

Background: MIF signals through the MAPK pathway to up-regulate proinflammatory cytokine synthesis.
Results: Pseudomonas aeruginosa-induced infection up-regulates MIF homotrimer formation and stimulates assembly of caveolin-1-rich lipid raft, thereby promoting MAPK signaling.
Conclusion: The homotrimeric form of MIF is the functionally active form of the molecule.
Significance: The discovery of the functionally active oligomeric form of MIF allows preferential pharmaceutical targeting.
Keywords: Cornea, Infectious Diseases, Inflammation, Innate Immunity, Pseudomonas aeruginosa, Macrophage Migration Inhibitory Factor
Abstract
Acute inflammation that arises during Pseudomonas aeruginosa-induced ocular infection can trigger tissue damage resulting in long term impairment of visual function, suggesting that the appropriate treatment strategy should include the use of anti-inflammatory agents in addition to antibiotics. We recently identified a potential target for modulation during ocular infection, macrophage migration inhibitory factor (MIF). MIF deficiency protected mice from inflammatory-mediated corneal damage resulting from acute bacterial keratitis. To gain a better understanding of the molecular mechanisms of MIF activity, we analyzed the oligomeric states and functional properties of MIF during infection. We found that in human primary corneal cells infected with P. aeruginosa, MIF is primarily in a homotrimeric state. Homotrimeric MIF levels correlated with the severity of infection in the corneas of infected mice, suggesting that the MIF homotrimers were the functionally active form of MIF. During infection, human primary corneal cells released more IL-8 when treated with recombinant, locked MIF trimers than when treated with lower MIF oligomers. MIF promoted P. aeruginosa–induced IL-8 responses via the formation of caveolin-1-rich “signaling hubs” in the corneal cells that led to elevated MAPK p42/p44 activation and sustained inflammatory signaling. These findings suggest that inhibiting homotrimerization of MIF or the functional activities of MIF homotrimers could have therapeutic benefits during ocular inflammation.
Introduction
Macrophage migration inhibitory factor (MIF),2 first described more than 50 years ago, owes its name to the observation that MIF, released by activated T-lymphocytes, leads to inhibition of macrophage migration in vitro (1, 2). Since its initial discovery, various biological activities have been attributed to MIF stemming from various phenotypic characteristics of the MIF-deficient mice, which were used in models of rheumatoid arthritis, diabetes, atherosclerosis, sepsis, asthma, and acute respiratory distress syndrome (3–7). All of these studies highlighted that MIF deficiency is associated with decreased levels of inflammatory responses, suggesting that inhibition of MIF by small molecules or antibodies could be therapeutically effective in settings of inflammatory damage. One of the major challenges in utilizing therapeutics, which modulate inflammation, is the resultant increased risk for infections. Interestingly, however, MIF-deficient mice appear protected from the consequences of acute P. aeruginosa lung and ocular infections, further strengthening the concept that reducing MIF activity may be more beneficial than harmful (8–10).
During an LPS challenge of mice, MIF induces sustained high production of a large panel of proinflammatory cytokines such as TNF-α, IFN-γ, IL-1β, IL-2, IL-6, IL-8, MIP-2, NO, Cyclooxygenase 2 (Cox2), products of the arachidonic acid pathway, and matrix metalloproteinases and thereby amplifies the proinflammatory responses (11–14). Although the molecular mechanisms underlying MIF-triggered inflammation are only partially elucidated, the significance of MIF-dependent pathways in tissue damage are based on the finding that MIF-deficient mice develop milder corneal damage from P. aeruginosa-induced keratitis, characterized by reduced bacterial levels and neutrophil infiltration and decreased overall inflammatory responses (8). These observations suggest that inhibition of MIF may be therapeutically beneficial in states of acute ocular inflammation (8).
MIF has both oxydoreductase and tautomerase enzymatic activities. Although the natural substrates for MIF remain obscure, small molecular inhibitors that target the enzymatically active sites of MIF hinder its biologic functions. For example, the tautomerase inhibitors that block the catalytically active proline residue either irreversibly, by forming covalent complexes (e.g. N-acetyl-p-benzoquinone imine (NAPQI) and 4-Iodo-6-phenylpyrimidine (4-IPP)), or by competing with the substrate (e.g. ISO-1 and OXIM11), lead to decreased levels of TNF-α elicited by LPS-challenged macrophages and increase mouse survival from sepsis (15). It is generally considered that inhibitors that target the tautomerase activity of MIF also affect the potency of binding to the CD74, a putative MIF receptor, thus revealing a connection between the tautomerase activity and receptor interaction (16, 17).
MIF induces the proinflammatory responses by forming a complex with the extracellular domain of the CD74 (18). Signal transduction is dependent upon CD74 and CD44 complex formation and translates in activation of MAPK p42/p44 (19, 20). Sustained activation of MAPK p44/p42 promotes cell survival and cell adhesion. Depending on the cell type, those complexes may include CXCR2, CXCR4, or CXCR7, additional MIF-recognizing receptors.
Keeping in mind that CD74 has a homotrimeric structure (18) and that MIF-CD74 interactions sustain inflammatory responses, it seems reasonable to hypothesize that MIF signaling through CD74 requires a complementary homotrimeric form of MIF. Crystallographic studies revealed that MIF crystallizes as a homotrimer with a molecular mass of 37.5 kDa (21–23); however, in certain cell lines, MIF oligomerization state is reported to be monomeric (molecular mass, 12.5 kDa) (24). Intriguingly, bovine MIF was reported to have multiple oligomeric forms but not homotrimers (25). Currently, it is not clear whether the functionally active form of MIF is the homotrimeric one or whether different oligomeric states of MIF have distinct functional properties: this may be attributed mostly to the requirement to produce stable recombinant MIF monomers and trimers.
We hypothesized that regulation of proinflammatory responses depends on the MIF oligomeric state leading to regulation of the assembly of signaling platforms. In the case of P. aeruginosa infection, these platforms could harbor specific pattern recognition receptors, thereby creating pathogen-specific signaling “hubs.” The half-life of these hubs could then determine the magnitude and the extent of the inflammatory responses. To define the oligomeric state of MIF that promotes inflammatory responses to P. aeruginosa, we compared the effects of “locked” MIF trimers versus MIF monomers on the inflammatory responses induced by P. aeruginosa corneal infection.
EXPERIMENTAL PROCEDURES
Mice
All studies were performed in accordance with the Harvard Medical School Institutional Animal Care and Use Committee guidelines. The experimental protocols were approved by the Institutional Animal Care and Use Committee of the Harvard Medical Area Office for Research Subject Protection and were consistent with the Association for Research in Vision and Ophthalmology guidelines for studies in animals.
C57BL6 or BALB/C mice were obtained from Harlan or Taconic Farms. Mice were housed and bred in the Channing Laboratory Animal Care Facilities.
