

Background: Secreted PCSK9 regulates LDL levels in plasma by mediating degradation of hepatic LDL receptors.
Results: LDL binds to PCSK9 in human plasma and in vitro and inhibits PCSK9 binding to cell surface LDL receptors.
Conclusion: A large proportion of circulating PCSK9 is bound to LDL in humans.
Significance: Regulatory mechanisms that affect activity of secreted PCSK9 represent novel targets for cholesterol-lowering therapies.
Keywords: Cholesterol Regulation, Lipid Metabolism, Lipoprotein Receptor, Low Density Lipoprotein (LDL), Protein-Protein Interactions
Abstract
Proprotein convertase subtilisin/kexin type-9 (PCSK9) is a secreted protein that binds to the epidermal growth factor-like-A domain of the low density lipoprotein receptor (LDLR) and mediates LDLR degradation in liver. Gain-of-function mutations in PCSK9 are associated with autosomal dominant hypercholesterolemia in humans. Size-exclusion chromatography of human plasma has shown PCSK9 to be partly associated with undefined high molecular weight complexes within the LDL size range. We used density gradient centrifugation to isolate LDL in plasma pooled from 5 normolipidemic subjects and report that >40% of total PCSK9 was associated with LDL. Binding of fluorophore-labeled recombinant PCSK9 to isolated LDL in vitro was saturable with a KD ∼ 325 nm. This interaction was competed >95% by excess unlabeled PCSK9, and competition binding curves were consistent with a one-site binding model. An N-terminal region of the PCSK9 prodomain (amino acids 31–52) was required for binding to LDL in vitro. LDL dose-dependently inhibited binding and degradation of cell surface LDLRs by exogenous PCSK9 in HuH7 cells. LDL also inhibited PCSK9 binding to mutant LDLRs defective at binding LDL. These data suggest that association of PCSK9 with LDL particles in plasma lowers the ability of PCSK9 to bind to cell surface LDLRs, thereby blunting PCSK9-mediated LDLR degradation.
Introduction
Elevated LDL-cholesterol (LDL-C)3 is a primary risk factor for the development of cardiovascular heart disease. Plasma clearance of LDL primarily occurs in liver through the action of the LDL receptor (LDLR), a cell surface protein that binds to the apolipoprotein B100 (apoB100) protein component of LDL with high affinity and promotes LDL particle uptake into cells via clathrin-mediated endocytosis (1). LDL is released from the LDLR within the acidic environment of the early endosome, after which the LDLR recycles to the cell surface.
The monogenic disorder autosomal dominant hypercholesterolemia derives from defective LDLR pathway clearance of plasma LDL-C and predominantly occurs as a result of mutations in LDLR (2) or APO-B100 (3). In rare cases autosomal dominant hypercholesterolemia results from point mutations of the gene encoding proprotein convertase subtilisin/kexin type-9 (PCSK9), a secreted serine protease (4). PCSK9 has been identified as a central regulator of plasma LDL-C levels though its ability to bind to LDLRs and mediate LDLR degradation in the liver (5, 6). Gain-of-function mutations in PCSK9 are associated with autosomal dominant hypercholesterolemia (7, 8); conversely, loss-of-function mutations in PCSK9 are associated with lowered levels of plasma LDL-C and decreased incidence of cardiovascular heart disease (9, 10).
PCSK9 is a member of the proprotein convertase (PC) family of serine proteases related to bacterial subtilisin and yeast kexin (8). PCSK9 is a modular protein consisting of a signal sequence followed by a prodomain, a subtilisin-like catalytic domain, and a C-terminal cysteine- and histidine-rich domain (11). Autocatalytic processing of PCSK9 in the endoplasmic reticulum results in release of the ∼14-kDa prodomain, which remains associated with the ∼60-kDa catalytic/C-terminal domains, masking the catalytic site in the mature secreted protein (8, 12–14). Although mature PCSK9 possesses inherent protease activity (13), this function is not required for LDLR degradation in response to exogenous PCSK9 in HepG2 cells (15) nor in mouse liver (16). Indeed, PCSK9 binds to the LDLR at a surface region of the catalytic domain that is >20 Å removed from the active site (17). The primary PCSK9 binding site on LDLR is located within the first of three epidermal growth factor-like repeats (EGF-A) of the EGF homology domain of the receptor, and this binding reaction is required for PCSK9-mediated LDLR degradation (18). In contrast to the ligand LDL, PCSK9 binding affinity to LDLR is dramatically increased at acidic pH (13, 18). Thus, PCSK9 fails to release from LDLR in the early endosomes and directs the receptor for degradation in late endosomes/lysosomes through an as yet undefined mechanism (18).
PCSK9 is mainly expressed in liver, with lower levels of expression in kidney, intestine, and brain (8). Like the LDLR, gene expression of PCSK9 is positively regulated by SREBP-2, a transcription factor that is activated in response to cellular cholesterol depletion (19–21). Cholesterol-lowering treatments with statins or ezetimibe have been shown to increase circulating PCSK9 levels in humans (22–24), which may limit their efficacy at lowering plasma LDL-C levels. Importantly, PCSK9 inhibition by either RNAi (25) or blocking antibodies (26) lowered plasma cholesterol levels and augmented the action of statins in mice and non-human primates and more recently in clinical trials in humans (27).
Plasma PCSK9 levels, as measured by ELISA, can vary widely within humans. For example, in one study of 3138 individuals, PCSK9 varied over an ∼100-fold range (33–2988 ng/ml; median = 487 ng/ml) (28). Nevertheless, a positive statistical correlation has been shown between levels of PCSK9 and plasma total cholesterol (29–31). Plasma PCSK9 has recently been shown to decrease with fasting in humans and transiently increase postprandially, mirroring markers of cholesterol synthesis (32), with its circulating levels following a diurnal rhythm (33).
It remains unclear whether the majority of plasma PCSK9 measurable by ELISA represents active or inactive forms of the protein. For instance there is evidence that a truncated form of PCSK9 found in human plasma samples results from proteolysis of PCSK9 by furin at a site in the catalytic domain that would remove a region of the protein required for LDLR binding (34). PCSK9 also displays considerable size heterogeneity in plasma samples, with evidence of oligomeric forms and/or association with large macromolecular complexes that may influence activity (35, 36).
Prompted by evidence of circulating PCSK9 association with large complexes, we investigated the potential interaction of PCSK9 with LDL in human plasma. We report that >40% of PCSK9 can be recovered in isolated plasma LDL derived from fasted normolipidemic subjects and that PCSK9 binds to LDL in vitro in a specific and saturable manner. Although LDL inhibited binding and degradation of cell surface LDLRs by exogenous PCSK9 in cultured hepatic cells, this effect did not require LDL binding to LDLRs, indicating the inhibitory effect is manifest on the PCSK9 molecule. These results raise the possibility that circulating PCSK9 bound to LDL has decreased binding activity toward cell surface LDLRs.
EXPERIMENTAL PROCEDURES
Materials
We obtained Lipofectamine 2000, fetal bovine serum (FBS), and newborn calf serum from Invitrogen, OptiprepTM density gradient medium (60% w/v iodixanol) from Axis-Shield, cholesterol and 25-hydroxycholesterol from Steroloids, and EDTA-free CompleteTM Protease Inhibitor Tablets from Roche Applied Science. All other reagents were from Sigma unless otherwise specified. Sodium mevalonate was prepared from mevalonic acid as described (37). Newborn calf lipoprotein-deficient serum (d > 1.215 g/ml) was prepared by ultracentrifugation (38). The LDLR cDNA expression vectors used in these studies were pLDLR17 (39) and LDLRΔR6 mutant constructed in pLDLR17 containing a deletion of residues 211–251 in the ligand binding domain (provided by R. Milne, University of Ottawa Heart Institute).
Antibodies
13D3, a monoclonal antibody developed against full-length human PCSK9 that recognizes an epitope in the catalytic domain, was a generous gift from J. Horton (University of Texas Southwestern Medical Center, Dallas, TX); rabbit anti-serum 1697 against full-length human PCSK9 was custom-produced by Biomatik (Cambridge, Ontario, Canada); rabbit anti-serum 3143 against the C-terminal 14 amino acids of LDLR was the kind gift of J. Herz (UT Southwestern Medical Center); 7H2, a monoclonal antibody recognizing non-reduced LDLR, and 1D1, a monoclonal antibody against human apoB, were kindly provided by R. Milne (University of Ottawa Heart Institute); goat anti-serum against apoB was from Millipore (catalogue no. 178467); a mouse anti-human transferrin receptor antibody was purchased from Invitrogen; biotin-labeled monoclonal anti-human IgG1 antibody, monoclonal anti-actin antibody (AC-10), and monoclonal anti-FLAG M2 antibody were from Sigma. Secondary IRDye-labeled goat anti-mouse and anti-rabbit IgG antibodies were from LI-COR Biosciences.
