

Background: Sarcolipin is a regulator of SERCA in skeletal and atrial muscle with inhibitory properties thought to be similar to phospholamban.
Results: Residues critical for SERCA inhibition reside in the luminal extension of sarcolipin.
Conclusion: The luminal extension of sarcolipin is a distinct and transferrable domain that encodes most of its inhibitory properties.
Significance: Sarcolipin and phospholamban use different inhibitory mechanisms to regulate SERCA.
Keywords: Calcium ATPase, Enzyme Kinetics, Membrane Proteins, Reconstitution of Membrane Transporters, Sarcoplasmic Reticulum (SR), Phospholamban, Sarcolipin
Abstract
The sarco(endo)plasmic reticulum calcium ATPase (SERCA) is regulated in a tissue-dependent manner via interaction with the short integral membrane proteins phospholamban (PLN) and sarcolipin (SLN). Although defects in SERCA activity are known to cause heart failure, the regulatory mechanisms imposed by PLN and SLN could have clinical implications for both heart and skeletal muscle diseases. PLN and SLN have significant sequence homology in their transmembrane regions, suggesting a similar mode of binding to SERCA. However, unlike PLN, SLN has a conserved C-terminal luminal tail composed of five amino acids (27RSYQY), which may contribute to a distinct SERCA regulatory mechanism. We have functionally characterized alanine mutants of the C-terminal tail of SLN using co-reconstituted proteoliposomes of SERCA and SLN. We found that Arg27 and Tyr31 are essential for SLN function. We also tested the effect of a truncated variant of SLN (Arg27stop) and extended chimeras of PLN with the five luminal residues of SLN added to its C terminus. The Arg27stop form of SLN resulted in loss of function, whereas the PLN chimeras resulted in superinhibition with characteristics of both PLN and SLN. Based on our results, we propose that the C-terminal tail of SLN is a distinct, essential domain in the regulation of SERCA and that the functional properties of the SLN tail can be transferred to PLN.
Introduction
Sarcolipin (SLN)3 was first described as a low molecular weight protein that co-purified with preparations of SERCA, and it was later named to reflect its origin as a proteolipid of the sarcoplasmic reticulum (SR) (1). SLN is the predominant regulator of SERCA and calcium homeostasis in fast twitch skeletal muscle where it may play an additional role in thermogenesis (2). However, SLN is also expressed with phospholamban (PLN) in the atria of the heart (3–6), which raises the possibility of an atrium-specific ternary complex that could lead to superinhibition of SERCA (7). SLN is a 31-residue type I integral membrane protein with a transmembrane domain and short cytoplasmic and luminal domains (1). PLN is a 52-residue type I integral membrane protein with a transmembrane domain and a longer cytoplasmic domain but no luminal domain (8). Given the homology between the transmembrane domains of these proteins, it was hypothesized that SLN binds to SERCA and alters the apparent calcium affinity of the enzyme in a manner similar to PLN (9). Inhibition would result from SLN and/or PLN binding to SERCA and stabilizing an E2 calcium-free state (10). Mixed evidence exists on whether these regulatory subunits dissociate (11) or remain bound to SERCA (12–14) during calcium transport.
Historically, SLN inhibition of SERCA has been less well characterized than PLN in part because of the assumption that SLN uses the same mechanism as PLN to inhibit SERCA. For PLN, SERCA inhibition is encoded by the transmembrane domain (15, 16), whereas reversal of inhibition via phosphorylation is enabled by the cytoplasmic domain (8). For SLN, the transmembrane domain contains residues involved in inhibitory function (9), the cytoplasmic domain contains a phosphorylation site (17), and the luminal domain is important for SR retention (18) and SERCA inhibition (19). However, there are important differences in the way that SLN inhibits SERCA. For example, some studies have found that SLN decreases the maximal activity (Vmax) of SERCA at saturating calcium concentrations, indicating that SLN inhibition is not reversed by calcium (19–21). In addition, two kinases have been reported to target SLN (17, 22), although it is not firmly established whether phosphorylation of SLN is an important physiological mechanism. Finally, it is well documented that the inhibitory PLN monomer can self-associate to form pentamers. In contrast, SLN is thought to exist primarily as a monomer, although evidence suggests that SLN can also oligomerize in detergent and membrane environments (23, 24).
The modeled site of interaction between SLN and the calcium-free state of SERCA was the same binding groove identified for PLN (M2, M4, M6, and M9). This was based on mutagenesis of both SERCA and SLN combined with functional measurements (9) and co-immunoprecipitation studies (7). Alanine-scanning mutagenesis of SLN revealed similarities and differences when compared with PLN. In general, mutagenesis of SLN had lesser effects on function and did not recapitulate the gain of function behavior associated with residues that destabilize the PLN pentamer. Nonetheless, key residues in both proteins were found to be important for physical association and function. Notably, mutation of Leu8 and Asn11 in SLN resulted in the expected loss of function seen for the comparable Leu31 and Asn34 of PLN, and mutation of the predicted phosphorylation site to glutamate (Thr5 to Glu) appeared to mimic phosphorylation and result in loss of function (17). Although the remaining sampled residues were neutral or loss of function, there are mixed observations on the functional importance of the unique luminal tail of SLN (Fig. 1) (9, 18, 19). The C-terminal sequences of PLN and SLN represent a marked difference between these two proteins where the hydrophobic Met50-Leu-Leu52 in PLN is replaced by the more polar Arg27-Ser-Tyr-Gln-Tyr31 in SLN. Importantly, this latter sequence is perfectly conserved among mammals (18).
FIGURE 1.

Sequence alignments for primary structures of SLN and PLN from representative species. The cytoplasmic, transmembrane, and luminal domains are indicated. Sequence variations in the luminal domains, as compared to human sequences, are indicated in bold. Consensus sequences for SLN and PLN were generated based on all known sequences (ClustalW).
Given the highly conserved nature of the SLN luminal tail and our incomplete understanding of its role in SERCA inhibition, we chose to investigate this domain by the co-reconstitution of SLN mutants with SERCA into proteoliposomes. Another motivating factor for this study was the observation that PLN and SLN can simultaneously bind to and regulate SERCA (7). Although superinhibition is thought to result from the tight fit of both PLN and SLN in the SERCA binding groove (M2, M4, M6, and M9), we hypothesized that the luminal domain of SLN may contribute to the strong inhibitory properties of the ternary complex. This prompted us to investigate chimeric PLN-SLN constructs. Herein, we provide new insights into the regulation of SERCA by the C-terminal domain of SLN. Alanine-scanning mutagenesis of this domain revealed at least partial loss of function associated with all residues (Arg27-Ser-Tyr-Gln-Tyr31), and the removal of the luminal tail in an Arg27stop construct also resulted in loss of function. Chimeric PLN variants possessing the luminal tail of SLN caused superinhibition of SERCA reminiscent of studies of the PLN-SLN-SERCA ternary complex (7). Finally, transferring the SLN luminal tail onto a generic transmembrane helix resulted in a chimera that completely mimicked SERCA inhibition by wild-type SLN. We conclude that the highly conserved C-terminal tail of SLN is a primary determinant for SERCA inhibition and that it is a distinct and transferrable functional domain.
EXPERIMENTAL PROCEDURES
Expression and Purification of Recombinant SLN
Recombinant SLN and PLN chimeras were expressed and purified as described previously (25) with the exception of an additional organic extraction step for SLN purification. Briefly, following protease digestion of the maltose-binding protein and SLN fusion protein, trichloroacetic acid was added to a final concentration of 6%. This mixture was incubated on ice for 20 min. The precipitate was collected by centrifugation at 4 °C and subsequently homogenized in a mixture of chloroform:isopropanol:water (4:4:1) and incubated at room temperature for 3 h. The organic phase, which was highly enriched in recombinant SLN, was removed, dried to a thin film under nitrogen gas, and resuspended in 7 m guanidine hydrochloride. Reverse-phase HPLC was performed as described (25), and the molecular mass was verified by MALDI-TOF mass spectrometry (Institute for Biomolecular Design, University of Alberta).
Synthetic Peptide Handling
Synthetic peptides (Arg27stop, Leu9, Leu9tail, and 27RSYQY) were purchased from Biomatik (Wilmington, DE; 95% purity grade, HPLC- and MS-verified). Unless otherwise specified, all synthetic peptides were acetylated at the N terminus and amidated at the C terminus. Except for 27RSYQY, which was solubilized in distilled H2O, all peptides were solubilized in 3:1 chloroform:trifluoroethanol at a concentration of ∼1 mg/ml. The peptide concentrations were verified by quantitative amino acid analysis.
