

Background: Hormone-sensitive lipase (HSL) is a multifunctional enzyme that is critically involved in regulating energy homeostasis.
Results: HSL catalyzes the hydrolysis of cholesteryl esters and plays an indispensable role in the regulation of Bt2cAMP-induced steroidogenic acute regulatory protein (StAR) expression.
Conclusion: The mode of action of HSL in cAMP/PKA-mediated regulation of steroidogenesis involves multiple signaling, including the LXR-regulatory pathway.
Significance: The present findings can be useful for better understanding a number of physiopathological processes.
Keywords: Cell Signaling, Cyclic AMP (cAMP), Molecular Cell Biology, Signal Transduction, Steroidogenesis, Testis, StAR Protein, Steroidogenic Tissues
Abstract
Hormone-sensitive lipase (HSL) catalyzes the hydrolysis of cholesteryl esters in steroidogenic tissues and, thus, facilitates cholesterol availability for steroidogenesis. The steroidogenic acute regulatory protein (StAR) controls the rate-limiting step in steroid biosynthesis. However, the modes of action of HSL in the regulation of StAR expression remain obscure. We demonstrate in MA-10 mouse Leydig cells that activation of the protein kinase A (PKA) pathway, by a cAMP analog Bt2cAMP, enhanced expression of HSL and its phosphorylation (P) at Ser-660 and Ser-563, but not at Ser-565, concomitant with increased HSL activity. Phosphorylation and activation of HSL coincided with increases in StAR, P-StAR (Ser-194), and progesterone levels. Inhibition of HSL activity by CAY10499 effectively suppressed Bt2cAMP-induced StAR expression and progesterone synthesis. Targeted silencing of endogenous HSL, with siRNAs, resulted in increased cholesteryl ester levels and decreased cholesterol content in MA-10 cells. Depletion of HSL affected lipoprotein-derived cellular cholesterol influx, diminished the supply of cholesterol to the mitochondria, and resulted in the repression of StAR and P-StAR levels. Cells overexpressing HSL increased the efficacy of liver X receptor (LXR) ligands on StAR expression and steroid synthesis, suggesting HSL-mediated steroidogenesis entails enhanced oxysterol production. Conversely, cells deficient in LXRs exhibited decreased HSL responsiveness. Furthermore, an increase in HSL was correlated with the LXR target genes, steroid receptor element-binding protein 1c and ATP binding cassette transporter A1, demonstrating HSL-dependent regulation of steroidogenesis predominantly involves LXR signaling. LXRs interact/cooperate with RXRs and result in the activation of StAR gene transcription. These findings provide novel insight and demonstrate the molecular events by which HSL acts to drive cAMP/PKA-mediated regulation of StAR expression and steroidogenesis in mouse Leydig cells.
Introduction
Steroid hormones are synthesized in steroidogenic tissues, and are required for life itself, in the case of adrenal steroids, and for maintaining reproductive function, in the case of gonadal steroids. The substrate for all steroid hormones is cholesterol, which can be derived from a number of sources, i.e. de novo synthesis of cellular cholesterol, lipoprotein-derived cholesteryl esters, and hydrolysis of cholesteryl esters stored in lipid droplets. Of the three cholesterol sources, lipoprotein-derived selective uptake of cholesteryl esters, via the scavenger receptor class B type 1 (SR-B1),2 provides the majority of cholesterol for steroidogenesis in mice (1, 2). Regardless of the source of cholesterol, the conversion of cholesteryl esters into free cholesterol serves as an important step in controlling cholesterol availability for steroidogenesis. The 30-kDa steroidogenic acute regulatory protein (StAR) mediates the rate-limiting and regulated step in steroid biosynthesis, i.e. the transport of cholesterol from the outer to the inner mitochondrial membrane (3–5). The expression of StAR protein is predominantly regulated by the cAMP/protein kinase A (PKA) signaling cascade in the adrenals and gonads, although several intracellular events have been demonstrated to be instrumental in this process (reviewed in Refs. 4, 6, and 7). An overwhelming amount of evidence indicates that the synthesis of StAR protein is tightly correlated with the synthesis of steroids in steroidogenic tissues. In the mouse StAR protein, two putative PKA phosphorylation sites (Ser-56 and Ser-194) have been identified, and mutations (Ser → Ala) in these sites demonstrated the importance of Ser-194 in the biological activity of StAR in steroid synthesis (8, 9). As such, whereas StAR plays an indispensable role in the regulation of cAMP/PKA-mediated steroid biosynthesis, a complete understanding of the regulation of its expression and function is not available.
Steroidogenic cells, as well as other tissues, possess a neutral cholesteryl ester hydrolase (NCEH) activity, which has been demonstrated to be the result of the activity of hormone-sensitive lipase (HSL) (10–12). HSL is a multifunctional lipase that plays an essential role in regulating intracellular cholesterol metabolism, and this process may contribute to a number of signaling processes in which cells utilize cholesterol, including steroidogenesis. The functional relevance of HSL in steroidogenic cells, especially in gonadal Leydig cells, in contrast to adipose tissue, is poorly understood, as the adipocyte form of HSL (HSLadi, ∼84 kDa in rat) was initially thought not to be expressed in Leydig cells (13). Instead, molecular analysis had identified a longer form of HSL in the testis (HSLtes, ∼130 kDa in rat), which was derived from the same gene but was structurally and functionally distinct from HSLadi (13, 14). Notably, later studies demonstrated the presence of the short form of HSL, similar to HSLadi, in different testicular compartments, including Leydig cells (15, 16).
Targeted disruption of HSL in mice results in the lack of NCEH activity in adrenals and testes accompanied with profound morphological alterations in these tissues, underscoring the relevance of HSL in a number of physiological functions (10, 12, 17, 18). Consequently, male mice homozygous for the mutant HSL allele (HSL−/−) were sterile as a result of oligospermia and not hypogonadism, indicating that the inactivation of HSL mainly affected spermatogenesis (10). Conversely, female mice were fertile, suggesting oogenesis was unaffected by HSL deficiency. Nonetheless, adrenal steroid biosynthesis was decreased in both male and female HSL-null mice (12, 17). The loss of NCEH activity in the testes of HSL−/− mice and its correlation to Leydig cell steroidogenesis remains unclear. However, it has been shown that the interaction of HSL with a number of proteins, including StAR and perilipin A (a lipid-droplet associated protein), is connected with its increased hydrolytic activity (19, 20).
Accumulating evidence indicates that oxysterols, endogenous oxidative metabolites of cholesterol, are involved in StAR expression and steroidogenesis (21–23). Oxysterols act as ligands for the liver X receptors, LXRα (NR1H3) and LXRβ (NR1H2), which are members of the nuclear receptor superfamily of transcription factors (23, 24). These receptors form obligate heterodimers with retinoid X receptors (RXRs), and have been shown to regulate the transcription of a number of genes involved in cholesterol utilization and metabolism, including SREBP-1c, ABCA1, apoE, and StAR (23–25). Both gonadal and adrenal tissues express LXRs as well as several LXR agonists, including 22R-hydroxycholesterol (22R-OH) and 25-OH (23, 26). Even so, the involvement of LXR signaling in HSL-mediated StAR expression and steroidogenesis has yet to be elucidated. Given the importance of the StAR protein in steroid biosynthesis and to elucidate a link between HSL and testicular function, studies regarding the role of HSL in Leydig cell steroidogenesis seem warranted. Utilizing MA-10 mouse Leydig tumor cells (a cell line that closely resembles its normal counterpart and has been widely used in studying physiological functions) as an in vitro model, the present studies provide evidence that HSL is primarily involved in catalyzing cholesteryl ester hydrolysis and, by so doing, plays an important role in the regulation of cAMP-mediated StAR expression and steroidogenesis through modulation of LXR pathways.
EXPERIMENTAL PROCEDURES
Reagents
CAY10499 was purchased from Cayman Chemical Co. (Ann Arbor, MI). 27-Hydroxycholesterol was obtained from Research Plus Inc. (Barnegat, NJ). T0901317, LG100268, H-89, protease inhibitor mixture, and dibutyryl cyclic AMP (Bt2cAMP) were purchased from Sigma.
Cells, Plasmids, Transfections, and Luciferase Assays
Mouse MA-10 Leydig tumor, KK-1 granulosa, and Y-1 adrenocortical (ATCC, Manassas, VA) cells were cultured in HEPES-buffered Waymouth's medium supplemented with 15% horse serum, DMEM/F-12 with high glucose plus 10% fetal bovine serum (FBS), and F-12K medium with 15% horse serum and 2.5% FBS, respectively, containing antibiotics, as described previously (9, 27, 28).
The 5′-flanking −254/−1 bp region of the mouse StAR promoter was synthesized using a PCR-based cloning strategy and inserted into the XhoI and HindIII cloning sites of the pGL3 basic vector (Promega, Madison, WI) that contains firefly luciferase as a reporter gene (27, 29). The pRL-SV40 plasmid containing the Renilla luciferase gene driven by the SV40 promoter was obtained from Promega. Mouse Srebp-1c promoter (−2.7 kb to +1 bp) driven luciferase reporter (30) and HSL expression plasmid (31) have been previously described. All clones were verified by restriction mapping and confirmed by automated sequencing on a PE Biosystems 310 Genetic Analyzer (ABI PRISMTM 310, PerkinElmer Life Sciences) at the Texas Tech University Biotechnology Core Facility.
MA-10 cells were cultured in either 35- or 60-mm plates to 60–70% confluence and cells were transfected using Lipofectamine 2000 reagent (Invitrogen), under optimized conditions (28, 32). The amount of DNA used in different transfections was equalized with pcDNA3.1 empty expression vector (Invitrogen).
For promoter analysis, transfection efficiency was normalized by co-transfecting 10–20 ng of pRL-SV40 vector. Luciferase activity in the cell lysates was determined by a Dual Luciferase reporter assay system (Promega), as described previously (27, 29). Following treatments, cells were washed with 0.01 m PBS, and 300 μl of the reporter lysis buffer was added to the cells. Cellular debris was pelleted by centrifugation at 12,000 × g for 10 min at 4 °C, and the supernatant was measured for relative light units (luciferase/Renilla) using a TD 20/20 Luminometer (Turner Designs, Sunnyvale, CA).