Protein Preparation
FLAG epitope-tagged recombinant human wild-type PCSK9 and PCSK9 expressing the gain-of-function D374Y mutation were produced in stably transfected HEK293S cells and purified as previously described (40) with the exceptions that suspension cells were cultured in UltraDOMATM medium (Lonza) supplemented with 10% (v/v) FBS, and final purification and buffer exchange was completed on a Superdex 200 10/300 GL column (GE Healthcare). Fluorescently labeled PCSK9 was prepared using the DyLight Antibody Labeling kit (Pierce) as per the manufacturer's instructions. The EGF-AB fragment of human LDLR (residues 293–372) was expressed as a glutathione S-transferase (GST) fusion in Rosetta-gami B cells (EMD Biosciences) and purified by glutathione chromatography followed by size-exclusion chromatography.
Cultured Cell Experiments
HuH7 human hepatoma cells (kindly provided by D. Figeys, University of Ottawa) were maintained in monolayer culture at 37 °C in 8–9% CO2 and were cultured in one of the following media: Medium A contained DMEM (4.5 g/liter glucose; Invitrogen) supplemented with 100 units/ml penicillin and 100 μg/ml streptomycin sulfate; Medium B contained Medium A supplemented with 10% FBS (v/v); sterol-depleting Medium C contained Medium A supplemented with 5% (v/v) newborn calf lipoprotein-deficient serum, 10 μm pravastatin, and 50 μm sodium mevalonate; sterol-supplemented Medium D contained Medium A with 5% (v/v) newborn calf lipoprotein-deficient serum, 10 μg/ml cholesterol, and 1 μg/ml 25-hydroxycholesterol.
LDLR Degradation Assays
HuH7 cells were cultured in Medium B to ∼50% confluency then cultured in sterol-depleting medium C for 18–24 h. Recombinant PCSK9-D374Y was incubated with LDL in medium C for 30 min at 37 °C, then the preconditioned medium was added to cells for 4 h at 37 °C. Whole cell extracts were prepared with Tris lysis buffer (50 mm Tris-Cl, pH 7.4, 150 mm NaCl, 1% Nonidet P-40 (EMD Biosciences), 0.5% sodium deoxycholate, 5 mm EDTA, 5 mm EGTA, CompleteTM protease inhibitor mixture, 1 mm phenylmethylsulfonyl fluoride (PMSF)), subjected to 8% SDS-PAGE, and transferred to nitrocellulose membranes (Bio-Rad) for immunoblot analysis. Secondary infrared dye (IRDye800)-labeled antibodies were used for detection on a LI-COR Odyssey infrared imaging system (LI-COR Biosciences). Band intensity was quantified using Odyssey 2.0 software.
PCSK9 Cellular Uptake Assays
HuH7 cells were grown and treated as described above with exception that 50 μm chloroquine was added to culture medium 30 min before and during 4 h of treatment with PCSK9-D347Y and LDL to inhibit lysosomal degradation of internalized PCSK9. In LDLR expression studies, HuH7 cells were transiently transfected with LDLR cDNA expression vectors using Lipofectamine 2000. Cells were then incubated overnight in sterol-supplemented medium D before treatments. Whole cell extracts were prepared for SDS-PAGE and immunoblot analysis as described above.
Ligand Blot Assays
HEK293 cells were transiently transfected with LDLR cDNA expression vectors with Lipofectamine 2000. Cells were resuspended on ice in sorbitol microsome buffer (50 mm HEPES-KOH, pH 7.2, 250 mm sorbitol, 10 mm KCl, 1.5 mm MgCl2, CompleteTM protease inhibitor mixture, 1 mm PMSF) and disrupted with 10 passages through a 23-gauge needle. Membranes were pelleted by centrifugation at 100,000 × g for 30 min then resuspended in pH 6/Triton X-100 buffer (40 mm Tris-maleate, pH 6.0, 100 mm NaCl, 2 mm CaCl2, 1 mm MgCl2, 1% Triton X-100, CompleteTM protease inhibitor mixture, 1 mm PMSF). Membrane proteins (35 μg/lane) were resolved on 8% SDS-PAGE under non-reducing conditions and transferred to nitrocellulose membranes. Membranes strips were blocked for 30 min (50 mm Tris-Cl, pH 7.4, 90 mm NaCl, 2 mm CaCl2, 2% (w/v) BSA, 0.05% (w/v) skim milk) then incubated for 1 h with 0.5 μg/ml DyLight680-labeled PCSK9-D374Y, 75 μg/ml LDL, or IgG-7H2 to native LDLR in blocking buffer followed by 3 × 10-min washes in blocking buffer containing 0.2% BSA. Blots were scanned on a LI-COR Odyssey infrared imaging system for visualization of LDLR-bound proteins either directly for dye-labeled PCSK9 or after immunoblot for apoB and using an IRDye800-labeled secondary antibody. Ligand blotting to LDLR EGF-AB protein was performed as above after slot-blotting of purified EGF-A protein to nitrocellulose using a Bio-Rad apparatus according to the manufacturer's instructions.
Plasma Collection and Lipoprotein Isolation
Blood samples were drawn from fasted healthy volunteers into evacuated tubes containing EDTA, and plasma was separated by low speed centrifugation. Protease inhibitors were added to the cleared plasma (1 mm PMSF and 50 units/liter aprotinin). VLDL (d = 1.006 g/ml), LDL (d = 1.019–1.065 g/ml), and HDL (d = 1.065–1.21 g/ml) were isolated from human plasma using sequential potassium bromide flotation ultracentrifugation (41) followed by extensive dialysis against phosphate-buffered saline (PBS) containing 0.25 mm EDTA. Protein concentrations in lipoprotein preparations were determined using a modified Lowry assay (42). LDL used for cell culture experiments was prepared with omission of protease inhibitors. Endogenous plasma PCSK9 was quantitatively recovered in the non-lipoprotein fraction after ultracentrifugation in high salt solutions (data not shown) and did not associate with any lipoprotein fraction.
Plasma Separation
Plasma samples were separated using a three-layered Optiprep gradient modified from a previously described protocol (43). A 12% Optiprep plasma solution was prepared by diluting 0.75 ml of plasma with 0.29 ml of 60% Optiprep and 0.26 ml of 10 mm Tris, pH 7.4. In Thinwall Ultra-Clear 2.2 ml tubes (Beckman Coulter), the plasma solution was layered below a 9% Optiprep solution (0.7 ml) prepared in 10 mm Tris, pH 7.4, and the tube was overlaid with 10 mm Tris, pH 7.4 (0.2 ml). The gradients were centrifuged in a TLS55 rotor (Beckman Coulter) at 100,000 × g for 18 h at 4 °C. Fractions were collected by aspiration from the upper layer.
Analysis of Optiprep Gradient Subfractions
Equivalent amounts of gradient fractions were adjusted to 0.5 ml in radioimmune precipitation assay buffer containing CompleteTM protease inhibitor mixture and 1 mm PMSF. Proteins of interest (PCSK9, apoB, IgG1) were precipitated overnight at 4 °C using antibody against either PCSK9 (1697) or apoB (178467) and protein A magnetic beads (Millipore). The beads were washed three times with radioimmune precipitation assay buffer and eluted in SDS-PAGE sample buffer for immunoblot analysis. To co-immunoprecipitate LDL and PCSK9, a total of 0.15 ml of the LDL fraction was brought up to a volume of 0.5 ml in 10 mm Tris, pH 7.4, 75 mm NaCl. Protein A beads and 10 μl of polyclonal antibody against apoB were added to the diluted fraction and incubated for 1 h at 4 °C. The beads were washed 3 times with 10 mm Tris, pH 7.4, and eluted in SDS-PAGE sample buffer for immunoblot analysis.
