented are mean ± S.E. where n ≥ 3.
TABLE 1.
Summary of kinetic parameters for acidic and neutral amino acid transport by wild type and mutant ASCT1
Substrates were tested at pH 7.5 and 5.5 in a NO3− containing buffer. EC50 values shown are μm unless otherwise stated, and Imax values are in nA. All values represent mean ± S.E. (n ≥ 3). A dash indicates the substrate did not generate currents; ND indicates a concentration response curve was not established.
| Substrate | ASCT1 |
A382T |
T459R |
A382T/T459R |
A382T/Q386E |
|||||
|---|---|---|---|---|---|---|---|---|---|---|
| EC50 | Imax | EC50 | Imax | EC50 | Imax | EC50 | Imax | EC50 | Imax | |
| pH 7.5 | ||||||||||
| l-Serine | 100 ± 20 | 1140 ± 30 | 120 ± 20 | 940 ± 60 | − | − | >1 mm | ND | 260 ± 20 | 415 ± 9 |
| l-Aspartate | − | − | 430 ± 20 | 220 ± 30 | 160 ± 30 | 1500 ± 200 | 2 ± 1 | 90 ± 50 | >1 mm | ND |
| d-aspartate | − | − | − | − | 390 ± 30 | 900 ± 200 | 0.8 ± 0.1 | 90 ± 20 | − | − |
| l-Glutamate | − | − | − | − | 400 ± 100 | 1600 ± 400 | 230 ± 30 | 400 ± 100 | − | − |
| l-Cysteate | − | − | − | − | 1.8 ± 0.2 | 530 ± 40 | 5 ± 1 | 240 ± 40 | − | − |
| pH 5.5 | ||||||||||
| l-Aspartate | >1 mm | ND | 40 ± 10 | 120 ± 30 | 80 ± 20 | 3350 ± 80 | 10 ± 4 | 240 ± 60 | 280 ± 30 | 340 ± 10 |
| d-aspartate | − | − | 250 ± 70 | 44 ± 8 | ND | − | ND | − | >1 mm | ND |
| l-Glutamate | − | − | − | − | 1000 ± 40 | 2000 ± 300 | >1 mm | 500 ± 200 | − | − |
| l-Cysteate | 450 ± 90 | 1600 ± 300 | 120 ± 20 | 240 ± 60 | 40 ± 10 | 800 ± 200 | 40 ± 10 | 190 ± 50 | 240 ± 20 | 300 ± 80 |
l-Serine application to oocytes expressing the A382T (TM7) mutant transporter at pH 7.5 generates outward currents with reduced amplitudes compared with ASCT1 (Fig. 2B, Table 1). The outward currents correlate with reduced levels of l-[3H]serine uptake into oocytes expressing A382T (Fig. 2F) compared with wild type ASCT1. However, the EC50 for the l-serine-activated anion current for A382T is similar to that of ASCT1 (Fig. 2D, Table 1). Thus, the rate of ASCT1-mediated neutral amino acid exchange is slightly impaired by the A382T mutation. More striking results are observed for the T459R (TM8) mutant transporter. No currents are generated by l-serine application to T459R expressing oocytes (Fig. 2B), and no significant l-[3H]serine uptake is observed (Fig. 2F), demonstrating that T459R is defective in the transport of neutral amino acids. Interestingly, combining both the A382T and T459R mutations in ASCT1 generates a transporter that produces outward currents in response to l-serine; however, the current sizes are markedly reduced compared with ASCT1 (Fig. 2B). The EC50 for the l-serine-activated anion conductance is increased for A382T/T459R compared with wild type ASCT1 and the A382T mutant (Fig. 2D, Table 1), and levels of l-[3H]serine uptake are not distinguishable from background (Fig. 2F), indicating that the double mutant has compromised neutral amino acid exchange.