Infection Model
Mice were anesthetized with ketamine and xylazine anesthetics (26). Three 0.5-cm scratches were made on the cornea and a 5-μl inoculum of P. aeruginosa delivered in onto the eye. Mice remained sedated for ∼30 min. The animals were followed for 48 h after which they were euthanized, and corneas were collected for analysis. Corneas were pooled from four individual animals and solubilized in 25 mm HEPES, pH 7.5, 50 mm NaCl, 4% sucrose, 1% trehalose, 1% Triton X-100, supplemented with Complete Mini protease inhibitor mixture (Roche Diagnostics).
Bacterial Strains, Cell Culture, Inocula, and in Vitro Infection Assays
P. aeruginosa strains 6294 and PA14 were used throughout these experiments. Bacteria were grown overnight at 37 °C on tryptic soy agar plates prior to experiments. Primary human corneal epithelial cells (HPCEC) were grown in six-well plates in either keratinocyte serum free medium (SFM) growth medium (Invitrogen) supplemented with penicillin, streptomycin, EGF, and pituitary gland extract, or in MEM/F12 mix supplemented with EGF, insulin, dimethyl sulfoxide, and cholera toxin as described in Ref. 27. Confluent monolayers of HPCEC were infected with different strains of P. aeruginosa at a multiplicity of infection of 20–40 for 1 h at 37 °C. Following treatment for 1 h with 300 μg/ml gentamicin, cells were supplemented with fresh growth medium. The tissue culture supernatants were collected at 6 and 24 h after infection to determine P. aeruginosa-induced cytokine levels. To monitor MAPK activity after P. aeruginosa infection, HPCEC were lyzed at 15, 30, 60, and 90 min after the onset of infection. Total cell lysates were prepared in radioimmune precipitation assay buffer (50 mm Tris-HCl, pH 7.5, 150 mm NaCl, 1% Triton X-100, 1% sodium deoxycholate, 1% SDS), supplemented with Complete Mini protease inhibitor mixture (Roche Diagnostics).
HPCEC
HPCEC were kindly provided by Dr. T. Zaidi and were harvested from corneal rings removed at surgery as described in Ref. 27. Cells were passaged in Keratinocyte SFM medium (Invitrogen), supplemented with EGF and pituitary extract. HPCEC were used within 5–7 passages. In addition, human primary corneal epithelial cells were purchased from CELLnTEC and maintained per the manufacturer's instructions.
Cytokine Analysis
Commercially available ELISA assays (R&D Systems) were used to determine IL-8 levels.
siRNA Experiments
Production of MIF by the primary HCEC was evaluated in the presence of MIF-specific siRNA treatments. Briefly, cells were transfected with MIF-specific or control siRNA (On-target plus SMARTpool; Dharmacon) by incubating with a transfection mixture that consisted of 50 nm siRNA (Dharmacon) and 6 μl of Oligofectamine (Invitrogen) in 1 ml of Opti-MEM (Invitrogen) for 48 h, after which the cells were maintained in regular growth medium. Epithelial cells were infected with P. aeruginosa (strain 6294) within 48 to 72 h after siRNA treatment. An additional non-siRNA-treated control was also included to determine whether there is an effect on inflammatory gene expressing from the control siRNA treatment. To monitor caveolin-1 function during infection, HPCEC were pretreated with caveolin-1-specific siRNA (ONtarget plus SMARTpool; Dharmacon) compared with control siRNA treatment.
Lipid Raft Preparations
HPCEC cells were grown to confluency and infected with P. aeruginosa (strain 6294 or PA14) at an multiplicity of infection of 20–40. Cells were washed twice with ice-cold PBS, scraped, pelleted by centrifugation at 1600 rpm (Beckman L8-M Ultracentrifuge) for 5 min at 4 °C. Pellets were resuspended in 500 μl of MBS, pH 6.5 (25 mm MES, 150 mm NaCl) containing Complete Mini protease inhibitor mixture. The cell suspension was then passed through a 22-gauge needle on a 1-ml syringe for five times to ensure homogenous suspension and 500 μl of 1% Triton-X-100 in MBS were added to the cell suspension. Cells were Dounce-homogenized on ice 20 times, and incubated for an additional 30 min to ensure complete lysis. The lysate was diluted with an equal volume of 80% sucrose/MBS, overlaid with 6.5 ml of 35% sucrose/MBS, and 2 ml of 5% sucrose/MBS. Samples were centrifuged in a Beckman SW40Ti swinging-bucket rotor (Beckman L8-M Ultracentrifuge) at 38,000 × g for 24 h at 4 °C. After centrifugation, 12 aliquots of 1-ml fractions were sequentially collected from the bottom of each tube. Aliquots of each fractions were mixed with NuPAGE sample buffer (Invitrogen), run on 4–12% NuPAGE gels (Invitrogen), and transferred, and protein present was analyzed by Western blotting.
Generation of Recombinant MIF N110C
A recombinant MIF (rMIF) construct substituting asparagines 110 with cysteine (N110C) was generated by overlapping PCR using human MIF cDNA (ATCC, IMAGE 2821346) sequence cloned into pET11b plasmid (Invitrogen). For PCR reaction 1, primer set 1 (primer 1, 5′-TAAGCTTTAATGCGGTAGTTTATCACAGTT-3′ and primer 3, 5′-CCAATG TGGGCTGGAACTGCTC-3′) was used to amplify MIF to generate fragment 1. Simultaneously, PCR reaction 2 used primer set 2 (primer 2, 5′-GGAGCAGTTCCAGCCCACATTG-3′ and primer 4, 5′-CACTATAGGGGAATTGTGAGCGGAT-3′) to amplify fragment 2. Both fragments 1 and 2 incorporated the N110C substitution. Following gel extraction (Qiagen), the purified PCR fragments 1 and 2 were used as templates, along with primers 1 and 4 to produce a 731-bp rMIF-N110C fragment. PCR reactions were run as follows: 35 cycles; 30 min at 95 °C, 45 min at 55 °C, and 45 min at 72 °C. The fragments were amplified with the proofreading Pfu polymerase (Stratagene). The resulting 731-bp PCR fragment was digested with BamH1 and XbaI, and the 348-bp fragment subcloned into pET11b for protein expression. The cDNA for the wild type rMIF and the rMIF N110C mutant were sequence-verified.