PCSK9 Binding to Lipoproteins in Vitro
DyLight800-labeled PCSK9 (1.25 μg/ml) was incubated with 0.5 mg/ml LDL or 1.0 mg/ml VLDL in the presence of 1% BSA. The reaction mixtures were incubated at 37 °C for 45 min and then separated using three-layered Optiprep gradients, as described under “Plasma Separation” above. The lipoprotein fractions were subjected SDS-PAGE and gels scanned directly to visualize DyLight800-labeled PCSK9 then transferred to nitrocellulose and immunoblotted for apoB as above. For binding to HDL, reaction mixtures were separated by FPLC on a Sephadex 200 10/300 GL column (GE Healthcare). Cholesterol was measured in column fractions using a kit (Biovision), and PCSK9 was measured by immunoblotting. PCSK9 binding to LDL was similarly determined using separation on a Superose 6 10/300 GL column.
Binding Studies
To generate saturation binding curves, isolated PCSK9-free LDL (0.5 mg/ml) was incubated with an increasing amount of purified PCSK9 (a mix of unlabeled and Dylight800-labeled PCSK9 at a 50:1 ratio) in Hepes buffer saline-calcium buffer (25 mm Hepes-KOH, pH 7.4, 150 mm NaCl, 2 mm CaCl2) containing 1% BSA for 1 h at 37 °C. 4× loading dye (10% Ficoll-400, 0.01% bromphenol blue) was added to the reactions, and the mixtures were resolved on 0.7% agarose gels (SeaKem LE) for 2 h at 40 V with electrode buffer of 90 mm Tris, pH 8.0, 80 mm borate, 2 mm calcium lactate. Gels were scanned directly using the LI-COR Odyssey infrared imaging system (LI-COR Biosciences), and bands representing DyLight800-PCSK9 bound to LDL were quantified using Odyssey 2.0 software (LI-COR Biosciences). The KD was determined by fitting data to a saturation curve by nonlinear regression using Prism 5 software (GraphPad software, Inc.). For competition binding, LDL was incubated as above with 100 nm DyLight800-labeled PCSK9 with increasing amounts of unlabeled PCSK9. The reaction mixtures were resolved on agarose gels as above, and Ki was determined by fitting the band intensity data to a sigmoidal dose-response curve using nonlinear regression.
Simultaneous Binding of PCSK9 to LDL and LDLR EGF-A Domain Protein
LDL (1 mg/ml), PCSK9-D374Y (a mix of unlabeled and Dylight680-labeled protein at a 30:1 ratio) (40 μg/ml), and/or DyLight800-labeled EGF-AB–GST (3 μg/ml) were incubated in various combinations (see Fig. 4 for details) and resolved on agarose gels as described above with the exception that gel and running buffer was 40 mm Tris acetate, pH 7.2, 2 mm CaCl2. Gels were transferred to nitrocellulose by capillary blotting, and dye-labeled PCSK9 and EGF-A proteins were visualized using the LI-COR Odyssey infrared imaging system.
FIGURE 4.
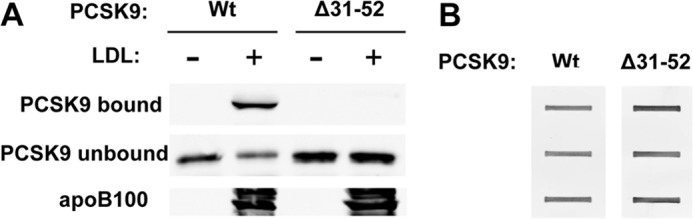
An N-terminal region of the prodomain is required for PCSK9 binding to LDL. A, purified FLAG-tagged wild-type PCSK9 or N-terminal truncated PCSK9 (Δ31–52) were incubated with isolated PCSK9-free LDL before separation in Optiprep gradients as described under “Experimental Procedures.” The LDL containing fractions was obtained, and associated PCSK9 was detected by SDS-PAGE and immunoblotting for the FLAG epitope tag. Unbound PCSK9 was detected after immunoprecipitation and immunoblotting. B, ligand blotting to LDLR EGF-A protein is shown. Purified recombinant LDLR EGF-AB domains expressed as a GST fusion protein was applied to nitrocellulose by slot-blotting. Blots were incubated with wild-type PCSK9 or Δ31–52 mutant PCSK9 followed by washing and detection by immunoblotting for the FLAG epitope tag.
Statistical Analysis
All presented values are the mean ± S.D. Statistical significance of differences between groups was determined using GraphPad Prism 5 software.
RESULTS
Binding of PCSK9 to LDL in Human Plasma
Flotation ultracentrifugation in iodixonal (Optiprep) gradients was used to assess PCSK9 association with lipoproteins in a pooled human plasma sample derived from 5 (2 male, 3 female) fasted normolipidemic subjects (total cholesterol 4.26 ± 0.8 mmol/liter; LDL-C 2.52 ± 0.8 mmol/liter; triglycerides 1.19 ± 0.5 mmol/liter). This method resolved the plasma into three collected fractions (light, medium, and heavy density) including a clearly visible LDL-containing zone (Fig. 1A), and PCSK9 was analyzed by immunoprecipitation and Western blotting. Although the majority of plasma PCSK9 (58 ± 3%, n = 5) was present in a heavy density fraction (non-lipoproteins and HDL), a large proportion (42 ± 3%) was recovered in the LDL-containing medium density fraction (Fig. 1B). This fraction contained the majority of apoB and undetectable levels of immunoglobulin G1, indicating little to no contaminating non-lipoprotein components. PCSK9 was not detectable in the light density fraction containing VLDL. PCSK9 was co-immunoprecipitated using a polyclonal anti-apoB antibody to bind LDL from the medium density fraction (Fig. 1C), confirming an association of PCSK9 with LDL. We were unable to detect LDLR extracellular domain by Western blotting in Optiprep fractions containing PCSK9 and LDL (data not shown); thus, shed LDLR-ECD is not quantitatively involved in PCSK9 binding to LDL in plasma.
FIGURE 1.

Association of PCSK9 with LDL in human plasma. A, plasma separation in Optiprep gradients and fractionation scheme is shown. The sample in the tube on the right was stained with Coomassie R-250 to visualize typical distribution of proteins in the gradient; the sample on the left is unstained, allowing direct view of LDL. B, shown are immunoprecipitation and immunoblotting of apoB, IgG1, and PCSK9 in gradient fractions collected after separation of a pooled plasma sample from five fasted normolipidemic subjects. Blots were scanned, and band intensity was quantified using the LI-COR Odyssey infrared imaging system. Graphic representations are the average and S.D. from five separate experiments. C, LDL was immunoprecipitated (IP) from the medium density fraction using anti-apoB polyclonal antibody. Samples obtained using just protein A beads (−Ab) and beads plus antibody (+Ab) were subjected to SDS-PAGE and immunoblotted for apoB and PCSK9. * denotes band consistent with a furin-cleaved form of PCSK9 (34).
Binding of PCSK9 to LDL, but Not VLDL or HDL, in Vitro
To further assess the interaction of PCSK9 with lipoproteins, we performed in vitro binding experiments using purified secreted recombinant human PCSK9 covalently labeled with infrared fluorescent dye (DyLight800) and lipoprotein fractions isolated from normolipidemic human plasma by sequential potassium bromide flotation ultracentrifugation. LDL isolated in this manner did not contain detectable levels of endogenous PCSK9 (data not shown), perhaps due to stripping of peripherally associated PCSK9 under high salt and high g forces. PCSK9 was incubated with equivalent concentrations of VLDL (d = 1.006 g/ml) and LDL (d = 1.019–1.065 g/ml) in buffer containing 1% w/v bovine serum albumin to saturate nonspecific binding sites, after which lipoprotein-PCSK9 complexes were separated from unbound PCSK9 by flotation ultracentrifugation in Optiprep gradients. After centrifugation, lipoprotein fractions were collected and analyzed by SDS-PAGE to visualize associated dye-labeled PCSK9. Under these conditions, PCSK9 did not bind to VLDL (Fig. 2A) but was robustly associated with LDL, in agreement with our findings in human plasma. Association of fluorophore-labeled PCSK9 with LDL was decreased >80% by co-incubation with a 500-fold excess of unlabeled PCSK9. Immunoblot analysis to compare LDL binding of DyLight800-labeled PCSK9 versus unlabeled PCSK9 showed no appreciable difference (Fig. 2B), indicating that covalently attached dye molecules did not affect PCSK9 ability to bind to LDL.
