

Background: Programs of cellular development and differentiation are controlled by enhancers.
Results: Human erythroid cell type-specific enhancers are marked by p300 and groups of transcription factors.
Conclusion: Enhancers are important regulators of species-specific erythroid cell structure and function.
Significance: Deciphering how nonpromoter regulatory elements control gene expression in erythroid cells is important for understanding inherited and acquired hematologic disease.
Keywords: Erythrocyte, Erythropoeisis, Genetic Diseases, Genetic Polymorphism, Genomics
Abstract
Identification of cell type-specific enhancers is important for understanding the regulation of programs controlling cellular development and differentiation. Enhancers are typically marked by the co-transcriptional activator protein p300 or by groups of cell-expressed transcription factors. We hypothesized that a unique set of enhancers regulates gene expression in human erythroid cells, a highly specialized cell type evolved to provide adequate amounts of oxygen throughout the body. Using chromatin immunoprecipitation followed by massively parallel sequencing, genome-wide maps of candidate enhancers were constructed for p300 and four transcription factors, GATA1, NF-E2, KLF1, and SCL, using primary human erythroid cells. These data were combined with gene expression analyses, and candidate enhancers were identified. Consistent with their predicted function as candidate enhancers, there was statistically significant enrichment of p300 and combinations of co-localizing erythroid transcription factors within 1–50 kb of the transcriptional start site (TSS) of genes highly expressed in erythroid cells. Candidate enhancers were also enriched near genes with known erythroid cell function or phenotype. Candidate enhancers exhibited moderate conservation with mouse and minimal conservation with nonplacental vertebrates. Candidate enhancers were mapped to a set of erythroid-associated, biologically relevant, SNPs from the genome-wide association studies (GWAS) catalogue of NHGRI, National Institutes of Health. Fourteen candidate enhancers, representing 10 genetic loci, mapped to sites associated with biologically relevant erythroid traits. Fragments from these loci directed statistically significant expression in reporter gene assays. Identification of enhancers in human erythroid cells will allow a better understanding of erythroid cell development, differentiation, structure, and function and provide insights into inherited and acquired hematologic disease.
Introduction
Erythrocytes are specialized cells that have evolved to efficiently carry out their primary functions of oxygen transport and delivery. Among the vertebrates, mammalian erythrocytes are unique. Mature mammalian erythrocytes are enucleate and lack most cellular organelles. Erythroid progenitor cells contain nuclei, but they are extruded, presumably to allow for additional hemoglobin content for more efficient oxygen transport. Lacking DNA, mature erythrocytes lack the capacity for cell division or RNA synthesis, and they have very limited capacity for self-repair. Thus, erythroid cells are highly specialized cells with a number of unique characteristics.
The regulation of programs controlling cellular development and differentiation vary temporally, between cell and tissue types, and between species. These programs are controlled by critical regulatory DNA sequences, cis-regulatory modules (CRMs),3 which include gene promoters, enhancers, silencers, and insulators. A detailed understanding of the structure and function of CRMs in varying cell types will provide insights into these regulatory programs and provide crucial information for predicting and understanding the phenotypic consequences of genetic variation in noncoding DNA. Recent studies utilizing genomic methodologies have shown that enhancers, a class of CRMs, are frequently associated with disease-associated genetic variants (1–6).
Enhancers have been classified into two groups, those marked by p300, the transcriptional co-activator also frequently found at gene promoters, and those that lack p300 and instead are occupied by cell and tissue type-specific transcription factors (7–12). In addition, specific patterns of enhancer-associated chromatin architecture have been described (13–26). Enhancers demonstrate tissue, cell, and developmental stage specificity. Phylogenetic conservation has been used as an indicator for functional conservation of enhancers throughout evolution (16, 27–32); however, varying degrees of sequence conservation between cell types in different species have been observed. In some human tissues, such as forebrain, enhancers are subject to stringent evolutionary constraint, whereas in others, such as heart, they are under weak evolutionary constraint (12, 16, 33–38). In some cell and tissue types, structural and functional conservation of enhancer elements is significantly lacking, even between species as close as mice and humans (34).
Mammalian genomes contain more enhancers than promoters, with enhancers subserving numerous roles in controlling gene regulation (6, 13, 39). Tissue-specific genes are more dependent on enhancer regulation and exhibit less promoter diversity than housekeeping genes, which are primarily regulated by their promoters with few enhancers in their genomic vicinity (1). Identification and characterization of enhancers that control programs of gene expression in highly specialized human erythroid cells will allow a better understanding of erythroid cell development, differentiation, structure, and function as well as provide insights into inherited and acquired hematologic disease.
This report describes the construction of genome-wide maps of candidate enhancers in primary human erythroid cells. Genome-wide maps of p300 and four erythroid transcription factors, GATA1, NF-E2, KLF1, and SCL, in human primary erythroid cell chromatin were constructed and analyzed with parallel gene expression analyses. Consistent with their predicted function, these regulatory elements were enriched near genes highly expressed in erythroid cells or involved in erythroid cell structure and function. Conservation analyses of candidate human enhancers revealed only moderate conservation with mouse and minimal conservation with nonplacental vertebrates. Fourteen candidate enhancers, representing 10 genetic loci, mapped to sites previously associated with biologically relevant erythroid cell traits in GWAS. Fragments from 9 of the 10 biologically relevant regions directed statistically significant expression in reporter gene assays.
EXPERIMENTAL PROCEDURES
Cell Culture and Selection
To obtain primary human erythroid cells, CD34+ cells were cultured and selected as described (41, 42). These cells represent the R3/R4 cell population of nucleated erythroid cells defined by Zhang et al. (43).
RNA Isolation and Preparation, Microarray Data Acquisition, and Analyses
RNA was prepared from primary human erythroid cells and prepared for microarray analyses as described (44, 45) and detailed in the supplemental Methods. Gene expression microarray quality control and data analyses are described in the supplemental Methods. Quantitative real-time PCR was performed to confirm expression levels of RNA transcripts with the primers in supplemental Table S1. Real-time PCR data were normalized as described (45). Triplicate analyses were performed for each target (44, 46).
Chromatin Immunoprecipitation
ChIP assays were performed as previously described with minor variations (see supplemental Methods) (44). After incubation, nuclei were sonicated with the Covaris S2 adaptive focused acoustics disrupter. Samples were immunoprecipitated with antibodies against GATA1 (sc-265, Santa Cruz Biotechnology, Inc., Santa Cruz, CA), NF-E2 (sc-22827, Santa Cruz Biotechnology, Inc.), KLF1 (ab2483, Abcam), SCL/Tal1 (sc-12984, Santa Cruz Biotechnology, Inc.), p300 (sc-585, Santa Cruz Biotechnology, Inc.), H3K4me2 (32356, Abcam), H3K4me3 (1012, Abcam), and nonspecific rabbit IgG (sc-2091, Santa Cruz Biotechnology, Inc.). Antibody-bound DNA-protein complexes were collected, washed, and eluted from the beads, and cross-linking of DNA-protein adducts was reversed. DNA was cleaned with the QIAquick PCR purification kit (Qiagen) according to the manufacturer's instructions.
Illumina High Throughput Sequencing and Data Analyses
DNA processing and high throughput sequencing were performed as described (44). Sequenced reads were mapped to the human genome (UCSC Genome Browser hg18 (47), NCBI Build 36 using the Eland short-read alignment program. The Model-based Alignment of ChIP-Seq (MACS) program was used to identify peaks with a p value of <10e−5 (48). Localization of binding sites relative to known genes was done using the ChIPseeqer package (49). Factor co-localization was determined using the Active Region Comparer. Motif finding was done using the Homer algorithm (50). Conservation of candidate enhancer regions between corresponding genomic regions of vertebrates was determined using the UCSC hg18 genome browser database (47) with the 44-way vertebrate and placental mammal PhastCons track (51).
The PhastCons conservation scores of regions surrounding promoters, exons, and distal and intergenic regions were compared with the PhastCons scores of randomized regions generated by combining the regions for all transcription factor binding sites and moving the regions to random locations in the genome outside of gaps in the known hg18 sequence using the BedTools ShuffleBed function. Conservation plots were generated using Cistrome (52). Conservation of human candidate enhancer regions was analyzed using the UCSC LiftOver tool. For LiftOver controls, sites were concatenated, randomly shuffled across the genome, and analyzed. The maximum PhastCons score for each candidate enhancer mapped to sites previously associated with biologically relevant erythroid cell traits in GWAS studies was determined using the Galaxy aggregate function (53, 54). The UCSC Genome Browser 7X regulatory potential table was used to determine the maximum regulatory potential (RP) scores for each region (54, 55).
Identification and Analysis of Biologically Relevant SNPs
The locations of SNPs shown to demonstrate highly significant linkage to erythroid cell-related traits were obtained from the UCSC Genome Browser database and the catalogue of GWAS compiled by NHGR, National Institutes of Health (4). Using BedTools software (see supplemental Methods), nonpromoter-related p300 peaks (TSS to ±1 kb) were intersected with erythroid-related SNPs, and overlap was identified. Similarly, peaks with two or more sites of erythroid transcription factor binding identified by Active Region Comparer were intersected with erythroid-related SNPs.
Validation of ChIP-seq Results
Primers were designed for representative binding regions for all five antibodies in target genes identified by the MACS program (supplemental Table S2). Immunoprecipitated DNA was analyzed by quantitative real-time PCR (iCycler, Bio-Rad) as described (44).