l-Serine application to the Q386E mutant generates anion currents that are reduced compared with wild type ASCT1 (Fig. 2C). Similarly, levels of l-[3H]serine uptake into Q386E expressing oocytes were reduced by ∼60% from that of wild type ASCT1 (Fig. 2F). However, the EC50 for the l-serine-activated current for Q386E is similar to the ASCT1 value (Fig. 2E, Table 1). These results demonstrate that although the EC50 for neutral amino acid transport is not altered by the Q386E mutation in ASCT1, the expression level of the transporter or rate of exchange may be affected. Combining A382T and Q386E mutations did not further alter the l-serine elicited anion currents (Fig. 2C). l-Serine is slightly less potent in this double mutant (Fig. 2E, Table 1), and l-[3H]serine uptake levels are reduced compared with Q386E alone (Fig. 2F). This suggests that neutral amino acid transport has been compromised by the addition of A382T to Q386E; however, A382T/Q386E remains a functional neutral amino acid exchanger.
Acidic Amino Acid Transport at pH 7.5 and 5.5
Application of l-aspartate or l-cysteate to ASCT1 expressing oocytes at pH 7.5 does not generate currents (Fig. 3A). However, at pH 5.5, l-cysteate activates the anion conductance of ASCT1 (Fig. 3A), which is consistent with previous studies, suggesting that it is a substrate for the transporter at pH < 6 (9). Despite generating similar outward currents to the neutral amino acids, the potency of l-cysteate activated anion currents for ASCT1 is lower than that of l-serine at pH 5.5 (Table 1, Fig. 3D). Application of l-aspartate to ASCT1 expressing oocytes also generates outward currents at pH 5.5; however, current amplitudes and potency are considerably lower than l-cysteate (Table 1, Fig. 3, A and D). The rate of l-[3H]aspartate uptake into oocytes expressing ASCT1 was also reduced compared with that of l-[3H]-serine uptake at pH 7.5 (Figs. 2F and 3C). l-Glutamate (up to 300 μm) does not generate any significant currents when applied to oocytes expressing ASCT1 at pH 7.5 or pH 5.5 (data not shown).
FIGURE 3.

Acidic amino acid transport by wild type and mutant ASCT1 transporters. A, current voltage relationships elicited by 300 μm l-cysteate (circles) and l-aspartate (triangles) in a NO3− containing buffer at pH 7.5 (closed symbols) and pH 5.5 (open symbols) in ASCT1. B and C, l-[3H]aspartate uptake into oocytes expressing wild type and mutant ASCT1 transporters. Oocytes expressing wild type ASCT1, A382T, T459R, and A382T/T459R, and uninjected oocytes were incubated in Cl− containing buffer with 10 μm l-[3H]aspartate at pH 7.5 (B) and pH 5.5 (C). D, l-serine (squares), l-cysteate (circles), and l-aspartate (triangles) concentration-response curves for ASCT1 at pH 5.5, +60 mV. Data are normalized to the predicted maximal current for each experiment. E, l-cysteate (circles) and l-aspartate (triangles) concentration-response curves for A382T at pH 7.5 (closed symbols) and pH 5.5 (open symbols), +60 mV. F and G, l-cysteate (circles), l-aspartate (triangles), and l-glutamate (inverse triangles) concentration-response curves for T459R (F) and A382T/T459R (G) at pH 7.5, +60 mV. Values are mean ± S.E. where n ≥ 3.
We then investigated the effects of the A382T, T459R, and Q386E mutations in ASCT1 on the acidic amino acid activated anion conductances and radiolabeled substrate uptake. In contrast to wild type ASCT1, l-aspartate activated the anion conductance of A382T at pH 7.5, although to a lesser extent than the neutral amino acids (Table 1). These small currents correlate with low levels of l-[3H]aspartate uptake (Fig. 3B). At pH 7.5, the EC50 for l-aspartate activated anion currents for A382T is 430 ± 20 μm (Fig. 3E, Table 1). This affinity increases considerably, to 36 ± 7 μm, when the pH is decreased to pH 5.5 (Fig. 3E). The rate of l-[3H]aspartate uptake also increases at pH 5.5 (Fig. 3C).