FIGURE 2.

Association of PCSK9 with isolated lipoproteins in vitro. A, purified recombinant PCSK9 covalently labeled with infrared fluorescent dye (DyLight800) was incubated with isolated human VLDL or PCSK9-free LDL as described under “Experimental Procedures.” Reaction mixtures were subjected to flotation ultracentrifugation in Optiprep density gradients, lipoprotein-containing fractions were collected, and portions were subjected to 4–20% SDS-PAGE. Gels were scanned directly using the LI-COR Odyssey infrared imaging system to visualize dye-labeled PCSK9 and transferred to nitrocellulose for immunoblot analysis of apoB100 using an infrared dye (IRDye680) labeled secondary antibody. B, equal amounts of unlabeled PCSK9 and DyLight800-labeled PCSK9 were incubated with LDL and separated in Optiprep gradients as in A. LDL-containing fractions were subjected to SDS-PAGE, and PCSK9 was detected by immunoblot analysis. C, purified FLAG-tagged PCSK9 was incubated with isolated lipoproteins and separated by FPLC on Superose 6 (LDL) or Superdex 200 (HDL) columns. Fractions were collected and analyzed for cholesterol to determine elution profile of lipoproteins and immunoblotted for the FLAG epitope tag to identify PCSK9 peaks. The peak elution volume of purified PCSK9 run on Superdex 200 in the absence of HDL is indicated by an arrow.
We were unable to assess PCSK9 association with HDL (d = 1.065–1.21 g/ml) using Optiprep gradients due to poor separation. For this purpose we incubated FLAG-tagged PCSK9 with PCSK9-free LDL or HDL and subjected these reaction mixtures to FPLC to separate free PCSK9 from PCSK9 bound to lipoproteins. Column fractions within the size range containing lipoproteins of interest were analyzed for cholesterol to indicate lipoprotein elution, and PCSK9 was detected by immunoblotting for the FLAG epitope tag. The peaks for LDL and PCSK9 showed precise overlap, indicating PCSK9 binding to LDL (Fig. 2C). Although HDL and free PCSK9 were not completely resolved by Superdex 200 chromatography, there was clear non-overlap of the respective peaks (Fig. 2C), indicating that PCSK9 does not bind to HDL in vitro.
PCSK9-LDL Binding Studies
For determination of the PCSK9-LDL binding characteristics, we turned to an electrophoretic mobility shift assay to separate LDL-bound PCSK9 from unbound PCSK9 in agarose gels. We first performed saturation binding experiments using a constant amount of PCSK9-free LDL incubated with increasing concentrations of Dylight800-labeled PCSK9. The binding of PCSK9 to LDL displayed typical saturation binding (Fig. 3A) with a KD value of 325 ± 68 nm (n = 4). We next generated competition binding curves in which the binding of fluorophore-labeled PCSK9 to LDL was competed with increasing concentrations of unlabeled PCSK9 (Fig. 3B). At the highest concentration of unlabeled PCSK9, binding of labeled PCSK9 was decreased >95%, indicating a high degree of specificity, and competition curves fit a one-site binding model (R2 > 0.988). As expected for a homologous competition assay, the Ki value of 399 ± 65 nm (n = 4) was in good agreement with the KD.
FIGURE 3.
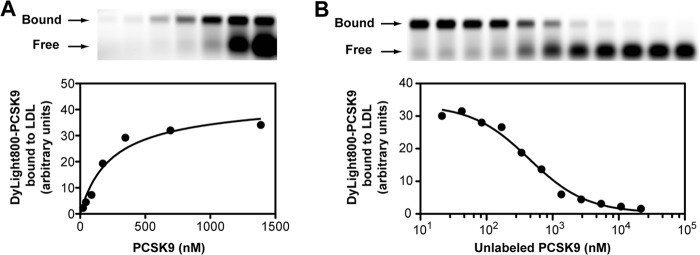
Binding studies of isolated LDL and recombinant PCSK9. A, isolated PCSK9-free human LDL was incubated for 60 min with increasing concentrations of Dylight800-labeled PCSK9 as described under “Experimental Procedures.” Reaction mixtures were resolved on 0.7% agarose gels, and gels were scanned on the LI-COR Odyssey infrared imaging system. Bands corresponding to LDL-bound PCSK9 were quantified, and a saturation binding curve was fitted using non-linear regression. B, Dylight800-labeled PCSK9 was incubated with LDL in the presence of increasing concentrations of unlabeled PCSK9. Reaction mixtures were resolved on agarose gels, and LDL bound dye-labeled PCSK9 was quantified as in A. Data were fit to a sigmoidal inhibition curve using non-linear regression. Shown are representative experiments repeated three other times with similar results.
A Region in the PCSK9 Prodomain Is Required for LDL Binding
To examine protein regions of PCSK9 involved in LDL binding, we assessed a potential role of the PCSK9 prodomain. It has been shown that deletion of amino acid 31 (the N-terminal residue of mature PCSK9) to amino acid 52 does not affect PCSK9 maturation and secretion but increases LDLR binding (17, 44). Purified recombinant FLAG-tagged wild-type PCSK9 or Δ31–52 mutant (Δ53-PCSK9) were incubated with isolated LDL followed by separation in Optiprep gradients. As expected, wild-type PCSK9 strongly associated with LDL, but no binding was observed between LDL and Δ53-PCSK9 (Fig. 4A). Ligand blotting showed normal binding of Δ53-PCSK9 to a purified protein consisting of the tandem EGF-A and EGF-B domains of the LDLR fused to GST (EGF-AB–GST) (Fig. 4B), indicating that the Δ53-PCSK9 protein was not misfolded.
LDL Does Not Interfere with PCSK9 Binding to the LDLR EGF-A Domain
PCSK9 has been shown to bind to the LDLR EGF-A domain through multiple interactions involving individual amino acid residues and surfaces in the PCSK9 catalytic domain encompassing a region from residue 153 (the N-terminal amino acid of the catalytic domain) to residue 381 (17). To assess whether large LDL particles could sterically interfere with this interaction, we determined the ability of LDL-bound PCSK9 to bind to the LDLR EGF-A domain. For this and subsequent experiments we used a mutant PCSK9 containing a gain-of-function mutation (D374Y) that confers increased binding to the LDLR EGF-A domain at neutral pH (40). This mutation did not affect binding to LDL (data not shown). LDL was incubated with DyLight800-labeled EGF-AB–GST and DyLight680-labeled PCSK9-D374Y emitting at a separate wavelength either alone or in combination. Reaction mixtures were then resolved on agarose gel electrophoresis, and dye-labeled protein co-migration with LDL was determined. As expected, PCSK9-D374Y shifted to a slower-migrating band in the presence of LDL, indicating LDL association (Fig. 5, lane 2). EGF-AB–GST was slightly faster migrating than PCSK9-D374Y and when incubated together these proteins did not form a complex, oligomeric or otherwise, that migrated near the LDL range but instead was intermediate between the two individual proteins (lane 5). EGF-AB–GST did not directly associate with LDL (lane 4) but co-migrated with LDL-associated PCSK9 when all three components were co-incubated (lane 6). We conclude that PCSK9 bound to LDL can simultaneously bind to the LDLR EGF-A domain, and the respective binding sites on PCSK9 for EGF-A and LDL do not physically overlap.
FIGURE 5.

Simultaneous binding of PCSK9-D374Y to LDL and LDLR EGF-A domain. Incubations were carried out for 1 h at 37 °C containing the indicated combinations of LDL, Dylight680-labeled PCSK9-D374Y, and Dylight800-labeled recombinant LDLR EGF-AB domain fragment as a fusion protein with GST as described under “Experimental Procedures.” Reaction mixtures were resolved in 0.7% agarose gels and transferred to nitrocellulose, and blots were scanned using the LI-COR Odyssey infrared imaging system using dual channel detection of dye-labeled proteins.