Silencing
Knockdown of endogenous HSL and LXRα/β was performed using small interfering RNAs (siRNAs) with either X-tremeGENE siRNA transfection reagent (Roche Diagnostics Corp.) or Lipofectamine 2000 reagent (Invitrogen), under optimized conditions (32, 33). Silencer negative control and mouse HSL (#1, 5′-GCUCAUCUCCUAUGACCUA-3′, and #2, 5′-GCAAGAGUAUGUCACGCUA-3′), mouse LXRα (5′-GGAGUGUCGACUUCGCAAA-3′), and LXRβ (5′-AGACAGAAUGCAUCACGUU-3′) siRNAs were obtained as annealed oligos (Ambion Inc., Austin, TX). MA-10 cells were transfected with different siRNAs at 50–100 nm, as specified in different experiments.
Immunoblotting
Immunoblotting studies were carried out using total cellular protein (32, 33). Briefly, cells were homogenized in lysis buffer, and equal amounts of protein (20–30 μg) were solubilized in sample buffer and loaded onto 6–12% SDS-PAGE (Mini Protean II System, Bio-Rad). The proteins were electrophoretically transferred onto ImmunoBlot PVDF membranes (Bio-Rad). Membranes were probed with primary antibodies that recognize, P-HSL (Ser-660, Ser-565, and Ser-563) and HSL (Cell Signaling, Beverly, MA), StAR (34), P-StAR (9), CYP11A1 (Chemicon, Temecula, CA), LXR α-β (Abcam Inc., Cambridge, MA), ABCA1 (Millipore, Temecula, CA), and β-actin (Applied Biosystems/Ambion). Following overnight incubation at 4 °C with primary antibodies, membranes were washed and incubated with appropriate horseradish peroxidase-conjugated secondary antibodies for 1 h at room temperature. Membranes were washed again, and immunodetection of different proteins was determined with the Chemiluminescence Imaging Western Lightning Kit (PerkinElmer Life Sciences). The membranes were exposed to x-ray films (Phenix Research Products, Candler, NC) and the intensity of immunospecific bands was quantified using a computer-assisted image analyzer (Visage 2000, BioImage, Ann Arbor, MI).
Quantitative Real-time PCR and RT-PCR
Total RNA was extracted from different groups using TRIzol reagent (Invitrogen). Semi-quantitative real-time PCR was performed using ABI 7000 Applied Biosystems (Foster City, CA), as described previously (32, 35). PCR primers used were the following: HSL (forward), 5′-CAGAAGGCACTAGGCGTGATG-3, HSL (reverse), 5′-GCTTGCGTCCACTTAGTTCCA-3′; StAR (forward), 5′-CCGGGTGGATGGGTCAA-3, StAR (reverse), 5′-CACCTCTCCCTGCTGGATGTA-3′; CYP11A1 (forward), 5′-CTCAGGGGTCATCAGTGATGACC-3′, CYP11A1 (reverse), 5′-GCTGCATGGTCCTTCCAGGTC-3′; 3β-HSD (forward), 5′-GCCACGAGGACGAGCATCGTG-3′, 3β-HSD (reverse), 5′-CATCTGAGATGTAGTGGAACTCTCC-3′; SR-B1 (forward), 5′-CATGGCTCAGAGAGTGACTACA-3′, SR-B1 (reverse), 5′-GCACGAAGGGATCGTCATAGCC-3′; perilipin (forward), 5′-GCCTCTCAGGATGAGAGCCATGAC-3′, perilipin (reverse), 5′-GTGCCCATTTCACTGCGGAGA-3′; and GAPDH (forward), 5′-GCAGTGGCAAAGTGGAGATTG-3′, GAPDH (reverse), 5′-GTGAGTGGAGTCATACTGGAACATG-3′. Quantitative PCR data were normalized against GAPDH and evaluated using the comparative Ct method (32, 35).
Mouse StAR and L19 cDNAs were amplified using a one-step RT-PCR procedure (32, 36). RT and PCR were run sequentially in the same assay, which included [α-32P]dCTP (PerkinElmer Life Sciences, Inc.) in the dNTP mixture. The primer pairs utilized were: StAR (forward), 5′-GACCTTGAAAGGCTCAGGAAGAAC-3′, StAR (reverse) 5′-TAGCTGAAGATGGACAGACTTGC-3′; L19 (forward), 5′-GAAATCGCCAATGCCAACTC-3′, and L19 (reverse) 5′-TCTTAGACCTGCGAGCCTCA-3′. The molecular sizes of StAR and L19 were determined on 1.2% agarose gels, which were vacuum dried and exposed to x-ray film (Phenix Research Products) for 1 to 5 h. Levels of StAR and L19 signals were quantified using Visage 2000. All PCR products were verified by sequencing.
PKA Activity
Determination of the PKA activity was assessed using the SimaTECT PKA assay system (Promega), as described previously (32, 35). Following treatments, cells were washed with 0.01 m cold PBS and collected in extraction buffer (25 mm Tris-HCl, pH 7.4, 0.5 mm EDTA, 0.5 mm EGTA, 10 mm β-mercaptoethanol, and 0.1% Triton X-100 containing protease inhibitor mixture). Cells were then sonicated for 2 cycles of 10-s pulses using a Tekmar Sonic Disruptor (Fisher Scientific, Pittsburgh, CA) and centrifuged at 14,000 × g for 5 min at 4 °C. The resulting supernatant (3–5 μl from each group) was added to 20 μl of kinase reaction mixture and incubated for 5 min at 30 °C. The reaction was stopped by adding 12.5 μl of termination buffer and processed according to the manufacturer's instructions. The recovered membranes, after washing, were analyzed by liquid scintillation counting (LS 6500, Beckman, Minnesota, MN).
HSL Activity
HSL activity was determined according to the procedures described previously (37), under optimized conditions. Cells obtained from different groups were washed with 0.01 m PBS, collected, resuspended in Tris buffer (25 mm Tris-HCl, pH 7.2, 1 mm EDTA, 20% glycerol containing protease inhibitor mixture), and sonicated for 5–6 cycles of 5-s pulses (Sonic Disruptor, Fisher Scientific). HSL activity was determined using [14C]cholesteryl oleate (PerkinElmer) as a substrate. Briefly, an aliquot of the cell extract (100 μg of protein) was mixed with assay buffer containing 50 mm sodium phosphate (pH 7.0), 0.5 mm EDTA, 0.5 mm DTT, 2.5% fatty acid-free BSA, and [14C]cholesteryl oleate (∼50,000 cpm), and the reaction was continued for 30 min at 37 °C. The reaction was stopped by adding 50 μl of 2 m NaOH to obtain a final pH of 11. After incubation, the released [14C]oleic acid was extracted with methanol:choloform:heptane (2:2:1, v/v) containing 100 μm unlabeled oleic acid as a carrier. Following vigorous mixing, the emulsion was centrifuged at 1,500 × g for 10 min, and 0.5 ml of the upper phase containing [14C]oleic acid was counted in liquid a scintillation counter (LS 6500, Beckman). One unit of enzyme activity is defined as 1 μmol of oleic acid released per min at 37 °C.
Assessment of Cholesteryl Esters and Cholesterol
Levels of cholesteryl esters and cholesterol were determined using the Amplex Red Cholesterol Assay Kit following the manufacturer's instructions (Invitrogen). The assay is a fluorescence-based enzyme-coupled reaction that can determine both cholesterol esters and free cholesterol. Briefly, after experimental treatments, cells were washed with 0.01 m PBS, collected, homogenized, and extracted with chloroform:isopropyl alcohol:Nonidet P-40 (7:11:0.5). The extract was then centrifuged at 13,000 × g for 10 min at 4 °C, the organic phase was transferred to another tube, the liquid was dried for 20–25 min at 50 °C, and the dried lipids were dissolved in 200–250 μl of cholesterol assay buffer. Extracted samples or cholesterol standard (10–20 μl) were mixed with Amplex Red working solution without (for measuring free cholesterol), or with (for measuring total cholesterol) cholesterol esterase in a final volume of 100 μl in a light protected condition. After incubation at 37 °C for 30 min, fluorescence was measured utilizing excitation at 560 nm and emission at 590 nm. Esterified cholesterol was calculated by subtracting the free cholesterol from the total cholesterol. Quantitation of all samples was determined in the linear range.
Statistical Analysis
All experiments were repeated at least three times. Statistical analysis was performed by analysis of variance using Statview (Abacus Concepts Inc., Berkeley, CA) followed by Fisher's protected least significant differences test. Data presented are the mean ± S.E., and p < 0.05 was considered statistically significant.
RESULTS
Assessment of the Role of HSL in Bt2cAMP-mediated StAR Expression and Steroidogenesis
The mechanism of action of HSL in the regulation of Bt2cAMP-mediated StAR expression and steroidogenesis was explored. MA-10 cells treated with Bt2cAMP (0.5 mm) for 6 h resulted in 5.3 ± 0.5- and 2.4 ± 0.3-fold increases in phosphorylation of HSL (P-HSL) at Ser-660 and Ser-563 over untreated cells, respectively (Fig. 1A). Bt2cAMP had no significant effect on endogenously activated P-HSL at Ser-565. This cAMP analog modestly, but consistently, elevated (1.9 ± 0.3-fold) total HSL protein, an increase that was not observed when a different Ab that detected HSL as a ∼84 kDa band was used (38). P-HSL and HSL appeared as a doublet, with 83-kDa major and 81-kDa minor species, respectively. Real-time PCR analysis revealed a 2.2 ± 0.3-fold increase in HSL mRNA over basal (Fig. 1B). Under similar experimental conditions, Bt2cAMP strongly elevated StAR protein (8.9 ± 0.9-fold), P-StAR (Ser-194), and StAR mRNA (17 ± 3.2-fold) levels, over their respective controls (Fig. 1, C and D). Inhibition of PKA activity by H-89 (20 μm; 9, 33) affected Bt2cAMP-induced P-HSL, HSL, and HSL mRNA expression, and coordinately repressed StAR, P-StAR, and StAR mRNA levels. P-StAR was not detected in control and H-89-treated cells. Expression of the CYP11A1 protein was unchanged in any of these treatments (Fig. 1C). Because Bt2cAMP markedly activated P-HSL at Ser-660, its relevance to steroidogenesis was studied in subsequent experiments.