Purification of rMIF and rN110C MIF
Wild type MIF and rMIF N110C proteins were produced in Escherichia coli BL21 Star(DE3) cells (Invitrogen), after induction with 1 mm isopropyl 1-thio-β-d-galactopyranoside for 3.5 h. Bacterial cells were pelleted by centrifugation in a Beckman Allegra 6R centrifuge (3,000 rpm, 15 min, ambient temperature). Approximately 1 g of bacterial pellet was resuspended in 5 ml of 50 mm Tris, pH 8.5, 50 mm KCl, 5 mm magnesium acetate, 0.1% sodium azide and sonicated. The lysate was centrifuged at 15,000 rpm for 20 min and then filtered through Millipore Steriflip vacuum filtration system with 0.2-μm membranes. The filtered lysate was loaded onto a 5-ml QHP HiTrap anion exchange column that was connected in series to a 5-ml Bio-Rad ceramic hydroxyapatite type II column, and then a 1-ml ToyoScreen phenyl 650 m hydrophobic interaction chromatography (phenyl HIC) column. The columns were equilibrated and washed with 25 mm HEPES, pH 7.5, 100 mm NaCl. The tandem arrangement of the columns was used to minimize the handling of the MIF protein. The first two columns functioned in a negative chromatography mode such that rMIF remained unbound. The rMIF was sufficiently hydrophobic such that high levels of salt (chaotropic or lyotropic) were not required to promote binding to the phenyl HIC column.
Purification of rMIF from HEK293 Cells
MIF cDNA was recloned into pcDNA3.3 (Invitrogen) using the pcDNA3.3 Topo TA cloning kit (Invitrogen) and used to transfect HEK 293 cells. Cell lysates were collected 7 days post-transfection, and rMIF monomers and trimers were purified as described above and in Ref. 28.
Size-exclusion Chromatography
Gel-filtration experiments were performed using a Thermo Accela System (PDA detection) using a TOSOH Bioasist G2SWXL column. 100 μg of the sample was analyzed using an isocratic gradient (25 mm HEPES, pH 7.3, 100 mm NaCl, 4% sucrose) with a flow rate of 1 ml/min with detection at 280 nm.
Immunohistochemical Analysis
Enucleated mouse eyes were fixed in 4% formalin, embedded into paraffin blocks, and cut into 4-μm sections. To visualize MIF protein in the ocular tissues, the sections were deparaffinized by a series of 15-min washes: two washes in 100% xylene, two washes in 90% ethanol, two washes in 75% ethanol, and two washes in water. Antigen was retrieved by boiling the washed sections in 10 mm sodium citrate at pH 6.0 for 30 min, followed by permeabilizing the tissue by incubating the sections in PBS containing 0.3% Triton X-100 for 15 min, blocking with PBS containing 0.3% Triton X-100 and 2% milk for 30 min and staining with a rabbit anti-rat MIF (Invitrogen) overnight in PBS containing 0.3% Triton X-100 and 2% milk at 4 °C. Sections were then washed three times in PBS and incubated for 1 h with secondary goat anti-rabbit IgG conjugated to Alexa Fluor 647, applied at a 1:200 dilution in PBS, 0.3% Triton X-100. Finally, sections were washed three times in PBS and counterstained with Sytox Green (Invitrogen) at a final concentration of 50 nm for 10 min at room temperature. Images were collected using an LSM 510 META laser scanning confocal microscope (Zeiss, Germany).
Statistical Analysis
Cytokine levels were compared among samples by two-tailed t tests (Prism 4, Graph Pad).
RESULTS
MIF Homotrimers Are Prevalent during P. aeruginosa Infection
With the exception of a single report on MIF expression by corneal epithelial cells as a consequence of either cytotoxic or invasive P. aeruginosa infection (29), no information is available on the cellular sources, signaling pathways, or mechanisms responsible for MIF release during ocular infection. To define key cell types responsible for MIF production in the cornea, immunohistochemical analysis was performed on mouse ocular tissues. In the resting corneas, the strongest MIF-specific signal was observed in the corneal epithelium (Fig. 1). However, MIF expression was not solely restricted to the corneal epithelium, demonstrating that multiple cell types express MIF. When rMIF was added simultaneously with the primary polyclonal anti-MIF during the staining, no specific signal was detected. Consistently, MIF-deficient corneas showed only background staining (data not shown).
FIGURE 1.

Immunochistochemical analysis for MIF in mouse corneas. Corneas from wild type C57BL6 mice were stained with rabbit anti-rat MIF polyclonal Ab, followed by goat anti-rat Alexa Fluor 647 secondary antibody in the presence or absence of rMIF and counterstained with the DNA-specific dye Sytox Green. The staining pattern indicates MIF enrichment in the corneal epithelium.
To characterize MIF protein distribution in HPCEC during infection, primary HPCEC were infected with P. aeruginosa 6294, and lipid raft fractions were purified based on Triton X-100 solubility. Western blotting analysis using polyclonal antibody to MIF revealed that in the non-infected primary HPCEC, MIF was predominantly localized within the cytoplasmic fractions (fractions 1–5, Fig. 2A), whereas infection induced enrichment of MIF within the lipid raft fractions (fractions 7–11, Fig. 2B). These changes correlated with the previously described enrichment of the scaffolding protein caveolin-1 into the lipid raft fractions in response to P. aeruginosa challenge and occurred within minutes after the onset of infection (30). Furthermore, we observed that infection induced enrichment for higher oligomeric forms of MIF, namely MIF trimers, in the lipid raft fractions.
FIGURE 2.

P. aeruginosa infection induces enrichment of MIF trimers in the caveolin-rich lipid raft structures. Non-infected (A) and P. aeruginosa 6294-infected (B) HPCEC were solubilized using Triton X-100 and subjected to sucrose density gradient centrifugation. The resulting fractions were loaded in a non-boiled, non-reduced form onto SDS-PAGE and analyzed for presence of MIF and caveolin-1 by Western blotting. Fractions 1–5 were enriched with cytoplasmic-derived proteins, whereas fractions 8–10 contained lipid raft-associated proteins. MIF containing fractions were overlaid with caveolin-1-containing fractions. Data are representative of four experiments. Western blotting images were analyzed by densitometry, and the levels of MIF monomers, trimers, and caveolin-1 were compared by measuring the intensity of the individual bands within the individual lanes. MOI, multiplicity of infection.
To question whether MIF trimers gave rise to MIF monomers, we overexpressed MIF in HEK293 cells under in minimal growth medium and purified rMIF monomers and trimers (Fig. 3). When denatured, the purified MIF trimers gave raise to MIF monomers (Fig. 3). Eukaryotic-derived MIF monomers and trimers had differential mobility on SDS-PAGE, which corresponded to the molecular masses for MIF monomers of 12.5 kDa and MIF trimers of 37.5 kDA (Fig. 3A). Consistently, native PAGE analysis of the purified rMIF material also revealed differential mobility pattern (Fig. 3B). rMIF monomers appeared to be retarded when compared with rMIF trimers. To confirm the data from the SDS-PAGE and native PAGE analysis, we analyzed the mobility properties of the two individual preparations using size-exclusion HPLC chromatography and observed that rMIF monomer preparation migrated slower than rMIF trimers (Fig. 3C). In addition, the rMIF trimer band was gel-excised, subjected to trypsin digest, sequenced, and yielded a sequence identical to that of human MIF.