LDL Antagonizes PCSK9 Binding to Cell Surface LDLRs and Subsequent LDLR Degradation
We next determined the effect of LDL on PCSK9-mediated LDLR degradation in HuH7 human hepatoma cells. Cells were cultured in lipoprotein-deficient medium containing a statin (pravastatin) to induce LDLR expression before the addition of PCSK9-D374Y in the absence or presence of increasing concentrations of PCSK9-free LDL. Agarose gel electrophoresis separation of the medium showed that PCSK9 association with LDL increased dose-dependently (Fig. 6A). LDL treatment alone resulted in a minor increase in LDLR levels in HuH7 cells (second through fourth lanes). In the absence of LDL, PCSK9-D374Y treatment (1.5 μg/ml for 4 h) resulted in a >50% decrease in LDLR protein levels (Fig. 6B, fifth lane). LDL inhibited PCSK9-mediated LDLR degradation in a dose-dependent manner, with ∼90% recovery of LDLR levels in the presence of 500 μg/ml of LDL (eighth lane). PCSK9 cell association and uptake has been shown to be highly dependent on binding to cell surface LDLRs (40). Similar to effects on LDLR degradation, LDL dose-dependently decreased cellular uptake of exogenous FLAG-tagged PCSK9-D374Y measured in the presence of 50 μm chloroquine to inhibit lysosomal degradation of PCSK9 (Fig. 6C), with >70% inhibition at the highest dose of LDL (500 μg/ml).
FIGURE 6.
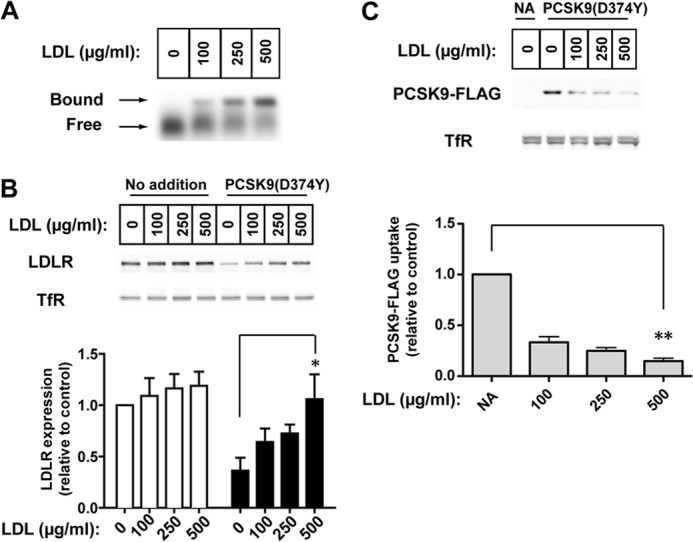
LDL inhibits LDLR degradation caused by exogenous purified PCSK9 and PCSK9 uptake in HuH7 human hepatoma cells. A, PCSK9-D374Y mixture (1.5 μg/ml; 20:1 ratio of unlabeled to Dylight680-labeled) was incubated for 45 min at 37 °C with increasing amounts of LDL in lipoprotein-deficient Medium B. An aliquot was subjected to agarose gel electrophoresis, and gels were scanned for fluorescent PCSK9. B, dose response for inhibition of PCSK9-mediated LDLR degradation by LDL is shown. HuH7 cells were cultured in sterol-depleting Medium C before incubation for 4 h with medium containing PCSK9-D374Y, and increasing amounts of LDL were prepared as in A. Protein in whole cell extracts were subjected to 8% SDS-PAGE followed by immunoblot analysis of LDLR and transferrin receptor (TfR). Secondary detection used an infrared dye (IRDye800)-labeled antibody. Blots were visualized and quantified using the LI-COR Odyssey infrared imaging system. LDLR levels were normalized to TfR expression. Graphical representations are the average and S.D. from five separate experiments. C, inhibition of LDLR-dependent PCSK9 cellular uptake by LDL is shown. Cells were cultured and treated with PCSK9 as in B but in the presence of 50 μm chloroquine to prevent lyososomal degradation of internalized PCSK9. Recombinant PCSK9-D374Y present in whole cell extracts was detected by immunoblotting for the FLAG epitope tag. NA, no addition; * indicates a statistical difference between columns with significance p < 0.05 by Student's t test; **, p < 0.005.
Inhibition of PCSK9-LDLR Binding Interaction by LDL Is Independent of LDL Binding to LDLR
LDL could inhibit PCSK9-mediated LDLR degradation through binding to the LDLR and affecting PCSK9-LDLR association by a competitive and/or allosteric mechanism. To assess this possibility, we transiently overexpressed in HuH7 cells either wild-type LDLR or an LDLR construct containing a deletion of repeat 6 of the ligand binding domain (LDLRΔR6). This deletion mimics a naturally occurring LDLR mutation associated with familial hypercholesterolemia (FH-Paris-1) resulting in defective LDL binding (2). Ligand blotting showed that LDL binding to LDLRΔR6 was decreased >90% compared with the wild-type LDLR, whereas PCSK9 binding to the mutant receptor was unaffected (Fig. 7A). We next tested the ability of LDL to inhibit LDLR-mediated cellular uptake of PCSK9 in transfected HuH7 cells in the presence of 50 μm chloroquine to inhibit lysosomal degradation of PCSK9. LDLR-transfected cells were treated with sterols (cholesterol/25-hydroxycholesterol) to suppress endogenous LDLR expression before incubation with FLAG-tagged PCSK9-D374Y (0.5 μg/ml; 4 h) in the absence or presence of LDL (500 μg/ml). PCSK9 uptake was negligible in vector-transfected cells treated with sterols (Fig. 7B, lane 2) and occurred at similar levels in cells expressing either wild-type LDLR or LDLRΔR6, indicating that PCSK9 binding and uptake into cells was mediated equally by either LDLR construct. Importantly, in cells expressing either wild-type LDLR or LDLRΔR6, PCSK9 uptake was inhibited >70% in the presence of LDL (fifth and eighth lanes). Thus, the inhibitory effect of LDL on LDLR-mediated PCSK9 uptake does not require LDL binding to the LDLR but is likely manifest through the LDL-PCSK9 interaction. It should be noted that at the near physiological concentrations used in these experiments, the apoB100 component of LDL is in >125-fold molar excess to PCSK9; thus, we do not expect that PCSK9 could affect LDL binding to LDLRs nor would PCSK9 cell uptake as a component of LDL contribute substantially to measured PCSK9 uptake.
FIGURE 7.
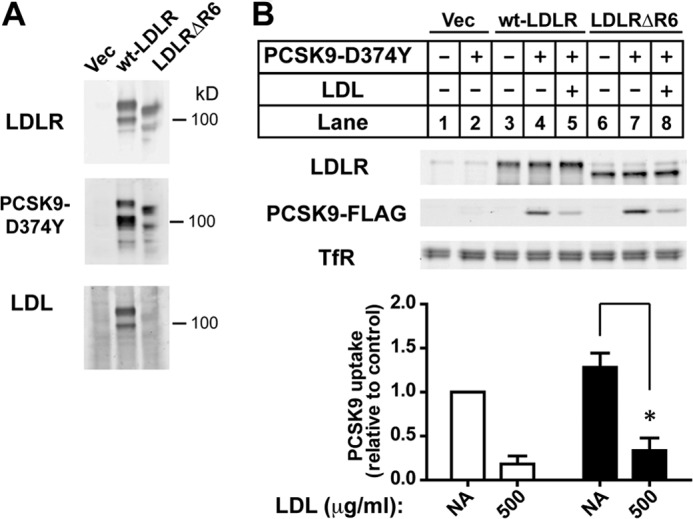
Inhibition of PCSK9 cellular uptake by LDL does not require LDL binding to the LDLR. A, ligand blots show binding of LDL and PCSK9-D374Y to immobilized wild-type and LDL binding-defective mutant LDLR. Total membrane extracts were prepared from HEK293 transiently expressing empty vector control (Vec), wild-type (wt) LDLR, or LDLR lacking repeat 6 of the ligand binding domain (LDLRΔR6) resolved on 8% SDS-PAGE under non-reducing conditions and transferred to nitrocellulose. Blots were incubated with a monoclonal antibody to LDLR (7H2), DyLight 680-labeled PCSK9-D374Y, or LDL as indicated. Bound proteins were visualized either directly (dye-labeled PCSK9) or after antibody detection as described under “Experimental Procedures.” B, HuH7 cells transiently overexpressing LDLR constructs used in A were cultured in sterol-supplemented Medium D before incubation with PCSK9-D374Y (0.5 μg/ml) in the absence or presence of LDL (500 μg/ml). Cells were cultured in the presence of 50 μm chloroquine to prevent lyososomal degradation of internalized PCSK9. Recombinant PCSK9 present in whole cell extracts was detected by immunoblotting for the FLAG epitope tag. Graphical representations are the average and S.D. from three separate experiments. NA, no addition; * indicates a statistical difference between columns with significance p < 0.005 by Student's t test.