FIGURE 1.

Effect of Bt2cAMP on P-HSL, HSL, StAR, P-StAR, CYP11A1, HSL mRNA, and StAR mRNA levels in MA-10 cells. Cells were pretreated without or with H-89 (20 μm) for 45 min, and then treated in the absence (Basal) or presence of Bt2cAMP (0.5 mm) for an additional 6 h, as indicated (A–D). Following treatments, cells were processed for either immunoblotting (A and C) or real-time PCR (B and D) analyses, as described under “Experimental Procedures.” Representative immunoblots illustrate P-HSL (Ser-660, Ser-565, Ser-563), HSL, StAR, and P-StAR (Ser-194) in different groups using 20–30 μg of total cellular protein. Immunoblots shown are representative of five independent experiments. Levels of StAR (B) and HSL (D) mRNAs were determined by real-time PCR and presented as fold-changes relative to respective controls. Results represent the mean ± S.E. of five independent experiments. β-Actin expression was assessed as loading controls. Letters above the bars indicate that these groups differ significantly at least at p < 0.05.
Cells treated with Bt2cAMP (0.1–1.0 mm) displayed increased P-HSL and HSL levels in a dose-dependent manner (Fig. 2A). Activation of HSL was evident (p < 0.05) with 0.25 mm Bt2cAMP, increased gradually thereafter, and reached a plateau between 0.5 and 1.0 mm (attaining a maximum of 5–6-fold over basal). Total HSL protein was optimally elevated ∼2.0-fold over untreated cells. The induction of StAR protein was significant (p < 0.05) in response to 0.1 mm Bt2cAMP, reaching a maximum of ∼8.6-fold with 0.75–1.0 mm, when compared with controls. Increasing doses of Bt2cAMP on P-StAR followed a similar pattern as those of StAR protein levels. Additionally, cells treated with Bt2cAMP (0.5 mm) for 0–12 h demonstrated increases in P-HSL and StAR in a temporal response manner (Fig. 2B). Induction of P-HSL was significant (p < 0.05) at 1 h, increased maximally between 4 and 6 h, began to decrease thereafter, but remained elevated over basal at 12 h. Expression of the StAR protein was elevated at 1 h, reached a maximum of ∼9-fold over basal between 4 and 8 h, and marginally decreased at 12 h. Dose- and time-dependent patterns in P-HSL in response to Bt2cAMP were parallel with StAR protein levels, demonstrating a correlation between HSL activation and StAR expression in MA-10 mouse Leydig cells.
FIGURE 2.

Dose- and time-dependent effects of Bt2cAMP on P-HSL, HSL, StAR, and P-StAR in MA-10 cells. Cells were treated in the absence or presence of Bt2cAMP with either increasing doses (0.1–1.0 mm) for 6 h or a fixed dose (0.5 mm) for 12 h. Following treatments, cells were subjected to immunoblotting analyses (A and B). Representative immunoblots show P-HSL (Ser-660), HSL, StAR, and P-StAR using 20–30 μg of total cellular protein (A and B). Immunoblots shown are representative of four independent experiments. β-Actin expression was assessed as loading controls. Integrated optical density values of P-HSL, HSL, StAR, and P-StAR in each band were quantified and normalized with the corresponding β-actin expression, and compiled data are presented (n = 4, ± S.E.).
In additional studies, Bt2cAMP (0.01–1.0 mm) elevated HSL activity in a dose-dependent manner, reaching a maximum of 3.4 ± 0.5-fold over untreated cells (Fig. 3A). Bt2cAMP-stimulated progesterone levels (367 ± 22-fold over basal with 1.0 mm) followed a similar pattern as those of HSL activity. Pretreatment with CAY10499 (a potent inhibitor of HSL; 10 μm) affected Bt2cAMP-induced HSL activity by 83 ± 11%, an event associated with a 66 ± 8% reduction in progesterone synthesis. This inhibitor also repressed (p < 0.05) HSL activity (1.46 ± 0.3 and 0.78 ± 0.09 pmol/min/μg of protein without and with CAY10499, respectively) and progesterone synthesis (1.14 ± 0.2 and 0.62 ± 0.05 ng/mg of protein without and with CAY10499, respectively) under basal conditions. However, CAY10499 had no significant effect on 22R-OH (20 μm)-stimulated progesterone levels (296 ± 19- and 308 ± 21-fold over basal in 22R-OH and 22R-OH plus CAY10499, respectively). This inhibitor had no effects on either basal or Bt2cAMP-induced PKA activity (Fig. 3B), demonstrating the suppressive effects of CAY10499 on Bt2cAMP-stimulated StAR and progesterone levels were due to inhibition of HSL activity and not PKA activity. Conversely, H-89 attenuated both basal and Bt2cAMP-induced PKA activity. Increasing doses of CAY10499 (1–20 μm) inhibited Bt2cAMP-stimulated P-HSL and HSL activity in a concentration-dependent manner (Fig. 3, C and D). Concurrently, CAY10499 affected Bt2cAMP-stimulated StAR, P-StAR, and progesterone levels. Whereas 1 μm CAY10499 (the lowest dose tested) attenuated these responses, its inhibitory effect was optimal at 10–20 μm. No noticeable changes were observed on CYP11A1 protein levels (Fig. 3C). These results indicate that CAY10499 inhibited the hydrolytic activity of HSL, diminished cholesterol availability, and resulted in the repression of Bt2cAMP-induced StAR expression and steroid biosynthesis.
FIGURE 3.

Effects of CAY10499 and H-89 on Bt2cAMP-treated HSL activity, P-HSL, StAR, P-StAR, and progesterone synthesis, and PKA activity. MA-10 cells were pretreated without or with CAY10499 (10 μm, A and B; or 1–20 μm, C and D) for 45 min, and then treated in the absence or presence of Bt2cAMP with either increasing doses (0.01–1.0 mm; A) or a fixed dose (0.5 mm; B-D) for an additional 4 (B) or 6 h (A, C, and D). Cells were then processed for determining HSL activity (A and D), PKA activity (B), and immunoblotting (C), as described under “Experimental Procedures.” [14C]Cholesteryl oleate was used as a substrate for determining HSL activity (A and D). [14C]Oleic acid released was measured and expressed in terms of HSL activity (pmol/min/μg of protein), which represent the mean ± S.E. five independent experiments (A and D). Representative immunoblots illustrate P-HSL (Ser-660), HSL, StAR, P-StAR, and CYP11A1 in different groups using 20–30 μg of total cellular protein (C). Immunoblots shown are representative of five independent experiments. Accumulation of progesterone in media was determined (±S.E., n = 5) and expressed as ng/mg of protein (A and D; right panels).
Role of HSL in Bt2cAMP Responsive StAR and Steroid Levels in Granulosa and Adrenal Cells
Involvement of HSL in Bt2cAMP-induced StAR protein and steroid levels was evaluated in mouse KK-1 granulosa and Y-1 adrenocortical cells (Fig. 4). Treatment with Bt2cAMP (0.5 mm) for 6 h demonstrated 3.7 ± 0.4- and 2.8 ± 0.3-fold increases in P-HSL over untreated KK-1 (Fig. 4A) and Y-1 (Fig. 4B) cells, respectively. HSL activity was increased 2.6 ± 0.4- and 2.1 ± 0.3-fold over the respective basal values. Bt2cAMP elevated StAR (4.2 ± 0.6- and 3.3 ± 0.5-fold) and progesterone (12.7 ± 5- and 5.4 ± 3-fold) levels in both KK-1 and Y-1 cells, when compared with their respective controls. CYP11A1 protein levels were unaltered. Induction of Bt2cAMP-mediated P-HSL and HSL activity was inhibited by CAY10499 in these cells (Fig. 4, A and B). These inhibitory effects were correlated with StAR, P-StAR (not illustrated), and progesterone levels, demonstrating that HSL plays important roles in controlling Bt2cAMP-mediated StAR expression and steroidogenesis both in gonadal and adrenal cells.
FIGURE 4.
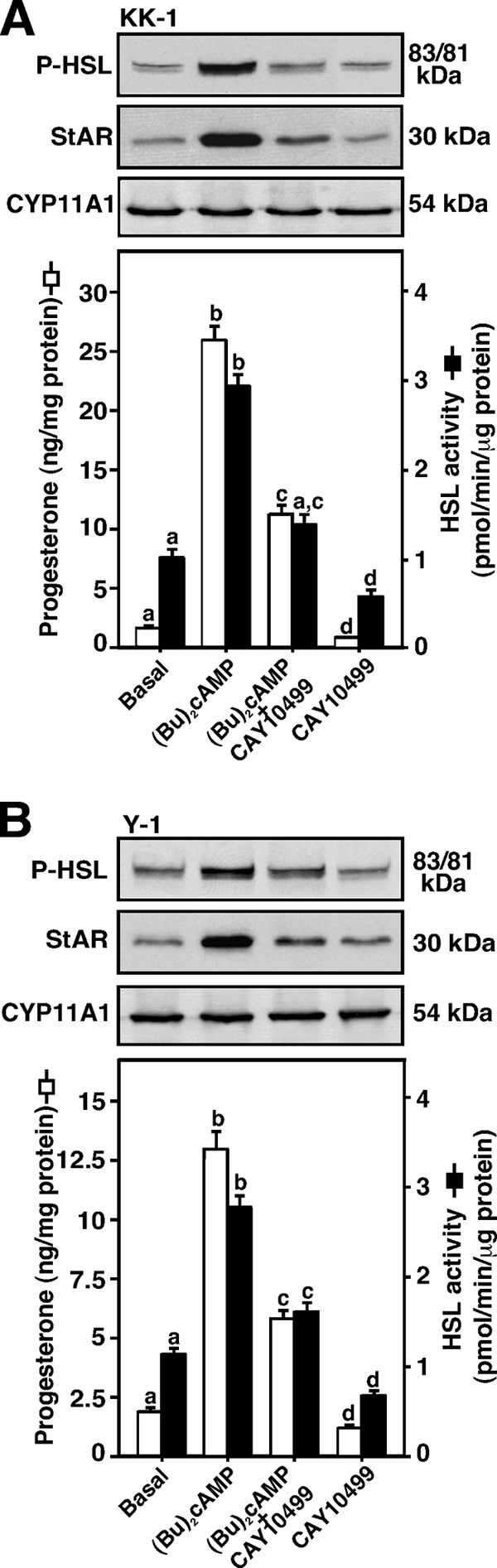
Effect of Bt2cAMP on P-HSL and HSL activity, and their correlation to StAR, CYP11A1, and progesterone levels. Mouse KK-1 granulosa (A) and Y-1 adrenocortical (B) cells were pretreated without or with CAY10499 (10 μm) for 45 min, and then treated in the absence (Basal) or presence of Bt2cAMP (0.5 mm) for an additional 6 h. Cells were then processed for either immunoblotting or determining HSL activity, as described under “Experimental Procedures” and in the legend of Fig. 3. Representative immunoblots illustrate P-HSL (Ser-660), StAR, and CYP11A1 in different groups using 20–30 μg of total cellular protein. Immunoblots shown are representative of four independent experiments. HSL activity in different groups was determined and expressed as picomole/min/μg of protein (A and B, right panels). Accumulation of progesterone (A and B, lower panels) in media was determined and expressed as nanogram/mg of protein. Data represent the mean ± S.E. of four independent experiments. Letters above the bars indicate that these groups differ significantly at least at p < 0.05.