FIGURE 3.

Purification and characterization of eukaryotic derived-rMIF. A, SDS-PAGE analysis of MIF oligomeric states. rMIF was overexpressed in HEK 293 cells, and the different rMIF oligomeric forms were purified. Lane 1, purified rMIF monomers; lane 2, purified rMIF trimers; lane 3, boiled rMIF trimers. B, native PAGE analysis of purified rMIF monomers and trimers. MIF bands were visualized by Western blotting using anti-MIF IgG recognizing both the monomers and trimers. C, HPLC profiles of MIF trimers (black peak) and MIF monomers (gray peak) separated via size-exclusion chromatography. mAU, microabsorbance units; expression of macrophage migration inhibitory factor during Pseudomonas keratitis.
To characterize MIF oligomeric states in the mouse corneas during infection in vivo, C57BL6 mice, which are highly sensitive to P. aeruginosa infection, and BALB/C mice, which are less susceptible to infection, were inoculated with 1 × 106 cfu of P. aeruginosa per eye, and corneal tissues were harvested, total cellular lysates were prepared and analyzed for MIF presence by SDS-PAGE, followed by Western blotting (31, 32). In the resting corneas, MIF existed predominantly in a monomeric state; however, infection of the C57BL6 mice with P. aeruginosa 6294 induced enrichment of higher molecular weight MIF-containing band (Fig. 4). In contrast, infected BALB/C mice demonstrated less disease and had less presence of trimeric MIF.
FIGURE 4.

Sensitivity to infection correlates with the presence of MIF trimers in the cornea. Corneas derived from non-infected and infected C57BL6 and BALB/C mice were harvested 48 h post-infection with P. aeruginosa 6294. Fifty micrograms of total corneal lysates were loaded onto SDS-PAGE in a non-boiled, non-reduced form and analyzed by Western blotting using rabbit anti-mouse MIF antisera. Lane 1, lysates from non-infected C57BL6 corneas; lane 2, lysates from infected C57BL6 corneas; lane 3, lysates from non-infected BALB/C corneas; lane 4, lysates from infected BALB/C corneas. The relative abundance of the individual bands was calculated by densitometry. Data are representative of three experiments.
Locked MIF Trimers Are Amplifiers of Proinflammatory Responses
Our current understanding of the biological activities of MIF is based on in vitro experiments using rMIF to stimulate MIF-dependent signaling pathways (33, 34). The E. coli-derived rMIF preparation contains different oligomeric forms of rMIF, raising the question of whether certain oligomeric forms specifically induce MIF-dependent functions. Because infection induced enrichment of the trimeric form of MIF in vivo (Fig. 4), we hypothesized that the elevated and sustained presence of the trimeric MIF is associated with an increase in the inflammatory responses and resultant tissue damage. To address this issue, we produced a mutant version of MIF, where the C-terminal Asn at position 110 was substituted to Cys. This mutant protein was reported previously to exist in a trimeric form due to an intersubunit disulfide bonding (35). Native PAGE comparison of the mobility profiles of E. coli-derived wild type rMIF and the N110C rMIF showed that the N110C mutant migrated faster than the rMIF, suggestive that the N110C MIF mutants existed predominantly in a higher oligomeric form, whereas the wild type rMIF existed in a monomerc form (Fig. 5). These observations agreed with the previously published results (35, 36).
FIGURE 5.
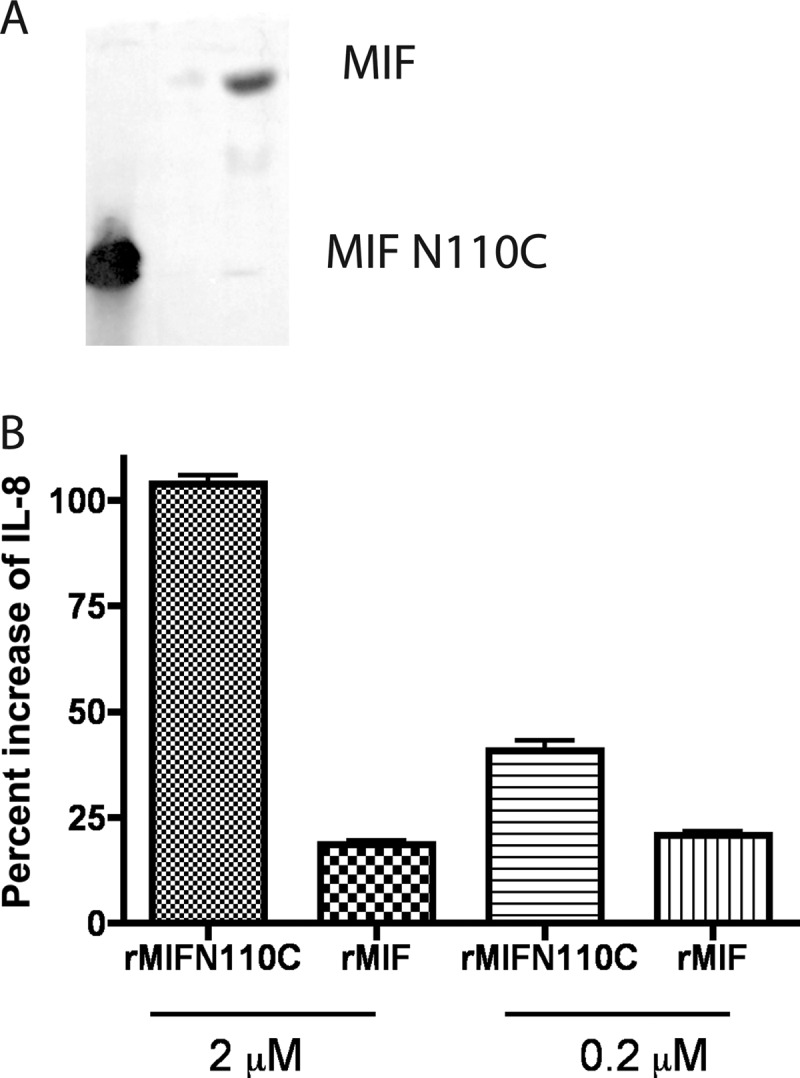
Reconstitution with locked MIF trimers increases IL-8 synthesis by HPCEC. A, native PAGE analysis of E. coli-derived rMIF N110C and rMIF. The N110C rMIF and rMIF monomers were separated on native PAGE and visualized by Western blotting analysis using anti-MIF IgG recognizing both MIF monomers and trimers. B, HPCEC were treated with MIF-specific siRNA to reduce MIF levels, infected with P. aeruginosa strain 6294 at multiplicity of infection 30, and reconstituted with either 2 or 0.2 μm of purified recombinant MIF N110C trimers or rMIF monomers. Tissue culture supernatants were collected at 6 h after infection and analyzed for IL-8 levels by ELISA. Data are presented as mean values ± S.D. and are representative of two independent experiments.