DISCUSSION
The current study investigates the association of PCSK9 with lipoproteins based upon previous size-exclusion chromatography studies demonstrating PCSK9 co-migration with LDL-sized complexes in human plasma samples (35, 36), a phenomenon that we have also observed. Herein, we confirm that PCSK9 is abundant within an LDL fraction isolated from human plasma using Optiprep density gradient separation. Indeed, >40% of total PCSK9 was present in the LDL fraction derived from a pooled plasma sample from fasted normolipidemic subjects (Fig. 1). In cultured HuH7 cells, LDL dose-dependently inhibited the ability of exogenous recombinant human PCSK9 to mediate degradation of LDLRs and inhibited LDLR-dependent PCSK9 uptake into cells (Fig. 6). This inhibitory effect was observed in cell culture at LDL concentrations below that typically found in human plasma. We propose a model in which a large proportion of PCSK9 in human plasma that is LDL-bound has diminished binding activity toward cell surface LDLR. Thus LDL inhibits PCSK9-mediated LDLR degradation and could also negatively affect PCSK9 clearance rates, as this has been shown in mouse studies to be dependent upon PCSK9 binding to LDLRs (16).
In vitro binding studies demonstrated a binding interaction between purified recombinant human PCSK9 and isolated human LDL but not VLDL or HDL (Fig. 2). The observed binding to LDL was specific, saturable, and consistent with a single molecular site (Fig. 3). The latter finding is highly suggestive that PCSK9 binds to the apoB100 protein moiety of LDL and not to lipid. If this is the case, it may explain the inability of PCSK9 to bind to VLDL. Previous studies have shown that numerous monoclonal antibody epitopes on apoB100 that are accessible in LDL are masked in larger triglyceride-rich VLDL particles (45). It is also established that large VLDL binds poorly to the LDLR. Upon reduction in size due to lipolysis, a conformational change in the C terminus of apoB100 unmasks the LDLR binding site (46). We were unable to conclusively demonstrate PCSK9 binding to apoB in LDL by covalent cross-linking due to a high level of nonspecific cross-links formed (data not shown). Proteomic studies have shown that isolated human LDL can contain numerous other protein species that we cannot rule out as contributing to PCSK9 binding (47). In support of a direct binding interaction between PCSK9 and apoB100 of LDL, it has recently been shown that overexpressed PCSK9 binds apoB in the secretory pathway of rat McArdle cells and affects apoB secretion (48).
Saturation binding experiments (Fig. 3) determined PCSK9 binding affinity to LDL to be in the high nm range (KD ∼ 325 nm), which falls within a range of binding affinities (170–840 nm) previously reported for the in vitro interaction of PCSK9 with the LDLR EGF-A domain at neutral pH (13, 14). Therefore, both PCSK9-interacting partners would be expected to bind at similar PCSK9 concentrations in vivo. Based upon the current data, we propose a model in which equilibrium binding of PCSK9 to LDL determines the proportion of plasma PCSK9 that is in an inhibited state (LDL-bound) versus an active state (unbound). When LDL-C is elevated, proportionally more PCSK9 would be bound to LDL and less active, leading to increased LDLR expression and plasma LDL-C clearance. Conversely, when LDL-C levels are low, a higher proportion of PCSK9 would be active, thus promoting liver LDLR degradation and limiting LDL-C clearance by the liver at the expense of peripheral tissues. In support of this theory, Grefhorst et al. (16) showed that recombinant PCSK9 infused into the circulation of mice to approximate steady-state physiological levels greatly diminished LDLRs in mouse liver while having no effect on LDLR expression in the adrenal glands. The innocuous nature of PCSK9 loss-of-function mutations in affecting human health (10, 49) suggests that such a protective role for PCSK9 may not be critical with a modern diet.
In a previous study, LDL was shown to inhibit the ability of PCSK9 to decrease LDLR-dependent cell uptake of DiI-LDL in HEK293 cells (50). These authors suggested that LDL could antagonize PCSK9 by reducing the accessibility of binding sites on cell surface LDLRs or by causing an allosteric conformational change in the LDLR. This seems unlikely in vivo because (i) apoB100 at a typical concentration of 100 mg/dl in plasma is in large molar excess to PCSK9 (>250-fold), and (ii) the measured binding affinity of LDL for the LDLR is >20-fold higher than that of PCSK9 in vitro (13, 51). A structural model based upon x-ray crystallization data also indicates that the respective binding sites for LDL and PCSK9 on the LDLR are not closely juxtaposed (17). In the current study we demonstrate that the inhibitory effect of LDL on PCSK9 is independent of LDL binding to LDLRs (Fig. 7). These data suggest an inhibitory mechanism whereby prior binding to LDL decreases the ability of PCSK9 to bind to the LDLR. Using an agarose gel co-migration assay, we determined that large LDL particles do not sterically interfere with the interaction between the PCSK9 catalytic domain and the LDLR EGF-A domain (Fig. 5). However, this assay does not determine the relative binding affinity of PCSK9 to the EGF-A domain, which could be decreased by LDL through an allosteric effect on the PCSK9 molecule. Deletion analysis revealed that an N-terminal region of the PCSK9 prodomain (amino acids 31–52) is required for LDL binding (Fig. 4). Interestingly, deletion of this region of the prodomain has been shown to increase PCSK9 binding affinity to the LDLR by >7-fold, suggesting its involvement in an allosteric conformation (17). More recently the LDLR binding determinant in this region has been mapped to a highly conserved acidic tract (amino acids 32–40) that may interact with positive elements, perhaps within the histidine-rich C-terminal domain of PCSK9 (44, 52). Therefore, it is possible that LDL affects an interaction involving the PCSK9 prodomain, resulting in decreased binding affinity for cell surface LDLRs.
PCSK9 has emerged in recent years as a major regulator of plasma LDL-C levels and associated cardiovascular heart disease risk. Efforts to inhibit plasma PCSK9 using anti-PCSK9 antibodies that block binding to the LDLR show extremely promising results in early phase clinical trials (27). Regulatory mechanisms that affect a distinction between active and inactive forms of PCSK9 represent another potential avenue for developing hypocholesterolemic therapeutics targeting plasma PCSK9. The further characterization of the effect of LDL association on PCSK9 activity could illuminate such a regulatory mechanism.
Acknowledgments
We thank Yves Marcel, Ross Milne, and Ruth McPherson for critical reading of the manuscript.
This work was supported by Heart and Stroke Foundation of Ontario Grant-in-aid 000120 (to T. A. L.).
- LDL-C
- LDL-cholesterol
- apo
- apolipoprotein
- LDLR
- low density lipoprotein receptor
- PCSK9
- proprotein convertase (PC) subtilisin/kexin type 9.
