

Background: Pentameric ligand-gated ion channels are modulated by general anesthetics.
Results: The crystal structure of ELIC in complex with bromoform reveals anesthetic binding in the channel pore and in novel sites in the transmembrane and extracellular domain.
Conclusion: General anesthetics allosterically modulate channel function via multisite binding.
Significance: Our data reveal detailed insight into multisite recognition of general anesthetics at the structural level.
Keywords: Anesthesia, Cys-loop Receptors, Ion Channels, Membrane Proteins, X-ray Crystallography, ELIC, Ligand-gated Ion Channels
Abstract
Pentameric ligand-gated ion channels (pLGICs), such as nicotinic acetylcholine, glycine, γ-aminobutyric acid GABAA/C receptors, and the Gloeobacter violaceus ligand-gated ion channel (GLIC), are receptors that contain multiple allosteric binding sites for a variety of therapeutics, including general anesthetics. Here, we report the x-ray crystal structure of the Erwinia chrysanthemi ligand-gated ion channel (ELIC) in complex with a derivative of chloroform, which reveals important features of anesthetic recognition, involving multiple binding at three different sites. One site is located in the channel pore and equates with a noncompetitive inhibitor site found in many pLGICs. A second transmembrane site is novel and is located in the lower part of the transmembrane domain, at an interface formed between adjacent subunits. A third site is also novel and is located in the extracellular domain in a hydrophobic pocket between the β7–β10 strands. Together, these results extend our understanding of pLGIC modulation and reveal several specific binding interactions that may contribute to modulator recognition, further substantiating a multisite model of allosteric modulation in this family of ion channels.
Introduction
General anesthetics, including alcohols and inhalational surgical agents, inhibit nerve signaling by interacting with proteins in the brain and spinal cord (1). One of the primary classes of neurological proteins modulated by general anesthetics is that of pentameric ligand-gated ion channels (pLGICs),5 which include ionotropic receptors for acetylcholine (ACh), Gly, and γ-aminobutyric acid (GABA) (2). Cation flux through nicotinic acetylcholine receptors (nAChRs) is generally excitatory and is inhibited by general anesthetics; conversely, chloride flux through Gly receptors (GlyRs) and GABA type A receptors (GABAARs) generally inhibits neuronal signaling, and many of these proteins are functionally enhanced by general anesthetics (3). An interesting exception to this rule is the ρ subtype of GABAAR (often referred to as GABACR), which conducts chloride but is inhibited by alcohols and anesthetics (4).
Several specific protein sites in pLGICs have been implicated in general anesthetic binding. Channels in this family are pentamers of identical or similar subunits, each of which contributes to a ligand binding extracellular domain, a pore-forming transmembrane domain, and a variable cytoplasmic domain (5). Mutagenesis (6), labeling (7), and molecular dynamics studies (8) of nAChRs have implicated the transmembrane domain in nAChR inhibition, possibly via obstruction of the channel pore. Conversely, a range of chimera and mutagenesis studies of GlyRs and GABAARs has implicated both intrasubunit and intersubunit transmembrane sites (9), as well as locations in the ligand binding (10) and intracellular (11) domains, in channel potentiation by alcohols and other general anesthetics. The range of implicated sites, as well as relatively high (high μm to low mm (12)) concentrations required to induce anesthesia, and the lack of distinctive pharmacophores for most of these agents (13) have led to the proposal that anesthetics may modulate channel function through simultaneous interactions with multiple distinct sites on any given receptor (2).
Recent crystal structures of homologs from bacteria and nematodes have revealed critical details of pLGIC structure. The ELIC structure is thought to present a nonconducting conformation, possibly corresponding to the closed state, whereas the Gloeobacter violaceus ligand-gated ion channel (GLIC) and glutamate-activated chloride channel (GluCl) structures represent a conducting conformation, possibly corresponding to the open state (14, 15). GLIC forms cation-selective channels that are activated by low pH and modulated by most alcohols (16) and other general anesthetics (17) in a manner similar to nAChRs and GABACRs; furthermore, its structure was recently determined in complex with the general anesthetics desflurane and propofol (18). Surprisingly, although general anesthetics are presumed to stabilize the closed or desensitized state(s) of a cation-selective ion channel (19), the known anesthetic-bound GLIC structures are associated with a presumed open state, superimposable with the modulator-free form solved in the presence of ligand (protons) (20). Thus, the structural consequences of binding modulators, particularly inhibitory agents such as general anesthetics, remain unclear. In this work, we sought to extend the observation of general anesthetic modulation of pLGICs to the model system of ELIC, which is a recently discovered GABA-activated bacterial pLGIC (21–24).
EXPERIMENTAL PROCEDURES
Protein Expression and Crystallization
ELIC was expressed and purified as described previously, but with minor modifications (21). In brief, ELIC was cloned into pET-11a expressed in the C43 Escherichia coli strain as an N-terminal fusion to maltose-binding protein. Membranes were solubilized with 2% Anagrade n-undecyl-β-d-maltoside (Anatrace), and the solubilized fraction was incubated with amylose resin (New England Biolabs). Affinity-bound protein was cleaved off with 3CV protease and further purified using size exclusion chromatography on a Superdex 200 10/300 GL (GE Healthcare). The running buffer was composed of 10 mm sodium phosphate, pH 8.0, 150 mm NaCl, and 0.15% n-undecyl-β-d-maltoside. Concentrated protein (10 mg/ml) was supplemented with E. coli lipids (Avanti Polar Lipids), and co-crystallization of ELIC with bromoform and bromoethanol (Sigma-Aldrich) was performed by sitting drop vapor diffusion at 4 °C. The crystallization buffer was composed of 200 mm ammonium sulfate, 50 mm N-(2-Acetamido)iminodiacetic acid (ADA), pH 6.5, and 9–12% PEG 4000. Crystals of ELIC in complex with bromoform were obtained at bromoform concentrations from 1 to 10 mm. Crystals were harvested after adding 30% glycerol as a cryoprotectant to the mother liquor. Crystals were cryo-cooled by immersion in liquid nitrogen. Crystals of ELIC in complex with bromoethanol did not diffract to sufficiently high resolution to allow structure determination.