Co-reconstitution of SERCA and Recombinant SLN
Routine procedures were used to purify SERCA1a from rabbit skeletal muscle SR vesicles and functionally reconstitute it into proteoliposomes with SLN. SERCA, SLN, egg yolk phosphatidylcholine, and egg yolk phosphatidic acid were solubilized with octaethylene glycol monododecyl ether (C12E8) to achieve final molar stoichiometries of 1 SERCA, 6 SLN, and 195 lipids. The co-reconstituted proteoliposomes containing SERCA and SLN were formed by the slow removal of detergent (with SM-2 Biobeads, Bio-Rad) followed by purification on a sucrose step gradient. The purified co-reconstituted proteoliposomes typically yield final molar stoichiometries of 1 SERCA, 4.5 SLN, and 120 lipids. This same procedure was used for the co-reconstitution of SERCA with PLN chimeras and synthetic transmembrane peptides. For the co-reconstitution of SERCA in the presence of 27RSYQY peptide, the peptide in aqueous solution was added to the reconstitution mixture at a molar ratio of 1 SERCA to 100 27RSYQY followed by detergent removal with SM-2 Biobeads to ensure incorporation of 27RSYQY inside the proteoliposomes.
Activity Assays
Calcium-dependent ATPase activities of the co-reconstituted proteoliposomes were measured by a coupled enzyme assay (26). The coupled enzyme assay reagents were of the highest purity available (Sigma-Aldrich). All co-reconstituted peptide constructs were compared with a negative control (SERCA reconstituted in the absence of SLN) and a matched positive control (SERCA co-reconstituted in the presence of either wild-type SLN, wild-type PLN, or Leu9 peptide). A minimum of three independent reconstitutions and activity assays were performed for each peptide, and the calcium-dependent ATPase activity was measured over a range of calcium concentrations (0.1–10 μm) for each assay. This method has been described in detail (27). The calcium concentration at half-maximal activity (KCa), Vmax, and Hill coefficient (nH) were calculated based on nonlinear least square fitting of the activity data to the Hill equation using Sigma Plot software (SPSS Inc., Chicago, IL). Errors were calculated as the S.E. for a minimum of three independent measurements. Comparison of KCa, Vmax, and nH parameters was carried out using between-subjects one-way analysis of variance followed by the Holm-Sidak test for pairwise comparisons (Sigma Plot).
Throughout “Results” and “Discussion,” we refer to SLN and PLN inhibition of SERCA. Inhibition is intended to reflect the effects that SLN and PLN have on the apparent calcium affinity of SERCA. The effects that SLN and PLN have on the maximal activity of SERCA are considered separately.
Kinetic Simulations
Reaction rate simulations have been described (28, 29) for the transport cycle of SERCA in the absence and presence of PLN inhibition, and we have adopted this approach to understand SERCA inhibition by wild-type SLN. As before, we performed a global nonlinear regression fit of the model of Cantilina et al. (28) to each plot of SERCA ATPase activity versus calcium concentration using Dynafit (Biokin Inc., Pullman, WA). Such fits were performed for co-reconstituted wild-type SLN, which was compared with positive (SERCA co-reconstituted with wild-type PLN) and negative (SERCA alone) control samples. All reaction rate constants were allowed to vary in the kinetic simulations, although the SERCA activity in the presence of SLN was best fit with modifications to only the three calcium binding steps in the reaction cycle (29). Agreement between the simulated and experimental data is indicated by the lowest residual sum of squares.
RESULTS
Wild-type PLN Versus Wild-type SLN
We first compared the reconstitution of SERCA in the absence and presence of recombinant human SLN (Fig. 2). The co-reconstitution method has been used extensively to study the functional regulation of SERCA by PLN (25, 27, 29–33), and the same approach was used herein for detailed characterization of SLN. The reconstituted proteoliposomes contain low lipid-to-protein ratios that mimic the native SR membranes, allowing the direct correlation of functional data (27, 29, 32, 33) with structural observations (34–36). As before, the proteoliposomes contained a lipid-to-protein molar ratio of ∼120:1 and a SERCA-to-SLN molar ratio of ∼4.5:1. For co-reconstitution of PLN, this molar ratio is similar to that found in cardiac SR (33, 37, 38); however, the SLN molar ratio used is higher than that found in skeletal SR (9). Although there is growing evidence that SLN can form higher order oligomers (23, 24, 39), the primary reason for using a higher ratio of SLN was to facilitate comparison with PLN variants studied previously (27, 29, 33) and herein. We measured the calcium-dependent ATPase activity of SERCA in the absence and presence of SLN. Proteoliposomes containing SERCA alone yielded a KCa of 0.46 μm and a Vmax of 4.1 μmol mg−1 min−1. Incorporation of wild-type SLN into proteoliposomes with SERCA resulted in a KCa of 0.80 μm calcium and a Vmax of 2.9 μmol mg−1 min−1 (Fig. 2 and Table 1). Thus, in the presence of SLN, SERCA had a lower apparent affinity for calcium (ΔKCa of 0.34) and a lower turnover rate (ΔVmax of −1.2). Comparative data for PLN indicated that it lowers the apparent calcium affinity of SERCA to a degree similar to that of SLN, and it has the opposite effect on Vmax. Nonetheless, the observed inhibitory activity of wild-type SLN in our system was consistent with previous observations (24, 30, 40) and served as a positive control for further studies.
FIGURE 2.

Functional data for wild-type SLN. A, topology models for wild-type SLN and wild-type PLN (white, cytosolic residues; gray, transmembrane residues; black, luminal residues). Shown are ATPase activity (B) and normalized ATPase activity (C) as a function of calcium concentration for SERCA alone (solid black line), SERCA in the presence of wild-type SLN (solid gray line), and SERCA in the presence of wild-type PLN (dashed line). The curves fitted to the experimental data are from a global nonlinear regression fit of SERCA reaction rate constants (28) to each plot of ATPase activity versus calcium concentration. The Vmax, KCa, and nH are given in Table 1. Each data point is the mean ± S.E. (error bars) (n ≥ 4). D, kinetic simulations highlighting the differences between PLN and SLN. Starting from values determined for wild-type PLN (Table 2 and Ref. 29), simulations were performed allowing all reaction rate constants to vary (solid black line), allowing only Brev to vary (Fit 1; solid gray line), or allowing only Bfor and Brev to vary (Fit 2; dashed line). The reaction rate constants are given in Table 2.
TABLE 1.
Kinetic parameters for SERCA in the absence and presence of various sarcolipin mutants, chimeras, and peptides
Between-subjects one-way analysis of variance followed by the Holm-Sidak test was used for pairwise comparisons against SERCA in the absence and presence of wild-type SLN. Note that the KCa value for SERCA in the presence of Arg27stop was not significantly different from the values determined for R27A, S28A, Y29A, Q30A, and Y31A.
| Vmax | KCa | nH | n | |
|---|---|---|---|---|
| μmol mg−1 min−1 | μm | |||
| SERCA | 4.1 ± 0.1 | 0.46 ± 0.02 | 1.7 ± 0.1 | 32 |
| WT SLN | 2.9 ± 0.1a | 0.80 ± 0.02a | 1.4 ± 0.1 | 10 |
| V26A | 2.9 ± 0.1a | 0.69 ± 0.03a | 1.4 ± 0.1 | 7 |
| R27A | 3.4 ± 0.1a,b | 0.51 ± 0.01b | 1.4 ± 0.1 | 6 |
| S28A | 3.5 ± 0.1a,b | 0.59 ± 0.03a,b | 1.5 ± 0.1 | 6 |
| Y29A | 3.2 ± 0.1a | 0.63 ± 0.03a,b | 1.3 ± 0.1 | 13 |
| Q30A | 2.6 ± 0.1a | 0.60 ± 0.04a,b | 1.4 ± 0.1 | 4 |
| Y31A | 4.6 ± 0.1a,b | 0.52 ± 0.02b | 1.3 ± 0.1 | 5 |
| Arg27stop | 3.4 ± 0.1a,b | 0.55 ± 0.02a,b | 1.6 ± 0.1 | 9 |
| WT PLN | 6.1 ± 0.1a,b | 0.88 ± 0.03a | 2.0 ± 0.1 | 9 |
| WT PLN + WT SLN | 3.4 ± 0.1a,b | 1.36 ± 0.07a,b | 1.7 ± 0.1 | 3 |
| cPLNlong | 2.9 ± 0.1a | 2.3 ± 0.09a,b | 1.6 ± 0.1 | 6 |
| cPLNshort | 2.2 ± 0.1a,b | 3.4 ± 0.20a,b | 1.9 ± 0.1 | 4 |
| 27RSYQY | 4.2 ± 0.1b | 0.41 ± 0.02b | 1.5 ± 0.1 | 5 |
| Leu9 | 4.4 ± 0.1b | 0.61 ± 0.05a,b | 1.6 ± 0.1 | 5 |
| Leu9tail | 3.0 ± 0.1a | 0.81 ± 0.05a | 1.5 ± 0.1 | 6 |
a p < 0.05 in the absence of wild-type SLN.
b p < 0.05 in the presence of wild-type SLN.