The T459R mutant generated more striking results with acidic amino acids. Application of l-aspartate, l-glutamate, and l-cysteate to T459R expressing oocytes at pH 7.5 all generated large currents (Table 1). l-Cysteate is the most potent of the acidic amino acids at T459R, followed by l-aspartate and l-glutamate (Fig. 3F, Table 1). Increasing extracellular K+ at pH 7.5 does not affect these currents (data not shown), suggesting that the transport process remains K+ independent. Decreasing the pH to 5.5 only slightly alters the EC50 values of the acidic amino acids, with l-cysteate remaining the most potent (Table 1). The rate of l-[3H]aspartate uptake into oocytes expressing T459R transporters was significantly greater than background (Fig. 3B); however, it was markedly reduced compared with the uptake rate of l-[3H]serine in wild type ASCT1 (Fig. 2F) despite the large anion currents observed.
The anion conductance of the combined mutation A382T/T459R is similarly stimulated by acidic amino acids, although with reduced amplitudes compared with T459R (Table 1). The potencies of l-cysteate, l-glutamate, and l-aspartate elicited currents for A382T/T459R are overall higher than for T459R, where the rank order of potency is l-aspartate, l-cysteate, and l-glutamate (Fig. 3G; Table 1). The combination of these two mutations results in lower EC50 values for acidic amino acids and suggests cooperativity between the threonine and arginine within the EAAT/GltPh binding site. Again, transport rates of l-[3H]aspartate uptake into oocytes expressing A382T/T459R transporters was markedly reduced (Fig. 3B) compared with the uptake rate of l-[3H]serine in wild type ASCT1 (Fig. 2F).
In contrast to wild type ASCT1, l-cysteate does not stimulate the anion conductance of the Q386E mutant at pH 5.5 (Fig. 4A). To investigate this further, we tested whether l-cysteate affects the rate of l-[3H]serine uptake. 1 mm l-cysteate inhibits l-[3H]serine uptake by wild type ASCT1 by 64 ± 3% at pH 5.5, and this level of inhibition is reduced to 14 ± 5% at pH 7.5 (Fig. 4B). However, for the Q386E mutant, 1 mm l-cysteate does not inhibit l-[3H]serine uptake at pH 5.5 beyond the 14 ± 5% observed in wild type ASCT1 at pH 7.5 (Fig. 4B). This indicates that acidic amino acid binding to the Q386E mutant at low pH is impaired. Surprisingly, the combination of A382T and Q386E within ASCT1 generated a transporter capable of acidic amino acid transport, which was similar in profile to the A382T mutant. l-Cysteate and l-aspartate stimulate the anion conductance of A382T/Q386E at pH 5.5 (Fig. 4A, Table 1), although they display relatively low affinities (Table 1). Furthermore, l-cysteate is also able to inhibit l-[3H]serine uptake into oocytes expressing A382T/Q386E at pH 5.5 by 66 ± 2% (Fig. 4B), which is comparable with that of wild type ASCT1. This indicates that the impairment of acidic amino acid transport by the Q386E mutation in ASCT1 can be rescued by the A382T mutation, suggesting cooperativity between the two residues. The equivalent glutamate residue in the EAATs has been implicated in proton binding, leading to the hypothesis that the threonine residue in the NMDGT motif aids protonation of the glutamate residue, without which the negative charge may repel acidic amino acids from the binding site.
FIGURE 4.

Q386E impairs acidic amino acid transport, which can be rescued by A382T. A, current voltage relationships elicited by 300 μm l-cysteate at +60 mV, in a NO3− containing buffer at pH 5.5, in Q386E (squares) and A382T/Q386E (circles). B, inhibition of ASCT1, Q386E, and A382T/Q386E mediated l-[3H]serine uptake by l-cysteate at pH 7.5 (open bars) and pH 5.5 (closed bars). Oocytes expressing ASCT1, Q386E, and A382T/Q386E were incubated in Cl− containing buffer at pH 7.5 and pH 5.5 with either 10 μm l-[3H]serine alone or 10 μm l-[3H]serine and 1 mm l-cysteate for 10min at room temperature. Uptake observed with 10 μm l-[3H]serine and 1 mm l-cysteate was calculated as a percentage of that seen with 10 μm l-[3H]serine incubation alone. Values presented are mean ± S.E. where n ≥ 3.