Structure Determination and Refinement
A diffraction data set to a resolution of 3.65 Å was used for structure determination and refinement (crystallographic statistics are shown in Table 1). Data integration was done in XDS, and scaling was done in SCALA. Model building and refinement were carried out by iterative cycles of manual rebuilding in COOT and refinement in PHENIX, using one Translation Libration Screw (TLS) body per subunit and five-fold Non-Crystallographic Symmetry (NCS) restraints. Anomalous difference density maps were calculated using reflections between 25 and 5 Å. The clearest anomalous peaks could be observed when data sets from three different crystals were merged to calculate anomalous maps (see statistics for the merged data set in Table 1). Because bromoform is a relatively symmetric and small molecule, we were not able to assign a unique binding pose at the present resolution limit, and therefore, we built a single bromine atom into each anomalous electron density peak. Model validation was done using MOLPROBITY, and all figures were prepared using PyMOL. The analysis of pore dimensions was carried out using HOLE (25).
TABLE 1.
Data statistics and refinement
SLS, Swiss Light Source; r.m.s.d., root mean square deviation.
| Data set 1 (crystal one) | Data set 2 (merged from three crystals) | |
|---|---|---|
| Crystallographic statistics | ||
| Beamline | X06A (SLS) | X06A (SLS) |
| Date of collection | September 13, 2011 | September 13, 2011 |
| Wavelength (Å) | 0.9193 | 0.9193 |
| Space group | P21 | P21 |
| a, b, c (Å) | 105.11, 266.25, 110.75 | 105.63, 266.33, 110.97 |
| β (°) | 109.78 | 108.63 |
| Resolution limits (Å) | 44.08–3.65 (3.85–3.65) | 25.00–5.00 (5.27–5.00) |
| Rmerge (%) | 10.6 (67.6) | 19.2 (33.3) |
| Rmeas | 12.4 (78.9) | 20.7 (35.8) |
| <I/σ> | 9.5 (2.1) | 12.1 (6.6) |
| Multiplicity | 3.8 (3.7) | 11.3 (11.5) |
| Completeness (%) | 98.9 (97.0) | 99.2 (100.0) |
| Total number of reflections | 237,239 (33495) | 282,159 (42154) |
| Number unique reflections | 62,852 (8988) | 24,931 (3680) |
| Anomalous completeness | 94.6 (92.6) | 99.2 (100.0) |
| Anomalous multiplicity | 1.9 (1.9) | 5.6 (5.6) |
| Refinement and model statistics | ||
| Number of residues in ASU | 3220 | |
| Number of atoms in ASU | 52,170 | |
| Rwork (%) | 22.87 | |
| Rfree (%) | 26.39 | |
| r.m.s.d. bond distance (Å) | 0.008 | |
| r.m.s.d. bond angle (°) | 1.335 | |
Mutagenesis and Two-electrode Voltage Clamp Recordings
For expression in Xenopus laevis oocytes, cDNAs encoding ELIC and human α1 GlyR were subcloned into pGEM-HE (26). All mutant receptors were created using a QuikChange method (Stratagene) and verified by sequencing (Agowa). The plasmids were linearized with NheI, and capped cRNA was synthesized using the T7 mMESSAGE-mMACHINE transcription kit (Ambion). Stage V-VI oocytes, prepared as described previously (23), were freshly harvested from the ovarian lobes of anesthetized frogs and subsequently injected with 30 nl of cRNA (∼100 ng/nl). The oocyte incubation solution contained (in mm): 96 NaCl, 2 KCl, 1.8 CaCl2, 2 MgCl2, and 5 HEPES (pH 7.4), supplemented with 50 mg/liter gentamicin sulfate.
Whole-cell currents from oocytes were recorded 1–2 days after injection using the two-electrode voltage clamp technique. For ELIC recordings, all electrophysiological experiments were conducted at a constant temperature (18 °C) using an Axoclamp 900A amplifier (Molecular Devices) controlled by a pClamp 10.2 data acquisition system (Molecular Devices). Data were sampled at 100 Hz and low pass-filtered at 10 Hz using a four-pole Bessel filter. Voltage and current electrodes were backfilled with 3 m KCl, and their resistances were kept as low as possible (<1 megaohms). Oocytes were clamped at −60 mV throughout all recordings. The bath perfusion solution contained (in mm): 96 NaCl, 2 KCl, 1.8 CaCl2, 1 MgCl2, 5 HEPES (pH 7.4). Current responses were separated by 180-s ligand-free intervals to avoid effects of desensitization. For constructing concentration-response curves, current responses were evoked by co-applications of GABA with modulator in between applications of GABA alone. To allow equilibration in the bath and to monitor direct effects on receptor function, co-applications of GABA with modulator were preceded by a 30-s preapplication of modulator alone. Solutions containing bromoform were prepared from a 1 m stock solution in dimethyl sulfoxide (DMSO) appropriately diluted into bath perfusion solution. The modulation was quantified by comparing current amplitudes in the presence and absence of modulator. Concentration-inhibition curves were fitted according to the Hill equation: y = 100/(1 + (IC50/[modulator])nH), where y is normalized current, IC50 is the concentration of modulator producing 50% inhibition, [modulator] is the modulator concentration, and nH is the Hill coefficient. Data analysis was carried out with Clampfit 10.2 (Molecular Devices) and Origin 7.0 (OriginLab).
For α1 GlyR recordings, we used a HiClamp apparatus (MultiChannel Systems, Reutlingen, Germany). The membrane potential of oocytes was clamped at a value of −80 mV, and currents evoked by Gly or compounds were recorded with a sampling frequency of 100 Hz. All recordings were performed at 18 °C, and cells were superfused with OR2 medium containing (in mm): 82.5 NaCl, 2.5 KCl, 1.8 CaCl2, 1 MgCl2, and 5 HEPES (pH 7.4). Concentration-activation curves were fitted according to the Hill equation: y = Imax/[1 + (EC50/[agonist])nH], where y is the normalized current amplitude, Imax is the maximal efficacy, EC50 is the agonist concentration at half-maximal efficacy, [agonist] is the agonist concentration, and nH is the Hill coefficient. Curve fitting was carried out using the least squares method in MATLAB. All numerical data are presented as means ± S.E. Statistical differences were assessed using a Student's t test.