Kinetic Simulations
In our previous studies, we have used kinetic reaction rate simulations to describe calcium transport by SERCA in the absence and presence of PLN (29, 33). The reaction scheme assumes that the binding of calcium to SERCA occurs as two steps linked by a slow structural transition that establishes cooperativity (E + Ca ↔ ECa ↔ E′Ca + Ca ↔ E′2Ca) (28, 41). The conformational change, represented by reaction rate constants Bfor and Brev, is the primary step affected by PLN that manifests as a decrease in the apparent calcium affinity of SERCA and increased cooperativity for calcium binding. To provide a mechanistic framework for the function of SLN, we used the same approach to gain further insight into the subtle differences between SLN and PLN regulation of SERCA (Fig. 2 and Table 2). Our kinetic analyses revealed that wild-type SLN targets the first two reaction steps, binding of the first calcium ion (Afor and Arev) and the subsequent conformational transition (Bfor and Brev).
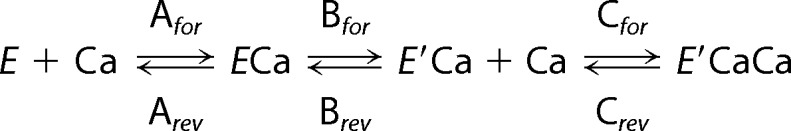 |
A dramatic increase in Arev was observed, indicating that SLN decreases the apparent calcium affinity of SERCA by driving the enzyme toward a calcium-free conformation. SLN also decreased the forward rate constant for the SERCA conformational change (Bfor), indicating that SLN lowers the maximal activity of SERCA by making this reaction step less favorable. To test whether our kinetic simulations for wild-type SLN were reliable, we ran additional simulations starting from the reaction rate constants determined for SERCA in the presence of wild-type PLN (28, 29). Holding all reaction rates constant and allowing only Bfor and Brev to vary, we attempted to force the simulation to fit the wild-type SLN experimental data with reaction rate constants that were similar to those found for wild-type PLN. These simulations resulted in poor fits to the experimental data (Fig. 2D).
TABLE 2.
Rate constants from kinetic simulations (s−1)
| Afor | Arev | Bfor | Brev | Cfor | Crev | Sum of squares | |
|---|---|---|---|---|---|---|---|
| SERCA | 190,000 | 400 | 30 | 40 | 1,810,000 | 16 | 0.002 |
| WT PLN | 190,000 | 400 | 45 | 25,500 | 250,000 | 16 | 0.004 |
| WT SLN | 156,530 | 64,736 | 22 | 135,140 | 1,810,000 | 16 | 0.001 |
| Alternate fits in Fig. 2Da | |||||||
| Fit 1 | 190,000 | 400 | 45 | 1,133,000 | 250,000 | 16 | 0.327 |
| Fit 2 | 190,000 | 400 | 22 | 60,562 | 250,000 | 16 | 0.03 |
a Only the values in bold were allowed to vary in the kinetic simulations.
It is interesting to compare the kinetic simulations for SLN and PLN. The primary effect of SLN is to make binding of the first calcium ion less favorable, thereby stabilizing a calcium-free conformation of SERCA. In contrast, the primary effect of PLN is to displace the SERCA ECa-E′Ca conformational equilibrium toward ECa, which has the additional consequence of enhancing cooperativity. In addition, our simulations indicate that the opposite effects that SLN and PLN have on the maximal activity of SERCA can be explained by an opposite effect on Bfor (SLN decreased Bfor and PLN increased Bfor). This latter observation is consistent with the notion that PLN (12, 14, 42, 43) and SLN (2) remain associated with SERCA at saturating calcium concentrations.
Alanine Substitutions in the Luminal Domain of SLN
The presence of the unique luminal domain in SLN as well as the high degree of conservation of its sequence could suggest that all of these amino acids might be required for regulation of SERCA. As a first step in examining the role of the luminal tail of SLN, we systematically mutated residues 26–31 of SLN to alanine, co-reconstituted each mutant with SERCA, and measured the calcium-dependent ATPase activity of the proteoliposomes (Fig. 3 and Table 1). For effective comparison of the calcium-dependent ATPase activity of SERCA in the presence of SLN mutants, we wished to confirm that each of the mutants was reconstituted into proteoliposomes with the same efficiency as wild-type SLN. To that end, we used quantitative gel electrophoresis to monitor the levels of SERCA and SLN in the proteoliposomes (27, 29). Incorporation of each of the SLN mutants did not significantly differ from wild type (Fig. 3A), indicating that any observed differences in SERCA function could be attributed to the SLN mutation.
FIGURE 3.

The effects of alanine mutation in the luminal domain of SLN on the KCa and Vmax of SERCA. A, SDS-PAGE of co-reconstituted proteoliposomes. The top gel shows the incorporation of SERCA (2 μg of co-reconstituted proteoliposomes were loaded onto a 10% gel and stained with Coomassie). The bottom gel shows the incorporation of wild-type and mutant SLN (2 μg of co-reconstituted proteoliposomes were loaded onto a 16% gel and stained with silver). KCa (B) and Vmax (C) values were determined from ATPase activity measurements for SERCA in the absence and presence of wild-type and mutant forms of SLN. Each data point is the mean ± S.E. (error bars) (n ≥ 4). The Vmax, KCa, and nH are given in Table 1. Asterisks indicate comparisons against wild-type SLN (p < 0.05).
Alanine substitution of any residue within the 27RSYQY motif had a negative impact on the ability of SLN to alter the apparent calcium affinity of SERCA, whereas substitution of the preceding residue, Val26, had a lesser effect. Although valine-to-alanine mutation is a conservative substitution, we could compare our results with mutation of the homologous residue in PLN (Val49; Fig. 1). The Val26-to-Ala mutant also served as an internal comparison for the luminal tail mutants described below. As expected, alanine substitution of Val26 had only a minor effect on the inhibitory properties of SLN, resulting in mild loss of function (KCa of 0.69 μm compared with 0.80 μm calcium for wild type; ΔKCa of 0.11). This compared well with findings for PLN where alanine substitution of the homologous position, Val49, has also been shown to have a mild effect on PLN function (29, 44). Thus, we concluded that Val26 is part of the transmembrane domain and not the tail region of SLN. For mutations within the luminal tail domain, the two most severe alanine substitutions were Arg27 to Ala and Tyr31 to Ala, which resulted in nearly complete loss of SERCA inhibition (KCa of 0.51 and 0.52 μm calcium; ΔKCa of 0.05 and 0.06, respectively). These mutants were determined to be the strongest loss of function mutations in the luminal domain of SLN, indicating the importance of a positively charged residue at the membrane surface and a more distal aromatic residue. Interestingly, these mutants had distinct effects on the maximal activity of SERCA. Given the ability of wild-type SLN to reduce the maximal activity of SERCA (Fig. 3 and Table 1), the Arg27-to-Ala mutant resulted in a slight loss of this behavior, and the Tyr31-to-Ala mutant resulted in a complete loss of this behavior. In fact, the Tyr31-to-Ala mutant had the opposite effect in that it caused a slight increase in the maximal activity of SERCA. Compare the Vmax values for SERCA alone (4.1 μmol mg−1 min−1), SERCA in the presence of wild-type SLN (2.9 μmol mg−1 min−1), SERCA in the presence of Arg27-to-Ala SLN (3.4 μmol mg−1 min−1), and SERCA in the presence of Tyr31 to Ala (4.6 μmol mg−1 min−1).
Alanine mutants of the remaining residues (Ser28, Tyr29, and Gln30) deviated from wild-type behavior with variable effects on the apparent calcium affinity and maximal activity of SERCA (Fig. 3). In terms of their effects on the apparent calcium affinity of SERCA, the mutants resulted in a comparable partial loss of function. Ranking these luminal tail residues in order of importance yielded Ser28 ≅ Tyr29 ≅ Gln30. In terms of the maximal activity of SERCA, the mutants had differential effects with the rank order of importance being Ser28 > Tyr29 > Gln30. Based on these observations, we concluded that alanine substitution at any position in the SLN luminal tail results in loss of function, indicating a crucial role of each of the five luminal residues in proper regulation of SERCA. Clearly, Tyr31 was the most essential residue for altering the apparent calcium affinity and depressing the maximal activity of SERCA. This suggests that, in addition to contributing to SERCA inhibition, the residues Ser28, Tyr29, and Gln30 may play a role in the proper positing of Tyr31.