Stereoselectivity of Aspartate Transport
The EAATs and GltPh are able to transport l-aspartate and d-aspartate with similar affinities (3, 20, 26), illustrating that the EAAT and GltPh binding sites are not stereo-selective for d- or l-aspartate. To investigate the level of similarity between the ASCT1 and EAAT substrate binding site, we examined the substrate stereo-selectivity of ASCT1 and the mutants. For ASCT1 at pH 5.5, l-aspartate elicits currents, whereas d-aspartate does not generate any significant currents. In both A382T and T459R, d-aspartate generates currents, but d-aspartate is considerably less potent than l-aspartate (Table 1). This suggests that structural differences in the binding pocket of ASCT1 may be causing aspartate to bind in an alternate conformation. Interestingly, the double mutant A382T/T459R exhibits more similar EC50 values for d- and l-aspartate (Table 1). This demonstrates that the binding of acidic amino acids to the A382T/T459R transporter more closely resembles that of the EAATs and GltPh compared with the single mutant transporters.
DISCUSSION
ASCT1, the EAATs, and GltPh have a high degree of amino acid sequence similarity, but their transport characteristics are strikingly different. In this study we have used GltPh as a structural model and sequence alignments of the SLC1 family to identify three candidate residues in ASCT1 that are expected to influence differences in substrate selectivity between ASCT1 and the other SLC1 family members. We describe the effect of replacing alanine 382, threonine 459 and glutamine 386 in ASCT1 with their corresponding EAAT residues.
Arginine 397 in TM8 and the NMDGT motif in the unwound region of TM7 of GltPh are crucial to binding of acidic amino acids in the EAATs and GltPh (18–20, 28). These residues are highly conserved throughout the glutamate transporter family, but in ASCT1 a threonine replaces the arginine in TM8 and an alanine residue replaces the threonine in the TM7 motif NMDGA (Fig. 1C). Changing this arginine in EAAT3 to residues such as cysteine or glutamate abolishes glutamate transport and introduces Na+-dependent serine exchange (18, 19). Our results complement these studies, by demonstrating that the reverse mutation in ASCT1 switches the substrate selectivity from neutral to acidic amino acids (Fig. 3). Furthermore, the introduction of a threonine in TM7 to complete the NMDGT motif increases the ability of ASCT1 to bind aspartate, particularly at pH 5.5. These results reinforce the importance of the TM7 threonine/alanine difference and the TM8 arginine/threonine difference in determining substrate selectivity in the SLC1 family of transporters. Acidic amino acids have a higher affinity for the combined mutant transporter A382T/T459R compared with the individual mutant transporters (Fig. 3, Table 1). This implies that there is co-operativity between the arginine and threonine residues within the binding site of the EAATs and GltPh and that multiple residues contribute to defining differences in substrate selectivity between the transporters.
Further analysis of the substrate selectivity of the double ASCT1 mutant A382T/T459R reveals an additional interesting observation. The double mutant A382T/T459R has high affinity for both d- and l-aspartate, but comparatively low affinity for l-glutamate (Table 1). This differential selectivity is comparable to that of GltPh (20, 26) but quite different to that of the EAATs, which have similar affinities for l-, d-aspartate and l-glutamate (3). This suggests that it is possible to create a GltPh-like substrate binding site in ASCT1 with just two mutations. It also suggests that while the arginine residue is likely to be the major determinant of acidic amino acid binding, the hydroxyl group of the threonine residue facilitates the interaction and also removes the stereoselectivity of the site for the d- and l-aspartate. The EAATs and GltPh differ at the third amino acid residue investigated in this study, but the triple mutant, A382T/T459R/Q386E displays the same substrate selectivity as the double mutant (data not shown). Thus, there remain additional molecular determinants that are required to confer the complete EAAT phenotype. There are no obvious candidate residues in close proximity to the substrate binding site.