Because reducing MIF levels in the primary HPCEC during P. aeruginosa-induced infection decreased the IL-8 and IL-6 responses mounted by the epithelial cells (8), we questioned the functional consequences of reconstituting MIF knocked down HPCEC with either N110C or rMIF wild type by measuring the response of corneal cells to equimolar concentrations of locked N110C rMIF trimers or rMIF preparation, containing predominantly monomeric MIF. When primary HPCEC were pretreated with MIF-specific siRNA to reduce endogenous MIF levels and reconstituted with either locked N110C rMIF trimers or rMIF monomers, the IL-8 release was significantly higher after treatment with rMIF trimers than with rMIF monomers.
MIF Regulates Caveolin-rich Platform Assembly and Signaling
To characterize the molecular mechanisms of MIF-induced modulation of bacteria-elicited inflammatory responses, primary HPCEC were treated with control siRNA or MIF-specific siRNA, and lipid raft fractions were prepared and analyzed by Western blotting for recruitment of signaling molecules described previously to be activated in response to infection (37, 38). As expected, caveolin-1, a scaffolding protein associated with the lipid rafts was partitioned between the cytoplasmic and the lipid raft fractions. The distribution of caveolin-1 changed after infection of control siRNA-treated cells. Increased levels of caveolin-1 were detected as early as 20 min after bacterial challenge. In contrast, when cells were treated with MIF siRNA to reduce endogenous MIF levels, caveolin-1 levels in the lipid-raft structures were lowered, indicating that MIF regulates caveolin-1 translocation (Fig. 6A). Much less pronounced caveolin-1 enrichment within the Triton X-lipid raft fractions was also observed during infection with ExoU expressing P. aeruginosa strain PA14, suggesting that both invasive and cytotoxic strains may induce protein redistribution to lipid rafts (Fig. 6B).
FIGURE 6.

MIF regulates the clustering of P. aeruginosa-induced caveolin-1-rich signaling hubs and downstream signaling. A, non-infected and infected with P. aeruginosa 6294 corneal cells were treated with control (CNTR) siRNA (top two panels) or MIF-specific siRNA (bottom two panels), solubilized using Triton X-100, and subjected to sucrose density gradient centrifugation, and the resulting fractions were analyzed for caveolin-1 by Western blotting. The graphs present the mean band intensities for the caveolin-1 bands for the individual fractions. B, non-infected and infected with P. aeruginosa PA14 HPCEC were solubilized using Triton X-100 and subjected to sucrose density gradient centrifugation, and the resulting fractions were analyzed for caveolin-1 by Western blotting. The mean band intensity values for the caveolin-1 bands were plotted for the individual fractions. C, HPCEC were treated with either CNTR siRNA or caveolin-1 (Cav-1) siRNA to reduce caveolin-1 levels prior to infection with P. aeruginosa 6294, tissue culture supernatants were collected at 6 h after the infection, and IL-8 levels were measured by ELISA. Data are shown as mean ± S.D. Data are representative out of three independent experiments.
To determine the impact of caveolin-1 on P. aeruginosa-induced HPCEC responses, levels of IL-8, major proinflammatory cytokine, were measured in cells pretreated with caveolin-1-sprecific siRNA. The reduction of caveolin-1 protein levels resulted in reduced IL-8 synthesis (Fig. 6C).
As an adaptor protein, caveolin-1 forms complexes with growth factor receptors, cell-substrate adhesion molecules, and kinases, facilitating inside-out and outside-in cell signaling. As the interaction of caveolin-1 with MAPK has been documented previously (38), we analyzed the role of caveolin-1 in regulating MAPK p42/p44 activation during P. aeruginosa infection. In primary HPCEC, P. aeruginosa infection induced rapid MAPK p42/p44 accumulation into the lipid rafts (Fig. 7), which was accompanied with MAPK p42/p44 phosphorylation. When cells were treated with caveolin-1-specific siRNA or MIF-specific siRNA, MAPK p42/p44 activation was ablated (Fig. 7), indicating that MIF regulates MAPKp42/p44 activation by modifying caveolin-1-rich platform assemblies in response to P. aeruginosa 6294 infection.
FIGURE 7.

Down-regulating caveolin-1 levels in HPCEC ablates MAPK p42/p44-dependent signaling. A, P. aeruginosa infection stimulated MAPK p42/p44 rapid recruitment to lipid rafts in caveolin-1-sufficient cells. HPCEC were infected with P. aeruginosa 6294 strain for 20 min, and lipid raft fractionations were carried out. Samples were analyzed for MAPK p42/p44 partitioning by Western blotting (top two panels). MAPK p42/p44 activation was detected by probing with phospho-specific p42/p44 Ab (bottom two panels). Western blot images were analyzed by densitometry and the presence of MAPK p42/p44 compared by measuring the intensity of the individual bands within the individual lanes. The graphs present relative intensities of the bands for the individual fractions. B, reducing MIF or caveolin-1 levels resulted in a decrease of MAPK p42/p44 activation elicited by P. aeruginosa 6294 infection. HPCEC were treated with control siRNA (CNTR), MIF-specific siRNA, or caveolin-1 (Cav-1)-specific siRNA, infected with P. aeruginosa strain 6294 at multiplicity of infection 30, total cellular lysates were prepared by lysis in radioimmune precipitation assay buffer at 15, 30, or 60 min post-infection. Equal levels of total protein were loaded onto 4–12% NuPAGE and analyzed for phospho-p42/p44 by Western blotting. The Western blot images were analyzed by densitometry, and the percent activation of MAPK p42/p44 was plotted as a function of time. MOI, multiplicity of infection.
DISCUSSION
Eye trauma and contact lens wear are the leading factors predisposing for inflammatory pathologies of the cornea related to infectious keratitis. Eye infections induced by P. aeruginosa can damage the entire cornea within 48 h and can ultimately cause corneal perforation. Treatment requires frequent application of broad spectrum antibiotics, administered as often as every 15–60 min for a period from 3–21 days to counteract the rapidly progressing acute bacterial infection. Although antibiotics are usually effective at limiting bacterial propagation, host inflammatory responses against the invading pathogens frequently result in a number of morbidities such as ocular tissue damage, delay of successful corneal healing, and fibrosis of portions of the cornea. Therefore, an optimal therapeutic strategy should include administering antibiotics and anti-inflammatory agents to inhibit bacterial proliferation and limit inflammation-mediated tissue damage. However, two recent clinical studies did not see a benefit from use of corticosteroids in the treatment of bacterial corneal ulcers in general or specifically for P. aeruginosa keratitis (39, 40). We are not surprised at these findings as MIF antagonizes the anti-inflammatory effects of glucocorticoids, having the ability to reverse the inhibitory effects of glucocorticoids on inflammatory cytokine production (14). Therefore, our results suggest that the failure to see a therapeutic benefit from glucocorticoids in bacterial keratitis could be due to MIF overproduction, Thus, to see a benefit from an anti-inflammatory therapy may require inhibition of MIF trimer formation.