Quenching of Intrinsic Tryptophan Fluorescence in Detergent-solubilized ELIC
Intrinsic tryptophan fluorescence quenching experiments were performed under equilibrium conditions using a FlexStation 3 microplate reader (Molecular Devices). 295 nm was used as the excitation wavelength, and fluorescence emission spectra were recorded from 320 to 370 nm. The fluorescence peak maximum for ELIC was near 340 nm, which was used as the emission wavelength for calculating quenching plots. The fluorescence quenching experiments were performed in the presence of 0–20 mm bromoform, and potassium bromide was used to evaluate nonspecific quenching. The quenched fluorescence was plotted as F/F0, where F0 and F are the fluorescence intensity in the absence and presence of bromoform, respectively.
RESULTS
Functional Effects of General Anesthetics on ELIC
To investigate whether ELIC is a useful model to study channel modulation by general anesthetics, we expressed ELIC in Xenopus oocytes and characterized the effects of various anesthetics, including chloroform, propofol, etomidate, and alcohols. To facilitate structural studies, we also tested the bromo analogs bromoform (Fig. 1a) and bromoethanol. Using the two-electrode voltage clamp technique, we applied the ELIC agonist GABA and compared current amplitudes in the absence and presence of anesthetics. We observed that ELIC is inhibited by micromolar concentrations of bromoform/chloroform and millimolar concentrations of alcohols (Fig. 1b). From concentration-inhibition curves, we calculated IC50 values of 125 ± 10 μm for bromoform (n = 4–10), 162 ± 26 μm for chloroform (n = 2–3), 125 ± 5 mm for ethanol (n = 3–4), and 72 ± 5 for mm bromoethanol (n = 3–9) (Fig. 1c). These results demonstrate that ELIC has pharmacological properties that are different from the related bacterial homolog GLIC and the more distant human pLGICs. For example, chloroform potentiates agonist-activated responses at GlyR (27) and GABAAR (28–30), whereas chloroform inhibits ELIC. In addition, methanol and ethanol potentiate agonist responses of GLIC (16) and human pLGICs (31, 32), whereas ethanol and bromoethanol inhibit ELIC. Despite these differences between ELIC and human pLGICs, our data demonstrate that certain anesthetics, including bromoform and chloroform, bind with IC50 values comparable with human pLGICs and suggest that ELIC is a potentially suitable model for structural studies.
FIGURE 1.

Modulation of ELIC by bromoform. a, chemical formulas of chloroform and bromoform. b, ELIC current traces were evoked by application of 5 mm GABA in the absence and presence of 200 μm bromoform or 100 mm bromoethanol. c, concentration-inhibition curves from the experiments shown in panel b. Squares are data for bromoform, and circles are data for bromoethanol (mean ± S.E.). Each data point represents the average of 3–10 experiments.
X-ray Crystal Structure of ELIC in Complex with Bromoform
We determined the crystal structure of ELIC in complex with bromoform, a chloroform analog, to take advantage of the anomalous diffraction signal generated by bromine atoms and facilitate the identification of bromoform binding sites in ELIC. Fig. 2, a–c, show a graphic representation of ELIC and an overlay with anomalous electron density shown as a magenta mesh. We clearly observe anomalous density at three different locations in ELIC, namely in the channel pore, at a site in the extracellular domain, and at a transmembrane site, which is at the interface between two neighboring subunits.
FIGURE 2.

X-ray crystal structure of ELIC in complex with bromoform. a, side view of ELIC in graphic representation and overlay with anomalous electron density, shown as magenta mesh at a contour level of 4σ. Each subunit of the pentamer is shown in a different color. Anomalous densities can be observed in the extracellular domain, transmembrane domain, and channel pore. b and c, extracellular and intracellular view of ELIC along the five-fold symmetry axis. The pore site is lined by the M2-helices of each of the five subunits. The transmembrane site is formed at the interface between two subunits, involving the M1- and M4-helix of one subunit and the M3-helix of the neighboring subunit. Bromoform molecules are represented as a single magenta sphere. d, comparison of the bromoform binding site in the closed channel pore of ELIC (left) and bromo-lidocaine binding site in the open channel pore of GLIC (right). Bromoform and bromo-lidocaine occupy overlapping binding sites, which are located in the hydrophobic part of the ion conduction pathway (hydrophobic residues are colored in yellow, hydrophilic residues are in green, and charged residues are in red). e, mutagenesis experiments in ELIC demonstrate that the inhibitory effect of 200 μm bromoform is almost completely eliminated in L9′S and strongly reduced in F16′S mutants. The inhibitory effect of bromoform was tested at an EC20 concentration of GABA, which was 0.5 mm for L9′S and 3 mm for wild type and F16′S. Error bars represent mean ± S.E. from 3–5 different experiments. Asterisks indicate significant difference from wild type (p < 0.05).
The strongest anomalous density can be observed in the middle of the channel pore at a position near 13′A of the pore-lining M2-helix (Fig. 2d, left), corresponding to the hydrophobic part of the channel pore (Fig. 2d, hydrophobic residues are shown in yellow, hydrophilic residues are shown in green, and charged residues are shown in red). This indicates that a bromoform molecule is bound close to the 13′A side chain and apparently stabilized between the bulkier 9′L and 16′F residues. This binding site overlaps with the recently described site for bromo-lidocaine in the open GLIC structure (Fig. 2d, right) (33), which likely represents the binding site for extensively studied noncompetitive inhibitors such as tricyclic antidepressants, chlorpromazine, lidocaine, and quinacrine (34–38). To investigate the functional importance of this hydrophobic site, we mutated 9′L and 16′F in ELIC to the hydrophilic Ser residue and compared the inhibitory effects of bromoform with wild type ELIC (Fig. 2e). We observed that the inhibition of ELIC by 200 μm bromoform is almost completely eliminated in the 9′S mutant (no further inhibition could be observed at bromoform concentrations up to 2 mm) and significantly reduced in the 16′S mutant. This result indicates that the hydrophobic region of the channel pore between 9′ and 16′ functionally contributes to the inhibitory effects of bromoform on ELIC and forms a possible binding site for general anesthetics.
Comparison of the bromoform-bound complex of ELIC with the previously published apoELIC structure (Protein Data Bank (PDB) code 2vl0 (21)) reveals that the pore-forming M2-helix undergoes a subtle conformational change upon binding of bromoform (Fig. 3a). This becomes especially clear upon comparison of the pore radius calculated for the two structures (Fig. 3b). The inward movement of the M2-helices causes the channel pore to adopt a more closed conformation than in the apo structure, particularly in the upper half (20′ to 13′) of the pore. We calculated that the pore radius in the bromoform-bound structure is reduced to 1.2 Å at 20′, 1.1 Å at 16′, and 2.5 Å at 13′, compared with 1.7, 1.5, and 3.3 Å in apoELIC (Fig. 3b). This conformational change in the M2-helices likely enables optimal hydrophobic interactions with the bromoform molecule bound near 13′A.