Silencing of HSL and Its Correlation to Steroidogenesis
The relevance of HSL on the steroidogenic response was further gauged with siRNAs specific to the mouse HSL gene to diminish endogenous HSL expression. As shown in Fig. 5A, MA-10 cells transfected with two different HSL siRNAs (HSL#1 and HSL#2) were capable of decreasing endogenous HSL protein levels ∼80%, when compared with a negative control siRNA (scrambled). Knockdown of HSL also markedly affected HSL activity. Bt2cAMP had little to no effect on HSL protein and HSL activity in HSL-depleted cells. The increase in Bt2cAMP-mediated StAR protein (∼8.3-fold over basal), in scrambled siRNA-transfected cells, was attenuated 56 ± 8% with HSL silencing. HSL siRNA#1 was more effective in decreasing Bt2cAMP-induced StAR protein. Knockdown of HSL also affected (p < 0.05) StAR expression under basal conditions. Induction of Bt2cAMP-mediated P-StAR levels was attenuated ∼50% in HSL-deficient cells. Expression of the CYP11A1 protein was unaffected with HSL siRNAs ruling out nonspecific silencing (Fig. 5A). Because HSL catalyzes the hydrolysis of cholesteryl esters, the levels of cholesteryl esters and cholesterol were determined next. It can be seen from Fig. 5B that cells transfected with two HSL siRNAs together (HSL#1 + 2) resulted in a 3.2 ± 0.3-fold increase in cholesteryl esters, but decreased the free cholesterol content by 48–60%. These results demonstrate that HSL deficiency affects cholesterol availability for steroidogenesis.
FIGURE 5.
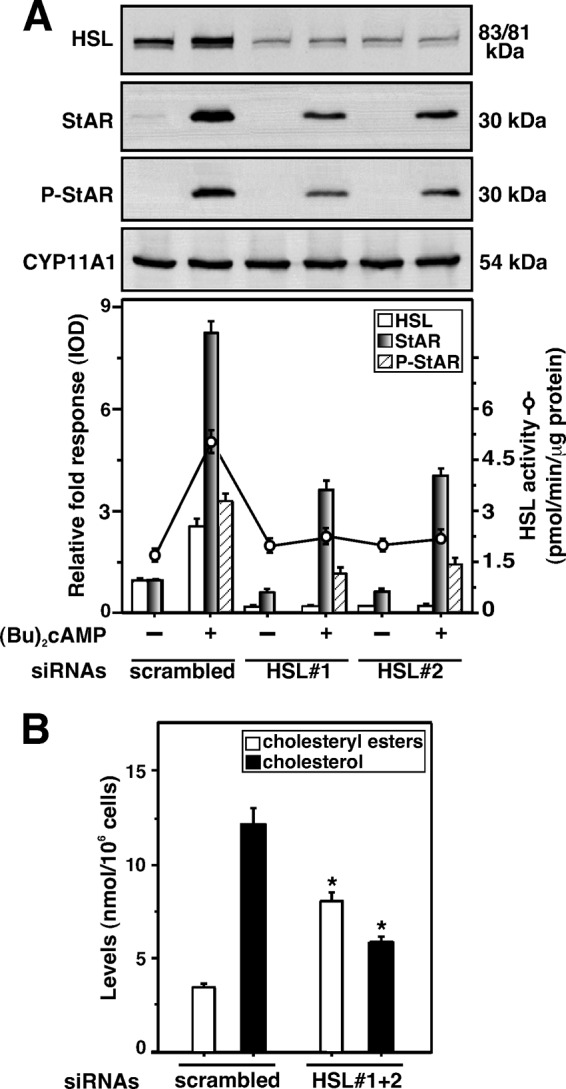
Transfection of HSL siRNAs in MA-10 cells and its relevance to HSL activity, HSL, StAR, P-StAR, cholesteryl esters, and cholesterol levels. Cells were transfected with a negative control siRNA (scrambled) and two HSL-specific siRNAs (HSL#1 and HSL#2) either at 100 nm individually (A) or together (HSL#1 + 2) at 50 nm each (B), as described under “Experimental Procedures.” Following 40 h of transfection, cells were treated without or with Bt2cAMP (0.5 mm) for an additional 6 h. Cells were then processed for either immunoblotting (A) or determining HSL activity (A, right panel). Representative immunoblots illustrate HSL, StAR, P-StAR, and CYP11A1 in different groups using 20–30 μg of total cellular protein. Immunoblots shown are representative of four independent experiments. Integrated optical density values of HSL, StAR, and P-StAR in each band were quantified, and compiled data (±S.E., n = 4) are presented (A, lower panel). Levels of cholesteryl esters and free cholesterol in the cell lysates were assessed using Amplex Red Cholesterol Assay Kit, as described under “Experimental Procedures,” and presented as nanomole/106 cells (B). Results represent the mean ± S.E. of four independent experiments. *, p < 0.05 versus scrambled.
To better understand these events, HSL-depleted MA-10 cells were analyzed for StAR, CYP11A1, 3β-HSD, SR-B1, and perilipin mRNA levels by real-time PCR (Fig. 6). The results presented in Fig. 6A illustrate that HSL siRNAs markedly diminished (p < 0.001) basal and Bt2cAMP-mediated HSL mRNA levels, when compared with scrambled cells. Knockdown of HSL suppressed Bt2cAMP-induced StAR mRNA levels by 62 ± 9% (Fig. 6B). CYP11A1 (Fig. 6C) and 3β-HSD (Fig. 6D) mRNAs were unaffected by HSL silencing, suggesting attenuation of HSL-dependent steroidogenesis does not affect these steroidogenic enzymes. Knockdown of HSL suppressed both basal and Bt2cAMP-induced SR-B1 mRNA (3.9 ± 0.5-fold over basal) between 42 and 58% (Fig. 6E). Depletion of HSL also affected (p < 0.05) basal and Bt2cAMP-induced perilipin mRNA levels (Fig. 6F), which supports previous observations indicating a physical interaction between HSL and perilipin (20).
FIGURE 6.

Silencing of HSL and its correlation to StAR, CYP11A1, 3β-HSD, SR-B1, and perilipin mRNA levels in MA-10 cells. Cells were transfected with a negative control siRNA (scrambled) and two HSL siRNAs (HSL#1 and HSL#2), as described under “Experimental Procedures” and in the legend of Fig. 5. Following 40 h of transfection, cells were treated in the absence or presence of Bt2cAMP (0.5 mm) for an additional 6 h. Total RNA was extracted from different groups, and mRNA levels for HSL (A), StAR (B), CYP11A1 (C), 3β-HSD (D), SR-B1 (E), and perilipin (F) were determined by real-time PCR and expressed as fold-changes relative to respective untreated cells. Results represent the mean ± S.E. of three independent experiments. Letters above the bars indicate that these groups differ significantly at least at p < 0.05.
HSL-mediated Regulation of StAR Expression and Steroid Synthesis Involves LXR Signaling
LXRs play key roles in the regulation of cholesterol and fatty acid homeostasis (24, 25). Oxysterols are ligands for LXRs that can bind to the −200/−185 bp region of the mouse StAR promoter (23). The hypothesis that HSL overexpression increases oxysterol production, and thereby can influence LXR target genes involved in StAR expression and steroidogenesis, was examined. MA-10 cells transfected with the −254/−1 StAR-Luc segment showed ∼2-fold increase in StAR reporter activity, in response to suboptimal doses of the LRX agonists, 27-hydroxycholesterol (27-HC, 0.25 μm) and T0901317 (T1317, 0.25 μm), over untreated cells (Fig. 7A). Overexpression of pcDNA3-HSL, within the context of the −254/−1 StAR-Luc, slightly increased basal but significantly elevated (p < 0.01) 27-HC- and T1317-mediated StAR promoter activity over the responses seen in mock-transfected (pcDNA3) cells. Cells overexpressing HSL showed an ∼2-fold increase in HSL protein expression (Fig. 7A, top panel). In contrast, HSL silencing demonstrated a decline (p < 0.05) in StAR promoter activity mediated by 27-HC and T1317. No significant effects of 27-HC and T1317 were observed on HSL protein levels. Additionally, both 27-HC and T1317 resulted in 2.2 ± 0.3- and 3.4 ± 0.4-fold increases in StAR protein levels, over untreated cells, respectively. Concurrently, these agonists showed 5 ± 2- and 9 ± 4-fold elevations in progesterone production (Fig. 7B). P-StAR was undetected in response to these agonists (data not shown). Overexpression of HSL coordinately elevated 27-HC- and T1317-induced StAR (4.3 ± 0.4- and 6.1 ± 0.5-fold over basal) and progesterone (17 ± 5- and 24 ± 8-fold over basal) levels, when compared with mock-transfected cells. Knockdown of HSL decreased 27-HC- and T1317-stimulated StAR protein and progesterone synthesis, levels similar to mock controls. Overexpression and silencing of HSL modestly altered (16–37%) basal expression of StAR expression and progesterone synthesis. These findings indicate that HSL-mediated steroidogenesis is influenced, at least in part, by LXR signaling. HSL overexpression enhances oxysterol production and, thereby the efficacy of LXR ligands involved in StAR protein and steroid synthesis, and these responses were affected by HSL silencing.