Reporter Gene Assays
Fourteen candidate enhancer regions were PCR-amplified using oligonucleotide primers immediately flanking the boundaries of the called peaks (supplemental Table S3). These fragments were cloned upstream of a SV40 promoter-firefly luciferase reporter cassette in the pGL2Promoter plasmid. The integrity of all test plasmids was confirmed by sequencing. The negative control plasmid contained a promoterless-firefly luciferase gene cassette, PGL2Basic (Promega), and the positive control plasmid contained a γ-globin gene promoter-firefly luciferase reporter gene cassette with the human β-globin gene HS2 enhancer cloned upstream of the γ-globin-luciferase cassette (56). 107 K562 cells (CCL 243, ATCC) were transfected by electroporation with a single pulse of 300 V at 950 microfarads with 15 μg of test plasmid and 0.3 μg of pRL-TK, a reporter plasmid expressing Renilla luciferase driven by the herpes simplex virus thymidine kinase promoter (Promega) as described (57). At least two preparations of each plasmid were tested in triplicate. Two days after transfection, cell extracts were analyzed using the Dual-Luciferase assay according to manufacturer's instructions (Promega). Firefly luciferase activity directed by each of the test plasmids, corrected for the Renilla luciferase activity of the co-transfection control, was normalized by firefly luciferase activity from the pGL2P control plasmid to obtain the -fold change. Statistical significance was determined as p < 0.05 by a one-tailed Student's t test.
Data Access
The raw data files generated by the ChIP-seq analyses and microarray assays have been submitted to the Gene Expression Omnibus (GEO) for use by other investigators (reference series GSE43626). The mRNA microarray experiments comply with MIAME (Minimum Information About a Microarray Experiment) standards (58).
RESULTS
mRNA Expression and p300 and Erythroid Transcription Factor ChIP-seq Analyses in Human Primary Erythroid Cells
Human primary erythroid cells, representing the R3/R4 populations of cells (43), were cultured from human CD34+ stem and progenitor cells. Transcriptome analyses were performed with erythroid cell mRNA hybridized to Illumina human v2 mRNA expression arrays. Levels of expression were assigned absent or present calls using the Illumina detection p values based on negative control hybridization probes. Of 19,707 transcripts examined, 8678 transcripts were expressed. Quantitative real-time PCR was performed to validate expression levels of representative mRNA transcripts assigned by the expression arrays (supplemental Table S4 and Fig. S1).
Using primary erythroid cell chromatin, ChIP-seq was performed utilizing antibodies specific for the transcriptional co-activator p300 and the erythroid transcription factors GATA1, NF-E2, KLF1, and SCL/Tal1 to generate genome-wide maps of factor binding. Genome-wide maps of H3K4me2 and H3K4me3 occupancy were similarly constructed. The MACS program was used to identify peaks with a cut-off of p < 10e−5 (supplemental Table S5). Validation of factor enrichment at selected peaks identified by ChIP-seq was performed by quantitative ChIP PCR for all five antibodies (supplemental Table S6).
Sites of p300 and Erythroid Transcription Factor Occupancy in Erythroid Cell Chromatin
The human genome was portioned into six bins relative to RefSeq genes corresponding to exons, introns, promoters, distal (−1 to −50 kb), downstream (+1 to +50 kb), and intergenic regions. Sites of factor occupancy were assigned to these bins, and percentages were calculated (Fig. 1). p300 and erythroid transcription factors were enriched in introns and distal regions (1–50 kb from a RefSeq gene) (Fig. 1). p300 occupancy was also enriched in promoters and exons, consistent with the alternate role of p300 as a transcriptional co-activator at gene promoters. Sites of erythroid transcription factor binding were also enriched in intergenic regions (>50 kb from a RefSeq gene; 17–18%) (Fig. 1). As in previous reports, erythroid transcription factor binding was very common in intron 1 of RefSeq genes (data not shown) (44, 59). These data suggest that transcription factors mark enhancers more commonly than p300 in erythroid cells because bona fide enhancers are expected to act in the genomic vicinity of their cognate genes (35).
FIGURE 1.
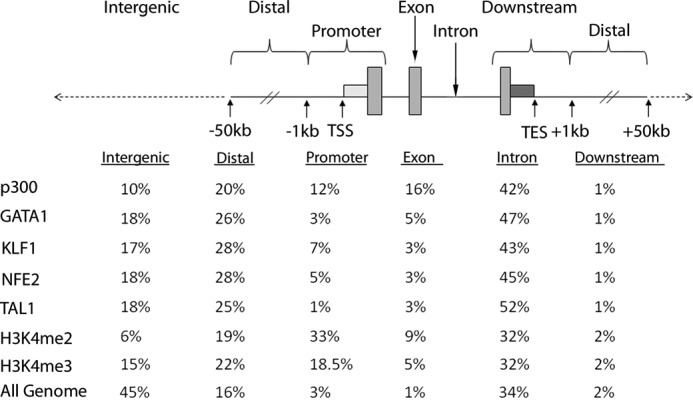
Distribution of p300 and erythroid transcription factor occupancy in human primary erythroid cell chromatin. The human genome was portioned into six bins relative to RefSeq genes. The data below represent the percentage of the human genome represented by each bin and the distribution of the p300 and erythroid transcription factor binding sites in each bin. TES, transcriptional end site.
Co-localization of p300 and Erythroid Transcription Factors
The co-localization of p300 and erythroid cell transcription factors was analyzed using Active Region Comparer. Erythroid transcription factors commonly co-localized, especially the combinations of KLF1 and NF-E2, GATA1 and KLF1, and GATA1 and NF-E2 (Table 1). Interestingly, three or more erythroid transcription factors co-localized frequently, ∼17% of the time. Co-localization of p300 with individual erythroid transcription factors was less frequent (Table 1). These data indicate that like other cell types studied, candidate erythroid cell enhancers are typically identified by p300 occupancy or co-localization of tissue-expressed transcription factors (60, 61).
TABLE 1.
Co-localization of p300 and erythroid transcription factor occupancy in erythroid cell chromatin
| Number of co-localizing peaks | Total number of peaks | Percentage of co-localizing peaks | Genesa | |
|---|---|---|---|---|
| GATA1 + KLF1 | 12,583 | 42,075 | 29.91 | 5843 |
| GATA1 + NFE2 | 11,553 | 41,817 | 27.63 | 5460 |
| GATA1 + TAL1 | 3562 | 46,143 | 7.72 | 1857 |
| GATA1 + p300 | 5293 | 65,624 | 8.06 | 2652 |
| KLF1 + NFE2 | 15,887 | 41,717 | 38.08 | 7192 |
| KLF1 + TAL1 | 6362 | 47,578 | 13.37 | 3050 |
| KLF1 + p300 | 5983 | 69,168 | 8.65 | 3354 |
| NFE2 + TAL1 | 4923 | 47,728 | 10.31 | 2462 |
| NFE2 + p300 | 5216 | 68,646 | 7.59 | 2933 |
| TAL1 + p300 | 383 | 69,815 | 0.54 | 270 |
| Erythroid transcription factor co-occupancy | ||||
| Any 2 of 4 | 24,614 | 68,742 | 35.81 | 9382 |
| Any 3 of 4 | 11,598 | 68,742 | 16.87 | 5410 |
| All 4 of 4 | 2353 | 68,742 | 3.43 | 1270 |
| Erythroid transcription factor co-occupancy with p300 | ||||
| Any 2 of 5 | 29,697 | 104,607 | 28.39 | 10,548 |
| Any 3 of 5 | 14,176 | 104,607 | 13.55 | 6455 |
| Any 4 of 5 | 4416 | 104,607 | 4.22 | 2375 |
| All 5 of 5 | 111 | 104,607 | 0.11 | 67 |
a The nearest gene with an annotated transcriptional start site within 50 kb of the identified peak.
The Homer program was utilized to identify overrepresented DNA motifs at sites of factor binding. Not surprisingly, related motifs were found among the erythroid transcription factors (e.g. GATA1 with PU.1, KLF1 with GATA1, NF-E2 with GATA1, and SCL with GATA1). These results are shown in supplemental Fig. S2.
Identification of Candidate Erythroid Enhancer Regions
Genomic studies have identified two classes of enhancers, those marked by p300 binding and those marked by binding of multiple cell- and tissue type-specific transcription factors. We defined candidate erythroid enhancers as regions of DNA marked by nonpromoter-associated p300 occupancy or nonpromoter binding of two or more erythroid transcription factors. Typically, cell- and tissue type-specific enhancers act over distances of tens to hundreds of kilobases (34). Thus, bona fide erythroid enhancers are expected to be enriched in the genomic vicinity of genes that are expressed and functional in erythroid cells (1, 13, 62). To determine whether erythroid enhancers are localized in this manner, gene expression in erythroid cells was correlated with sites of occupancy of p300 and erythroid transcription factors. To exclude gene promoters, localization of p300 or erythroid transcription factors within 1 kb of annotated TSSs was excluded from the analyses. There was a statistically significant higher erythroid expression of genes with p300 binding sites within 1–50 kb of the TSS compared with expression of genes with p300 binding sites >50 kb from a TSS (Fig. 2, p < 2.2e−16; supplemental Table S7). Similar to p300, there was statistically significant higher expression of genes with erythroid transcription factor binding sites within 1–50 kb of the TSS compared with expression of genes with binding sites >50 kb from a TSS (Fig. 2). This was true when combinations of two, three, or four co-localizing erythroid transcription factors were analyzed (p < 2.2e−16 for all three combinations, respectively). As expected (6), H3K4me3 occupancy was uncommonly found at sites of candidate enhancers (supplemental Table S7).