FIGURE 3.

Bromoform stabilizes the ELIC pore in a closed conformation. a, ribbon representation of the pore-lining M2-helix in the apoELIC structure (blue, PDB code 2vl0) and the bromoform-bound structure (red). The view is along the five-fold symmetry axis looking down on the channel pore from the extracellular domain. The dashed lines are distance measurements between 13′A Cα atoms of different subunits. b, pore radius analysis for apoELIC (blue), bromoform-bound ELIC (red), and GLIC, which likely corresponds to an open pore conformation (PDB code 3eam).
In the extracellular domain, we find anomalous density behind the β7–β10 strands where a hydrophobic site is formed by residues Leu-128 and Ile-195 (Fig. 2b). In the lower part of the transmembrane domain, we find anomalous density in a pre-existing pocket formed at the protein-lipid interface, between the M1- and M4-helices of one subunit and the M3-helix of a neighboring subunit (Fig. 2c). This intersubunit bromoform site in ELIC is different from the previously identified binding site for propofol and desflurane in GLIC (18), which is located farther up the transmembrane domain at an intrasubunit pocket (Fig. 4a). The intersubunit bromoform site is formed at five pre-existing cavities in the ELIC pentamer (Fig. 4, b and c). Such cavities are absent in the GLIC and Caenorhabditis elegans GluCl structures (14), where the protein surface forms a lipid-exposed groove, rather than a pocket. Detailed analysis of the amino acid side chains contributing to the transmembrane pocket shows that this site is lined by several aromatic residues (Fig. 4d, Trp-221 and Trp-225 in M1 and Phe-275 in M3) as well as several hydrophobic residues (Ala-218 in M1; Ile-278, Leu-279, and Ile-282 in M3; and Leu-303 and Pro-306 in M4).
FIGURE 4.

Molecular recognition of bromoform at an intersubunit transmembrane site. a and b, Comparison of the propofol binding site in GLIC (a) and the bromoform binding site of ELIC (b) in a surface representation. Propofol binds in an intrasubunit pocket in the upper half of the transmembrane domain. Bromoform binds in an intersubunit pocket farther down the transmembrane domain. The transmembrane domains of two neighboring subunits are shown in orange and blue. The propofol and bromoform pockets are highlighted in yellow. The extracellular domain is shown in white. The inset shows a more detailed view of the intersubunit transmembrane site. The magenta mesh represents anomalous electron density contoured at 4σ. c, cross-section through a surface representation of ELIC. The intersubunit bromoform site is formed at a pre-existing cavity, which is occupied by a bromoform molecule (single bromine atoms are shown as magenta spheres) in all five sites. d, detailed view of the amino acid residues in ELIC that form the intersubunit bromoform site (highlighted in yellow). The site is formed at the interface between M1 and M4 of one subunit (blue) and M3 of the neighboring subunit (orange).
To further probe the importance of this novel intersubunit transmembrane site, we took advantage of the presence of two tryptophan residues (Trp-221 and Trp-225 in ELIC) and investigated the possible quenching of intrinsic tryptophan fluorescence in the presence of bromoform. We observe that increasing concentrations of bromoform progressively decrease intrinsic tryptophan fluorescence of detergent-solubilized ELIC (Fig. 5) and that this effect is strongly reduced in the W221Y+W225Y mutant. These data demonstrate that the intersubunit cleft inside the membrane forms a binding site for bromoform. Further experiments in oocytes supported these data; at an EC20 GABA concentration, L9′S receptors were not significantly inhibited by bromoform (described above), but L9′S+F275A and L9′S+W225A mutant receptors showed significant inhibition, with EC20 currents reducing to 78 ± 4 (n = 3) and 81 ± 6% (n = 3), respectively, in the presence of 200 μm bromoform.
FIGURE 5.
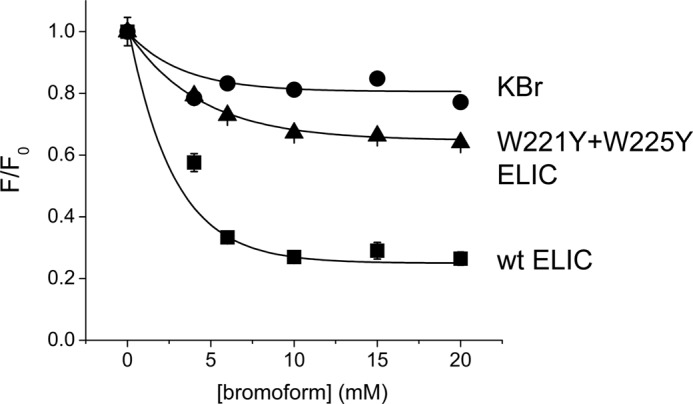
Quenching of intrinsic tryptophan fluorescence in ELIC by bromoform. Increasing concentrations of bromoform cause quenching of intrinsic fluorescence in ELIC. This effect is strongly reduced in the double mutant W221Y/W225Y. Each data point is the average of 3 experiments. Error bars indicate S.E.
Using an alignment of ELIC with sequences of the related GLIC channel and human pLGICs, we found that amino acid residues forming the transmembrane bromoform site in ELIC are highly conserved in GLIC and anionic pLGICs (GABAA/CRs and GlyRs), but not in cationic family members (nAChRs and 5-HT3Rs) (Fig. 6). The high degree of residue conservation indicates that the intersubunit pocket not only exists in ELIC, but also is likely to exist in anionic pLGICs, where it potentially could mediate functional effects in eukaryotic inhibitory receptors.
FIGURE 6.

Conservation of bromoform binding residues at a transmembrane intersubunit site in pLGICs. Alignment of ELIC, GLIC, and selected sequences of human GlyR, GABAA/CR, nAChR, and 5-HT3Rs is shown. Numbers at the top of the alignment correspond to ELIC residues, and numbers at the bottom correspond to α1 GlyR residues. Residues are colored in shades of blue using an identity threshold of 50%. Yellow residues correspond to the highlighted residues in the intersubunit bromoform site in ELIC (Fig. 3d). Aromatic residues at these positions as well as Pro-307 are strongly conserved in GlyR and GABAA/CR, but not in nAChR and 5-HT3Rs. The dashed line separates inhibitory (top) from excitatory (bottom) receptors.