Our recent findings were supportive of the concept that targeting MIF could have therapeutic benefits in reducing inflammatory damage during bacterial keratitis, using a model of bacterial keratitis in mice induced by scratch injury, representative of corneal trauma, demonstrating that inhibiting MIF in the presence of antibiotic treatment was protective during acute infection induced by P. aeruginosa (8). Here, we extended our understanding of the molecular basis for the role of MIF in the pathogenesis of keratitis by examining whether different oligomeric forms of MIF exist in vivo. Using recombinant monomeric or trimeric human MIF, we found that the locked trimeric MIF isoform was more efficient at inducing IL-8 production by primary HPCEC, suggesting that the homotrimers are an active MIF isoform mediating stimulation of proinflammatory functions by MIF (Fig. 5). We also observed that the predominant form of MIF during infection of the sensitive C57BL6 strain is the higher oligomeric form of MIF. Our results are consistent with the recently published screens for MIF inhibitors (35) and strengthen the scientific rationale for screening for MIF inhibitors that target the trimeric state of the molecule to achieve a decrease in proinflammatory responses where deemed necessary.
It is of future interest to determine how dynamic are the different isoforms, what drives trimer assembly, whether trimer assembly is concentration dependent, and whether there are any isoform-specific functions. To this end, we expect that the homotrimeric MIF breaks down to monomers in a concentration- and pH-dependent manner with the trimeric form being predominant at higher concentrations.
To gain additional insight into other factors involved in MIF-regulated cell signaling events, we analyzed the dynamics and protein composition of lipid rafts formed in response to P. aeruginosa-induced infection. P. aeruginosa uses caveolin-1-enriched endocytic pathways to gain entry into lung and corneal epithelial cells (37, 42). Caveolin-1 is found in the lipid raft microdomains when isolated into Triton X-100 insoluble fractions (43, 44). Reducing MIF levels resulted in a decrease in caveolin-1 enrichment into lipid raft fractions, and this effect translated into a decrease in MAPK p42/p44 signaling in response to infection. Thus, another function of MIF appears to be an ability to promote assembly of “signaling platforms” within lipid rafts by regulating caveolin-1 enrichment. This observation is consistent with a previously suggested role for MIF in regulating lipid raft composition in lung adenocarcinoma cells (38, 45).
Furthermore, the ability of MIF to regulate caveolin-1-dependent complexes were documented in primary HPCEC by examining the MAPK activation state, which is dependent on the contextual expression of caveolin-1 where either inhibition or activation of signaling pathways is observed (46). The MAPK p42/p44 is localized to caveolin-rich fractions (38). However, when infected with P. aeruginosa, the primary HPCEC with siRNA-reduced levels of caveolin-1 failed to increase MAPK phosphorylation, suggesting that during infection the composition of caveolin-1-enriched “signaling hubs” changes to transmit an outside-in MAPK activation signal. These observations are intriguing, given that P. aeruginosa invasive strains such as 6294 utilize MAPK p42/p44 to gain entry into the epithelial host and suggest that caveolin-rich platforms are facilitating p42/p44 activation (47). Unlike infection with the 6294, infection with the cytotoxic PA14 strain yielded milder changes of the caveolin-1 enrichment, suggesting that MIF-dependent regulation may impact the pathogenesis of those strains differently. We have also not tested how differences of the levels of Type III secretion system or other virulence factors produced by P. aeruginosa inside the host cell could impact caveolin assemblies or provide alternative explanations to the observed phenotype.
Because MAPK signaling stimulates transcription of IL-8 (48), decreasing the levels of caveolin-1 in P. aeruginosa-infected cells resulted in the expected reduction of IL-8 production (Fig. 6), suggesting that in the infected primary HCEC, caveolin-1 sustains MAPK activation. Because of its multiple interacting partners, caveolin-1 is implicated in regulating acute inflammatory injury, cellular proliferation, and fibrosis (41). To determine which of these functions are regulated by MIF is an important future research direction that will bring novel understanding of the molecular mechanisms behind MIF-induced signaling.
In conclusion, we found that MIF exists in multiple oligomeric forms, which appear dynamic in nature. The trimeric form of MIF is the functionally active, and its formation is favored by P. aeruginosa infection in corneal epithelial cells and the infected murine corneal epithelium, associated with MIF promotion of the assembly of caveolin-rich lipid rafts, leading to host inflammatory mediator production in response to infection (Fig. 8). MIF rapid response and activation of inflammation are important in the pathogenesis of corneal damage from infection and are a strong target for intervention, consistent with the recent findings that glucocorticoid therapy of bacterial keratitis is not therapeutic (39, 40) potentially due to the inhibitory effects of MIF on these anti-inflammatory drugs. These observations provide a scientific rationale for future drug screens for novel MIF-neutralizing agents, wherein targeting the oligomeric state could be a key to successful interventions to obviate the negative outcomes from inflammation that result in loss of visual acuity following microbial keratitis.
FIGURE 8.

Model. MIF homotrimers regulate epithelial responses to P. aeruginosa-induced infection in an autocrine manner by promoting the assembly of caveolin-1-rich signaling hubs. Infection stimulated MAPK p42/p44 recruitment to the caveolin-rich structures, and MAPK activation was prolonged by MIF to support proinflammatory gene expression.
This work was supported by National Eye Institute Grants 1R21EY019944 (to M. G.), R01 EY022054 (to M. G.), and R01EY016144 (to G. P.).
- MIF
- macrophage migration inhibitory factor
- HPCEC
- primary human corneal epithelial cell(s)
- rMIF
- recombinant MIF.