It should be noted that the SLN luminal tail was examined in a previous study using HEK-293 cells and co-expression with SERCA (9). Alanine substitution of the luminal tail residues was found to have a minimal impact on SERCA activity, which contrasts with our findings described above. However, it was later shown that mutations in the SLN tail affect the retention of SLN in the endoplasmic reticulum of HEK-293 cells (18) such that improper trafficking of SLN may have been an unappreciated factor in the previous study. In addition, the previous study measured calcium transport activity of SERCA, whereas we have measured ATPase activity. Recent evidence suggests that SLN may play a role in thermogenesis by uncoupling SERCA ATPase activity from calcium transport (2).
Removal of the SLN Luminal Tail (Arg27stop)
Because all of the alanine substitutions in the SLN luminal domain created defects in SERCA regulation, we next chose to remove the luminal tail and test the inhibitory capacity of the transmembrane domain of SLN alone. For comparative purposes, recall that ∼80% of the inhibitory activity of PLN is encoded by its transmembrane domain (29). To this end, a truncated variant of SLN, Arg27stop, missing the last five C-terminal amino acids (27RSYQY) was generated by chemical synthesis. During synthesis, the C terminus was amidated to avoid a free carboxyl at the end of the transmembrane domain. Incorporation of Arg27stop into proteoliposomes with SERCA proceeded normally, and the final SERCA-to-peptide ratio was comparable with that for wild-type SLN (data not shown). The effect of this peptide on SERCA activity was measured in the same fashion as the alanine mutants described above. As one might expect given the results from alanine mutagenesis, the complete removal of the luminal tail resulted in major loss of SLN function (Fig. 4 and Table 1). The KCa of SERCA was 0.55 μm calcium in the presence of Arg27stop compared with 0.80 μm calcium in the presence of wild-type SLN (∼26% of wild-type inhibitory capacity). Note that the KCa value for Arg27stop is not significantly different from the alanine substitutions discussed above. Arg27stop also resulted in a partial recovery of the maximal activity with a Vmax value halfway between that of SERCA alone and SERCA in the presence of wild-type SLN. The large change in the inhibitory potency of Arg27stop agrees with the alanine substitution data and further demonstrates the necessity of the luminal domain of SLN in SERCA regulation. Interestingly, the luminal tail of SLN rather than the transmembrane domain appeared to encode most of the inhibitory properties of SLN. This contrasts with PLN where the inhibitory properties are encoded by the transmembrane domain. This was a surprising finding given that SLN and PLN have homologous transmembrane domains, which are thought to interact with the same site on SERCA and use a similar mechanism of inhibition.
FIGURE 4.

Removing the luminal tail of SLN. A, topology model for Arg27stop SLN (white, cytosolic residues; gray, transmembrane residues). KCa (B) and Vmax (C) values were determined from ATPase activity measurements for SERCA in the absence and presence of Arg27stop SLN. Each data point is the mean ± S.E. (error bars) (n ≥ 4). The Vmax, KCa, and nH are given in Table 1. Asterisks indicate comparisons against SERCA in the absence and presence of wild-type SLN (p < 0.05).
Adding the SLN Luminal Tail to PLN
If the luminal domain encodes the inhibitory properties of SLN, we wondered what would happen if the luminal tail were transferred to the homologous PLN. To test this idea, two chimeric peptides were constructed from wild-type PLN, one with the five luminal residues of SLN (27RSYQY) added to the C terminus (cPLNlong) and one with the luminal residues added after Val49 of PLN (cPLNshort). This latter construct placed the luminal tail of SLN at the homologous position in PLN (Fig. 1 and Fig. 5). The calcium-dependent ATPase activity was measured for SERCA in the presence of the chimeras where SERCA alone served as a negative control and SERCA in the presence of wild-type PLN served as a positive control (Fig. 5 and Table 1). Including wild-type PLN in proteoliposomes with SERCA resulted in the expected decrease in the apparent calcium affinity of SERCA (ΔKCa of 0.42 μm calcium) and increase in the maximal activity of SERCA (from 4.1 to 6.1 μmol mg−1 min−1). Compared with wild-type PLN, including the chimeras in proteoliposomes resulted in superinhibition of SERCA (ΔKCa values of 1.84 μm calcium for cPLNlong and 2.94 μm calcium for cPLNshort). Importantly, both chimeras also resulted in a decrease in the maximal activity of SERCA that was comparable with wild-type SLN. This was in marked contrast to the increase in SERCA maximal activity observed with wild-type PLN in the co-reconstituted proteoliposomes (27, 29, 33). Thus, adding the SLN luminal tail to PLN had a synergistic effect on the apparent calcium affinity of SERCA and an SLN-like effect on the maximal activity of SERCA. Although both PLN chimeras were superinhibitory, the cPLNshort construct resulted in a much larger shift in the apparent calcium affinity of SERCA. The potent inhibition by cPLNshort may be due to better positioning of the SLN luminal tail and the fact that this chimera appears to be monomeric by SDS-PAGE (Fig. 5D).
FIGURE 5.

Transferring the luminal tail of SLN to PLN. A, topology models of cPLNlong and cPLNshort chimeras (white, cytosolic residues; gray, transmembrane residues; black, luminal residues). Shown are ATPase activity (B) and normalized ATPase activity (C) as a function of calcium concentration for SERCA alone (solid black line), SERCA in the presence of wild-type PLN (solid gray line), and SERCA in the presence of cPLNlong (dashed black line) and cPLNshort (dotted black line). Each data point is the mean ± S.E. (error bars) (n ≥ 4). The Vmax, KCa, and nH are given in Table 1. D, SDS-PAGE of cPLNlong (left lane) and cPLNshort (right lane) chimeras (5 μg per lane; 16% gel). Pentameric and monomeric PLN chimeras are indicated.
The SLN Luminal Tail Is a Distinct Functional Domain
The experiments thus far seemed to indicate that the luminal domain of SLN encodes most of its inhibitory activity. To test the inhibitory properties of this domain in isolation, we synthesized a peptide corresponding to 27RSYQY with an acetylated N terminus. Previous work by others has demonstrated that such a peptide can decrease the maximal activity of SERCA albeit under excess peptide conditions (∼1000-fold (19)). Herein, the soluble peptide was co-reconstituted with SERCA such that the peptide was on the interior of the proteoliposomes with access to the luminal region of SERCA, and excess peptide on the exterior of the proteoliposomes was removed by sucrose gradient purification. Although SERCA was treated with up to a 100-fold molar excess of peptide prior to reconstitution, this had no effect on SERCA activity (Table 1). These data indicate that the 27RSYQY peptide by itself did not result in the robust SERCA regulation observed for wild-type SLN or the PLN-SLN chimeras. Similar findings have been reported for the cytoplasmic domain of PLN, which does not appear to regulate SERCA as an isolated, soluble domain (16, 45).
The transmembrane helix of SLN tethers the luminal domain to the membrane surface and provides a high local concentration relative to SERCA. As such, a soluble peptide may be a poor mimic of this domain. To bring the luminal domain to the membrane surface, we tethered the 27RSYQY motif to a model transmembrane helix. We have previously characterized a synthetic leucine-alanine peptide that contains all of the naturally occurring leucine residues in the transmembrane domain of PLN with all other residues substituted with alanine (designated Leu9). A variety of generic transmembrane peptides including Leu9 have been found to be weak inhibitors of SERCA (25, 31). To test the action of the luminal tail of SLN on SERCA activity, we synthesized a peptide that contained the Leu9 sequence with the five luminal residues of SLN at its C terminus (Fig. 6; designated Leu9tail). Because the Leu9 peptide has been characterized (31), it served as a positive control for our studies of the Leu9tail peptide. In the co-reconstituted proteoliposomes, Leu9 had a slight effect on the apparent calcium affinity of SERCA (ΔKCa of 0.15 μm calcium; ∼36% of wild-type PLN inhibitory activity) and a small effect on the maximal activity of SERCA. Inclusion of the SLN luminal tail significantly increased the inhibitory capacity of the Leu9 peptide (ΔKCa of 0.35 μm calcium compared with ΔKCa of 0.34 μm for wild-type SLN) and reduced the maximal activity of SERCA to the same degree as wild-type SLN. Compare the Vmax values for SERCA alone (4.1 μmol mg−1 min−1), SERCA in the presence of Leu9 (4.4 μmol mg−1 min−1), SERCA in the presence of wild-type SLN (2.9 μmol mg−1 min−1), and SERCA in the presence of Leu9tail (3.0 μmol mg−1 min−1). These observations support the notion that the luminal tail encodes much of the inhibitory properties of SLN and that it is a distinct and transferrable regulatory domain.