An unexpected observation from this study was that although acidic amino acids bind to the mutant transporters and activate anion conductances of similar amplitude to the serine-activated conductance of ASCT1, the transport rates of l-aspartate by the mutant transporters are markedly reduced compared with that of l-serine transport by wild type ASCT1 (Fig. 3). This highlights that the extent of anion conductance activation varies for different substrates and further re-inforces the notion that distinct conformational states support the transport and channel functions of members of this family of transporters (29–31).
Glutamate 404 in TM7 of EAAT2 (Q386 in ASCT1) has been proposed to form a K+/H+ binding site (24, 25) and is highly conserved throughout the EAAT family. In ASCTs and GltPh this residue is a glutamine and both of these transporters lack H+ and K+ coupling, which supports the proposal that E404 plays important roles in K+ and/or H+ binding. When this glutamate residue is mutated to a glutamine in EAAT3, the transporter functions as an electroneutral exchanger of acidic amino acids where there is no net transport of substrate (25). Interestingly, this homoexchange is pH independent indicating that H+ coupling has been impaired. While ASCT-mediated transport is not coupled to H+, ASCT1 does exhibit pH-dependent changes in substrate selectivity, with acidic amino acids able to bind at low pH (9). pH sensitivity of neutral amino acid transport was engineered in ASCT2 by generating the Q392E mutation (25), further supporting the role of this glutamate residue in H+ coupling. While replacing Q386 with a glutamate in ASCT1 did not introduce K+ and/or H+ coupling, acidic amino acid binding is impaired, with l-cysteate no longer being a substrate at low pH (Fig. 4). We hypothesize that the introduced glutamate residue is not protonated in ASCT1, thereby retaining its negative charge and repelling negatively charged amino acid substrates from the binding site. In EAAT3, the side chain of the equivalent glutamate residue has been shown to have an apparent pKa of 8 (32), and therefore would be protonated at physiological pH. We suggest that neighboring residues are influencing the proton binding capacity of the glutamate side chain, to shift the pKa from ∼4 to 8 within the transporter. A potential candidate is the threonine at the end of the NMDGT motif in TM7. This threonine is in close proximity to the side chain of the glutamate residue (13) (Fig. 1B), where the hydroxyl group may assist in stabilizing a H+ bound to glutamate. While acidic amino acids have no effect on the Q386E mutant, the double mutant A382T/Q386E does support acidic amino acid activated anion conductances (Fig. 4), which is consistent with the proposed role of the threonine residue in stabilizing a proton on the glutamate residue. Without the additional polarity from the hydroxyl group of threonine, the pKa of the glutamate side chain may remain closer to its natural pKa, thereby retaining its negative charge and impeding entry of acidic amino acids into the Q386E binding site. This implies that both a threonine and glutamate residue are required for H+ binding in the EAATs.
This study has contributed to our understanding of the structural basis for the differences in substrate selectivity between the members of the glutamate transporter family; ASCT1, the EAATs and GltPh. A further aspect that remains unresolved in this family is the structural basis for K+ binding and coupling to counter-transport that appears to be unique to the EAATs. A similar approach to the one described in this study may be useful in delineating the mechanistic details of counter-transport.
Acknowledgments
We thank Cheryl Handford for expert technical assistance and all who maintain the University of Sydney Xenopus laevis colony.
This work was supported by Discovery Grant DP1092729 from the Australian Research Council.
- ASCT
- alanine, serine, cysteine transporter
- EAAT
- excitatory amino acid transporter
- SLC1
- solute carrier family 1
- TM7
- transmembrane domain 7
- HP1
- hairpin loop 1
- GltPh
- prokaryotic aspartate transporter.