REFERENCES
- 1. Goldstein J. L., Brown M. S. (2009) The LDL receptor. Arterioscler. Thromb. Vasc. Biol. 29, 431–438 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Hobbs H. H., Brown M. S., Goldstein J. L. (1992) Molecular genetics of the LDL receptor gene in familial hypercholesterolemia. Hum. Mutat. 1, 445–466 [DOI] [PubMed] [Google Scholar]
- 3. Innerarity T. L., Mahley R. W., Weisgraber K. H., Bersot T. P., Krauss R. M., Vega G. L., Grundy S. M., Friedl W., Davignon J., McCarthy B. J. (1990) Familial defective apolipoprotein B-100. A mutation of apolipoprotein B that causes hypercholesterolemia. J. Lipid Res. 31, 1337–1349 [PubMed] [Google Scholar]
- 4. Abifadel M., Rabès J. P., Devillers M., Munnich A., Erlich D., Junien C., Varret M., Boileau C. (2009) Mutations and polymorphisms in the proprotein convertase subtilisin kexin 9 (PCSK9) gene in cholesterol metabolism and disease. Hum. Mutat. 30, 520–529 [DOI] [PubMed] [Google Scholar]
- 5. Horton J. D., Cohen J. C., Hobbs H. H. (2009) PCSK9. A convertase that coordinates LDL catabolism. J. Lipid Res. 50, S172–S177 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Seidah N. G. (2009) PCSK9 as a therapeutic target of dyslipidemia. Expert. Opin. Ther. Targets 13, 19–28 [DOI] [PubMed] [Google Scholar]
- 7. Abifadel M., Varret M., Rabès J. P., Allard D., Ouguerram K., Devillers M., Cruaud C., Benjannet S., Wickham L., Erlich D., Derré A., Villéger L., Farnier M., Beucler I., Bruckert E., Chambaz J., Chanu B., Lecerf J. M., Luc G., Moulin P., Weissenbach J., Prat A., Krempf M., Junien C., Seidah N. G., Boileau C. (2003) Mutations in PCSK9 cause autosomal dominant hypercholesterolemia. Nat. Genet. 34, 154–156 [DOI] [PubMed] [Google Scholar]
- 8. Seidah N. G., Benjannet S., Wickham L., Marcinkiewicz J., Jasmin S. B., Stifani S., Basak A., Prat A., Chretien M. (2003) The secretory proprotein convertase neural apoptosis-regulated convertase 1 (NARC-1). Liver regeneration and neuronal differentiation. Proc. Natl. Acad. Sci. U.S.A. 100, 928–933 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Cohen J., Pertsemlidis A., Kotowski I. K., Graham R., Garcia C. K., Hobbs H. H. (2005) Low LDL cholesterol in individuals of African descent resulting from frequent nonsense mutations in PCSK9. Nat. Genet. 37, 161–165 [DOI] [PubMed] [Google Scholar]
- 10. Cohen J. C., Boerwinkle E., Mosley T. H., Jr., Hobbs H. H. (2006) Sequence variations in PCSK9, low LDL, and protection against coronary heart disease. N. Engl. J. Med. 354, 1264–1272 [DOI] [PubMed] [Google Scholar]
- 11. Benjannet S., Rhainds D., Essalmani R., Mayne J., Wickham L., Jin W., Asselin M. C., Hamelin J., Varret M., Allard D., Trillard M., Abifadel M., Tebon A., Attie A. D., Rader D. J., Boileau C., Brissette L., Chrétien M., Prat A., Seidah N. G. (2004) NARC-1/PCSK9 and its natural mutants. Zymogen cleavage and effects on the low density lipoprotein (LDL) receptor and LDL cholesterol. J. Biol. Chem. 279, 48865–48875 [DOI] [PubMed] [Google Scholar]
- 12. Naureckiene S., Ma L., Sreekumar K., Purandare U., Lo C. F., Huang Y., Chiang L. W., Grenier J. M., Ozenberger B. A., Jacobsen J. S., Kennedy J. D., DiStefano P. S., Wood A., Bingham B. (2003) Functional characterization of Narc 1, a novel proteinase related to proteinase K. Arch. Biochem. Biophys. 420, 55–67 [DOI] [PubMed] [Google Scholar]
- 13. Cunningham D., Danley D. E., Geoghegan K. F., Griffor M. C., Hawkins J. L., Subashi T. A., Varghese A. H., Ammirati M. J., Culp J. S., Hoth L. R., Mansour M. N., McGrath K. M., Seddon A. P., Shenolikar S., Stutzman-Engwall K. J., Warren L. C., Xia D., Qiu X. (2007) Structural and biophysical studies of PCSK9 and its mutants linked to familial hypercholesterolemia. Nat. Struct. Mol. Biol. 14, 413–419 [DOI] [PubMed] [Google Scholar]
- 14. Piper D. E., Jackson S., Liu Q., Romanow W. G., Shetterly S., Thibault S. T., Shan B., Walker N. P. (2007) The crystal structure of PCSK9. A regulator of plasma LDL-cholesterol. Structure 15, 545–552 [DOI] [PubMed] [Google Scholar]
- 15. McNutt M. C., Lagace T. A., Horton J. D. (2007) Catalytic activity is not required for secreted PCSK9 to reduce low density lipoprotein receptors in HepG2 cells. J. Biol. Chem. 282, 20799–20803 [DOI] [PubMed] [Google Scholar]
- 16. Grefhorst A., McNutt M. C., Lagace T. A., Horton J. D. (2008) Plasma PCSK9 preferentially reduces liver LDL receptors in mice. J. Lipid Res. 49, 1303–1311 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Kwon H. J., Lagace T. A., McNutt M. C., Horton J. D., Deisenhofer J. (2008) Molecular basis for LDL receptor recognition by PCSK9. Proc. Natl. Acad. Sci. U.S.A. 105, 1820–1825 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Zhang D.-W., Lagace T. A., Garuti R., Zhao Z., McDonald M., Horton J. D., Cohen J. C., Hobbs H. H. (2007) Binding of proprotein convertase subtilisin/kexin type 9 to epidermal growth factor-like repeat A of low density lipoprotein receptor decreases receptor recycling and increases degradation. J. Biol. Chem. 282, 18602–18612 [DOI] [PubMed] [Google Scholar]
- 19. Maxwell K. N., Soccio R. E., Duncan E. M., Sehayek E., Breslow J. L. (2003) Novel putative SREBP and LXR target genes identified by microarray analysis in liver of cholesterol-fed mice. J. Lipid Res. 44, 2109–2119 [DOI] [PubMed] [Google Scholar]
- 20. Horton J. D., Shah N. A., Warrington J. A., Anderson N. N., Park S. W., Brown M. S., Goldstein J. L. (2003) Combined analysis of oligonucleotide microarray data from transgenic and knockout mice identifies direct SREBP target genes. Proc. Natl. Acad. Sci. U.S.A. 100, 12027–12032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Dubuc G., Chamberland A., Wassef H., Davignon J., Seidah N. G., Bernier L., Prat A. (2004) Statins up-regulate PCSK9, the gene encoding the proprotein convertase neural apoptosis-regulated convertase-1 implicated in familial hypercholesterolemia. Arterioscler. Thromb. Vasc. Biol. 24, 1454–1459 [DOI] [PubMed] [Google Scholar]
- 22. Careskey H. E., Davis R. A., Alborn W. E., Troutt J. S., Cao G., Konrad R. J. (2008) Atorvastatin increases human serum levels of proprotein convertase subtilisin/kexin type 9. J. Lipid Res. 49, 394–398 [DOI] [PubMed] [Google Scholar]
- 23. Mayne J., Dewpura T., Raymond A., Cousins M., Chaplin A., Lahey K. A., Lahaye S. A., Mbikay M., Ooi T. C., Chrétien M. (2008) Plasma PCSK9 levels are significantly modified by statins and fibrates in humans. Lipids Health Dis. 7, 22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Davignon J., Dubuc G. (2009) Statins and ezetimibe modulate plasma proprotein convertase subtilisin kexin-9 (PCSK9) levels. Trans. Am. Clin. Climatol. Assoc. 120, 163–173 [PMC free article] [PubMed] [Google Scholar]
- 25. Frank-Kamenetsky M., Grefhorst A., Anderson N. N., Racie T. S., Bramlage B., Akinc A., Butler D., Charisse K., Dorkin R., Fan Y., Gamba-Vitalo C., Hadwiger P., Jayaraman M., John M., Jayaprakash K. N., Maier M., Nechev L., Rajeev K. G., Read T., Röhl I., Soutschek J., Tan P., Wong J., Wang G., Zimmermann T., de Fougerolles A., Vornlocher H. P., Langer R., Anderson D. G., Manoharan M., Koteliansky V., Horton J. D., Fitzgerald K. (2008) Therapeutic RNAi targeting PCSK9 acutely lowers plasma cholesterol in rodents and LDL cholesterol in nonhuman primates. Proc. Natl. Acad. Sci. U.S.A. 105, 11915–11920 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Chan J. C., Piper D. E., Cao Q., Liu D., King C., Wang W., Tang J., Liu Q., Higbee J., Xia Z., Di Y., Shetterly S., Arimura Z., Salomonis H., Romanow W. G., Thibault S. T., Zhang R., Cao P., Yang X. P., Yu T., Lu M., Retter M. W., Kwon G., Henne K., Pan O., Tsai M. M., Fuchslocher B., Yang E., Zhou L., Lee K. J., Daris M., Sheng J., Wang Y., Shen W. D., Yeh W. C., Emery M., Walker N. P., Shan B., Schwarz M., Jackson S. M. (2009) A proprotein convertase subtilisin/kexin type 9 neutralizing antibody reduces serum cholesterol in mice and nonhuman primates. Proc. Natl. Acad. Sci. U.S.A. 106, 9820–9825 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Stein E. A., Mellis S., Yancopoulos G. D., Stahl N., Logan D., Smith W. B., Lisbon E., Gutierrez M., Webb C., Wu R., Du Y., Kranz T., Gasparino E., Swergold G. D. (2012) Effect of a monoclonal antibody to PCSK9 on LDL cholesterol. N. Engl. J. Med. 366, 1108–1118 [DOI] [PubMed] [Google Scholar]