Structure-guided Mutagenesis of Bromoform Interactions in Human α1 Glycine Receptors
The aromatic residues forming the intersubunit bromoform site are strongly conserved in anionic pLGICs, but not in cationic pLGICs, which indicates that this site may have a specific role in GABAA/CRs and GlyRs. To investigate the possible functional contribution of this site to the general anesthetic modulation of eukaryote receptors, we chose the human α1 GlyR as a model receptor for mutagenesis studies. The α1 GlyR is a homopentameric receptor, which simplifies interpretation of mutagenesis experiments, and its modulation by general anesthetics, including chloroform, has been studied extensively (27, 39–42). We mutated the homologous residues Phe-295 and Tyr-301 in the human α1 GlyR, which correspond to Phe-275 and Ile-282 in the M3-helix of ELIC (Fig. 7a). We chose these residues because they are strictly conserved aromatic residues in anionic, but not in cationic, eukaryote pLGICs (Fig. 6). Using two-electrode voltage clamp recordings from Xenopus oocytes, we measured ligand-activated currents for a range of glycine concentrations (from 3 μm to 3 mm, Fig. 7b) and compared the functional effects of wild type with F295A and Y301A receptors in the absence and presence of 200 μm bromoform.
FIGURE 7.

Mutagenesis of the intersubunit bromoform binding site in the human α1 glycine receptor. a, graphic representation of the intersubunit bromoform binding site in the transmembrane domain of ELIC. Interacting residues are shown in yellow sticks. The bromoform molecule is shown as a single magenta sphere. b, two-electrode voltage clamp recordings from oocytes expressing wild type α1 GlyRs, F295A, and Y301A mutants. Each current trace is shown in a specific color to facilitate comparison of the range of glycine concentrations: 3 μm (green), 10 μm (red), 30 μm (blue), 100 μm (yellow), 300 μm (magenta), 1 mm (cyan), and 3 mm (black). To reach saturation for Tyr-301 receptors, we also applied 10 mm (orange). Each oocyte was exposed to the same range of glycine concentrations in the presence of 200 μm bromoform (BrF). The insets for WT and F295A show a magnified view of traces obtained at low glycine concentrations. The asterisk indicates potentiation by bromoform. c, concentration-activation curves for WT, F295A, and Y301A GlyRs in the absence of bromoform. d–f, concentration-activation curves in the absence (gray line, −BrF) and presence (black line, +BrF) of 200 μm bromoform for WT (d), F295A (e), and Y301A (f) receptors. Error bars in panels c–f indicate S.E.
From the concentration-activation curve for wild type receptors (Fig. 7c), we calculated an EC50 value of 98 ± 4 μm and a Hill coefficient (nH) of 3.8 ± 0.2 (n = 5), which is in agreement with other studies (43). When compared with wild type receptors, we observe that the concentration-activation curve of F295A receptors is significantly shifted to the left (Fig. 7c) with an EC50 value of 22 ± 1 μm and nH of 3.1 ± 0.2 (n = 7, p < 0.05). In contrast, the concentration-activation curve of Y301A receptors is shifted to the right (Fig. 7c) with an EC50 value of 688 ± 35 μm and nH of 2.25 ± 0.09 (n = 5). These results are indicative of an important role of these residues in channel gating as the F295A mutation appears to lower the energy barrier for glycine-induced channel opening, whereas the Y301A mutation is compatible with a glycine-induced increase of the energy barrier for channel opening.
In the presence of bromoform (Fig. 7d), we observe for wild type and F295A receptors a small but significant increase in EC50 values (112 ± 6 μm, n = 5 for wild type, p < 0.05 and 26 ± 2 μm, n = 7 for F295A receptors, p < 0.05). In addition, bromoform reduced the maximal current amplitude at saturating glycine concentrations (Imax) to 76.6 ± 0.9% (n = 5) for wild type and 76 ± 6% (n = 7) for F295A receptors, respectively. Bromoform also reduces the slope of the concentration-activation curve (nH) to 2.8 ± 0.1 for wild type (n = 5, p < 0.05) and 2.2 ± 0.1 (n = 7, p < 0.05) for F295A receptors. At low glycine concentrations, however, bromoform caused a significant potentiation of F295A receptors when compared with wild type receptors (Fig. 7b, inset, asterisk), indicating that this residue may play a role in bromoform modulation. The evidence is stronger, however, for a role of Tyr-301 as the EC50 value of Y301A in the presence of bromoform was increased to 1240 ± 60 μm (p < 0.05), with Imax being reduced to 64 ± 6%. Low glycine concentrations (3–100 μm, indicated with asterisk) again resulted in bromoform potentiation in Y301A receptors. Thus, as reported previously for anesthetic effects in GABA receptors (4, 44), changes caused by bromoform exposure can be diametrically opposed and depend on agonist concentration. The interpretation of our data obtained from two-electrode voltage clamp recordings on mutant GlyR is difficult because the macroscopic effect on channel current likely depends on the action of bromoform at multiple binding sites, as suggested by the ELIC crystal structure. In addition, it is possible that the mutations either have direct effects on bromoform recognition or alternatively, cause allosteric effects that indirectly alter channel modulation by bromoform. Despite these limitations, our results support the hypothesis that the transmembrane binding site for bromoform, observed in the ELIC crystal structure, is also present in GlyRs as mutation of Phe-295 and Tyr-301 alters bromoform modulation.
DISCUSSION
Similar to nAChRs (45), GABACRs (4), and GLIC (16, 17), ELIC exhibited inhibition by alcohols and other general anesthetics. These results are consistent with the expanding theme of pLGIC sensitivity to general anesthetic modulation (2) and support the relevance of ELIC as a model for pLGIC function as well as structure. Furthermore, consistent with evidence from other pLGICs, ELIC bound the anesthetic derivative bromoform at multiple sites, including a transmembrane site at the protein-lipid interface. The low affinity and therapeutic index of inhalational anesthetics require caution in interpreting binding observed at high concentrations of modulators (1); however, some bromoform-bound ELIC crystals were grown with as little as 1 mm of the agent, close to the EC50 both for inhibition of ELIC and for chloroform anesthesia in humans (12). This similar potency, combined with the presence of equivalent sites in other pLGICs, suggests that anesthetic inhibition of ELIC and related channels arises from binding to one or more of the sites we observe, and by extrapolation, anesthetic inhibition in related channels is due to their binding to the equivalent sites in these proteins.