FIGURE 2.
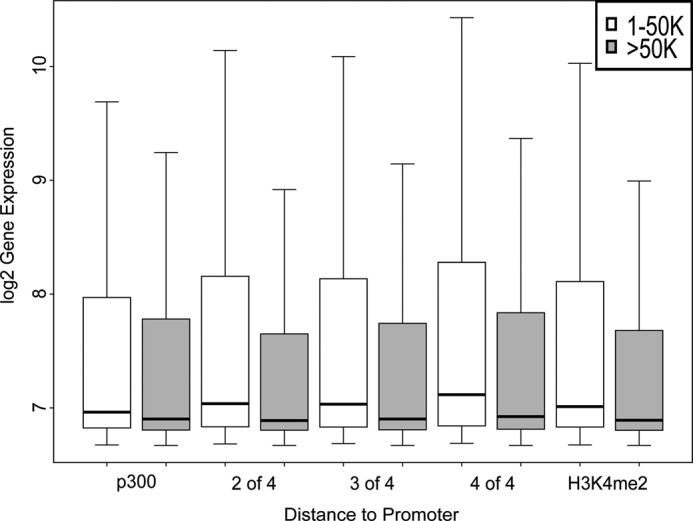
Gene expression and p300 and erythroid transcription factor occupancy in human primary erythroid cells. Gene expression levels were determined in primary human erythroid cell mRNA using Illumina microarrays. Expression levels of genes with p300 and erythroid transcription factor binding sites between 1 and 50 kb from the transcription start site (white boxes) were compared with the expression levels of genes with binding sites >50 kb away (gray boxes). Combinations of the four erythroid transcription factors studied (any two of the four, any three of the four, and all four) are shown. Error bars, S.D.
We also examined whether candidate enhancers were enriched near genes with known erythroid cell structure or function. We performed an unsupervised statistical enrichment analysis of functional gene annotations (63). Candidate erythroid enhancers identified by two of four erythroid transcription factors were associated with genes linked to erythroid cell-related phenotypes (Table 2 and supplemental Table S6). Candidate enhancers identified by p300 were not associated with genes linked to erythroid cell-related phenotypes (data not shown). Analyzing genes with candidate enhancers identified by 2 of 4 erythroid transcription factors by Gene Ontology (GO) annotation identified biological processes involved in erythroid cell function, including K-Cl cotransporter activity, myosin binding, glucose transport, and cellular iron homeostasis (supplemental Table S8).
TABLE 2.
Top enriched annotations of putative target genes near candidate human erythroid enhancers
Unsupervised enrichment analysis of annotated genes in the proximity of candidate enhancer regions identified by at least two of four erythroid transcription factors. The top enriched Mouse Genome Informatics phenotype ontology terms showing highly significant enrichment of genes implicated in erythroid cell-related phenotypes are shown. Only terms that showed significant enrichment and had a binomial -fold enrichment of ≥2 were considered.
| Top enriched phenotypes | Binomial false discovery rate Q value | Binomial -fold enrichment |
|---|---|---|
| -fold | ||
| Abnormal hemoglobin | 1.16e − 21 | 2.06971 |
| Decreased susceptibility to autoimmune disorder | 1.41e − 21 | 2.214783 |
| Abnormal acute inflammation | 1.09e − 20 | 2.15709 |
| Abnormal circulating tumor necrosis factor level | 7.86e − 20 | 2.713094 |
| Reticulocytosis | 2.71e − 19 | 2.839544 |
| Abnormal platelet physiology | 1.10e − 18 | 2.234179 |
| Abnormal hemoglobin content | 1.89e − 18 | 2.045812 |
| Increased circulating tumor necrosis factor level | 2.75e − 18 | 3.24539 |
| Joint inflammation | 5.02e − 17 | 2.171807 |
| Decreased hemoglobin content | 1.18e − 16 | 2.219195 |
| Abnormal iron homeostasis | 1.95e − 16 | 2.908582 |
| Abnormal iron level | 2.65e − 16 | 3.071508 |
| Hemolytic anemia | 2.99e − 16 | 4.024983 |
| Abnormal reticulocyte morphology | 4.10e − 16 | 2.325927 |
| Microcytosis | 1.60e − 15 | 4.88007 |
| Abnormal erythroid progenitor cell morphology | 5.59e − 15 | 2.192923 |
To further determine whether candidate enhancers identified by two of four erythroid transcription factors were associated with genes with erythroid cell function, the number of genes induced during erythroid differentiation associated with candidate enhancers (1–50 kb) was compared with the number of genes with randomized candidate enhancers (1–50 kb). The number of genes induced during erythroid differentiation was significantly higher than the number of genes from randomized enhancer locations (p < 0.01).
In addition, candidate enhancers were associated with genes in the GO term categories erythrocyte differentiation and erythrocyte homeostasis (p < 0.01 and p < 0.01, respectively) and were not associated with genes in the GO term categories muscle differentiation and neuron differentiation (p = 0.50 and p = 0.91, respectively).
Conservation Analyses of Candidate Enhancer Regions
Evolutionary constraint in regions of noncoding DNA has served as a proxy for functional constraint in the identification of candidate enhancer regions. However, recent studies have demonstrated that many enhancers are rapidly evolving, and in some species, many enhancers are both evolutionarily young and species-specific (33, 36). We investigated conservation of candidate erythroid enhancers between humans, mice, chickens, frogs, and zebrafish at different levels of stringency using the UCSC Genome Browser LiftOver tool, a computational tool that utilizes BLAT algorithm alignments to identify orthologous sequences between species (47, 64). Conservation was analyzed for candidate enhancers located in distal, intergenic, and intron regions, avoiding the high degree of conservation typically found between gene promoters and exons. Even at lower stringency (50% minimum ratio of bases that must remap), there was very high conservation between humans and mice for p300, all four erythroid transcription factors, and the combination of two of four erythroid transcription factors compared with randomly shuffled control sequences (Table 3). There was a very large falloff of conservation between human and lower nonmammalian species with nucleated circulating erythrocytes for the erythroid transcription factors, even at low stringency (50% minimum ratio of bases that must remap).
TABLE 3.
Evolutionary conservation at sites of p300 and erythroid transcription factor occupancy in distal, intergenic, and intron regions in erythroid cell chromatin
Conservation of human candidate enhancer regions was analyzed using the UCSC LiftOver tool at stringency levels of 75 or 50% of bases in the region that must remap.
| p300 | GATA1 | NFE2 | KLF1 | SCL | 2 of 4 erythroid transcription factors | Shuffled control | |
|---|---|---|---|---|---|---|---|
| 75% stringency | |||||||
| Mouse | 37.0 | 28.0 | 29.7 | 30.4 | 39.0 | 31.6 | 23.3 |
| Chicken | 3.1 | 0.8 | 0.9 | 0.8 | 1.9 | 1.0 | 1.1 |
| Frog | 2.5 | 0.7 | 0.6 | 0.5 | 0.6 | 0.7 | 0.6 |
| Zebrafish | 1.5 | 0.6 | 0.5 | 0.3 | 0.8 | 0.5 | 0.6 |
| 50% stringency | |||||||
| Mouse | 52.1 | 47.8 | 49.3 | 50.7 | 54.2 | 50.6 | 35.7 |
| Chicken | 6.1 | 2.3 | 2.1 | 2.2 | 3.4 | 2.4 | 2.2 |
| Frog | 5.2 | 2.2 | 1.6 | 1.6 | 1.4 | 1.8 | 1.2 |
| Zebrafish | 3.6 | 1.9 | 1.4 | 1.1 | 1.3 | 1.4 | 1.3 |
Conservation plots using PhastCons conservation scores with the 44-way vertebrate and placental mammal PhastCons track were constructed for binding regions of p300 and the four erythroid transcription factors. Strong conservation for p300 and all of the erythroid transcription factors was present in gene promoters and exons. However, there was weak constraint for p300 and the erythroid transcription factors, with the exception of SCL/Tal1, at distal and intergenic sites (Fig. 3).
FIGURE 3.

Conservation plots. The average PhastCons score in the 1000 bases surrounding the center of TF-bound regions is shown. The PhastCons score is a measure of the phylogenetic conservation within mammalian genomes. Promoter (left top), exon (left bottom), intergenic (right top), and distal (right bottom) regions were analyzed separately. Intronic regions are not shown.
Candidate Enhancer Regions and Biologically Relevant Single Nucleotide Polymorphisms
We explored whether candidate erythroid enhancers are enriched in regions associated with biologically relevant erythroid cell traits. We collected a data set of erythroid-associated noncoding SNPs (see “Experimental Procedures”) from the GWAS catalogue of NHGRI, National Institutes of Health (4). Currently, the functional significance of the overwhelming majority of these SNPs is unknown. SNP locations were compared with the sites of p300 or erythroid transcription factor occupancy. Fourteen SNPs associated with erythroid cell phenotypes were identified (Table 4 and supplemental Fig. S3), with four of the biologically relevant SNPs located in intron 2 of the BCL11A gene on chromosome 2. p300 occupancy was found at six SNPs, three without erythroid transcription factors and once each with two, three, and four co-localizing erythroid factors, respectively. Nine of the fourteen SNPs had co-occupancy with the combination of erythroid factors GATA1, NF-E2, and KLF1.
TABLE 4.