- 28. Lakoski S. G., Lagace T. A., Cohen J. C., Horton J. D., Hobbs H. H. (2009) Genetic and metabolic determinants of plasma PCSK9 levels. J. Clin. Endocrinol. Metab. 94, 2537–2543 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Mayne J., Raymond A., Chaplin A., Cousins M., Kaefer N., Gyamera-Acheampong C., Seidah N. G., Mbikay M., Chrétien M., Ooi T. C. (2007) Plasma PCSK9 levels correlate with cholesterol in men but not in women. Biochem. Biophys. Res. Commun. 361, 451–456 [DOI] [PubMed] [Google Scholar]
- 30. Welder G., Zineh I., Pacanowski M. A., Troutt J. S., Cao G., Konrad R. J. (2010) High-dose atorvastatin causes a rapid sustained increase in human serum PCSK9 and disrupts its correlation with LDL cholesterol. J. Lipid Res. 51, 2714–2721 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Lambert G., Ancellin N., Charlton F., Comas D., Pilot J., Keech A., Patel S., Sullivan D. R., Cohn J. S., Rye K.-A., Barter P. J. (2008) Plasma PCSK9 concentrations correlate with LDL and total cholesterol in diabetic patients and are decreased by fenofibrate treatment. Clin. Chem. 54, 1038–1045 [DOI] [PubMed] [Google Scholar]
- 32. Browning J. D., Horton J. D. (2010) Fasting reduces plasma proprotein convertase, subtilisin/kexin type 9, and cholesterol biosynthesis in humans. J. Lipid Res. 51, 3359–3363 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Persson L., Cao G., Ståhle L., Sjöberg B. G., Troutt J. S., Konrad R. J., Gälman C., Wallén H., Eriksson M., Hafström I., Lind S., Dahlin M., Amark P., Angelin B., Rudling M. (2010) Circulating proprotein convertase subtilisin kexin type 9 has a diurnal rhythm synchronous with cholesterol synthesis and is reduced by fasting in humans. Arterioscler. Thromb. Vasc. Biol. 30, 2666–2672 [DOI] [PubMed] [Google Scholar]
- 34. Benjannet S., Rhainds D., Hamelin J., Nassoury N., Seidah N. G. (2006) The proprotein convertase (PC) PCSK9 is inactivated by furin and/or PC5/6A. Functional consequences of natural mutations and post-translational modifications. J. Biol. Chem. 281, 30561–30572 [DOI] [PubMed] [Google Scholar]
- 35. Fan D., Yancey P. G., Qiu S., Ding L., Weeber E. J., Linton M. F., Fazio S. (2008) Self-association of human PCSK9 correlates with its LDLR-degrading activity. Biochemistry 47, 1631–1639 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Luo Y., Warren L., Xia D., Jensen H., Sand T., Petras S., Qin W., Miller K. S., Hawkins J. (2009) Function and distribution of circulating human PCSK9 expressed extrahepatically in transgenic mice. J. Lipid Res. 50, 1581–1588 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Brown M. S., Faust J. R., Goldstein J. L., Kaneko I., Endo A. (1978) Induction of 3-hydroxy-3-methylglutaryl coenzyme A reductase activity in human fibroblasts incubated with compactin (ML-236B), a competitive inhibitor of the reductase. J. Biol. Chem. 253, 1121–1128 [PubMed] [Google Scholar]
- 38. Goldstein J. L., Basu S. K., Brown M. S. (1983) Receptor-mediated endocytosis of low-density lipoprotein in cultured cells. Methods Enzymol. 98, 241–260 [DOI] [PubMed] [Google Scholar]
- 39. Russell D. W., Brown M. S., Goldstein J. L. (1989) Different combinations of cysteine-rich repeats mediate binding of low density lipoprotein receptor to two different proteins. J. Biol. Chem. 264, 21682–21688 [PubMed] [Google Scholar]
- 40. Lagace T. A., Curtis D. E., Garuti R., McNutt M. C., Park S. W., Prather H. B., Anderson N. N., Ho Y. K., Hammer R. E., Horton J. D. (2006) Secreted PCSK9 decreases the number of LDL receptors in hepatocytes and in livers of parabiotic mice. J. Clin. Invest. 116, 2995–3005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Havel R. J., Eder H. A., Bragdon J. H. (1955) The distribution and chemical composition of ultracentrifugally separated lipoproteins in human serum. J. Clin. Invest. 34, 1345–1353 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Peterson G. L. (1977) A simplification of the protein assay method of Lowry et al., which is more generally applicable. Anal. Biochem. 83, 346–356 [DOI] [PubMed] [Google Scholar]
- 43. Yee M. S., Pavitt D. V., Tan T., Venkatesan S., Godsland I. F., Richmond W., Johnston D. G. (2008) Lipoprotein separation in a novel iodixanol density gradient, for composition, density, and phenotype analysis. J. Lipid Res. 49, 1364–1371 [DOI] [PubMed] [Google Scholar]
- 44. Benjannet S., Saavedra Y. G., Hamelin J., Asselin M. C., Essalmani R., Pasquato A., Lemaire P., Duke G., Miao B., Duclos F., Parker R., Mayer G., Seidah N. G. (2010) Effects of the prosegment and pH on the activity of PCSK9. Evidence for additional processing events. J. Biol. Chem. 285, 40965–40978 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Marcel Y. L., Hogue M., Weech P. K., Davignon J., Milne R. W. (1988) Expression of apolipoprotein B epitopes in lipoproteins. Relationship to conformation and function. Arteriosclerosis 8, 832–844 [DOI] [PubMed] [Google Scholar]
- 46. Boren J., Lee I., Zhu W., Arnold K., Taylor S., Innerarity T. L. (1998) Identification of the low density lipoprotein receptor-binding site in apolipoprotein B100 and the modulation of its binding activity by the carboxyl terminus in familial defective apo-B100. J. Clin. Invest. 101, 1084–1093 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Karlsson H., Leanderson P., Tagesson C., Lindahl M. (2005) Proteomics 5, 551–565 [DOI] [PubMed] [Google Scholar]
- 48. Sun H., Samarghandi A., Zhang N., Yao Z., Xiong M., Teng B. B. (2012) Proprotein convertase subtilisin/kexin type 9 interacts with apolipoprotein B and prevents its intracellular degradation, irrespective of the low-density lipoprotein receptor. Arterioscler. Thromb. Vasc. Biol. 32, 1585–1595 [DOI] [PubMed] [Google Scholar]
- 49. Zhao Z., Tuakli-Wosornu Y., Lagace T. A., Kinch L., Grishin N. V., Horton J. D., Cohen J. C., Hobbs H. H. (2006) Molecular characterization of loss-of-function mutations in PCSK9 and identification of a compound heterozygote. Am. J. Hum. Genet. 79, 514–523 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Fisher T. S., Lo Surdo P., Pandit S., Mattu M., Santoro J. C., Wisniewski D., Cummings R. T., Calzetta A., Cubbon R. M., Fischer P. A., Tarachandani A., De Francesco R., Wright S. D., Sparrow C. P., Carfi A., Sitlani A. (2007) Effects of pH and low density lipoprotein (LDL) on PCSK9-dependent LDL receptor regulation. J. Biol. Chem. 282, 20502–20512 [DOI] [PubMed] [Google Scholar]
- 51. Schneider W. J., Beisiegel U., Goldstein J. L., Brown M. S. (1982) Purification of the low density lipoprotein receptor, an acidic glycoprotein of 164,000 molecular weight. J. Biol. Chem. 257, 2664–2673 [PubMed] [Google Scholar]
- 52. Holla Ø. L., Laerdahl J. K., Strøm T. B., Tveten K., Cameron J., Berge K. E., Leren T. P. (2011) Removal of acidic residues of the prodomain of PCSK9 increases its activity toward the LDL receptor. Biochem. Biophys. Res. Commun. 406, 234–238 [DOI] [PubMed] [Google Scholar]