FIGURE 6.
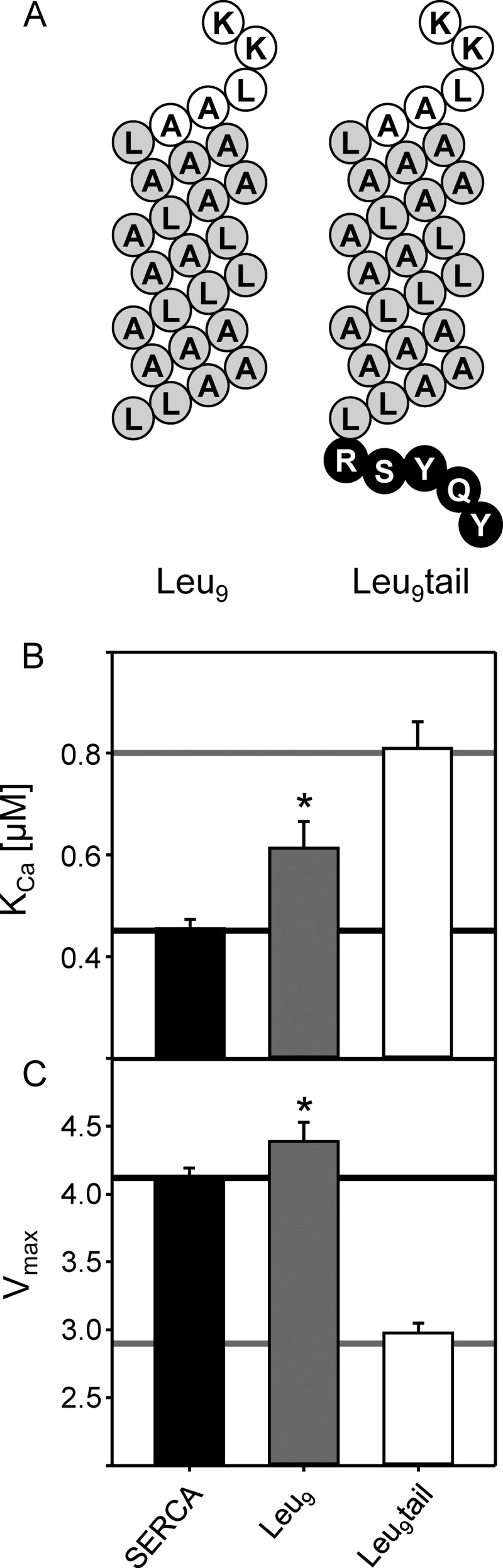
Transferring the luminal tail of SLN to a generic transmembrane helix. A, topology model of Leu9 and Leu9tail (white, cytosolic residues; gray, transmembrane residues; black, luminal residues). KCa (B) and Vmax (C) values were determined from ATPase activity measurements for SERCA in the absence and presence of Leu9 and Leu9tail. For comparative purposes, the black lines indicate the values for SERCA alone, and the gray lines indicate values for SERCA in the presence of wild-type SLN. Notice that Leu9tail closely recapitulates wild-type SLN. Each data point is the mean ± S.E. (error bars) (n ≥ 4). The Vmax, KCa, and nH are given in Table 1. Asterisks indicate comparisons against wild-type SLN (p < 0.05).
DISCUSSION
Wild-type PLN Versus Wild-type SLN
Based on the available evidence, it was reasonable to assume that SLN and PLN would use similar inhibitory mechanisms to regulate SERCA. As such, SLN and PLN could represent redundant regulatory subunits separated by distinct tissue distributions with SLN primarily in skeletal muscle and PLN in cardiac and smooth muscle. As an endogenous inhibitor of SERCA, SLN plays a central role in regulating calcium transport in skeletal muscle. However, SLN is co-expressed in atrial muscle along with PLN (3–6), which raises questions about why redundant regulatory mechanisms would be required in this tissue. As has been shown for PLN, SLN alters the apparent calcium affinity of SERCA albeit to a lesser degree (Fig. 2 and Ref. 9). PLN is highly conserved among mammalian species with a transmembrane domain that encodes SERCA inhibition and a cytoplasmic domain that is a target for regulation. SLN is also highly conserved with a transmembrane domain analogous to that in PLN, a short cytoplasmic domain that is a target for regulation, and a unique, highly conserved luminal domain. Given these similarities and differences between SLN and PLN, it is important to understand the regulatory mechanism of SLN with potential relevance for heart and skeletal muscle diseases.
Because this is our first detailed characterization of SLN, it is important to consider aspects of the experimental system, namely co-reconstituted proteoliposomes containing SERCA1a and recombinant SLN. Such proteoliposomes have been extensively used by us (27, 29, 32–36, 46) and others (16, 47–52) for the detailed characterization of PLN structure and function. They contain SERCA-to-lipid molar ratios that approximate SR membranes, and the inclusion of PLN in these proteoliposomes has the expected effect on the apparent calcium affinity of SERCA (28). At these low lipid-to-protein ratios, PLN has an additional effect on the maximal activity of SERCA (27, 29, 51). As shown herein, SLN is readily incorporated into the proteoliposomes, and we observed the expected effect on the apparent calcium affinity of SERCA (Fig. 3) (9). In contrast to PLN, SLN has an opposite effect on the maximal activity of SERCA.
Using the proteoliposomes described above, we found that wild-type SLN altered the apparent calcium affinity of SERCA at a level equivalent to ∼80% of the inhibitory capacity of wild-type PLN. Interestingly, the SERCA inhibition by wild-type SLN is comparable with what is observed for peptides encoding only the transmembrane domain of PLN (31, 53, 54). Nonetheless, in contrast to what is observed for PLN, SLN decreased the maximal activity of SERCA (Fig. 2 and Table 1). Given these differential effects on SERCA activity, we used kinetic simulations to identify SERCA reaction steps that may be altered by SLN. In this regard, PLN is known to alter a SERCA conformational change that follows binding of the first calcium ion, thereby establishing cooperativity for binding of a second calcium ion (28, 29). Surprisingly, we found that SLN does not fit this kinetic model. Instead, SLN stabilizes a calcium-free conformation of SERCA by altering the binding of the first calcium ion. Combined with the observed decrease in the maximal activity of SERCA, we concluded that SLN uses an inhibitory mechanism that is distinct from that used by PLN.
SLN Structural Elements Involved in SERCA Inhibition
Because SLN appeared to use a unique mechanism to regulate SERCA, it was important to identify the structural features that encode this behavior. There are nine invariant residues in SLN that mainly occur in the transmembrane domain (Fig. 1), and only four of these residues are invariant in the homologue PLN. The short cytoplasmic domain of SLN is variable across a wide range of species, whereas the luminal domain exhibits a high degree of conservation particularly among mammals. Alanine-scanning mutagenesis of the luminal domain revealed that Arg27 and Tyr31 are the two most essential residues for SERCA regulation; both residues are required for the effect on the apparent calcium affinity of SERCA, and Tyr31 is required for the effect on the maximal activity of SERCA (Fig. 3). The interaction of Tyr31 with SERCA has been recognized (19), although another study did not identify luminal residues as important for SLN inhibitory function (9). Nonetheless, we found that mutation of either Arg27 or Tyr31 strongly suppressed SERCA inhibition by SLN. One might predict that Arg27 could reside at the membrane interface and therefore aid in positioning of SLN relative to SERCA and the lipid bilayer. Alanine substitution would remove the positively charged side chain and extend the hydrophobic surface of the transmembrane domain of SLN, thereby causing misalignment of SLN within the binding groove of SERCA (M2, M4, M6, and M9).
It is interesting to notice that the nature of the critical residues, Arg27 and Tyr31, suggest that cation-π or π-π interactions might be involved in the SERCA-SLN inhibitory complex. By analogy with PLN, the C terminus of SLN is thought to interact with the luminal end of the M2 transmembrane segment of SERCA (55). Toward the luminal end of the M1-M2 region, there are five aromatic residues, Phe73, Trp77, and Phe78 on M1 and Phe88 and Phe92 on M2. Phe88 and Phe92 on M2 flank an interaction site identified for PLN where Val89 of SERCA was cross-linked to Val49 of PLN (55, 56). Val49 of PLN is equivalent to Val26 of SLN; thus, Arg27, Tyr29, and Tyr31 of SLN may be proximal to Phe88 and Phe92 of SERCA (Fig. 7). Arg27 may form a cation-π interaction with Phe92, whereas Tyr29 and/or Tyr31 may form a cation-π or π-stacking interactions with Phe88. Given that the M1-M2 region of SERCA undergoes large structural rearrangements as the enzyme transitions from the calcium-free E2 state to the calcium-bound E1 state, these molecular interactions could explain the important inhibitory role of the luminal residues of SLN.