Candidate erythroid enhancers at sites of biologically relevant single nucleotide polymorphisms
| Enhancer | SNP ID | Chromosome | Gene(s) | P300 | GATA1 | NFE2 | KLF1 | SCL | H3K4me2 | H3K4me3 |
|---|---|---|---|---|---|---|---|---|---|---|
| Q1 | rs6684514 | chr11 | BGLAP, PAQR6,a SMG5, TMEM79, C1orf85,a VHLL,a CCT3, C1orf182a | × | ||||||
| Q2 | rs4910742 | chr11 | HBB, HBE | × | × | × | × | |||
| Q3 | rs1427407 | chr2 | BCL11A (intron 9) | × | × | |||||
| Q4 | rs6706648 | chr2 | BCL11A (intron 9) | × | × | |||||
| Q5 | rs7565301 | chr2 | BCL11A (intron 9) | × | × | × | ||||
| Q6 | rs7606173 | chr2 | BCL11A (intron 9) | × | × | × | × | × | × | × |
| Q7 | rs6092477 | chr20 | RAE1, RBM38, CTCFLa | × | × | |||||
| Q8 | rs7775698 | chr6 | HBS1L,a MYB | × | × | |||||
| Q9 | rs643381 | chr6 | CITED2 | × | × | × | ||||
| Q10 | rs4516970 | chr6 | WTAP, SOD2 | × | × | × | ||||
| Q11 | rs12718597 | chr7 | IKZF1 (intron 9) | × | × | × | × | × | ||
| Q12 | rs4737009 | chr8 | ANK1 (intron 9) | × | × | × | × | |||
| Q13 | rs385893 | chr9 | AK3 | × | × | × | ||||
| Q14 | rs8176746 | chr9 | ABO | × |
a Little or no expression in erythroid cell mRNA..
Recent studies have used PhastCons analyses and RP scores to predict whether or not a region of DNA contains a functional CRM (54, 59, 65). PhastCons uses a hidden Markov model method on aligned genomic sequences to estimate a probability that any nucleotide is conserved (66). The UCSC Genome Browser was used to determine 44-way placental mammal PhastCons scores for each of the 14 candidate biologically relevant enhancer regions. Twelve of 14 enhancer regions had maximal PhastCons scores of >0.8, suggesting that they contain a functional CRM (Table 5). An alternative way to predict the presence of CRMs is the RP score, which evaluates whether regions of DNA sequence have patterns more similar to those of regulatory elements or neutral DNA. Positive RP scores (RP scores of >0) (54) indicate conserved regions that contain a functional CRM. All 14 enhancer regions had positive maximal RP scores.
TABLE 5.
Evolutionary conservation at sites of biologically relevant candidate erythroid cell enhancers
Maximum PhastCons and regulatory potential scores were calculated for each biologically relevant erythroid cell enhancer. Regions with PhastCons scores of >0.8 and regulatory potential scores of >0 were considered to have conserved regions predicted to contain a cis-regulatory module.
| Candidate enhancer region | Maximum PhastCons score | Maximum regulatory potential score |
|---|---|---|
| Q1 | 1 | 0.548 |
| Q2 | 1 | 0.408 |
| Q3 | 1 | 0.265 |
| Q4 | 0.998 | 0.503 |
| Q5 | 0.986 | 0.293 |
| Q6 | 1 | 0.393 |
| Q7 | 0.19 | 0.092 |
| Q8 | 1 | 0.322 |
| Q9 | 1 | 0.245 |
| Q10 | 0.979 | 0.045 |
| Q11 | 0.045 | 0.284 |
| Q12 | 1 | 0.379 |
| Q13 | 0.999 | 0.438 |
| Q14 | 1 | 0.598 |
Reporter Gene Assay of Biologically Relevant Enhancers in Erythroid Cells
Individual reporter gene plasmids were prepared with the biologically relevant enhancer elements cloned upstream of a human γ-globin gene promoter-luciferase reporter gene cassette. These plasmids were transfected into human K562 cells, which have features of human erythroid cells. After 2 days, the cells were harvested, and luciferase activity was analyzed. The luciferase reporter gene was driven by the human γ-globin gene promoter, which is expressed in K562 cells. Activity from test plasmids was normalized to that directed by the human γ-globin gene promoter-luciferase reporter gene control plasmid. Twelve of the 14 candidate enhancers mapped to biologically relevant SNPs directed statistically significant (p < 0.05) reporter gene activity (Fig. 4 and supplemental Table S9). A cluster of four of these SNPs, all linked with levels of hemoglobin F, were located in intron 2 of the BCL11A gene. Fragments containing three of four of these SNPs directed statistically significant reporter gene activity, suggesting that the other SNPs required intact chromatin for their function, they were in linkage disequilibrium, they were nonfunctional, or they were associated with other functions.
FIGURE 4.

Activity of candidate erythroid cell enhancers in luciferase reporter gene assays. Individual reporter gene plasmids were prepared with the biologically relevant enhancer elements, labeled Q1–Q14, corresponding to Table 4, cloned upstream of a human γ globin gene promoter-luciferase reporter gene cassette. These plasmids were transfected into human K562 cells as described. After 2 days, the cells were harvested, and luciferase activity was analyzed. Activity from test plasmids was normalized to that directed by the human γ-globin gene promoter-luciferase reporter gene control plasmid. Relative luciferase activity was expressed as that obtained from the test plasmids versus the activity obtained from the SV40 promoter-luciferase reporter gene plasmid pGL2P plasmid taking into account the transfection efficiency. The data are the means ± S.D. (error bars) of at least six independent transfection experiments. The positive control plasmid contained the β-globin gene HS2 enhancer cloned upstream of a γ-globin gene promoter-firefly luciferase reporter gene cassette. The negative control plasmid contained a promoterless luciferase reporter gene cassette.
DISCUSSION
Mammalian erythroid cells are an excellent example of the complexity in temporal, developmental, and differentiation stage-specific changes exhibited by a single cell type. Mammalian erythroid cells originate from hematopoietic stem and progenitor cells. In the embryo and fetus, erythroid cells have differing developmental origins, with the primitive erythroid cell lineage developing from yolk sac-derived erythroid progenitors and the definitive cell lineage maturing from two different developmentally regulated stem and progenitor cell populations (67–70). These cells have different programs of regulation, with variation in spatial, temporal, and site-specific differentiation. Indeed, altered programs of erythropoiesis are activated throughout the life of the organism, such as occurs after blood loss, oxidative stress, or other organismal stress.
Our conservation analyses revealed that erythroid cell enhancers, like heart enhancers, are under weak evolutionary constraint, particularly when comparing placental mammals and nonplacental vertebrates. They also indicated that many candidate erythroid enhancers are species-specific and evolutionarily young. Mammalian erythrocytes are among the most highly specialized cells known, having evolved to an enucleate cell endowed with a highly redundant cell membrane. These changes, which increase surface area and cytoplasmic volume ratios, are primarily attributed to the need for additional hemoglobin content for oxygen transport, making cellular oxygen diffusion more efficient. As homeotherms evolved, oxygen demands increased, and organisms evolved to meet these demands. Birds developed a flow-through respiratory system, significantly more efficient than mammalian respiratory systems. It has been suggested that mammals diverged at this point, developing enucleate erythrocytes with increased oxygen carrying capacity to adapt to increased oxygen demands (71–74). Because discrete changes in CRMs may alter gene expression, generating potential for the genesis of novel species-specific traits (75, 76), identification of gene expression changes occurring over short evolutionary distances can suggest the origin of species-specific traits. Thus, comparative studies of enhancers in human and nonplacental vertebrates will probably provide novel information about the evolution of erythrocyte structure and function.
Our understanding of enhancer structure and function continues to expand. Previous studies, such as those of the globin gene loci (77–81), the GATA1 and SCL/Tal1 gene loci (82–93), and the erythropoietin gene locus (83), characterized enhancers as distantly located, positively acting cis-regulatory elements (77, 80). Recent studies have shown that enhancers have additional, complex roles in cellular gene regulation. These include roles in determining nuclear organization (6), transcription initiation and release of RNA polymerase II from promoter pausing (18), transcriptional competence (11), insulator activity (95, 96), development, and cell fate determination (11, 19, 97). Recent data indicate that the secondary enhancers synergize with primary enhancers to fine tune gene expression (98, 99). Noncoding RNAs have also been linked to enhancer function (100–106), and intergenic enhancers have been shown to act as alternate, tissue-specific promoters generating abundant, spliced, multiexonic poly(A)+ RNAs (107).
Rapid advances in genomic technologies, including genome-wide association studies, functional genomics, and high throughput gene expression analyses, are increasing our knowledge of gene regulation and its role in determining complex traits (75, 108). GWAS have identified a catalogue of polymorphisms associated with phenotypic traits, with most of these polymorphic variants located in noncoding regions of the genome. In parallel, functional genomics studies, particularly ChIP-seq-based analyses, have identified regions of DNA with regulatory potential on a genome-wide scale (109–112). Catalogs of genome-wide erythroid transcription factor occupancy in erythroid cells (113–124), which localize and define cis regulatory elements, are essential for our understanding of the mechanisms of phenotypic variation in inherited and acquired disease (35). Other studies of erythroid enhancers have demonstrated the role of intragenic enhancers as alternative promoters (107) and the combinatorial assembly of developmental stage-specific enhancers in regulating gene expression during erythropoiesis (115). Our data demonstrate the role of cell-expressed transcription factors and p300 in marking erythroid cell enhancers, reveal the lack of evolutionary constraint of human erythroid enhancers, and show a significant link of enhancers with human erythroid cell phenotypes. Ongoing synthesis of the data obtained from complementary lines of investigation is beginning to unravel the complex mechanisms of genetic variation in disease susceptibility (125).