The strongest bromoform anomalous signal occupied a pore-blocking site near the 13′A residue in the M2-helix. These data are consistent with high occupancy of bromoform, although the occupancy was amplified by its position on the five-fold noncrystallographic symmetry axis imposed to refine the structure. Binding of inhibitors in the channel pore is consistent with mutagenesis, labeling, and molecular dynamics studies of nAChRs (6–8). The anesthetic position is also equivalent to that observed in a recent co-crystal structure of GLIC with the local anesthetic derivative bromo-lidocaine (33); moreover, molecular dynamics studies of GLIC binding to the volatile anesthetics isoflurane and propofol (46, 47) support binding in the channel pore, in addition to other sites. Given the steric occlusion to ion conductance that would result from occupancy of the pore by an agent the size of bromoform, binding at this position seems likely to contribute to inhibition of ELIC. This is confirmed by our mutagenesis study in ELIC, which demonstrates that the pore mutants L9′S and F16′S display less inhibition by bromoform.
A previous study has revealed binding of isoflurane to an otherwise dehydrated, presumed nonconducting state of the GLIC pore (46). This work is consistent with the present structure, which shows bromoform binding to a presumed closed state of the channel. Willenbring et al. (46) proposed that the small, hydrophobic nature of most general anesthetics allows them to pass the channel hydrophobic gate even in a conformation that is unable to pass water or ions. Anesthetics may, in fact, preferentially bind to the closed state, which is better represented by the ELIC pore than the open state represented by GLIC. The presence of detergent molecules bound in the pore of the known GLIC crystal structures (20) could prevent observation of anesthetics in equivalent sites in the known anesthetic co-crystal structures (18).
A second bromoform site occupied the intersubunit cleft, facing the membrane and close to the intracellular side in the transmembrane domain. The general anesthetics halothane and thiopental quenched tryptophan fluorescence in an equivalent site in a recent study of GLIC (48). The intersubunit binding site is surrounded by hydrophobic aromatic residues that are conserved in GlyRs and GABAARs, but not in nAChRs, consistent with a subtype-specific role for this site in modulation or in another critical aspect of channel gating. The membrane-facing intersubunit interface also binds the allosteric activator ivermectin in recent co-crystal structures with the C. elegans glutamate-gated chloride channel (14), further supporting a role for intersubunit binding in allosteric modulation of channel function. A proximal intersubunit cavity, closer to the extracellular side than the pocket observed here, has been heavily implicated in anesthetic potentiation of GlyRs and most GABAARs (9). This “higher” intersubunit cavity was also recently shown to mediate alcohol potentiation of the bacterial channel GLIC (16). Blockage of this alternative intersubunit site, for example by substituting isoleucine at the aligned residues Ser-270 in α1 and Ser-265 in β1 GABAAR subunits, resulted in channel inhibition by ethanol and enflurane (49), suggesting that potentiation via this site masked an independent mechanism of inhibition, perhaps via one or more alternative binding sites.
Notably, the intrasubunit transmembrane binding site occupied by desflurane and propofol in recent co-crystal structures with GLIC (18) was not occupied by bromoform in our ELIC co-crystal structure. This result may indicate differences in binding modes for the two channel types; it may also reflect the limitations of co-crystallization with a channel “locked” in a particular functional state. The presumed preferential binding of inhibitors to the closed state(s) of an ion channel (19) supports the relevance of the binding modes observed with the closed state ELIC structure.
In summary, cumulative evidence indicates the existence of different binding sites in pLGICs for general anesthetics. One site is located in the upper part of the transmembrane domain in an intrasubunit pocket between the M1- and M3-helix of a single subunit, which corresponds to the binding site for propofol and desflurane (Fig. 8, green sphere) (18). A second transmembrane site is located at the subunit interface formed between the M1-helix of one subunit and the M3-helix of a neighboring subunit (Fig. 8, yellow sphere). This site corresponds to the binding site for alcohols, certain general anesthetics, and ivermectin (14). Our study unveils a novel transmembrane anesthetic binding site located farther down the transmembrane domain at an interface formed between the M1- and M4-helix of one subunit and the M3-helix of a neighboring subunit (Fig. 8, magenta sphere (IS)). In addition, we provide the first experimental evidence that certain anesthetics, such as chloroform and bromoform, can bind in the hydrophobic portion of the channel pore (Fig. 8, magenta sphere (PS)). Together, these studies substantiate the view of multisite allosteric modulation in the family of pentameric ligand-gated ion channels.
FIGURE 8.

Overview of different general anesthetic binding sites revealed in crystal structures of pLGICs. A graphic representation of two neighboring subunits of the ELIC pentamer is shown. The different spheres correspond to different binding site for general anesthetics: the propofol-desflurane site (green) (18), the alcohol-ivermectin site (yellow) (14), and the three bromoform sites identified in this study (magenta). ES, extracellular site; PS, pore site; IS, intersubunit site).
Acknowledgments
We thank local contacts at beamline X06A of the Swiss Light Source for assistance during data collection, Neil Harrison for discussing bromoform effects on GABAA receptors, and several members of the Neurocypres consortium for discussion.
This work was supported by Grants KU Leuven OT/08/048 and FWO-Vlaanderen G.0739.09, G.0743.10, and G.0939.11 (to C. U.), the European Union Seventh Framework Programme under grant agreement HEALTH-F2-2007-202088 (to C. U., D. B., and S. C. R. L.), and the Wellcome Trust (to S. C. R. L. and K. L. P.).
The atomic coordinates and structure factors (code 3zkr) have been deposited in the Protein Data Bank (http://wwpdb.org/).
- pLGIC
- pentameric ligand-gated ion channel
- ELIC
- E. chrysanthemi ligand-gated ion channel
- GLIC
- G. violaceus ligand-gated ion channel
- ACh
- acetylcholine
- nAChR
- nicotinic acetylcholine receptor
- GlyR
- Gly receptor
- GABAAR
- GABA type A receptor
- GABACR
- GABA type C receptor
- 5-HT3R
- 5-hydroxytryptamine type 3 receptor.