FIGURE 7.
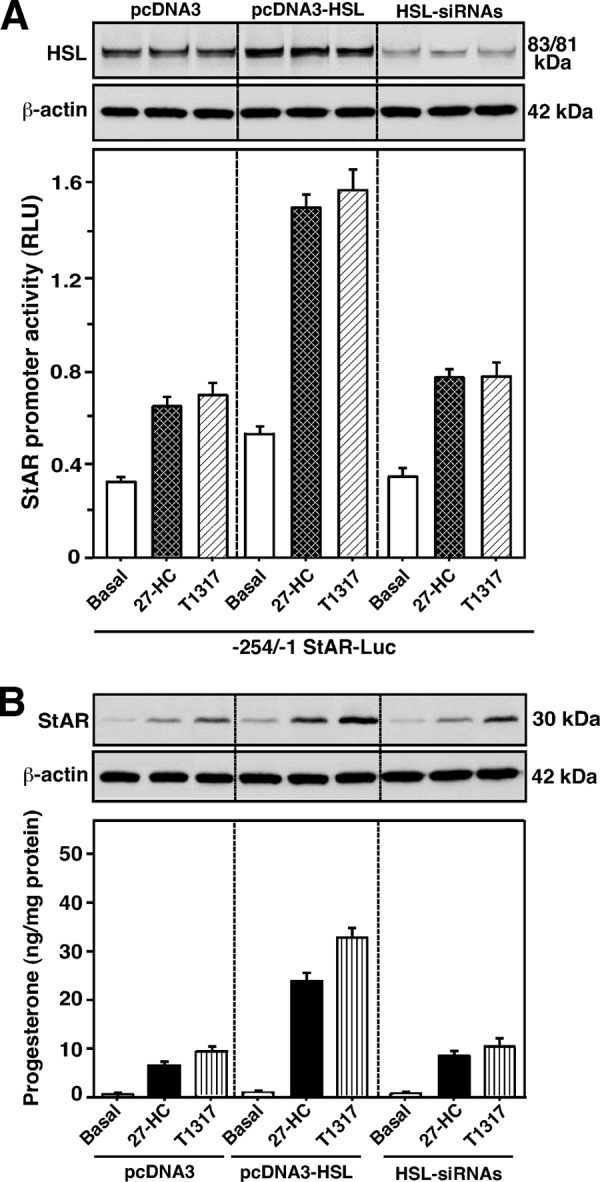
Overexpression and silencing of HSL on 27-HC- and T1317-responsive StAR promoter activity, HSL, StAR, and progesterone synthesis in MA-10 cells. Cells were transfected with pcDNA3 (2.0 μg), pcDNA3-HSL (2.0 μg), or two HSL-siRNAs together (50 nm each; A and B), within the context of the −254/−1 bp StAR luciferase reporter segment (−254/−1 StAR-Luc; 1.0 μg, A), in the presence of pRL-SV40, as described under “Experimental Procedures.” Following 36 (A) or 40 h (B) of transfection, cells were treated without (Basal) or with suboptimal doses of 27-HC (0.25 μm) and T1317 (0.25 μm) for an additional 6 h. A, luciferase activity in the cell lysates was determined and expressed as StAR promoter activity, relative light units (RLU) (luciferase/Renilla), which represent the mean ± S.E. of four independent experiments. Cells were also utilized for determining HSL (A) and StAR protein (B) levels by immunoblotting. Representative immunoblots show expression of HSL (A, upper panel) and StAR (B) in different groups using 20–30 μg of total cellular protein. Immunoblots shown are representative of four independent experiments. β-Actin expression was assessed as loading controls. Accumulation of progesterone in media was determined and expressed as nanogram/mg of protein (B, bottom panel). Results represent the mean ± S.E. of four independent experiments.
To gain more insight into these mechanisms, the role of LXR-target genes, SREBP-1c and ABCA1, in HSL-mediated StAR expression was evaluated (Fig. 8). MA-10 cells transfected with pcDNA3-HSL, in the presence of the −2.7 kb/+1 Srebp-1c promoter-driven luciferase reporter plasmid, demonstrated concentration-dependent increases in Srebp-1c promoter activity in response to 0.25 μm T1317 (Fig. 8A). The induction of T1317 responsive Srebp-1c promoter activity was maximally 3.1 ± 0.4-fold when compared at 0.5 μg of cDNA in the absence of T1317, demonstrating that HSL enhances Srebp-1c gene expression. Involvement of LXR signaling in HSL-mediated StAR expression was further verified with ABCA1, which is known to play an important role in cholesterol efflux (23). Treatment of MA-10 cells transfected with pcDNA3-HSL with a suboptimal dose of Bt2cAMP (0.1 mm) demonstrated increases in Abca1 protein levels (attaining a maximum of 4.7 ± 0.4-fold over pcDNA3-transfected cells) in a concentration-dependent manner (Fig. 8B). Overexpression of HSL in the cells was verified by determining P-HSL that, in turn, mirrored Abca1 levels. The increases in Bt2cAMP-mediated P-HSL (3.6 ± 0.4-fold) and ABCA1 levels were parallel with StAR protein (5.8 ± 0.5-fold) and P-StAR (3.1 ± 0.4-fold), when compared with the responses seen in respective mock-transfected cells. Additionally, the relevance of LXR signaling in HSL-dependent StAR expression and steroidogenesis was evaluated in cells lacking LXR proteins (Fig. 8, C and D). MA-10 cells transfected with LXRα and LXRβ siRNAs together demonstrated ∼70% decreases in endogenous LXRα/β protein levels, when compared with scrambled siRNA-transfected cells. Bt2cAMP (0.5 mm) demonstrated a 2–3-fold increase in LXRα/β protein expression in scrambled cells. No apparent effect of Bt2cAMP was observed in LXRα/β-silenced cells. Knockdown of LXRα/β affected (p < 0.05) both basal and Bt2cAMP-induced P-HSL. Whereas CAY10499 had no effect on Bt2cAMP-treated LXRα/β protein levels, it inhibited P-HSL between 65 and 78% in both groups. Decreases in LXRα/β and P-HSL levels affected Bt2cAMP-induced StAR, P-StAR, and progesterone levels. These results corroborate the data presented in Fig. 7 and demonstrate that HSL-mediated regulation of StAR expression and steroid synthesis involves the LXR regulatory pathway in MA-10 cells.
FIGURE 8.

Assessment of LXR signaling on HSL function and their correlation to P-HSL, Abca1, StAR, P-StAR, and progesterone synthesis in MA-10 cells. Cells were transfected with either pcDNA3 (2.0 μg) or pcDNA3-HSL (0.5–2.5 μg), within the context of the −2.7 kb/+1 Srebp-1c promoter-driven luciferase reporter plasmid (1.0 μg; A), in the presence and absence of pRL-SV40. Cells were also transfected with a negative control siRNA (scrambled) and mouse LXRα+β siRNAs together (100 nm each), as described under “Experimental Procedures.” Following 36 (A) or 40 h (B–D) of transfection, cells were pretreated without or with CAY10499 (10 μm; C and D) for 45 min, and then incubated in the absence or presence of T1317 (0.25 μm; A) and Bt2cAMP (0.1 mm, B; 0.5 mm, C and D) for an additional 6 h, as indicated. A, luciferase activity in the cell lysates was determined and expressed as Srebp-1c promoter activity, relative light units (RLU) (luciferase/Renilla). Cells were also processed for immunoblotting (B and C). Representative immunoblots illustrate P-HSL (Ser-660), Abca1, LXRα/β, StAR, and P-StAR in different groups using 20–30 μg of total cellular protein (B and C). β-Actin expression was assessed as loading controls. Accumulation of progesterone in media was determined and expressed as nanogram/mg of protein (D). Data represent the mean ± S.E. of three independent experiments. Letters above the bars indicate that these groups differ significantly at least at p < 0.05.
The LXR receptors form heterodimers with RXRs and have been demonstrated to control cholesterol utilization and metabolism (23–25). Contribution of these phenomena in StAR expression and steroid synthesis was evaluated in MA-10 cells. As shown in Fig. 9A, cells treated with T1317 (0.5 μm) and LG100268 (an RXR agonist, 0.5 μm, LG) documented 3.4 ± 0.5- and 2.6 ± 0.3-fold increases in StAR mRNA levels over untreated cells, respectively. Treatment of these agonists together showed an 8.2 ± 0.9-fold increase in StAR mRNA expression. Addition of a subthreshold concentration of Bt2cAMP (0.1 mm), to T1317 plus LG treatment, markedly increased (∼10-fold over basal) StAR mRNA expression, a level similar to the maximum stimulatory effect of Bt2cAMP (1.0 mm). T1317 and LG, individually, were capable of elevating (p < 0.05) StAR protein levels over untreated cells (Fig. 9B). P-StAR was undetected in response to T1317 and LG. Combined effects of these agonists, in the presence of 0.1 mm Bt2cAMP, strikingly activated StAR and P-StAR levels. The individual effect of T1317 and LG was also enhanced (p < 0.01) in combination with 0.1 mm Bt2cAMP (data not shown). Concurrently, T1317 and LG demonstrated 12 ± 4- and 5 ± 2-fold increases in progesterone production over untreated cells, respectively (Fig. 9C). These agonists together resulted in a 31 ± 7-fold increase in progesterone production. Treatment with 0.1 mm Bt2cAMP, individually, significantly elevated (p < 0.05) StAR mRNA, StAR protein, P-StAR, and progesterone levels, when compared with the respective basal values. Inclusion of 0.1 mm Bt2cAMP, to T1317 and LG treatment, synergistically activated progesterone synthesis concomitant with StAR and P-StAR levels. Collectively, these findings indicate that LXRs functionally cooperate/interact with RXRs in the regulation of StAR transcription and steroidogenesis in mouse Leydig cells.
FIGURE 9.