Identification of critical cis-regulatory elements in erythroid cells will also be extremely useful in the genetic diagnosis of patients with hematologic disease. In some cases of inherited disease, deleterious coding region mutations have been identified on one allele, but the causative mutation in trans has not been identified. For instance, erythroid cells from a subset of patients with recessively inherited, α-spectrin-linked anemia have decreased α-spectrin mRNA levels and diminished α-spectrin protein synthesis, leading to abnormal, spectrin-deficient erythrocytes (126–128). The precise genetic basis (or bases) (i.e. the mutations on one or both alleles) of decreased spectrin mRNA accumulation in these cases is not known, even after mutation screening of the promoter and coding exons of the α-spectrin gene. Similarly, in congenital dyserythropoietic anemia type II, a recessively inherited disorder due to mutations in the SEC23B gene, a number of patients exhibit all of the phenotypic characteristics of congenital dyserythropoietic anemia type II, but a SEC23B mutation has only been identified on one allele (40, 94). Both of these genes have candidate enhancers in the genomic vicinity, making these regions excellent candidates for disease-associated mutations in these patients.
This work was supported, in whole or in part, by National Institutes of Health Grants K12HD000850, HL65448, and DK62039.

This article contains supplemental Methods, Tables S1–S9, and Figs. S1–S3.
- CRM
- cis-regulatory module
- TSS
- transcriptional start site
- ChIP-seq
- chromatin immunoprecipitation-sequencing
- GWAS
- genome-wide association studies
- RP
- regulatory potential
- H3K4me2 and H3K4me3
- histone H3 Lys-4 di- and trimethylated, respectively.
REFERENCES
- 1. Ernst J., Kheradpour P., Mikkelsen T. S., Shoresh N., Ward L. D., Epstein C. B., Zhang X., Wang L., Issner R., Coyne M., Ku M., Durham T., Kellis M., Bernstein B. E. (2011) Mapping and analysis of chromatin state dynamics in nine human cell types. Nature 473, 43–49 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Noonan J. P., McCallion A. S. (2010) Genomics of long-range regulatory elements. Annu. Rev. Genomics Hum. Genet. 11, 1–23 [DOI] [PubMed] [Google Scholar]
- 3. Kleinjan D. A., van Heyningen V. (2005) Long-range control of gene expression. Emerging mechanisms and disruption in disease. Am. J. Hum. Genet. 76, 8–32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Hindorff L. A., Sethupathy P., Junkins H. A., Ramos E. M., Mehta J. P., Collins F. S., Manolio T. A. (2009) Potential etiologic and functional implications of genome-wide association loci for human diseases and traits. Proc. Natl. Acad. Sci. U.S.A. 106, 9362–9367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. VanderMeer J. E., Ahituv N. (2011) cis-regulatory mutations are a genetic cause of human limb malformations. Dev. Dyn. 240, 920–930 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Bulger M., Groudine M. (2011) Functional and mechanistic diversity of distal transcription enhancers. Cell 144, 327–339 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Chen C. Y., Morris Q., Mitchell J. A. (2012) Enhancer identification in mouse embryonic stem cells using integrative modeling of chromatin and genomic features. BMC Genomics 13, 152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Junion G., Spivakov M., Girardot C., Braun M., Gustafson E. H., Birney E., Furlong E. E. (2012) A transcription factor collective defines cardiac cell fate and reflects lineage history. Cell 148, 473–486 [DOI] [PubMed] [Google Scholar]
- 9. Koch F., Fenouil R., Gut M., Cauchy P., Albert T. K., Zacarias-Cabeza J., Spicuglia S., de la Chapelle A. L., Heidemann M., Hintermair C., Eick D., Gut I., Ferrier P., Andrau J. C. (2011) Transcription initiation platforms and GTF recruitment at tissue-specific enhancers and promoters. Nat. Struct. Mol. Biol. 18, 956–963 [DOI] [PubMed] [Google Scholar]
- 10. Szutorisz H., Dillon N., Tora L. (2005) The role of enhancers as centres for general transcription factor recruitment. Trends Biochem. Sci. 30, 593–599 [DOI] [PubMed] [Google Scholar]
- 11. Xu J., Watts J. A., Pope S. D., Gadue P., Kamps M., Plath K., Zaret K. S., Smale S. T. (2009) Transcriptional competence and the active marking of tissue-specific enhancers by defined transcription factors in embryonic and induced pluripotent stem cells. Genes Dev. 23, 2824–2838 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Gotea V., Visel A., Westlund J. M., Nobrega M. A., Pennacchio L. A., Ovcharenko I. (2010) Homotypic clusters of transcription factor binding sites are a key component of human promoters and enhancers. Genome Res. 20, 565–577 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Heintzman N. D., Hon G. C., Hawkins R. D., Kheradpour P., Stark A., Harp L. F., Ye Z., Lee L. K., Stuart R. K., Ching C. W., Ching K. A., Antosiewicz-Bourget J. E., Liu H., Zhang X., Green R. D., Lobanenkov V. V., Stewart R., Thomson J. A., Crawford G. E., Kellis M., Ren B. (2009) Histone modifications at human enhancers reflect global cell-type-specific gene expression. Nature 459, 108–112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Heintzman N. D., Ren B. (2009) Finding distal regulatory elements in the human genome. Curr. Opin. Genet. Dev. 19, 541–549 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Heintzman N. D., Stuart R. K., Hon G., Fu Y., Ching C. W., Hawkins R. D., Barrera L. O., Van Calcar S., Qu C., Ching K. A., Wang W., Weng Z., Green R. D., Crawford G. E., Ren B. (2007) Distinct and predictive chromatin signatures of transcriptional promoters and enhancers in the human genome. Nat. Genet. 39, 311–318 [DOI] [PubMed] [Google Scholar]
- 16. Visel A., Blow M. J., Li Z., Zhang T., Akiyama J. A., Holt A., Plajzer-Frick I., Shoukry M., Wright C., Chen F., Afzal V., Ren B., Rubin E. M., Pennacchio L. A. (2009) ChIP-seq accurately predicts tissue-specific activity of enhancers. Nature 457, 854–858 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Ghisletti S., Barozzi I., Mietton F., Polletti S., De Santa F., Venturini E., Gregory L., Lonie L., Chew A., Wei C.-L., Ragoussis J., Natoli G. (2010) Identification and characterization of enhancers controlling the inflammatory gene expression program in macrophages. Immunity 32, 317–328 [DOI] [PubMed] [Google Scholar]
- 18. Ong C. T., Corces V. G. (2011) Enhancer function. New insights into the regulation of tissue-specific gene expression. Nat. Rev. Genet. 12, 283–293 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Ong C. T., Corces V. G. (2012) Enhancers. Emerging roles in cell fate specification. EMBO Rep. 13, 423–430 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Creyghton M. P., Cheng A. W., Welstead G. G., Kooistra T., Carey B. W., Steine E. J., Hanna J., Lodato M. A., Frampton G. M., Sharp P. A., Boyer L. A., Young R. A., Jaenisch R. (2010) Histone H3K27ac separates active from poised enhancers and predicts developmental state. Proc. Natl. Acad. Sci. U.S.A. 107, 21931–21936 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Schmidl C., Klug M., Boeld T. J., Andreesen R., Hoffmann P., Edinger M., Rehli M. (2009) Lineage-specific DNA methylation in T cells correlates with histone methylation and enhancer activity. Genome Res. 19, 1165–1174 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. He H. H., Meyer C. A., Shin H., Bailey S. T., Wei G., Wang Q., Zhang Y., Xu K., Ni M., Lupien M., Mieczkowski P., Lieb J. D., Zhao K., Brown M., Liu X. S. (2010) Nucleosome dynamics define transcriptional enhancers. Nat. Genet. 42, 343–347 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Wang Z., Zang C., Rosenfeld J. A., Schones D. E., Barski A., Cuddapah S., Cui K., Roh T.-Y., Peng W., Zhang M. Q., Zhao K. (2008) Combinatorial patterns of histone acetylations and methylations in the human genome. Nat. Genet. 40, 897–903 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Hon G., Wang W., Ren B. (2009) Discovery and annotation of functional chromatin signatures in the human genome. PLoS Comput. Biol. 5, e1000566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Rada-Iglesias A., Bajpai R., Swigut T., Brugmann S. A., Flynn R. A., Wysocka J. (2011) A unique chromatin signature uncovers early developmental enhancers in humans. Nature 470, 279–283 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Cui K., Zang C., Roh T. Y., Schones D. E., Childs R. W., Peng W., Zhao K. (2009) Chromatin signatures in multipotent human hematopoietic stem cells indicate the fate of bivalent genes during differentiation. Cell Stem Cell 4, 80–93 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Li Q., Ritter D., Yang N., Dong Z., Li H., Chuang J. H., Guo S. (2010) A systematic approach to identify functional motifs within vertebrate developmental enhancers. Developmental Biology 337, 484–495 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Visel A., Prabhakar S., Akiyama J. A., Shoukry M., Lewis K. D., Holt A., Plajzer-Frick I., Afzal V., Rubin E. M., Pennacchio L. A. (2008) Ultraconservation identifies a small subset of extremely constrained developmental enhancers. Nat. Genet. 40, 158–160 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Pennacchio L. A., Rubin E. M. (2001) Genomic strategies to identify mammalian regulatory sequences. Nat. Rev. Genet. 2, 100–109 [DOI] [PubMed] [Google Scholar]