REFERENCES
- 1. Bloom B. R., Bennett B. (1966) Mechanism of a reaction in vitro associated with delayed-type hypersensitivity. Science 153, 80–82 [DOI] [PubMed] [Google Scholar]
- 2. David J. R. (1966) Delayed hypersensitivity in vitro: its mediation by cell-free substances formed by lymphoid cell-antigen interaction. Proc. Natl. Acad. Sci. U.S.A. 56, 72–77 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Leech M., Metz C., Hall P., Hutchinson P., Gianis K., Smith M., Weedon H., Holdsworth S. R., Bucala R., Morand E. F. (1999) Macrophage migration inhibitory factor in rheumatoid arthritis: evidence of proinflammatory function and regulation by glucocorticoids. Arthritis Rheum. 42, 1601–1608 [DOI] [PubMed] [Google Scholar]
- 4. Mikulowska A., Metz C. N., Bucala R., Holmdahl R. (1997) Macrophage migration inhibitory factor is involved in the pathogenesis of collagen type II-induced arthritis in mice. J. Immunol. 158, 5514–5517 [PubMed] [Google Scholar]
- 5. Lan H. Y., Bacher M., Yang N., Mu W., Nikolic-Paterson D. J., Metz C., Meinhardt A., Bucala R., Atkins R. C. (1997) The pathogenic role of macrophage migration inhibitory factor in immunologically induced kidney disease in the rat. J. Exp. Med. 185, 1455–1465 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Brown F. G., Nikolic-Paterson D. J., Hill P. A., Isbel N. M., Dowling J., Metz C. M., Atkins R. C. (2002) Urine macrophage migration inhibitory factor reflects the severity of renal injury in human glomerulonephritis. J. Am. Soc. Nephrol. 13, S7–13 [PubMed] [Google Scholar]
- 7. Yabunaka N., Nishihira J., Mizue Y., Tsuji M., Kumagai M., Ohtsuka Y., Imamura M., Asaka M. (2000) Elevated serum content of macrophage migration inhibitory factor in patients with type 2 diabetes. Diabetes Care 23, 256–258 [DOI] [PubMed] [Google Scholar]
- 8. Gadjeva M., Nagashima J., Zaidi T., Mitchell R. A., Pier G. B. (2010) Inhibition of macrophage migration inhibitory factor ameliorates ocular Pseudomonas aeruginosa-induced keratitis. PLoS Pathog 6, e1000826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Adamali H., Armstrong M. E., McLaughlin A. M., Cooke G., McKone E., Costello C. M., Gallagher C. G., Leng L., Baugh J. A., Fingerle-Rowson G., Bucala R. J., McLoughlin P., Donnelly S. C. (2012) Macrophage migration inhibitory factor enzymatic activity, lung inflammation, and cystic fibrosis. Am. J. Respir. Crit. Care Med. 186, 162–169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Roger T., David J., Glauser M. P., Calandra T. (2001) MIF regulates innate immune responses through modulation of Toll-like receptor 4. Nature 414, 920–924 [DOI] [PubMed] [Google Scholar]
- 11. Bernhagen J., Calandra T., Mitchell R. A., Martin S. B., Tracey K. J., Voelter W., Manogue K. R., Cerami A., Bucala R. (1993) MIF is a pituitary-derived cytokine that potentiates lethal endotoxaemia. Nature 365, 756–759 [DOI] [PubMed] [Google Scholar]
- 12. Bernhagen J., Calandra T., Bucala R. (1994) The emerging role of MIF in septic shock and infection. Biotherapy 8, 123–127 [DOI] [PubMed] [Google Scholar]
- 13. Calandra T., Bernhagen J., Mitchell R. A., Bucala R. (1994) The macrophage is an important and previously unrecognized source of macrophage migration inhibitory factor. J. Exp. Med. 179, 1895–1902 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Calandra T., Bucala R. (1995) Macrophage migration inhibitory factor: a counter-regulator of glucocorticoid action and critical mediator of septic shock. J. Inflamm. 47, 39–51 [PubMed] [Google Scholar]
- 15. Al-Abed Y., Dabideen D., Aljabari B., Valster A., Messmer D., Ochani M., Tanovic M., Ochani K., Bacher M., Nicoletti F., Metz C., Pavlov V. A., Miller E. J., Tracey K. J. (2005) ISO-1 binding to the tautomerase active site of MIF inhibits its pro-inflammatory activity and increases survival in severe sepsis. J. Biol. Chem. 280, 36541–36544 [DOI] [PubMed] [Google Scholar]
- 16. Jorgensen W. L., Trofimov A., Du X., Hare A. A., Leng L., Bucala R. (2011) Benzisothiazolones as modulators of macrophage migration inhibitory factor. Bioorg. Med. Chem. Lett. 21, 4545–4549 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Jorgensen W. L., Gandavadi S., Du X., Hare A. A., Trofimov A., Leng L., Bucala R. (2010) Receptor agonists of macrophage migration inhibitory factor. Bioorg. Med. Chem. Lett. 20, 7033–7036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Leng L., Metz C. N., Fang Y., Xu J., Donnelly S., Baugh J., Delohery T., Chen Y., Mitchell R. A., Bucala R. (2003) MIF signal transduction initiated by binding to CD74. J. Exp. Med. 197, 1467–1476 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Shi X., Leng L., Wang T., Wang W., Du X., Li J., McDonald C., Chen Z., Murphy J. W., Lolis E., Noble P., Knudson W., Bucala R. (2006) CD44 is the signaling component of the macrophage migration inhibitory factor-CD74 receptor complex. Immunity 25, 595–606 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Gore Y., Starlets D., Maharshak N., Becker-Herman S., Kaneyuki U., Leng L., Bucala R., Shachar I. (2008) Macrophage migration inhibitory factor induces B cell survival by activation of a CD74-CD44 receptor complex. J. Biol. Chem. 283, 2784–2792 [DOI] [PubMed] [Google Scholar]
- 21. Lolis E., Bucala R. (1996) Crystal structure of macrophage migration inhibitory factor (MIF), a glucocorticoid-induced regulator of cytokine production, reveals a unique architecture. Proc. Assoc. Am. Phys. 108, 415–419 [PubMed] [Google Scholar]
- 22. Sun H. W., Swope M., Cinquina C., Bedarkar S., Bernhagen J., Bucala R., Lolis E. (1996) The subunit structure of human macrophage migration inhibitory factor: evidence for a trimer. Protein Eng. 9, 631–635 [DOI] [PubMed] [Google Scholar]
- 23. Sun H. W., Bernhagen J., Bucala R., Lolis E. (1996) Crystal structure at 2.6-A resolution of human macrophage migration inhibitory factor. Proc. Natl. Acad. Sci. U.S.A. 93, 5191–5196 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Potolicchio I., Santambrogio L., Strominger J. L. (2003) Molecular interaction and enzymatic activity of macrophage migration inhibitory factor with immunorelevant peptides. J. Biol. Chem. 278, 30889–30895 [DOI] [PubMed] [Google Scholar]
- 25. Cherepkova O. A., Lutova E. M., Gurvits B. Y. (2005) Charge heterogeneity of bovine brain macrophage migration inhibitory factor. Neurochem. Res. 30, 151–158 [DOI] [PubMed] [Google Scholar]