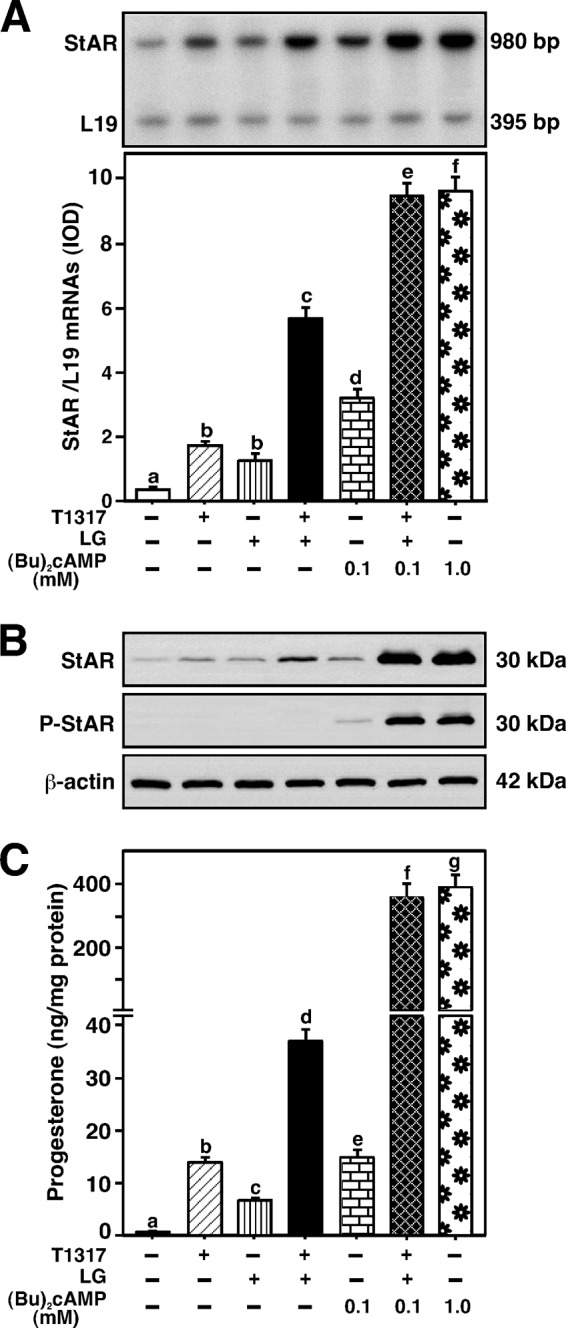
Effects of LXR and RXR agonists on Bt2cAMP-induced StAR mRNA, StAR protein, P-StAR, and progesterone synthesis. MA-10 cells were treated without or with T1317 (0.5 μm), LG100268 (0.5 μm, LG), Bt2cAMP (either 0.1 or 1.0 mm), or a combination, for 6 h, as indicated (A-C). Cells were then subjected to total RNA extraction (A) or cellular protein preparation (B) for determining StAR mRNA and StAR/P-StAR levels, by quantitative RT-PCR and immunoblotting, respectively. Representative autoradiogram (A) and immunoblots (B) illustrate expression of StAR mRNA, StAR protein, and P-StAR in different groups. RT-PCR and immunoblotting analyses were, respectively, run with 1 μg of total RNA and 20–30 μg of total cellular protein. Integrated optical density values of StAR mRNA expression in each band were quantified and normalized with the corresponding L19 bands, and presented as StAR/L19 mRNAs (A, bottom panel). β-Actin expression was assessed as loading controls. Accumulation of progesterone in media was determined and expressed as nanogram/mg of protein (C). Results represent the mean ± S.E. of four independent experiments. Letters above the bars indicate that these groups differ significantly at least at p < 0.05.
DISCUSSION
Steroid hormones are essential for maintenance of normal reproductive function and bodily homeostasis. The biosynthesis of steroid hormones is predominantly regulated by trophic hormones, where several factors and processes play permissible roles (reviewed in Refs. 4–6). The interaction of trophic hormones with specific receptors increase intracellular cAMP levels, which in turn, activate cAMP-dependent PKA and results in the phosphorylation of proteins involved in steroidogenesis. There is now a wealth of information indicating that the acute regulation of steroid biosynthesis is dictated principally by the StAR protein, a rapidly synthesized mitochondrial phosphoprotein whose expression, activation, and removal largely involve cAMP-dependent mechanisms in the adrenals and gonads. The contributions of a multifunctional enzyme, HSL, which catalyzes the hydrolysis of cholesteryl esters, were explored to determine its potential role in the regulation of StAR protein expression and, thus, steroid biosynthesis. The present studies expand our understanding of the manner in which HSL regulates cAMP-mediated StAR expression and steroidogenesis in steroidogenic cells.
One of the hallmarks of HSL function is its regulation by reversible phosphorylation. Hormonal control of HSL activity is primarily mediated by the phosphorylation of several serine residues, i.e. Ser-563, Ser-659, and Ser-660 (sequences corresponding to rat HSL), by cAMP-dependent PKA in vitro as well as in vivo (11, 39, 40). Additionally, phosphorylation of Ser-565 (41) by several kinases and Ser-600 (42) by MAPK/ERK, respectively, has been demonstrated, suggesting different phosphorylation sites may have discrete functions. Our current data demonstrate that activation of PKA, by Bt2cAMP, enhanced phosphorylation of HSL at Ser-660 and Ser-563 (but not Ser-565), and this phosphorylation was concomitant with its hydrolytic activity in MA-10 Leydig cells. These effects coincided with increases in StAR expression and progesterone synthesis, and were qualitatively similar in both mouse KK-1 granulosa and Y-1 adrenocortical cells. Selective inhibition of HSL activity by CAY10499, an endocannabinoid that covalently binds HSL (43), or blockade of PKA signaling by H-89, markedly affected Bt2cAMP-induced StAR and steroid levels. CAY10499 inhibited neither PKA activity nor 22R-OH-treated progesterone synthesis, demonstrating the repression of Bt2cAMP responsive steroidogenesis was due to HSL inhibition. CYP11A1 protein levels were unaltered by any of these treatments, suggesting that the acute effect on Bt2cAMP-mediated StAR and steroid levels does not involve increased transcription/translation of genes encoding steroidogenic enzymes (6, 7). These observations imply that activation of HSL by cAMP/PKA signaling plays an important role in controlling StAR expression and steroidogenesis in gonadal and adrenal cells. In the present study, whereas phosphorylation of Ser-660 and Ser-563, by Bt2cAMP, was found to trigger the hydrolytic activity of HSL, involvement of additional site(s) including Ser-659, cannot be excluded. Studies have demonstrated that mutation of Ser-563 → Ala markedly affected HSL activity (44). Increasing evidence indicates that Ser-565 phosphorylation does not activate the enzyme (reviewed in Ref. 45). However, phosphorylation of Ser-563 and -565 has been suggested to be mutually exclusive due to steric hindrance. Conspicuously, activation of the MAPK/ERK pathway has been demonstrated to phosphorylate HSL at Ser-600 and to subsequently increase its activity in regulating adipocyte lipolysis (42). Hence, it is conceivable that phosphorylation of different serine residues may cause conformational alterations of HSL for fine-tuning the regulatory events associated with the hydrolytic activity of HSL involved in influencing steroidogenesis.
HSL is the primary NCEH in steroidogenic tissues and HSL/NCEH activity is nearly abolished in the adrenals and testes of HSL−/− mice (10, 12, 17, 18, 46). Our results demonstrate that knockdown of HSL attenuated both HSL peptide and HSL activity by 80–90% in MA-10 cells. Depletion of HSL resulted in a significant decline in free cholesterol content that was accompanied with ∼50% reduction in Bt2cAMP-induced StAR and P-StAR levels, demonstrating a correlation between HSL action and StAR expression. Furthermore, interference with HSL activity resulted in a significant rise in cholesterol ester levels. These results are in support of previous findings that demonstrated that inhibition of cholesterol ester hydrolase increases esterified cholesterol in the testis, but decreases serum testosterone and testicular steroidogenesis (47). In addition, suppression of HSL/NCEH activity by DNA antisense to HSL elevates esterified cholesterol and attenuates cholesterol content in Chinese hamster ovary cells (48). HSL−/− male mice exhibit several testicular anomalies, including decreased weight, vacuolated seminiferous tubules, reduced spermatids, and sterility (10, 17, 18, 46). However, circulating steroid hormone levels were normal in these mice. The mechanism accounting for persistent steroidogenesis with little to no HSL/NCEH activity, in HSL-null mice, remains unclear, and may involve one or more compensatory event(s), including acyl-CoA:cholesterol acyltransferase activity and/or de novo cholesterol synthesis. The functional importance of HSL, in the present study, was assessed utilizing CAY10499 and siRNAs and demonstrated that interference with HSL activity effectively represses Bt2cAMP-induced StAR expression and steroidogenesis in different steroidogenic cells. Noteworthy, HSL also mediates retinyl ester hydrolysis (49), and recently we observed that the induction of retinoic acid, and ligands for both retinoic acid receptor- and RXR (α, β, and γ)-mediated StAR expression was significantly diminished in HSL-depleted MA-10 cells.3 Also, retinoic acid-mediated steroidogenesis was demonstrated to be predominantly increased by RXRα, which can bind to the StAR promoter and control its transcription. Therefore, it is reasonable to speculate that repression of Bt2cAMP-induced expression and phosphorylation of StAR, in conjunction with HSL deficiency was, at least in part, due to perturbed retinoid metabolism.