- 30. Pennacchio L. A., Ahituv N., Moses A. M., Prabhakar S., Nobrega M. A., Shoukry M., Minovitsky S., Dubchak I., Holt A., Lewis K. D., Plajzer-Frick I., Akiyama J., De Val S., Afzal V., Black B. L., Couronne O., Eisen M. B., Visel A., Rubin E. M. (2006) In vivo enhancer analysis of human conserved non-coding sequences. Nature 444, 499–502 [DOI] [PubMed] [Google Scholar]
- 31. Pheasant M., Mattick J. S. (2007) Raising the estimate of functional human sequences. Genome Res. 17, 1245–1253 [DOI] [PubMed] [Google Scholar]
- 32. Hon G., Ren B., Wang W. (2008) ChromaSig. A probabilistic approach to finding common chromatin signatures in the human genome. PLoS Comput. Biol. 4, e1000201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Blow M. J., McCulley D. J., Li Z., Zhang T., Akiyama J. A., Holt A., Plajzer-Frick I., Shoukry M., Wright C., Chen F., Afzal V., Bristow J., Ren B., Black B. L., Rubin E. M., Visel A., Pennacchio L. A. (2010) ChIP-Seq identification of weakly conserved heart enhancers. Nat. Genet. 42, 806–810 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. May D., Blow M. J., Kaplan T., McCulley D. J., Jensen B. C., Akiyama J. A., Holt A., Plajzer-Frick I., Shoukry M., Wright C., Afzal V., Simpson P. C., Rubin E. M., Black B. L., Bristow J., Pennacchio L. A., Visel A. (2012) Large-scale discovery of enhancers from human heart tissue. Nat. Genet. 44, 89–93 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Visel A., Rubin E. M., Pennacchio L. A. (2009) Genomic views of distant-acting enhancers. Nature 461, 199–205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Schmidt D., Wilson M. D., Ballester B., Schwalie P. C., Brown G. D., Marshall A., Kutter C., Watt S., Martinez-Jimenez C. P., Mackay S., Talianidis I., Flicek P., Odom D. T. (2010) Five-vertebrate ChIP-seq reveals the evolutionary dynamics of transcription factor binding. Science 328, 1036–1040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Loots G. G., Locksley R. M., Blankespoor C. M., Wang Z. E., Miller W., Rubin E. M., Frazer K. A. (2000) Identification of a coordinate regulator of interleukins 4, 13, and 5 by cross-species sequence comparisons. Science 288, 136–140 [DOI] [PubMed] [Google Scholar]
- 38. Margulies E. H., Cooper G. M., Asimenos G., Thomas D. J., Dewey C. N., Siepel A., Birney E., Keefe D., Schwartz A. S., Hou M., Taylor J., Nikolaev S., Montoya-Burgos J. I., Löytynoja A., Whelan S., Pardi F., Massingham T., Brown J. B., Bickel P., Holmes I., Mullikin J. C., Ureta-Vidal A., Paten B., Stone E. A., Rosenbloom K. R., Kent W. J., Bouffard G. G., Guan X., Hansen N. F., Idol J. R., Maduro V. V., Maskeri B., McDowell J. C., Park M., Thomas P. J., Young A. C., Blakesley R. W., Muzny D. M., Sodergren E., Wheeler D. A., Worley K. C., Jiang H., Weinstock G. M., Gibbs R. A., Graves T., Fulton R., Mardis E. R., Wilson R. K., Clamp M., Cuff J., Gnerre S., Jaffe D. B., Chang J. L., Lindblad-Toh K., Lander E. S., Hinrichs A., Trumbower H., Clawson H., Zweig A., Kuhn R. M., Barber G., Harte R., Karolchik D., Field M. A., Moore R. A., Matthewson C. A., Schein J. E., Marra M. A., Antonarakis S. E., Batzoglou S., Goldman N., Hardison R., Haussler D., Miller W., Pachter L., Green E. D., Sidow A. (2007) Analyses of deep mammalian sequence alignments and constraint predictions for 1% of the human genome. Genome Res. 17, 760–774 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Bulger M., Groudine M. (2010) Enhancers. The abundance and function of regulatory sequences beyond promoters. Dev. Biol. 339, 250–257 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Iolascon A., Russo R., Esposito M. R., Asci R., Piscopo C., Perrotta S., Fénéant-Thibault M., Garçon L., Delaunay J. (2010) Molecular analysis of 42 patients with congenital dyserythropoietic anemia type II. New mutations in the SEC23B gene and a search for a genotype-phenotype relationship. Haematologica 95, 708–715 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Panzenböck B., Bartunek P., Mapara M. Y., Zenke M. (1998) Growth and differentiation of human stem cell factor/erythropoietin-dependent erythroid progenitor cells in vitro. Blood 92, 3658–3668 [PubMed] [Google Scholar]
- 42. Migliaccio A. R., Migliaccio G., Di Baldassarre A., Eddleman K. (2002) Circulating hematopoietic progenitor cells in a fetus with α thalassemia. Comparison with the cells circulating in normal and non-thalassemic anemia fetuses and implications for in utero transplantations. Bone Marrow Transplant. 30, 75–80 [DOI] [PubMed] [Google Scholar]
- 43. Zhang J., Socolovsky M., Gross A. W., Lodish H. F. (2003) Role of Ras signaling in erythroid differentiation of mouse fetal liver cells. Functional analysis by a flow cytometry-based novel culture system. Blood 102, 3938–3946 [DOI] [PubMed] [Google Scholar]
- 44. Steiner L. A., Maksimova Y., Schulz V., Wong C., Raha D., Mahajan M. C., Weissman S. M., Gallagher P. G. (2009) Chromatin architecture and transcription factor binding regulate expression of erythrocyte membrane protein genes. Mol. Cell Biol. 29, 5399–5412 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Steiner L. A., Schulz V. P., Maksimova Y., Wong C., Gallagher P. G. (2011) Patterns of histone H3 lysine 27 monomethylation and erythroid cell type-specific gene expression. J. Biol. Chem. 286, 39457–39465 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Vandesompele J., De Preter K., Pattyn F., Poppe B., Van Roy N., De Paepe A., Speleman F. (2002) Accurate normalization of real-time quantitative RT-PCR data by geometric averaging of multiple internal control genes. Genome Biol. 3, RESEARCH0034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Karolchik D., Kuhn R. M., Baertsch R., Barber G. P., Clawson H., Diekhans M., Giardine B., Harte R. A., Hinrichs A. S., Hsu F., Kober K. M., Miller W., Pedersen J. S., Pohl A., Raney B. J., Rhead B., Rosenbloom K. R., Smith K. E., Stanke M., Thakkapallayil A., Trumbower H., Wang T., Zweig A. S., Haussler D., Kent W. J. (2008) The UCSC Genome Browser Database. 2008 update. Nucleic Acids Res. 36, D773–D779 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Zhang Y., Liu T., Meyer C. A., Eeckhoute J., Johnson D. S., Bernstein B. E., Nusbaum C., Myers R. M., Brown M., Li W., Liu X. S. (2008) Model-based analysis of ChIP-Seq (MACS). Genome Biol. 9, R137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Giannopoulou E. G., Elemento O. (2011) An integrated ChIP-seq analysis platform with customizable workflows. BMC Bioinformatics 12, 277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Bailey T. L., Elkan C. (1994) Fitting a mixture model by expectation maximization to discover motifs in biopolymers. Proc. Int. Conf. Intell. Syst. Mol. Biol. 2, 28–36 [PubMed] [Google Scholar]
- 51. Siepel A., Bejerano G., Pedersen J. S., Hinrichs A. S., Hou M., Rosenbloom K., Clawson H., Spieth J., Hillier L. W., Richards S., Weinstock G. M., Wilson R. K., Gibbs R. A., Kent W. J., Miller W., Haussler D. (2005) Evolutionarily conserved elements in vertebrate, insect, worm, and yeast genomes. Genome Res. 15, 1034–1050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Liu T., Ortiz J. A., Taing L., Meyer C. A., Lee B., Zhang Y., Shin H., Wong S. S., Ma J., Lei Y., Pape U. J., Poidinger M., Chen Y., Yeung K., Brown M., Turpaz Y., Liu X. S. (2011) Cistrome. An integrative platform for transcriptional regulation studies. Genome Biol. 12, R83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Giardine B., Riemer C., Hardison R. C., Burhans R., Elnitski L., Shah P., Zhang Y., Blankenberg D., Albert I., Taylor J., Miller W., Kent W. J., Nekrutenko A. (2005) Galaxy. A platform for interactive large-scale genome analysis. Genome Res. 15, 1451–1455 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. King D. C., Taylor J., Elnitski L., Chiaromonte F., Miller W., Hardison R. C. (2005) Evaluation of regulatory potential and conservation scores for detecting cis-regulatory modules in aligned mammalian genome sequences. Genome Res. 15, 1051–1060 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Elnitski L., Miller W., Hardison R. (1997) Conserved E boxes function as part of the enhancer in hypersensitive site 2 of the β-globin locus control region. Role of basic helix-loop-helix proteins. J. Biol. Chem. 272, 369–378 [DOI] [PubMed] [Google Scholar]
- 56. Jane S. M., Ney P. A., Vanin E. F., Gumucio D. L., Nienhuis A. W. (1992) Identification of a stage selector element in the human γ-globin gene promoter that fosters preferential interaction with the 5′ HS2 enhancer when in competition with the β-promoter. EMBO J. 11, 2961–2969 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Gallagher P. G., Liem R. I., Wong E., Weiss M. J., Bodine D. M. (2005) GATA-1 and Oct-1 are required for expression of the human α-hemoglobin-stabilizing protein gene. J. Biol. Chem. 280, 39016–39023 [DOI] [PubMed] [Google Scholar]