- 26. Preston M. J., Fleiszig S. M., Zaidi T. S., Goldberg J. B., Shortridge V. D., Vasil M. L., Pier G. B. (1995) Rapid and sensitive method for evaluating Pseudomonas aeruginosa virulence factors during corneal infections in mice. Infect Immun. 63, 3497–3501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Zaidi T. S., Lyczak J., Preston M., Pier G. B. (1999) Cystic fibrosis transmembrane conductance regulator-mediated corneal epithelial cell ingestion of Pseudomonas aeruginosa is a key component in the pathogenesis of experimental murine keratitis. Infect Immun. 67, 1481–1492 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Zaidi T., Reidy T., D'Ortona S., Fichorova R., Pier G., Gadjeva M. (2011) CD74 deficiency ameliorates Pseudomonas aeruginosa-induced ocular infection. Sci. Rep. 1, 58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Thakur A., Xue M. L., Wang W., Lloyd A., Wakefield D., Willcox M. D. (2001) Expression of macrophage migration inhibitory factor during Pseudomonas keratitis. Clin. Exp. Ophthalmology 29, 179–182 [DOI] [PubMed] [Google Scholar]
- 30. Kowalski M. P., Dubouix-Bourandy A., Bajmoczi M., Golan D. E., Zaidi T., Coutinho-Sledge Y. S., Gygi M. P., Gygi S. P., Wiemer E. A., Pier G. B. (2007) Host resistance to lung infection mediated by major vault protein in epithelial cells. Science 317, 130–132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. McClellan S. A., Lighvani S., Hazlett L. D. (2006) IFN-γ: regulation of nitric oxide in the P. aeruginosa-infected cornea. Ocul. Immunol. Inflamm. 14, 21–28 [DOI] [PubMed] [Google Scholar]
- 32. Hazlett L. D. (2004) Corneal response to Pseudomonas aeruginosa infection. Prog. Retin. Eye Res. 23, 1–30 [DOI] [PubMed] [Google Scholar]
- 33. Bernhagen J., Krohn R., Lue H., Gregory J. L., Zernecke A., Koenen R. R., Dewor M., Georgiev I., Schober A., Leng L., Kooistra T., Fingerle-Rowson G., Ghezzi P., Kleemann R., McColl S. R., Bucala R., Hickey M. J., Weber C. (2007) MIF is a noncognate ligand of CXC chemokine receptors in inflammatory and atherogenic cell recruitment. Nat. Med. 13, 587–596 [DOI] [PubMed] [Google Scholar]
- 34. Meyer-Siegler K. L., Iczkowski K. A., Leng L., Bucala R., Vera P. L. (2006) Inhibition of macrophage migration inhibitory factor or its receptor (CD74) attenuates growth and invasion of DU-145 prostate cancer cells. J. Immunol. 177, 8730–8739 [DOI] [PubMed] [Google Scholar]
- 35. Ouertatani-Sakouhi H., El-Turk F., Fauvet B., Cho M. K., Pinar Karpinar D., Le Roy D., Dewor M., Roger T., Bernhagen J., Calandra T., Zweckstetter M., Lashuel H. A. (2010) Identification and characterization of novel classes of macrophage migration inhibitory factor (MIF) inhibitors with distinct mechanisms of action. J. Biol. Chem. 285, 26581–26598 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. El-Turk F., Cascella M., Ouertatani-Sakouhi H., Narayanan R. L., Leng L., Bucala R., Zweckstetter M., Rothlisberger U., Lashuel H. A. (2008) The conformational flexibility of the carboxy terminal residues 105–114 is a key modulator of the catalytic activity and stability of macrophage migration inhibitory factor. Biochemistry 47, 10740–10756 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Bajmoczi M., Gadjeva M., Alper S. L., Pier G. B., Golan D. E. (2009) Cystic fibrosis transmembrane conductance regulator and caveolin-1 regulate epithelial cell internalization of Pseudomonas aeruginosa. Am. J. Physiol. Cell Physiol 297, C263–277 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Engelman J. A., Chu C., Lin A., Jo H., Ikezu T., Okamoto T., Kohtz D. S., Lisanti M. P. (1998) Caveolin-mediated regulation of signaling along the p42/44 MAP kinase cascade in vivo. A role for the caveolin-scaffolding domain. FEBS Lett. 428, 205–211 [DOI] [PubMed] [Google Scholar]
- 39. Deleted in proof.
- 40. Srinivasan M., Mascarenhas J., Rajaraman R., Ravindran M., Lalitha P., Glidden D. V., Ray K. J., Hong K. C., Oldenburg C. E., Lee S. M., Zegans M. E., McLeod S. D., Lietman T. M., Acharya N. R. (2012) Corticosteroids for bacterial keratitis: the Steroids for Corneal Ulcers Trial (SCUT). Arch. Ophthalmol. 130, 143–150 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Pavlides S., Tsirigos A., Vera I., Flomenberg N., Frank P. G., Casimiro M. C., Wang C., Fortina P., Addya S., Pestell R. G., Martinez-Outschoorn U. E., Sotgia F., Lisanti M. P. (2010) Loss of stromal caveolin-1 leads to oxidative stress, mimics hypoxia and drives inflammation in the tumor microenvironment, conferring the “reverse Warburg effect”: a transcriptional informatics analysis with validation. Cell Cycle 9, 2201–2219 [DOI] [PubMed] [Google Scholar]
- 42. Zaidi T., Bajmoczi M., Zaidi T., Golan D. E., Pier G. B. (2008) Disruption of CFTR-dependent lipid rafts reduces bacterial levels and corneal disease in a murine model of Pseudomonas aeruginosa keratitis. Invest. Ophthalmol. Vis. Sci. 49, 1000–1009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Soong G., Reddy B., Sokol S., Adamo R., Prince A. (2004) TLR2 is mobilized into an apical lipid raft receptor complex to signal infection in airway epithelial cells. J. Clin. Invest. 113, 1482–1489 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Sargiacomo M., Sudol M., Tang Z., Lisanti M. P. (1993) Signal transducing molecules and glycosyl-phosphatidylinositol-linked proteins form a caveolin-rich insoluble complex in MDCK cells. J. Cell Biol. 122, 789–807 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Swant J. D., Rendon B. E., Symons M., Mitchell R. A. (2005) Rho GTPase-dependent signaling is required for macrophage migration inhibitory factor-mediated expression of cyclin D1. J. Biol. Chem. 280, 23066–23072 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Jin Y., Lee S. J., Minshall R. D., Choi A. M. (2011) Caveolin-1: a critical regulator of lung injury. Am. J. Physiol. Lung Cell Mol Physiol 300, L151–160 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Evans D. J., Maltseva I. A., Wu J., Fleiszig S. M. (2002) Pseudomonas aeruginosa internalization by corneal epithelial cells involves MEK and ERK signal transduction proteins. FEMS Microbiol. Lett. 213, 73–79 [DOI] [PubMed] [Google Scholar]
- 48. Bezzerri V., Borgatti M., Finotti A., Tamanini A., Gambari R., Cabrini G. (2011) J. Immunol. 187, 6069–6081 [DOI] [PubMed] [Google Scholar]