In mice, HDL-derived selective uptake of cholesteryl esters, via SR-B1, provides most of the cholesterol for steroidogenesis, with lesser contributions from LDL and de novo synthesis (1, 50). We (9, 51), and others (2, 52), have demonstrated a striking correlation between hormonal induction of SR-B1 and steroid synthesis in different steroidogenic cell models. In addition, high levels of StAR expression and steroid production have been reported to be associated with constitutive expression of HSL and SR-B1 in R2C rat Leydig cells (51). The present results provide evidence that knockdown of HSL diminished SR-B1 expression and coordinately repressed free cholesterol, StAR, and P-StAR levels in MA-10 cells. Concomitantly, suppression of perilipin mRNA levels was observed to occur with HSL deficiency, demonstrating that interaction of perilipin and HSL may play a role in StAR expression (20). Noteworthy, two perilipin isoforms (A and C) have been shown to be associated with cholesteryl ester droplets in the adrenal and Leydig cells (53). Both HSL and perilipin are phosphorylated by PKA and this post-translational event facilitates the translocation of HSL from the cytosol to lipid droplets, the site of its action (53, 54). We also observed that depletion of HSL affected HDL (when compared with LDL) and mediated StAR protein levels, either individually or in combination with Bt2cAMP (data not shown). This demonstrates the importance of HSL in processing HDL-derived cholesteryl esters for steroid biosynthesis as shown in previous studies (12). It has been reported that overexpression of SR-B1 increases the uptake of HDL-derived cholesteryl esters and thereby the steroidogenic potential in Y1 cells (2). In human granulosa cells, silencing of SR-B1 decreases basal and forskolin-responsive StAR and steroid levels without altering HSL activation (52). Nonetheless, an inverse correlation between SR-B1 protein expression and ACTH-induced corticosterone synthesis in the adrenals of HSL−/− mice has been demonstrated (12). These seeming contradictions could be due to differences in environmental conditions (in vitro versus in vivo), tissue specificity (Leydig, adrenal, and granulosa), receptor-effector coupling, and/or the presence of as yet unidentified factor(s) that play a role in steroidogenesis. Overall, HSL deficiency affects SR-B1 mRNA levels, diminishes the uptake of HDL-derived cholesteryl esters, and results in insufficient availability of cholesterol for steroidgenesis. As such, it appears unlikely that a single mechanism is involved in the HSL-mediated regulation of StAR expression, as interference with HSL activity affects multiple processes involved in the steroidogenic pathway.
An intriguing aspect of the present findings is the involvement of LXR signaling in the regulation of HSL-dependent StAR expression and steroidogenesis. LXRs bind to oxysterol ligands, activate transcription of target genes, and play important roles in controlling intracellular cholesterol trafficking, metabolism, and balance (23, 24, 55). The functional relevance of the LXR pathway was assessed by different approaches demonstrating that HSL-mediated regulation of steroidogenesis involves modulation of the LXR-target genes, Srebp-1c and abca1, in mouse Leydig cells. Pharmacologic activation of LXR utilizing potent agonists that mimic activation by oxysterols, resulted in significant increases in StAR expression and steroid synthesis following HSL overexpression. Conversely, HSL silencing decreased these steroidogenic responses mediated by LXR agonists. An increase in HSL levels was capable of enhancing Srebp-1c promoter activity as well as Abca1 protein expression, which mirrored StAR and P-StAR levels. Noteworthy, however, StAR expression has been influenced by several oxysterols (21, 23). These include 25-hydroxycholesterol that is produced by testicular macrophages and stimulates Leydig cell steroidogenesis (26). The identification of oxysterol(s) and their relative contribution to HSL-mediated activation of StAR expression and steroid synthesis is currently underway. It is conceivable that LXR activation enhances plasma membrane cholesterol trafficking and efflux and modulates cholesterol esterification, thus contributing to the effects of LXR agonists in controlling the steroidogenic response. Previous studies have shown that LXRs and/or their interaction with RXRs regulate the expression of Srebp-1c, Abca1, and StAR for maintaining adrenal cholesterol balance (23, 56). In macrophages, the increase in cholesteryl ester hydrolysis by HSL has been correlated with LXR-mediated Abca1 expression (57). Also, the regulation of hCG- and growth factor-stimulated androstenedione synthesis is connected with abca1 mRNA levels in theca-interstitial cells (58). Mice lacking LXRα and LXRβ display reduced fertility, and it is further reported that LXRs are critically involved in regulating cholesterol homeostasis and male reproductive function (59, 60). Studies have also demonstrated that LXR plays key roles in the regulation of rennin and c-myc gene transcription by cAMP signaling (61). In accordance, our present findings demonstrate that knockdown of LXR-α and -β in MA-10 cells significantly affected, but not abolished, Bt2cAMP-induced P-HSL, StAR, P-StAR, and progesterone levels. These imply that HSL is capable of influencing steroidogenesis in cells lacking LXRs. Nevertheless, LXRs were found to interact/cooperate with RXRs and result in synergistic activation of Bt2cAMP-mediated StAR expression and steroid synthesis. Noteworthy, the marked elevation in the steroidogenic response concomitant with moderate increases in StAR and steroid levels by LXR and RXR agonists was due to a requirement for PKA-dependent post-translational modification. Regulation of StAR and steroid hormones in the gonads and adrenals is mediated by a plethora of signaling pathways, and it is likely that LXRs might influence a number of these processes. Based on the present findings, it is plausible that HSL-mediated regulation of StAR expression, by cAMP/PKA signaling, involves a number of signaling pathways, including the LXR regulatory pathway, which opens a new gateway to steroidogenesis.
Taken together, HSL, through its action on the hydrolysis of cholesteryl esters, plays a key role in the regulation of Bt2cAMP-mediated StAR expression and steroid biosynthesis, which involves LXR signaling, and demonstrates a direct molecular mechanism to explain the findings presented in this investigation. The present results also provide a link between HSL-LXR signaling and ABCA1, a key component in atherosclerosis. The association of the LXR regulatory pathway in HSL-dependent steroidogenesis can be useful for better understanding a number of pathophysiological processes connected with abnormal cholesterol utilization, storage, and metabolism.
Acknowledgments
We thank Drs. H. Park (University of Seoul, Seoul, Republic of Korea) and S. Azhar (Veterans Affairs Palo Alto Health Care System, Palo Alto, CA) for the generous gifts of SREBP-1c and HSL plasmids, respectively. We also thank Dr. M. Ascoli (University of Iowa College of Medicine, Iowa City, Iowa) for the gift of MA-10 cells. The superb technical assistance of Yuping Sun is acknowledged.
This work was supported, in whole or in part, by National Institutes of Health Grant HD-17481 (to D. M. S.), Robert A. Welch Foundation Grant B1–0028 (to D. M. S.), and the Université Paris Diderot-Paris 7.
P. R. Manna and D. M. Stocco, unpublished observations.
- SR-B1
- scavenger receptor class B type 1
- StAR
- steroidogenic acute regulatory protein
- HSL
- hormone-sensitive lipase
- NCEH
- neutral cholesteryl ester hydrolase
- Bt2cAMP
- dibutyryl cAMP
- MAPK/ERK
- mitogen-activated protein kinase/extracellular signal-regulated kinase
- CYP11A1
- cytochrome P450 side chain cleavage enzyme
- 3β-HSD
- 3β-hydroxysteroid dehydrogenase enzyme
- LXR
- liver X receptor
- 22R-OH
- 22R-hydroxycholesterol
- 27-HC
- 27-hydroxycholesterol
- T1317
- T0901317
- LG
- RXR
- retinoid X receptor
- SREBP-1c
- sterol regulatory element-binding protein 1c
- ABCA1
- ATP-binding cassette transporter 1.
REFERENCES
- 1. Glass C., Pittman R. C., Weinstein D. B., Steinberg D. (1983) Dissociation of tissue uptake of cholesterol ester from that of apoprotein A-I of rat plasma high density lipoprotein. Selective delivery of cholesterol ester to liver, adrenal, and gonad. Proc. Natl. Acad. Sci. U.S.A. 80, 5435–5439 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Reaven E., Nomoto A., Cortez Y., Azhar S. (2006) Consequences of over-expression of rat scavenger receptor, SR-BI, in an adrenal cell model. Nutr. Metab. 3, 43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Clark B. J., Wells J., King S. R., Stocco D. M. (1994) The purification, cloning, and expression of a novel luteinizing hormone-induced mitochondrial protein in MA-10 mouse Leydig tumor cells. Characterization of the steroidogenic acute regulatory protein (StAR). J. Biol. Chem. 269, 28314–28322 [PubMed] [Google Scholar]
- 4. Stocco D. M., Wang X., Jo Y., Manna P. R. (2005) Multiple signaling pathways regulating steroidogenesis and steroidogenic acute regulatory protein expression. More complicated than we thought. Mol. Endocrinol. 19, 2647–2659 [DOI] [PubMed] [Google Scholar]
- 5. Miller W. L. (2007) StAR search. What we know about how the steroidogenic acute regulatory protein mediates mitochondrial cholesterol import. Mol. Endocrinol. 21, 589–601 [DOI] [PubMed] [Google Scholar]
- 6. Manna P. R., Stocco D. M. (2005) Regulation of the steroidogenic acute regulatory protein expression. Functional and physiological consequences. Curr. Drug Targets Immune Endocr. Metabol. Disord 5, 93–108 [DOI] [PubMed] [Google Scholar]
- 7. Miller W. L., Bose H. S. (2011) Early steps in steroidogenesis. Intracellular cholesterol trafficking. J. Lipid Res. 52, 2111–2135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Arakane F., King S. R., Du Y., Kallen C. B., Walsh L. P., Watari H., Stocco D. M., Strauss J. F., 3rd. (1997) Phosphorylation of steroidogenic acute regulatory protein (StAR) modulates its steroidogenic activity. J. Biol. Chem. 272, 32656–32662 [DOI] [PubMed] [Google Scholar]