- 58. Brazma A., Hingamp P., Quackenbush J., Sherlock G., Spellman P., Stoeckert C., Aach J., Ansorge W., Ball C. A., Causton H. C., Gaasterland T., Glenisson P., Holstege F. C., Kim I. F., Markowitz V., Matese J. C., Parkinson H., Robinson A., Sarkans U., Schulze-Kremer S., Stewart J., Taylor R., Vilo J., Vingron M. (2001) Minimum information about a microarray experiment (MIAME). Toward standards for microarray data. Nat. Genet. 29, 365–371 [DOI] [PubMed] [Google Scholar]
- 59. Cheng Y., King D. C., Dore L. C., Zhang X., Zhou Y., Zhang Y., Dorman C., Abebe D., Kumar S. A., Chiaromonte F., Miller W., Green R. D., Weiss M. J., Hardison R. C. (2008) Transcriptional enhancement by GATA1-occupied DNA segments is strongly associated with evolutionary constraint on the binding site motif. Genome Res. 18, 1896–1905 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Zhou V. W., Goren A., Bernstein B. E. (2011) Charting histone modifications and the functional organization of mammalian genomes. Nat. Rev. Genet. 12, 7–18 [DOI] [PubMed] [Google Scholar]
- 61. Hardison R. C., Taylor J. (2012) Genomic approaches towards finding cis-regulatory modules in animals. Nat. Rev. Genet. 13, 469–483 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Bernstein B. E., Kamal M., Lindblad-Toh K., Bekiranov S., Bailey D. K., Huebert D. J., McMahon S., Karlsson E. K., Kulbokas E. J., 3rd, Gingeras T. R., Schreiber S. L., Lander E. S. (2005) Genomic maps and comparative analysis of histone modifications in human and mouse. Cell 120, 169–181 [DOI] [PubMed] [Google Scholar]
- 63. McLean C. Y., Bristor D., Hiller M., Clarke S. L., Schaar B. T., Lowe C. B., Wenger A. M., Bejerano G. (2010) GREAT improves functional interpretation of cis-regulatory regions. Nat. Biotechnol. 28, 495–501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Kent W. J. (2002) BLAT. The BLAST-like alignment tool. Genome Res. 12, 656–664 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Kim T. H., Abdullaev Z. K., Smith A. D., Ching K. A., Loukinov D. I., Green R. D., Zhang M. Q., Lobanenkov V. V., Ren B. (2007) Analysis of the vertebrate insulator protein CTCF-binding sites in the human genome. Cell 128, 1231–1245 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Siepel A., Haussler D. (2004) Combining phylogenetic and hidden Markov models in biosequence analysis. J. Comput. Biol. 11, 413–428 [DOI] [PubMed] [Google Scholar]
- 67. England S. J., McGrath K. E., Frame J. M., Palis J. (2011) Immature erythroblasts with extensive ex vivo self-renewal capacity emerge from the early mammalian fetus. Blood 117, 2708–2717 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. McGrath K. E., Frame J. M., Fromm G. J., Koniski A. D., Kingsley P. D., Little J., Bulger M., Palis J. (2011) A transient definitive erythroid lineage with unique regulation of the β-globin locus in the mammalian embryo. Blood 117, 4600–4608 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Palis J. (2008) Ontogeny of erythropoiesis. Curr. Opin. Hematol. 15, 155–161 [DOI] [PubMed] [Google Scholar]
- 70. Palis J., Malik J., McGrath K. E., Kingsley P. D. (2010) Primitive erythropoiesis in the mammalian embryo. Int. J. Dev. Biol. 54, 1011–1018 [DOI] [PubMed] [Google Scholar]
- 71. Glomski C. A., Tamburlin J. (1990) The phylogenetic odyssey of the erythrocyte. II. The early or invertebrate prototypes. Histol. Histopathol. 5, 513–525 [PubMed] [Google Scholar]
- 72. Glomski C. A., Tamburlin J., Chainani M. (1992) The phylogenetic odyssey of the erythrocyte. III. Fish, the lower vertebrate experience. Histol. Histopathol. 7, 501–528 [PubMed] [Google Scholar]
- 73. Glomski C. A., Tamburlin J., Hard R., Chainani M. (1997) The phylogenetic odyssey of the erythrocyte. IV. The amphibians. Histol Histopathol. 12, 147–170 [PubMed] [Google Scholar]
- 74. Nikinmaa M. (1997) Oxygen and carbon dioxide transport in vertebrate erythrocytes. An evolutionary change in the role of membrane transport. J. Exp. Biol. 200, 369–380 [DOI] [PubMed] [Google Scholar]
- 75. Wray G. A. (2007) The evolutionary significance of cis-regulatory mutations. Nat. Rev. Genet. 8, 206–216 [DOI] [PubMed] [Google Scholar]
- 76. Carroll S. B. (2008) Evo-devo and an expanding evolutionary synthesis. A genetic theory of morphological evolution. Cell 134, 25–36 [DOI] [PubMed] [Google Scholar]
- 77. Higgs D. R., Vernimmen D., Wood B. (2008) Long-range regulation of α-globin gene expression. Adv. Genet. 61, 143–173 [DOI] [PubMed] [Google Scholar]
- 78. Higgs D. R., Weatherall D. J. (2009) The α thalassaemias. Cell. Mol. Life Sci. 66, 1154–1162 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Sankaran V. G., Xu J., Orkin S. H. (2010) Advances in the understanding of haemoglobin switching. Br. J. Haematol. 149, 181–194 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Fromm G., Bulger M. (2009) A spectrum of gene regulatory phenomena at mammalian β-globin gene loci. Biochem. Cell Biol. 87, 781–790 [DOI] [PubMed] [Google Scholar]
- 81. Harju S., McQueen K. J., Peterson K. R. (2002) Chromatin structure and control of β-like globin gene switching. Exp. Biol. Med. (Maywood) 227, 683–700 [DOI] [PubMed] [Google Scholar]
- 82. Suzuki M., Moriguchi T., Ohneda K., Yamamoto M. (2009) Differential contribution of the Gata1 gene hematopoietic enhancer to erythroid differentiation. Mol. Cell Biol. 29, 1163–1175 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Suzuki N., Obara N., Pan X., Watanabe M., Jishage K., Minegishi N., Yamamoto M. (2011) Specific contribution of the erythropoietin gene 3′ enhancer to hepatic erythropoiesis after late embryonic stages. Mol. Cell Biol. 31, 3896–3905 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Nishimura S., Takahashi S., Kuroha T., Suwabe N., Nagasawa T., Trainor C., Yamamoto M. (2000) A GATA box in the GATA-1 gene hematopoietic enhancer is a critical element in the network of GATA factors and sites that regulate this gene. Mol. Cell Biol. 20, 713–723 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. McDevitt M. A., Fujiwara Y., Shivdasani R. A., Orkin S. H. (1997) An upstream, DNase I hypersensitive region of the hematopoietic-expressed transcription factor GATA-1 gene confers developmental specificity in transgenic mice. Proc. Natl. Acad. Sci. U.S.A. 94, 7976–7981 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Valverde-Garduno V., Guyot B., Anguita E., Hamlett I., Porcher C., Vyas P. (2004) Differences in the chromatin structure and cis-element organization of the human and mouse GATA1 loci. Implications for cis-element identification. Blood 104, 3106–3116 [DOI] [PubMed] [Google Scholar]
- 87. Onodera K., Takahashi S., Nishimura S., Ohta J., Motohashi H., Yomogida K., Hayashi N., Engel J. D., Yamamoto M. (1997) GATA-1 transcription is controlled by distinct regulatory mechanisms during primitive and definitive erythropoiesis. Proc. Natl. Acad. Sci. U.S.A. 94, 4487–4492 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Wozniak R. J., Boyer M. E., Grass J. A., Lee Y., Bresnick E. H. (2007) Context-dependent GATA factor function. Combinatorial requirements for transcriptional control in hematopoietic and endothelial cells. J. Biol. Chem. 282, 14665–14674 [DOI] [PubMed] [Google Scholar]
- 89. Smith A. M., Sanchez M. J., Follows G. A., Kinston S., Donaldson I. J., Green A. R., Göttgens B. (2008) A novel mode of enhancer evolution. The Tal1 stem cell enhancer recruited a MIR element to specifically boost its activity. Genome Res. 18, 1422–1432 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Ogilvy S., Ferreira R., Piltz S. G., Bowen J. M., Göttgens B., Green A. R. (2007) The SCL +40 enhancer targets the midbrain together with primitive and definitive hematopoiesis and is regulated by SCL and GATA proteins. Mol. Cell Biol. 27, 7206–7219 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Göttgens B., Nastos A., Kinston S., Piltz S., Delabesse E. C., Stanley M., Sanchez M. J., Ciau-Uitz A., Patient R., Green A. R. (2002) Establishing the transcriptional programme for blood. The SCL stem cell enhancer is regulated by a multiprotein complex containing Ets and GATA factors. EMBO J. 21, 3039–3050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Delabesse E., Ogilvy S., Chapman M. A., Piltz S. G., Gottgens B., Green A. R. (2005) Transcriptional regulation of the SCL locus. Identification of an enhancer that targets the primitive erythroid lineage in vivo. Mol. Cell Biol. 25, 5215–5225 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Dhami P., Bruce A. W., Jim J. H., Dillon S. C., Hall A., Cooper J. L., Bonhoure N., Chiang K., Ellis P. D., Langford C., Andrews R. M., Vetrie D. (2010) Genomic approaches uncover increasing complexities in the regulatory landscape at the human SCL (TAL1) locus. PLoS ONE 5, e9059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Russo R., Esposito M. R., Asci R., Gambale A., Perrotta S., Ramenghi U., Forni G. L., Uygun V., Delaunay J., Iolascon A. (2010) Mutational spectrum in congenital dyserythropoietic anemia type II. Identification of 19 novel variants in SEC23B gene. Am. J. Hematol. 85, 915–920 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Core L. J., Lis J. T. (2009) Paused Pol II captures enhancer activity and acts as a potent insulator. Genes Dev. 23, 1606–1612 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Maksimenko O., Golovnin A., Georgiev P. (2008) Enhancer-promoter communication is regulated by insulator pairing in a Drosophila model bigenic locus. Mol. Cell Biol. 28, 5469–5477 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Borok M. J., Tran D. A., Ho M. C., Drewell R. A. (2010) Dissecting the regulatory switches of development. Lessons from enhancer evolution in Drosophila. Development 137, 5–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Guerrero L., Marco-Ferreres R., Serrano A. L., Arredondo J. J., Cervera M. (2010) Secondary enhancers synergise with primary enhancers to guarantee fine-tuned muscle gene expression. Dev. Biol. 337, 16–28 [DOI] [PubMed] [Google Scholar]