FIGURE 7.

Model showing the proximity of the luminal domain of SLN (Arg27, Tyr29, and Tyr31 are magenta) to the luminal end of M1-M2 of SERCA (particularly Phe88 and Phe92, which are in blue). SERCA is in the E2 conformation (60). SLN is shown as a continuous α-helix, although we anticipate that the luminal tail may be partially unwound in the SERCA-SLN inhibitory complex. Based on models for the SERCA-PLN inhibitory complex (46, 61), SLN was aligned in the M2, M4, M6, and M9 binding groove of SERCA. The positions of Arg27, Tyr29, and Tyr31 are the approximate positions in the molecular model constructed by Asahi et al. (7).
The Luminal Extension of SLN Is a Distinct and Transferrable Regulatory Domain
There are several roles that have been suggested for the luminal domain of SLN, including SR retention and functional interaction with SERCA. For PLN, SR retention occurs via the diarginine motif in its cytoplasmic domain as well as through the direct interaction with SERCA (57). The luminal 27RSYQY sequence of SLN has been shown to be involved in SR retention, although the interaction with SERCA may also be a retention mechanism (18). There have been mixed reports on the inhibitory contributions of the SLN luminal domain (9, 19). Co-expression of SERCA and SLN mutants in HEK-293 cells revealed minor contributions of the C-terminal residues where loss of function occurred only when both Tyr29 and Tyr31 were mutated (9). Endoplasmic reticulum retention may have been affected in this study (18). Another study highlighted the importance of the two luminal aromatic residues in regulating SERCA. Using solid-state NMR combined with functional measurements, a soluble 27RSYQY peptide was shown to interact with SERCA and lower its maximal activity (19). There was no effect of this peptide on the apparent calcium affinity of SERCA. In contrast, our results indicate that the luminal domain of SLN alters both the maximal activity and apparent calcium affinity of SERCA. Given these disparate observations, we sought another experimental approach to elucidate the regulatory capacity of the SLN luminal domain.
To test the functional contributions of this domain, two PLN-SLN chimeras were constructed. The first construct possessed the 27RSYQY sequence added to the C terminus of wild-type PLN. In the context of the chimera, the full-length PLN sequence was expected to retain wild-type functional properties, and the functional effects of the SLN luminal domain were expected to be additive. The second construct possessed the 27RSYQY sequence added after Val49 of PLN, effectively replacing the C-terminal 50MLL sequence. In the context of this chimera, the shortened PLN sequence might alter its functional properties; however, the SLN luminal domain would be optimally positioned for interaction with SERCA. To our surprise, both chimeras turned out to be potent superinhibitors of SERCA where the luminal domain of SLN had a synergistic effect on PLN inhibitory function. In fact, the chimeras were reminiscent of the superinhibition observed for the ternary SERCA-PLN-SLN complex (7) thought to exist in the atria. These observations are consistent with the mutagenesis data described above and support the notion that the luminal domain of SLN possesses inhibitory activity. Although the chimeras were reminiscent of the ternary complex, they were much more potent inhibitors of SERCA. Comparing the values in Table 1, the ΔKCa values were 0.9 μm calcium for the ternary complex, 1.84 μm calcium for the long chimera (cPLNlong), and 2.94 μm calcium for the short chimera (cPLNshort). In the ternary complex, PLN and SLN are thought to interact with SERCA as well as each other (7), and this may create steric restrictions that impact the proper positioning of critical regulatory domains, the transmembrane domain of PLN and the luminal domain of SLN. However, in the context of the chimeras, both the transmembrane domain of PLN and the luminal domain of SLN may be properly positioned in the SERCA binding groove (M2, M4 M6, and M9 (7)). Because these two domains use distinct mechanisms to regulate SERCA, the combined effect could result in the observed superinhibition. It is also noteworthy that by SDS-PAGE the short chimera is largely monomeric, which could contribute to the superinhibition seen for this construct.
Although the PLN-SLN chimeras were consistent with a functional role for the luminal domain of SLN, this was one of several potential explanations for the observed superinhibition. There are a variety of ways to convert PLN into a superinhibitor of SERCA that might not reflect a functional contribution from the luminal 27RSYQY sequence. PLN superinhibition can result from mutation (27, 58), depolymerization of the PLN pentamer (44, 59), and reverse engineering of PLN peptides (25, 31). If the luminal region of SLN is an independent functional and structural domain, it should be able to regulate SERCA in the absence of either the SLN or PLN transmembrane domain. However, a soluble 27RSYQY peptide by itself did not alter SERCA activity over a range of excess concentrations. This may not be surprising because the luminal 27RSYQY sequence is normally tethered to the SR membrane. To further test the luminal domain as an independent functional entity, we tethered the 27RSYQY sequence to a model transmembrane peptide designated Leu9 (31). Using a reverse engineering approach, Leu9 was derived from the PLN transmembrane sequence where the nine leucine residues were retained and all other residues were mutated to alanine. This peptide is a weak inhibitor of SERCA with ∼36% of wild-type PLN inhibitory activity. Adding the luminal tail of SLN to Leu9 generated a construct that was indistinguishable from wild-type SLN (Table 1 and Fig. 6). Compare the KCa and Vmax values for SERCA in the presence of Leu9tail (0.81 μm calcium and 3.0 μmol mg−1 min−1) and SERCA in the presence of wild-type SLN (0.80 μm calcium and 2.9 μmol mg−1 min−1). These data clearly demonstrate that the luminal region of SLN is a distinct structural and functional domain.
In summary, critical determinants for SLN inhibitory function are found in its unique and conserved luminal tail. The structural elements that may contribute to the formation of a SERCA-SLN inhibitory complex include Arg27, Tyr29, and Tyr31 of SLN and a series of aromatic residues at the base of M1-M2 of SERCA (particularly Phe88 and Phe92). These molecular interactions account for much of the inhibitory function of SLN, and this contrasts sharply with what is known for PLN. The transmembrane domain of PLN encodes ∼80% of its inhibitory activity, whereas ∼75% of the inhibitory activity of SLN is encoded by the luminal tail. These distinct structural elements that are used by PLN and SLN to regulate SERCA translate into different inhibitory mechanisms (28, 29). PLN slows a SERCA conformational transition that follows binding of the first calcium ion and establishes cooperativity for binding of a second calcium ion. SLN alters binding of the first calcium ion, thereby stabilizing a calcium-free conformation of SERCA. Given that these functional properties can be transferred to PLN or a generic transmembrane helix, we conclude that the luminal tail of SLN is a distinct, essential, and transferrable regulatory domain.
This work was supported in part by Canadian Institutes of Health Research Grant MOP53306.
- SLN
- sarcolipin
- SERCA
- sarco(endo)plasmic reticulum calcium ATPase
- PLN
- phospholamban
- SR
- sarcoplasmic reticulum
- KCa
- calcium concentration at half-maximal activity
- nH
- Hill coefficient
- Vmax
- maximal activity.