REFERENCES
- 1. Christensen H. N., Liang M., Archer E. G. (1967) A distinct Na+-requiring transport system for alanine, serine, cysteine, and similar amino acids. J. Biol. Chem. 242, 5237–5246 [PubMed] [Google Scholar]
- 2. Christensen H. (1990) Role of amino acid transport and countertransport in nutrition and metabolism. Physiol. Rev. 70, 43–77 [DOI] [PubMed] [Google Scholar]
- 3. Arriza J. L., Kavanaugh M. P., Fairman W. A., Wu Y. N., Murdoch G. H., North R. A., Amara S. G. (1993) Cloning and expression of a human neutral amino acid transporter with structural similarity to the glutamate transporter gene family. J. Biol. Chem. 268, 15329–15332 [PubMed] [Google Scholar]
- 4. Shafqat S., Tamarappoo B. K., Kilberg M. S., Puranam R. S., McNamara J. O., Guadaño-Ferraz A., Fremeau R. T., Jr. (1993) Cloning and expression of a novel Na (+)-dependent neutral amino acid transporter structurally related to mammalian Na+/glutamate cotransporters. J. Biol. Chem. 268, 15351–15355 [PubMed] [Google Scholar]
- 5. Utsunomiya-Tate N., Endou H., Kanai Y. (1996) Cloning and functional characterization of a system ASC-like Na+-dependent neutral amino acid transporter. J. Biol. Chem. 271, 14883–14890 [DOI] [PubMed] [Google Scholar]
- 6. Bröer A., Brookes N., Ganapathy V., Dimmer K. S., Wagner C. A., Lang F., Bröer S. (1999) The astroglial ASCT2 amino acid transporter as a mediator of glutamine efflux. J. Neurochem. 73, 2184–2194 [PubMed] [Google Scholar]
- 7. Sakai K., Shimizu H., Koike T., Furuya S., Watanabe M. (2003) Neutral amino acid transporter ASCT1 is preferentially expressed in L-Ser-synthetic/storing glial cells in the mouse brain with transient expression in developing capillaries. J. Neurosci. 23, 550–560 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Fuchs B. C., Bode B. P. (2005) Amino acid transporters ASCT2 and LAT1 in cancer: partners in crime? Semin. Cancer Biol. 15, 254–266 [DOI] [PubMed] [Google Scholar]
- 9. Tamarappoo B. K., McDonald K. K., Kilberg M. S. (1996) Expressed human hippocampal ASCT1 amino acid transporter exhibits a pH-dependent change in substrate specificity. Biochim. Biophys. Acta 1279, 131–136 [DOI] [PubMed] [Google Scholar]
- 10. Howard A., Goodlad R. A., Walters J. R., Ford D., Hirst B. H. (2004) Increased expression of specific intestinal amino acid and peptide transporter mRNA in rats fed by TPN is reversed by GLP-2. J. Nutr. 134, 2957–2964 [DOI] [PubMed] [Google Scholar]
- 11. Fairman W. A., Vandenberg R. J., Arriza J. L., Kavanaugh M. P., Amara S. G. (1995) An excitatory amino-acid transporter with properties of a ligand-gated chloride channel. Nature 375, 599–603 [DOI] [PubMed] [Google Scholar]
- 12. Arriza J. L., Eliasof S., Kavanaugh M. P., Amara S. G. (1997) Excitatory amino acid transporter 5, a retinal glutamate transporter coupled to a chloride conductance. Proc. Natl. Acad. Sci. U.S.A. 94, 4155–4160 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Yernool D., Boudker O., Jin Y., Gouaux E. (2004) Structure of a glutamate transporter homologue from Pyrococcus horikoshii. Nature 431, 811–818 [DOI] [PubMed] [Google Scholar]
- 14. Levy L. M., Warr O., Attwell D. (1998) Stoichiometry of the glial glutamate transporter GLT-1 expressed inducibly in a Chinese hamster ovary cell line selected for low endogenous Na+-dependent glutamate uptake. J. Neurosci. 18, 9620–9628 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Zerangue N., Kavanaugh M. (1996) Flux coupling in a neuronal glutamate transporter. Nature 383, 634–637 [DOI] [PubMed] [Google Scholar]
- 16. Ryan R. M., Mitrovic A. D., Vandenberg R. J. (2004) The chloride permeation pathway of a glutamate transporter and its proximity to the glutamate translocation pathway. J. Biol. Chem. 279, 20742–20751 [DOI] [PubMed] [Google Scholar]