- 99. Hong J. W., Hendrix D. A., Levine M. S. (2008) Shadow enhancers as a source of evolutionary novelty. Science 321, 1314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Mattick J. S. (2010) Linc-ing Long noncoding RNAs and enhancer function. Dev. Cell 19, 485–486 [DOI] [PubMed] [Google Scholar]
- 101. Ørom U. A., Derrien T., Beringer M., Gumireddy K., Gardini A., Bussotti G., Lai F., Zytnicki M., Notredame C., Huang Q., Guigo R., Shiekhattar R. (2010) Long noncoding RNAs with enhancer-like function in human cells. Cell 143, 46–58 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Ørom U. A., Shiekhattar R. (2011) Long non-coding RNAs and enhancers. Curr. Opin. Genet. Dev. 21, 194–198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Orom U. A., Shiekhattar R. (2011) Noncoding RNAs and enhancers. Complications of a long-distance relationship. Trends Genet. 27, 433–439 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Yoo E. J., Cooke N. E., Liebhaber S. A. (2012) An RNA-independent linkage of noncoding transcription to long-range enhancer function. Mol. Cell Biol. 32, 2020–2029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Kim T.-K., Hemberg M., Gray J. M., Costa A. M., Bear D. M., Wu J., Harmin D. A., Laptewicz M., Barbara-Haley K., Kuersten S., Markenscoff-Papadimitriou E., Kuhl D., Bito H., Worley P. F., Kreiman G., Greenberg M. E. (2010) Widespread transcription at neuronal activity-regulated enhancers. Nature 465, 182–187 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Wang D., Garcia-Bassets I., Benner C., Li W., Su X., Zhou Y., Qiu J., Liu W., Kaikkonen M. U., Ohgi K. A., Glass C. K., Rosenfeld M. G., Fu X. D. (2011) Reprogramming transcription by distinct classes of enhancers functionally defined by eRNA. Nature 474, 390–394 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Kowalczyk M. S., Hughes J. R., Garrick D., Lynch M. D., Sharpe J. A., Sloane-Stanley J. A., McGowan S. J., De Gobbi M., Hosseini M., Vernimmen D., Brown J. M., Gray N. E., Collavin L., Gibbons R. J., Flint J., Taylor S., Buckle V. J., Milne T. A., Wood W. G., Higgs D. R. (2012) Intragenic enhancers act as alternative promoters. Mol. Cell 45, 447–458 [DOI] [PubMed] [Google Scholar]
- 108. Epstein D. J. (2009) Cis-regulatory mutations in human disease. Brief Funct. Genomic Proteomic 8, 310–316 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Park P. J. (2009) ChIP-seq. Advantages and challenges of a maturing technology. Nat. Rev. Genet. 10, 669–680 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Farnham P. J. (2009) Insights from genomic profiling of transcription factors. Nat. Rev. Genet. 10, 605–616 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Barski A., Zhao K. (2009) Genomic location analysis by ChIP-Seq. J. Cell Biochem. 107, 11–18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Kadauke S., Blobel G. A. (2009) Chromatin loops in gene regulation. Biochim. Biophys. Acta 1789, 17–25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Pilon A. M., Ajay S. S., Kumar S. A., Steiner L. A., Cherukuri P. F., Wincovitch S., Anderson S. M., NISC Comparative Sequencing Center, Mullikin J. C., Gallagher P. G., Hardison R. C., Margulies E. H., Bodine D. M. (2011) Genome-wide ChIP-Seq reveals a dramatic shift in the binding of the transcription factor erythroid Kruppel-like factor during erythrocyte differentiation. Blood 118, e139–e148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Tallack M. R., Whitington T., Yuen W. S., Wainwright E. N., Keys J. R., Gardiner B. B., Nourbakhsh E., Cloonan N., Grimmond S. M., Bailey T. L., Perkins A. C. (2010) A global role for KLF1 in erythropoiesis revealed by ChIP-seq in primary erythroid cells. Genome Res. 20, 1052–1063 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Xu J., Shao Z., Glass K., Bauer D. E., Pinello L., Van Handel B., Hou S., Stamatoyannopoulos J. A., Mikkola H. K., Yuan G. C., Orkin S. H. (2012) Combinatorial assembly of developmental stage-specific enhancers controls gene expression programs during human erythropoiesis. Dev. Cell 23, 796–811 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Wilson N. K., Miranda-Saavedra D., Kinston S., Bonadies N., Foster S. D., Calero-Nieto F., Dawson M. A., Donaldson I. J., Dumon S., Frampton J., Janky R., Sun X. H., Teichmann S. A., Bannister A. J., Göttgens B. (2009) The transcriptional program controlled by the stem cell leukemia gene Scl/Tal1 during early embryonic hematopoietic development. Blood 113, 5456–5465 [DOI] [PubMed] [Google Scholar]
- 117. Yu M., Riva L., Xie H., Schindler Y., Moran T. B., Cheng Y., Yu D., Hardison R., Weiss M. J., Orkin S. H., Bernstein B. E., Fraenkel E., Cantor A. B. (2009) Insights into GATA-1-mediated gene activation versus repression via genome-wide chromatin occupancy analysis. Mol. Cell 36, 682–695 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Fujiwara T., O'Geen H., Keles S., Blahnik K., Linnemann A. K., Kang Y. A., Choi K., Farnham P. J., Bresnick E. H. (2009) Discovering hematopoietic mechanisms through genome-wide analysis of GATA factor chromatin occupancy. Mol. Cell 36, 667–681 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119. Kadauke S., Udugama M. I., Pawlicki J. M., Achtman J. C., Jain D. P., Cheng Y., Hardison R. C., Blobel G. A. (2012) Tissue-specific mitotic bookmarking by hematopoietic transcription factor GATA1. Cell 150, 725–737 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. Chlon T. M., Doré L. C., Crispino J. D. (2012) Cofactor-mediated restriction of GATA-1 chromatin occupancy coordinates lineage-specific gene expression. Mol. Cell 47, 608–621 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121. Doré L. C., Chlon T. M., Brown C. D., White K. P., Crispino J. D. (2012) Chromatin occupancy analysis reveals genome-wide GATA factor switching during hematopoiesis. Blood 119, 3724–3733 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Palii C. G., Perez-Iratxeta C., Yao Z., Cao Y., Dai F., Davison J., Atkins H., Allan D., Dilworth F. J., Gentleman R., Tapscott S. J., Brand M. (2011) Differential genomic targeting of the transcription factor TAL1 in alternate haematopoietic lineages. EMBO J. 30, 494–509 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123. Kassouf M. T., Hughes J. R., Taylor S., McGowan S. J., Soneji S., Green A. L., Vyas P., Porcher C. (2010) Genome-wide identification of TAL1's functional targets. Insights into its mechanisms of action in primary erythroid cells. Genome Res. 20, 1064–1083 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124. Cheng Y., Wu W., Kumar S. A., Yu D., Deng W., Tripic T., King D. C., Chen K. B., Zhang Y., Drautz D., Giardine B., Schuster S. C., Miller W., Chiaromonte F., Zhang Y., Blobel G. A., Weiss M. J., Hardison R. C. (2009) Erythroid GATA1 function revealed by genome-wide analysis of transcription factor occupancy, histone modifications, and mRNA expression. Genome Res. 19, 2172–2184 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125. Ko M., Sohn D. H., Chung H., Seong R. H. (2008) Chromatin remodeling, development and disease. Mutat. Res. 647, 59–67 [DOI] [PubMed] [Google Scholar]
- 126. Eber S., Lux S. E. (2004) Hereditary spherocytosis. Defects in proteins that connect the membrane skeleton to the lipid bilayer. Semin. Hematol. 41, 118–141 [DOI] [PubMed] [Google Scholar]
- 127. Gallagher P. G., Tse W. T., Marchesi S. L., Zarkowsky H. S., Forget B. G. (1991) A defect in α-spectrin mRNA accumulation in hereditary pyropoikilocytosis. Trans. Assoc. Am. Physicians 104, 32–39 [PubMed] [Google Scholar]
- 128. Agre P., Casella J. F., Zinkham W. H., McMillan C., Bennett V. (1985) Partial deficiency of erythrocyte spectrin in hereditary spherocytosis. Nature 314, 380–383 [DOI] [PubMed] [Google Scholar]