REFERENCES
- 1. Franks N. P. (2006) Molecular targets underlying general anaesthesia. Br. J. Pharmacol. 147, Suppl. 1, S72–S81 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Forman S. A., Miller K. W. (2011) Anesthetic sites and allosteric mechanisms of action on Cys-loop ligand-gated ion channels. Can. J. Anaesth. 58, 191–205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Campagna J. A., Miller K. W., Forman S. A. (2003) Mechanisms of actions of inhaled anesthetics. N. Engl. J. Med. 348, 2110–2124 [DOI] [PubMed] [Google Scholar]
- 4. Mihic S. J., Harris R. A. (1996) Inhibition of rho1 receptor GABAergic currents by alcohols and volatile anesthetics. J. Pharmacol. Exp. Ther. 277, 411–416 [PubMed] [Google Scholar]
- 5. Baenziger J. E., Corringer P. J. (2011) 3D structure and allosteric modulation of the transmembrane domain of pentameric ligand-gated ion channels. Neuropharmacology 60, 116–125 [DOI] [PubMed] [Google Scholar]
- 6. Forman S. A., Miller K. W., Yellen G. (1995) A discrete site for general anesthetics on a postsynaptic receptor. Mol. Pharmacol. 48, 574–581 [PubMed] [Google Scholar]
- 7. Pratt M. B., Husain S. S., Miller K. W., Cohen J. B. (2000) Identification of sites of incorporation in the nicotinic acetylcholine receptor of a photoactivatible general anesthetic. J. Biol. Chem. 275, 29441–29451 [DOI] [PubMed] [Google Scholar]
- 8. Brannigan G., LeBard D. N., Hénin J., Eckenhoff R. G., Klein M. L. (2010) Multiple binding sites for the general anesthetic isoflurane identified in the nicotinic acetylcholine receptor transmembrane domain. Proc. Natl. Acad. Sci. U.S.A. 107, 14122–14127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Olsen R. W., Li G. D. (2011) GABAA receptors as molecular targets of general anesthetics: identification of binding sites provides clues to allosteric modulation. Can. J. Anaesth. 58, 206–215 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Wallner M., Olsen R. W. (2008) Physiology and pharmacology of alcohol: the imidazobenzodiazepine alcohol antagonist site on subtypes of GABAA receptors as an opportunity for drug development? Br. J. Pharmacol. 154, 288–298 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Moraga-Cid G., Yevenes G. E., Schmalzing G., Peoples R. W., Aguayo L. G. (2011) A Single phenylalanine residue in the main intracellular loop of α1 γ-aminobutyric acid type A and glycine receptors influences their sensitivity to propofol. Anesthesiology 115, 464–473 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Lopes C. M., Franks N. P., Lieb W. R. (1998) Actions of general anaesthetics and arachidonic pathway inhibitors on K+ currents activated by volatile anaesthetics and FMRFamide in molluscan neurones. Br. J. Pharmacol. 125, 309–318 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Sear J. W. (2009) What makes a molecule an anaesthetic? Studies on the mechanisms of anaesthesia using a physicochemical approach. Br. J. Anaesth. 103, 50–60 [DOI] [PubMed] [Google Scholar]
- 14. Hibbs R. E., Gouaux E. (2011) Principles of activation and permeation in an anion-selective Cys-loop receptor. Nature 474, 54–60 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Hilf R. J., Dutzler R. (2009) A prokaryotic perspective on pentameric ligand-gated ion channel structure. Curr. Opin. Struct. Biol. 19, 418–424 [DOI] [PubMed] [Google Scholar]
- 16. Howard R. J., Murail S., Ondricek K. E., Corringer P. J., Lindahl E., Trudell J. R., Harris R. A. (2011) Structural basis for alcohol modulation of a pentameric ligand-gated ion channel. Proc. Natl. Acad. Sci. U.S.A. 108, 12149–12154 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Weng Y., Yang L., Corringer P. J., Sonner J. M. (2010) Anesthetic sensitivity of the Gloeobacter violaceus proton-gated ion channel. Anesth. Analg. 110, 59–63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Nury H., Van Renterghem C., Weng Y., Tran A., Baaden M., Dufresne V., Changeux J. P., Sonner J. M., Delarue M., Corringer P. J. (2011) X-ray structures of general anaesthetics bound to a pentameric ligand-gated ion channel. Nature 469, 428–431 [DOI] [PubMed] [Google Scholar]
- 19. Léna C., Changeux J. P. (1993) Allosteric modulations of the nicotinic acetylcholine receptor. Trends Neurosci. 16, 181–186 [DOI] [PubMed] [Google Scholar]
- 20. Bocquet N., Nury H., Baaden M., Le Poupon C., Changeux J. P., Delarue M., Corringer P. J. (2009) X-ray structure of a pentameric ligand-gated ion channel in an apparently open conformation. Nature 457, 111–114 [DOI] [PubMed] [Google Scholar]
- 21. Hilf R. J., Dutzler R. (2008) X-ray structure of a prokaryotic pentameric ligand-gated ion channel. Nature 452, 375–379 [DOI] [PubMed] [Google Scholar]
- 22. Zimmermann I., Dutzler R. (2011) Ligand activation of the prokaryotic pentameric ligand-gated ion channel ELIC. PLoS Biol. 9, e1001101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Thompson A. J., Alqazzaz M., Ulens C., Lummis S. C. (2012) The pharmacological profile of ELIC, a prokaryotic GABA-gated receptor. Neuropharmacology 63, 761–767 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Spurny R., Ramerstorfer J., Price K., Brams M., Ernst M., Nury H., Verheij M., Legrand P., Bertrand D., Bertrand S., Dougherty D. A., de Esch I. J., Corringer P. J., Sieghart W., Lummis S. C., Ulens C. (2012) Pentameric ligand-gated ion channel ELIC is activated by GABA and modulated by benzodiazepines. Proc. Natl. Acad. Sci. U.S.A. 109, E3028–E3034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Smart O. S., Neduvelil J. G., Wang X., Wallace B. A., Sansom M. S. (1996) HOLE: a program for the analysis of the pore dimensions of ion channel structural models. J. Mol. Graph. 14, 354–360, 376 [DOI] [PubMed] [Google Scholar]