- 17. Zerangue N., Kavanaugh M. P. (1996) ASCT-1 is a neutral amino acid exchanger with chloride channel activity. J. Biol. Chem. 271, 27991–27994 [DOI] [PubMed] [Google Scholar]
- 18. Conradt M., Stoffel W. (1995) Functional analysis of the high affinity, Na+-dependent glutamate transporter GLAST-1 by site-directed mutagenesis. J. Biol. Chem. 270, 25207–25212 [DOI] [PubMed] [Google Scholar]
- 19. Bendahan A., Armon A., Madani N., Kavanaugh M. P., Kanner B. I. (2000) Arginine 447 plays a pivotal role in substrate interactions in a neuronal glutamate transporter. J. Biol. Chem. 275, 37436–37442 [DOI] [PubMed] [Google Scholar]
- 20. Boudker O., Ryan R. M., Yernool D., Shimamoto K., Gouaux E. (2007) Coupling substrate and ion binding to extracellular gate of a sodium-dependent aspartate transporter. Nature 445, 387–393 [DOI] [PubMed] [Google Scholar]
- 21. Huang Z., Tajkhorshid E. (2010) Identification of the Third Na+ Site and the Sequence of Extracellular Binding Events in the Glutamate Transporter. Biophys. J. 99, 1416–1425 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Bastug T., Heinzelmann G., Kuyucak S., Salim M., Vandenberg R. J., Ryan R. M. (2012) Position of the Third Na+ Site in the Aspartate Transporter GltPh and the Human Glutamate Transporter, EAAT1. PLoS One 7, e33058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Tao Z., Rosental N., Kanner B. I., Gameiro A., Mwaura J., Grewer C. (2010) Mechanism of cation binding to the glutamate transporter EAAC1 probed with mutation of the conserved amino acid residue T101. J. Biol. Chem. 285, 17725–17733 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Kavanaugh M. P., Bendahan A., Zerangue N., Zhang Y., Kanner B. I. (1997) Mutation of an amino acid residue influencing potassium coupling in the glutamate transporter GLT-1 induces obligate exchange. J. Biol. Chem. 272, 1703–1708 [DOI] [PubMed] [Google Scholar]
- 25. Grewer C., Watzke N., Rauen T., Bicho A. (2003) Is the glutamate residue Glu-373 the proton acceptor of the excitatory amino acid carrier 1? J. Biol. Chem. 278, 2585–2592 [DOI] [PubMed] [Google Scholar]
- 26. Ryan R. M., Compton E. L., Mindell J. A. (2009) Functional characterization of a Na+-dependent aspartate transporter from Pyrococcus horikoshii. J. Biol. Chem. 284, 17540–17548 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Poulsen M. V., Vandenberg R. J. (2001) Niflumic acid modulates uncoupled substrate-gated conductances in the human glutamate transporter EAAT4. J. Physiol. 534, 159–167 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Seal R. P., Leighton B. H., Amara S. G. (2000) A model for the topology of excitatory amino acid transporters determined by the extracellular accessibility of substituted cysteines. Neuron 25, 695–706 [DOI] [PubMed] [Google Scholar]
- 29. Borre L., Kavanaugh M. P., Kanner B. I. (2002) Dynamic equilibrium between coupled and uncoupled modes of a neuronal glutamate transporter. J. Biol. Chem. 277, 13501–13507 [DOI] [PubMed] [Google Scholar]
- 30. Seal R. P., Shigeri Y., Eliasof S., Leighton B. H., Amara S. G. (2001) Sulfhydryl modification of V449C in the glutamate transporter EAAT1 abolishes substrate transport but not the substrate-gated anion conductance. Proc. Natl. Acad. Sci. U.S.A. 98, 15324–15329 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Ryan R. M., Vandenberg R. J. (2002) Distinct conformational states mediate the transport and anion channel properties of the glutamate transporter EAAT-1. J. Biol. Chem. 277, 13494–13500 [DOI] [PubMed] [Google Scholar]
- 32. Watzke N., Rauen T., Bamberg E., Grewer C. (2000) On the mechanism of proton transport by the neuronal excitatory amino acid carrier 1. J. Gen. Physiol. 116, 609–622 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. DeLano W. L. (2010) The PyMOL Molecular Graphics System, version 3.1, Schrödinger, LLC, New York [Google Scholar]