REFERENCES
- 1. Wawrzynow A., Theibert J. L., Murphy C., Jona I., Martonosi A., Collins J. H. (1992) Sarcolipin, the “proteolipid” of skeletal muscle sarcoplasmic reticulum, is a unique, amphipathic, 31-residue peptide. Arch. Biochem. Biophys. 298, 620–623 [DOI] [PubMed] [Google Scholar]
- 2. Bal N. C., Maurya S. K., Sopariwala D. H., Sahoo S. K., Gupta S. C., Shaikh S. A., Pant M., Rowland L. A., Bombardier E., Goonasekera S. A., Tupling A. R., Molkentin J. D., Periasamy M. (2012) Sarcolipin is a newly identified regulator of muscle-based thermogenesis in mammals. Nat. Med. 18, 1575–1579 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Minamisawa S., Wang Y., Chen J., Ishikawa Y., Chien K. R., Matsuoka R. (2003) Atrial chamber-specific expression of sarcolipin is regulated during development and hypertrophic remodeling. J. Biol. Chem. 278, 9570–9575 [DOI] [PubMed] [Google Scholar]
- 4. Babu G. J., Bhupathy P., Carnes C. A., Billman G. E., Periasamy M. (2007) Differential expression of sarcolipin protein during muscle development and cardiac pathophysiology. J. Mol. Cell. Cardiol. 43, 215–222 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Odermatt A., Taschner P. E., Scherer S. W., Beatty B., Khanna V. K., Cornblath D. R., Chaudhry V., Yee W. C., Schrank B., Karpati G., Breuning M. H., Knoers N., MacLennan D. H. (1997) Characterization of the gene encoding human sarcolipin (SLN), a proteolipid associated with SERCA1: absence of structural mutations in five patients with Brody disease. Genomics 45, 541–553 [DOI] [PubMed] [Google Scholar]
- 6. Vangheluwe P., Schuermans M., Zádor E., Waelkens E., Raeymaekers L., Wuytack F. (2005) Sarcolipin and phospholamban mRNA and protein expression in cardiac and skeletal muscle of different species. Biochem. J. 389, 151–159 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Asahi M., Sugita Y., Kurzydlowski K., De Leon S., Tada M., Toyoshima C., MacLennan D. H. (2003) Sarcolipin regulates sarco(endo)plasmic reticulum Ca2+-ATPase (SERCA) by binding to transmembrane helices alone or in association with phospholamban. Proc. Natl. Acad. Sci. U.S.A. 100, 5040–5045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Simmerman H. K., Collins J. H., Theibert J. L., Wegener A. D., Jones L. R. (1986) Sequence analysis of phospholamban. Identification of phosphorylation sites and two major structural domains. J. Biol. Chem. 261, 13333–13341 [PubMed] [Google Scholar]
- 9. Odermatt A., Becker S., Khanna V. K., Kurzydlowski K., Leisner E., Pette D., MacLennan D. H. (1998) Sarcolipin regulates the activity of SERCA1, the fast-twitch skeletal muscle sarcoplasmic reticulum Ca2+-ATPase. J. Biol. Chem. 273, 12360–12369 [DOI] [PubMed] [Google Scholar]
- 10. Waggoner J. R., Huffman J., Froehlich J. P., Mahaney J. E. (2007) Phospholamban inhibits Ca-ATPase conformational changes involving the E2 intermediate. Biochemistry 46, 1999–2009 [DOI] [PubMed] [Google Scholar]
- 11. Chen Z., Akin B. L., Jones L. R. (2010) Ca2+ binding to site I of the cardiac Ca2+ pump is sufficient to dissociate phospholamban. J. Biol. Chem. 285, 3253–3260 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Negash S., Yao Q., Sun H., Li J., Bigelow D. J., Squier T. C. (2000) Phospholamban remains associated with the Ca2+- and Mg2+-dependent ATPase following phosphorylation by cAMP-dependent protein kinase. Biochem. J. 351, 195–205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Traaseth N. J., Thomas D. D., Veglia G. (2006) Effects of Ser16 phosphorylation on the allosteric transitions of phospholamban/Ca2+-ATPase complex. J. Mol. Biol. 358, 1041–1050 [DOI] [PubMed] [Google Scholar]
- 14. Karim C. B., Zhang Z., Howard E. C., Torgersen K. D., Thomas D. D. (2006) Phosphorylation-dependent conformational switch in spin-labeled phospholamban bound to SERCA. J. Mol. Biol. 358, 1032–1040 [DOI] [PubMed] [Google Scholar]
- 15. Sasaki T., Inui M., Kimura Y., Kuzuya T., Tada M. (1992) Molecular mechanism of regulation of Ca2+ pump ATPase by phospholamban in cardiac sarcoplasmic reticulum. Effects of synthetic phospholamban peptides on Ca2+ pump ATPase. J. Biol. Chem. 267, 1674–1679 [PubMed] [Google Scholar]
- 16. Reddy L. G., Jones L. R., Cala S. E., O'Brian J. J., Tatulian S. A., Stokes D. L. (1995) Functional reconstitution of recombinant phospholamban with rabbit skeletal Ca2+-ATPase. J. Biol. Chem. 270, 9390–9397 [DOI] [PubMed] [Google Scholar]
- 17. Bhupathy P., Babu G. J., Ito M., Periasamy M. (2009) Threonine-5 at the N-terminus can modulate sarcolipin function in cardiac myocytes. J. Mol. Cell. Cardiol. 47, 723–729 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Gramolini A. O., Kislinger T., Asahi M., Li W., Emili A., MacLennan D. H. (2004) Sarcolipin retention in the endoplasmic reticulum depends on its C-terminal RSYQY sequence and its interaction with sarco(endo)plasmic Ca2+-ATPases. Proc. Natl. Acad. Sci. U.S.A 101, 16807–16812 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Hughes E., Clayton J. C., Kitmitto A., Esmann M., Middleton D. A. (2007) Solid-state NMR and functional measurements indicate that the conserved tyrosine residues of sarcolipin are involved directly in the inhibition of SERCA1. J. Biol. Chem. 282, 26603–26613 [DOI] [PubMed] [Google Scholar]
- 20. Tupling A. R., Asahi M., MacLennan D. H. (2002) Sarcolipin overexpression in rat slow twitch muscle inhibits sarcoplasmic reticulum Ca2+ uptake and impairs contractile function. J. Biol. Chem. 277, 44740–44746 [DOI] [PubMed] [Google Scholar]
- 21. Babu G. J., Bhupathy P., Petrashevskaya N. N., Wang H., Raman S., Wheeler D., Jagatheesan G., Wieczorek D., Schwartz A., Janssen P. M., Ziolo M. T., Periasamy M. (2006) Targeted overexpression of sarcolipin in the mouse heart decreases sarcoplasmic reticulum calcium transport and cardiac contractility. J. Biol. Chem. 281, 3972–3979 [DOI] [PubMed] [Google Scholar]
- 22. Gramolini A. O., Trivieri M. G., Oudit G. Y., Kislinger T., Li W., Patel M. M., Emili A., Kranias E. G., Backx P. H., Maclennan D. H. (2006) Cardiac-specific overexpression of sarcolipin in phospholamban null mice impairs myocyte function that is restored by phosphorylation. Proc. Natl. Acad. Sci. U.S.A 103, 2446–2451 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Autry J. M., Rubin J. E., Pietrini S. D., Winters D. L., Robia S. L., Thomas D. D. (2011) Oligomeric Interactions of Sarcolipin and the Ca-ATPase. J. Biol. Chem. 286, 31697–31706 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Hellstern S., Pegoraro S., Karim C. B., Lustig A., Thomas D. D., Moroder L., Engel J. (2001) Sarcolipin, the shorter homologue of phospholamban, forms oligomeric structures in detergent micelles and in liposomes. J. Biol. Chem. 276, 30845–30852 [DOI] [PubMed] [Google Scholar]
- 25. Afara M. R., Trieber C. A., Ceholski D. K., Young H. S. (2008) Peptide inhibitors use two related mechanisms to alter the apparent calcium affinity of the sarcoplasmic reticulum calcium pump. Biochemistry 47, 9522–9530 [DOI] [PubMed] [Google Scholar]
- 26. Warren G. B., Toon P. A., Birdsall N. J., Lee A. G., Metcalfe J. C. (1974) Reconstitution of a calcium pump using defined membrane components. Proc. Natl. Acad. Sci. U.S.A 71, 622–626 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Trieber C. A., Douglas J. L., Afara M., Young H. S. (2005) The effects of mutation on the regulatory properties of phospholamban in co-reconstituted membranes. Biochemistry 44, 3289–3297 [DOI] [PubMed] [Google Scholar]
- 28. Cantilina T., Sagara Y., Inesi G., Jones L. R. (1993) Comparative studies of cardiac and skeletal sarcoplasmic reticulum ATPases: effect of phospholamban antibody on enzyme activation. J. Biol. Chem. 268, 17018–17025 [PubMed] [Google Scholar]
- 29. Trieber C. A., Afara M., Young H. S. (2009) Effects of phospholamban transmembrane mutants on the calcium affinity, maximal activity, and cooperativity of the sarcoplasmic reticulum calcium pump. Biochemistry 48, 9287–9296 [DOI] [PubMed] [Google Scholar]