- 26. Liman E. R., Tytgat J., Hess P. (1992) Subunit stoichiometry of a mammalian K+ channel determined by construction of multimeric cDNAs. Neuron 9, 861–871 [DOI] [PubMed] [Google Scholar]
- 27. Beckstead M. J., Phelan R., Mihic S. J. (2001) Antagonism of inhalant and volatile anesthetic enhancement of glycine receptor function. J. Biol. Chem. 276, 24959–24964 [DOI] [PubMed] [Google Scholar]
- 28. Kash T. L., Jenkins A., Harrison N. L. (2003) Molecular volume determines the activity of the halogenated alkane bromoform at wild-type and mutant GABAA receptors. Brain Res. 960, 36–41 [DOI] [PubMed] [Google Scholar]
- 29. Jenkins A., Greenblatt E. P., Faulkner H. J., Bertaccini E., Light A., Lin A., Andreasen A., Viner A., Trudell J. R., Harrison N. L. (2001) Evidence for a common binding cavity for three general anesthetics within the GABAA receptor. J. Neurosci. 21, RC136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Harris R. A., Mihic S. J., Dildy-Mayfield J. E., Machu T. K. (1995) Actions of anesthetics on ligand-gated ion channels: role of receptor subunit composition. FASEB J. 9, 1454–1462 [DOI] [PubMed] [Google Scholar]
- 31. Mascia M. P., Machu T. K., Harris R. A. (1996) Enhancement of homomeric glycine receptor function by long-chain alcohols and anaesthetics. Br. J. Pharmacol. 119, 1331–1336 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Nakahiro M., Arakawa O., Nishimura T., Narahashi T. (1996) Potentiation of GABA-induced Cl− current by a series of n-alcohols disappears at a cutoff point of a longer-chain n-alcohol in rat dorsal root ganglion neurons. Neurosci. Lett. 205, 127–130 [DOI] [PubMed] [Google Scholar]
- 33. Hilf R. J., Bertozzi C., Zimmermann I., Reiter A., Trauner D., Dutzler R. (2010) Structural basis of open channel block in a prokaryotic pentameric ligand-gated ion channel. Nat. Struct. Mol. Biol. 17, 1330–1336 [DOI] [PubMed] [Google Scholar]
- 34. Gumilar F., Arias H. R., Spitzmaul G., Bouzat C. (2003) Molecular mechanisms of inhibition of nicotinic acetylcholine receptors by tricyclic antidepressants. Neuropharmacology 45, 964–976 [DOI] [PubMed] [Google Scholar]
- 35. Revah F., Galzi J. L., Giraudat J., Haumont P. Y., Lederer F., Changeux J. P. (1990) The noncompetitive blocker [3H]chlorpromazine labels three amino acids of the acetylcholine receptor γ subunit: implications for the α-helical organization of regions MII and for the structure of the ion channel. Proc. Natl. Acad. Sci. U.S.A. 87, 4675–4679 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Charnet P., Labarca C., Leonard R. J., Vogelaar N. J., Czyzyk L., Gouin A., Davidson N., Lester H. A. (1990) An open-channel blocker interacts with adjacent turns of α-helices in the nicotinic acetylcholine receptor. Neuron 4, 87–95 [DOI] [PubMed] [Google Scholar]
- 37. Pascual J. M., Karlin A. (1998) Delimiting the binding site for quaternary ammonium lidocaine derivatives in the acetylcholine receptor channel. J. Gen. Physiol. 112, 611–621 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Yu Y., Shi L., Karlin A. (2003) Structural effects of quinacrine binding in the open channel of the acetylcholine receptor. Proc. Natl. Acad. Sci. U.S.A. 100, 3907–3912 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Beckstead M. J., Weiner J. L., Eger E. I., 2nd, Gong D. H., Mihic S. J. (2000) Glycine and γ-aminobutyric acidA receptor function is enhanced by inhaled drugs of abuse. Mol. Pharmacol. 57, 1199–1205 [PubMed] [Google Scholar]
- 40. Daniels S., Roberts R. J. (1998) Post-synaptic inhibitory mechanisms of anaesthesia; glycine receptors. Toxicol. Lett. 100–101, 71–76 [DOI] [PubMed] [Google Scholar]
- 41. Beckstead M. J., Phelan R., Trudell J. R., Bianchini M. J., Mihic S. J. (2002) Anesthetic and ethanol effects on spontaneously opening glycine receptor channels. J. Neurochem. 82, 1343–1351 [DOI] [PubMed] [Google Scholar]
- 42. Jenkins A., Lobo I. A., Gong D., Trudell J. R., Solt K., Harris R. A., Eger E. I., 2nd. (2008) General anesthetics have additive actions on three ligand gated ion channels. Anesth. Analg. 107, 486–493 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Rajendra S., Lynch J. W., Schofield P. R. (1997) The glycine receptor. Pharmacol. Ther. 73, 121–146 [DOI] [PubMed] [Google Scholar]
- 44. Harrison N. L., Kugler J. L., Jones M. V., Greenblatt E. P., Pritchett D. B. (1993) Positive modulation of human γ-aminobutyric acid type A and glycine receptors by the inhalation anesthetic isoflurane. Mol. Pharmacol. 44, 628–632 [PubMed] [Google Scholar]
- 45. Wachtel R. E. (1995) Relative potencies of volatile anesthetics in altering the kinetics of ion channels in BC3H1 cells. J. Pharmacol. Exp. Ther. 274, 1355–1361 [PubMed] [Google Scholar]
- 46. Willenbring D., Liu L. T., Mowrey D., Xu Y., Tang P. (2011) Isoflurane alters the structure and dynamics of GLIC. Biophys. J. 101, 1905–1912 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. LeBard D. N., Hénin J., Eckenhoff R. G., Klein M. L., Brannigan G. (2012) General anesthetics predicted to block the GLIC pore with micromolar affinity. PLoS Comput. Biol. 8, e1002532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Chen Q., Cheng M. H., Xu Y., Tang P. (2010) Anesthetic binding in a pentameric ligand-gated ion channel: GLIC. Biophys. J. 99, 1801–1809 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Mihic S. J., Ye Q., Wick M. J., Koltchine V. V., Krasowski M. D., Finn S. E., Mascia M. P., Valenzuela C. F., Hanson K. K., Greenblatt E. P., Harris R. A., Harrison N. L. (1997) Sites of alcohol and volatile anaesthetic action on GABAA and glycine receptors. Nature 389, 385–389 [DOI] [PubMed] [Google Scholar]