- 30. Douglas J. L., Trieber C. A., Afara M., Young H. S. (2005) Rapid, high-yield expression and purification of Ca2+-ATPase regulatory proteins for high-resolution structural studies. Protein Expr. Purif. 40, 118–125 [DOI] [PubMed] [Google Scholar]
- 31. Afara M. R., Trieber C. A., Glaves J. P., Young H. S. (2006) Rational design of peptide inhibitors of the sarcoplasmic reticulum calcium pump. Biochemistry 45, 8617–8627 [DOI] [PubMed] [Google Scholar]
- 32. Ceholski D. K., Trieber C. A., Holmes C. F., Young H. S. (2012) Lethal, hereditary mutants of phospholamban elude phosphorylation by protein kinase A. J. Biol. Chem. 287, 26596–26605 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Ceholski D. K., Trieber C. A., Young H. S. (2012) Hydrophobic imbalance in the cytoplasmic domain of phospholamban is a determinant for lethal dilated cardiomyopathy. J. Biol. Chem. 287, 16521–16529 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Young H. S., Jones L. R., Stokes D. L. (2001) Locating phospholamban in co-crystals with Ca2+-ATPase by cryoelectron microscopy. Biophys. J. 81, 884–894 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Stokes D. L., Pomfret A. J., Rice W. J., Glaves J. P., Young H. S. (2006) Interactions between Ca2+-ATPase and the pentameric form of phospholamban in two-dimensional co-crystals. Biophys. J. 90, 4213–4223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Glaves J. P., Trieber C. A., Ceholski D. K., Stokes D. L., Young H. S. (2011) Phosphorylation and mutation of phospholamban alter physical interactions with the sarcoplasmic reticulum calcium pump. J. Mol. Biol. 405, 707–723 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Ferrington D. A., Yao Q., Squier T. C., Bigelow D. J. (2002) Comparable levels of Ca-ATPase inhibition by phospholamban in slow-twitch skeletal and cardiac sarcoplasmic reticulum. Biochemistry 41, 13289–13296 [DOI] [PubMed] [Google Scholar]
- 38. Negash S., Chen L. T., Bigelow D. J., Squier T. C. (1996) Phosphorylation of phospholamban by cAMP-dependent protein Kinase enhances interactions between Ca-ATPase polypeptide chains in cardiac sarcoplasmic reticulum membranes. Biochemistry 35, 11247–11259 [DOI] [PubMed] [Google Scholar]
- 39. Becucci L., Guidelli R., Karim C. B., Thomas D. D., Veglia G. (2009) The role of sarcolipin and ATP in the transport of phosphate ion into the sarcoplasmic reticulum. Biophys. J. 97, 2693–2699 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Buck B., Zamoon J., Kirby T. L., DeSilva T. M., Karim C., Thomas D., Veglia G. (2003) Overexpression, purification, and characterization of recombinant Ca-ATPase regulators for high-resolution solution and solid-state NMR studies. Protein Expr. Purif. 30, 253–261 [DOI] [PubMed] [Google Scholar]
- 41. Inesi G., Kurzmack M., Lewis D. (1988) Kinetic and equilibrium characterization of an energy-transducing enzyme and its partial reactions. Methods Enzymol. 157, 154–190 [DOI] [PubMed] [Google Scholar]
- 42. Fowler C., Huggins J. P., Hall C., Restall C. J., Chapman D. (1989) The effects of calcium, temperature, and phospholamban phosphorylation on the dynamics of the calcium-stimulated ATPase of canine cardiac sarcoplasmic reticulum. Biochim. Biophys. Acta 980, 348–356 [DOI] [PubMed] [Google Scholar]
- 43. Bidwell P., Blackwell D. J., Hou Z., Zima A. V., Robia S. L. (2011) Phospholamban binds with differential affinity to calcium pump conformers. J. Biol. Chem. 286, 35044–35050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Kimura Y., Kurzydlowski K., Tada M., MacLennan D. H. (1997) Phospholamban inhibitory function is enhanced by depolymerization. J. Biol. Chem. 272, 15061–15064 [DOI] [PubMed] [Google Scholar]
- 45. Lockwood N. A., Tu R. S., Zhang Z., Tirrell M. V., Thomas D. D., Karim C. B. (2003) Structure and function of integral membrane protein domains resolved by peptide-amphiphiles: application to phospholamban. Biopolymers 69, 283–292 [DOI] [PubMed] [Google Scholar]
- 46. Seidel K., Andronesi O. C., Krebs J., Griesinger C., Young H. S., Becker S., Baldus M. (2008) Structural characterization of Ca2+-ATPase-bound phospholamban in lipid bilayers by solid-state nuclear magnetic resonance (NMR) spectroscopy. Biochemistry 47, 4369–4376 [DOI] [PubMed] [Google Scholar]
- 47. Reddy L. G., Jones L. R., Pace R. C., Stokes D. L. (1996) Purified, reconstituted cardiac Ca2+-ATPase is regulated by phospholamban but not by direct phosphorylation with Ca2+/calmodulin-dependent protein kinase. J. Biol. Chem. 271, 14964–14970 [DOI] [PubMed] [Google Scholar]
- 48. Thomas D. D., Reddy L. G., Karim C. B., Li M., Cornea R., Autry J. M., Jones L. R., Stamm J. (1998) Direct spectroscopic detection of molecular dynamics and interactions of the calcium pump and phospholamban. Ann. N.Y. Acad. Sci. 853, 186–194 [DOI] [PubMed] [Google Scholar]
- 49. Reddy L. G., Jones L. R., Thomas D. D. (1999) Depolymerization of phospholamban in the presence of calcium pump: a fluorescence energy transfer study. Biochemistry 38, 3954–3962 [DOI] [PubMed] [Google Scholar]
- 50. Karim C. B., Paterlini M. G., Reddy L. G., Hunter G. W., Barany G., Thomas D. D. (2001) Role of cysteine residues in structural stability and function of a transmembrane helix bundle. J. Biol. Chem. 276, 38814–38819 [DOI] [PubMed] [Google Scholar]
- 51. Reddy L. G., Cornea R. L., Winters D. L., McKenna E., Thomas D. D. (2003) Defining the molecular components of calcium transport regulation in a reconstituted membrane system. Biochemistry 42, 4585–4592 [DOI] [PubMed] [Google Scholar]
- 52. Reddy L. G., Autry J. M., Jones L. R., Thomas D. D. (1999) Co-reconstitution of phospholamban mutants with the Ca-ATPase reveals dependence of inhibitory function on phospholamban structure. J. Biol. Chem. 274, 7649–7655 [DOI] [PubMed] [Google Scholar]
- 53. Karim C. B., Marquardt C. G., Stamm J. D., Barany G., Thomas D. D. (2000) Synthetic null-cysteine phospholamban analogue and the corresponding transmembrane domain inhibit the Ca-ATPase. Biochemistry 39, 10892–10897 [DOI] [PubMed] [Google Scholar]
- 54. Kimura Y., Kurzydlowski K., Tada M., MacLennan D. H. (1996) Phospholamban regulates the Ca2+-ATPase through intramembrane interactions. J. Biol. Chem. 271, 21726–21731 [DOI] [PubMed] [Google Scholar]
- 55. Morita T., Hussain D., Asahi M., Tsuda T., Kurzydlowski K., Toyoshima C., Maclennan D. H. (2008) Interaction sites among phospholamban, sarcolipin, and the sarco(endo)plasmic reticulum Ca2+-ATPase. Biochem. Biophys. Res. Commun. 369, 188–194 [DOI] [PubMed] [Google Scholar]
- 56. Chen Z., Akin B. L., Stokes D. L., Jones L. R. (2006) Cross-linking of C-terminal residues of phospholamban to the Ca2+ pump of cardiac sarcoplasmic reticulum to probe spatial and functional interactions within the transmembrane domain. J. Biol. Chem. 281, 14163–14172 [DOI] [PubMed] [Google Scholar]
- 57. Sharma P., Ignatchenko V., Grace K., Ursprung C., Kislinger T., Gramolini A. O. (2010) Endoplasmic reticulum protein targeting of phospholamban: a common role for an N-terminal di-arginine motif in ER retention? PLoS One 5, e11496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Kimura Y., Asahi M., Kurzydlowski K., Tada M., MacLennan D. H. (1998) Phospholamban domain Ib mutations influence functional interactions with the Ca2+-ATPase isoform of cardiac sarcoplasmic reticulum. J. Biol. Chem. 273, 14238–14241 [DOI] [PubMed] [Google Scholar]
- 59. Cornea R. L., Jones L. R., Autry J. M., Thomas D. D. (1997) Mutation and phosphorylation change the oligomeric structure of phospholamban in lipid bilayers. Biochemistry 36, 2960–2967 [DOI] [PubMed] [Google Scholar]
- 60. Toyoshima C., Nomura H. (2002) Structural changes in the calcium pump accompanying the dissociation of calcium. Nature 418, 605–611 [DOI] [PubMed] [Google Scholar]
- 61. Toyoshima C., Asahi M., Sugita Y., Khanna R., Tsuda T., MacLennan D. (2003) Modeling of the inhibitory interaction of phospholamban with the Ca2+ ATPase. Proc. Natl. Acad. Sci. U.S.A. 100, 467–472 [DOI] [PMC free article] [PubMed] [Google Scholar]