- 9. Manna P. R., Chandrala S. P., King S. R., Jo Y., Counis R., Huhtaniemi I. T., Stocco D. M. (2006) Molecular mechanisms of insulin-like growth factor-I mediated regulation of the steroidogenic acute regulatory protein in mouse leydig cells. Mol. Endocrinol. 20, 362–378 [DOI] [PubMed] [Google Scholar]
- 10. Osuga J., Ishibashi S., Oka T., Yagyu H., Tozawa R., Fujimoto A., Shionoiri F., Yahagi N., Kraemer F. B., Tsutsumi O., Yamada N. (2000) Targeted disruption of hormone-sensitive lipase results in male sterility and adipocyte hypertrophy, but not in obesity. Proc. Natl. Acad. Sci. U.S.A. 97, 787–792 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Holm C. (2003) Molecular mechanisms regulating hormone-sensitive lipase and lipolysis. Biochem. Soc. Trans. 31, 1120–1124 [DOI] [PubMed] [Google Scholar]
- 12. Kraemer F. B., Shen W. J., Harada K., Patel S., Osuga J., Ishibashi S., Azhar S. (2004) Hormone-sensitive lipase is required for high-density lipoprotein cholesteryl ester-supported adrenal steroidogenesis. Mol. Endocrinol. 18, 549–557 [DOI] [PubMed] [Google Scholar]
- 13. Holst L. S., Langin D., Mulder H., Laurell H., Grober J., Bergh A., Mohrenweiser H. W., Edgren G., Holm C. (1996) Molecular cloning, genomic organization, and expression of a testicular isoform of hormone-sensitive lipase. Genomics 35, 441–447 [DOI] [PubMed] [Google Scholar]
- 14. Holm C., Kirchgessner T. G., Svenson K. L., Fredrikson G., Nilsson S., Miller C. G., Shively J. E., Heinzmann C., Sparkes R. S., Mohandas T. (1988) Hormone-sensitive lipase. Sequence, expression, and chromosomal localization to 19 cent-q13.3. Science 241, 1503–1506 [DOI] [PubMed] [Google Scholar]
- 15. Mairal A., Melaine N., Laurell H., Grober J., Holst L. S., Guillaudeux T., Holm C., Jégou B., Langin D. (2002) Characterization of a novel testicular form of human hormone-sensitive lipase. Biochem. Biophys. Res. Commun. 291, 286–290 [DOI] [PubMed] [Google Scholar]
- 16. Kabbaj O., Yoon S. R., Holm C., Rose J., Vitale M. L., Pelletier R. M. (2003) Relationship of the hormone-sensitive lipase-mediated modulation of cholesterol metabolism in individual compartments of the testis to serum pituitary hormone and testosterone concentrations in a seasonal breeder, the mink (Mustela vison). Biol. Reprod. 68, 722–734 [DOI] [PubMed] [Google Scholar]
- 17. Li H., Brochu M., Wang S. P., Rochdi L., Côté M., Mitchell G., Gallo-Payet N. (2002) Hormone-sensitive lipase deficiency in mice causes lipid storage in the adrenal cortex and impaired corticosterone response to corticotropin stimulation. Endocrinology 143, 3333–3340 [DOI] [PubMed] [Google Scholar]
- 18. Wang S. P., Chung S., Soni K., Bourdages H., Hermo L., Trasler J., Mitchell G. A. (2004) Expression of human hormone-sensitive lipase (HSL) in postmeiotic germ cells confers normal fertility to HSL-deficient mice. Endocrinology 145, 5688–5693 [DOI] [PubMed] [Google Scholar]
- 19. Shen W. J., Patel S., Natu V., Hong R., Wang J., Azhar S., Kraemer F. B. (2003) Interaction of hormone-sensitive lipase with steroidogenic acute regulatory protein: facilitation of cholesterol transfer in adrenal. J. Biol. Chem. 278, 43870–43876 [DOI] [PubMed] [Google Scholar]
- 20. Shen W. J., Patel S., Miyoshi H., Greenberg A. S., Kraemer F. B. (2009) Functional interaction of hormone-sensitive lipase and perilipin in lipolysis. J. Lipid Res. 50, 2306–2313 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Christenson L. K., McAllister J. M., Martin K. O., Javitt N. B., Osborne T. F., Strauss J. F., 3rd. (1998) Oxysterol regulation of steroidogenic acute regulatory protein gene expression. Structural specificity and transcriptional and posttranscriptional actions. J. Biol. Chem. 273, 30729–30735 [DOI] [PubMed] [Google Scholar]
- 22. King S. R., Matassa A. A., White E. K., Walsh L. P., Jo Y., Rao R. M., Stocco D. M., Reyland M. E. (2004) Oxysterols regulate expression of the steroidogenic acute regulatory protein. J. Mol. Endocrinol. 32, 507–517 [DOI] [PubMed] [Google Scholar]
- 23. Cummins C. L., Volle D. H., Zhang Y., McDonald J. G., Sion B., Lefrançois-Martinez A. M., Caira F., Veyssière G., Mangelsdorf D. J., Lobaccaro J. M. (2006) Liver X receptors regulate adrenal cholesterol balance. J. Clin. Invest. 116, 1902–1912 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Repa J. J., Berge K. E., Pomajzl C., Richardson J. A., Hobbs H., Mangelsdorf D. J. (2002) Regulation of ATP-binding cassette sterol transporters ABCG5 and ABCG8 by the liver X receptors α and β. J. Biol. Chem. 277, 18793–18800 [DOI] [PubMed] [Google Scholar]
- 25. Volle D. H., Lobaccaro J. M. (2007) Role of the nuclear receptors for oxysterols LXRs in steroidogenic tissues: beyond the “foie gras,” the steroids and sex? Mol. Cell Endocrinol. 265, 183–189 [DOI] [PubMed] [Google Scholar]
- 26. Lukyanenko Y. O., Chen J. J., Hutson J. C. (2001) Production of 25-hydroxycholesterol by testicular macrophages and its effects on Leydig cells. Biol. Reprod. 64, 790–796 [DOI] [PubMed] [Google Scholar]
- 27. Manna P. R., Dyson M. T., Eubank D. W., Clark B. J., Lalli E., Sassone-Corsi P., Zeleznik A. J., Stocco D. M. (2002) Regulation of steroidogenesis and the steroidogenic acute regulatory protein by a member of the cAMP response-element binding protein family. Mol. Endocrinol. 16, 184–199 [DOI] [PubMed] [Google Scholar]
- 28. Manna P. R., Huhtaniemi I. T., Stocco D. M. (2009) Mechanisms of protein kinase C signaling in the modulation of 3′,5′-cyclic adenosine monophosphate-mediated steroidogenesis in mouse gonadal cells. Endocrinology 150, 3308–3317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Manna P. R., Eubank D. W., Stocco D. M. (2004) Assessment of the role of activator protein-1 on transcription of the mouse steroidogenic acute regulatory protein gene. Mol. Endocrinol. 18, 558–573 [DOI] [PubMed] [Google Scholar]
- 30. Lee Y. S., Lee H. H., Park J., Yoo E. J., Glackin C. A., Choi Y. I., Jeon S. H., Seong R. H., Park S. D., Kim J. B. (2003) Twist2, a novel ADD1/SREBP1c interacting protein, represses the transcriptional activity of ADD1/SREBP1c. Nucleic Acids Res. 31, 7165–7174 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Sztalryd C., Komaromy M. C., Kraemer F. B. (1995) Overexpression of hormone-sensitive lipase prevents triglyceride accumulation in adipocytes. J. Clin. Invest. 95, 2652–2661 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Manna P. R., Soh J. W., Stocco D. M. (2011) The involvement of specific PKC isoenzymes in phorbol ester-mediated regulation of steroidogenic acute regulatory protein expression and steroid synthesis in mouse Leydig cells. Endocrinology 152, 313–325 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Manna P. R., Dyson M. T., Jo Y., Stocco D. M. (2009) Role of dosage-sensitive sex reversal, adrenal hypoplasia congenita, critical region on the X chromosome, gene 1 in protein kinase A- and protein kinase C-mediated regulation of the steroidogenic acute regulatory protein expression in mouse Leydig tumor cells. Mechanism of action. Endocrinology 150, 187–199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Bose H. S., Whittal R. M., Baldwin M. A., Miller W. L. (1999) The active form of the steroidogenic acute regulatory protein, StAR, appears to be a molten globule. Proc. Natl. Acad. Sci. U.S.A. 96, 7250–7255 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Dyson M. T., Kowalewski M. P., Manna P. R., Stocco D. M. (2009) The differential regulation of steroidogenic acute regulatory protein-mediated steroidogenesis by type I and type II PKA in MA-10 cells. Mol. Cell. Endocrinol. 300, 94–103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Manna P. R., Tena-Sempere M., Huhtaniemi I. T. (1999) Molecular mechanisms of thyroid hormone-stimulated steroidogenesis in mouse leydig tumor cells. Involvement of the steroidogenic acute regulatory (StAR) protein. J. Biol. Chem. 274, 5909–5918 [DOI] [PubMed] [Google Scholar]
- 37. Fredrikson G., Strålfors P., Nilsson N. O., Belfrage P. (1981) Hormone-sensitive lipase from adipose tissue of rat. Methods Enzymol. 71, 636–646 [DOI] [PubMed] [Google Scholar]
- 38. Manna P. R., Dyson M. T., Stocco D. M. (2009) Regulation of the steroidogenic acute regulatory protein gene expression. Present and future perspectives. Mol. Hum. Reprod. 15, 321–333 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Osterlund T. (2001) Structure-function relationships of hormone-sensitive lipase. Eur. J. Biochem. 268, 1899–1907 [DOI] [PubMed] [Google Scholar]
- 40. Krintel C., Mörgelin M., Logan D. T., Holm C. (2009) Phosphorylation of hormone-sensitive lipase by protein kinase A in vitro promotes an increase in its hydrophobic surface area. FEBS J. 276, 4752–4762 [DOI] [PubMed] [Google Scholar]
- 41. Garton A. J., Yeaman S. J. (1990) Identification and role of the basal phosphorylation site on hormone-sensitive lipase. Eur. J. Biochem. 191, 245–250 [DOI] [PubMed] [Google Scholar]
- 42. Greenberg A. S., Shen W. J., Muliro K., Patel S., Souza S. C., Roth R. A., Kraemer F. B. (2001) Stimulation of lipolysis and hormone-sensitive lipase via the extracellular signal-regulated kinase pathway. J. Biol. Chem. 276, 45456–45461 [DOI] [PubMed] [Google Scholar]
- 43. Savinainen J. R., Yoshino M., Minkkilä A., Nevalainen T., Laitinen J. T. (2010) Characterization of binding properties of monoglyceride lipase inhibitors by a versatile fluorescence-based technique. Anal. Biochem. 399, 132–134 [DOI] [PubMed] [Google Scholar]
- 44. Shen W. J., Patel S., Natu V., Kraemer F. B. (1998) Mutational analysis of structural features of rat hormone-sensitive lipase. Biochemistry 37, 8973–8979 [DOI] [PubMed] [Google Scholar]
- 45. Yeaman S. J. (2004) Hormone-sensitive lipase. New roles for an old enzyme. Biochem. J. 379, 11–22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Chung S., Wang S. P., Pan L., Mitchell G., Trasler J., Hermo L. (2001) Infertility and testicular defects in hormone-sensitive lipase-deficient mice. Endocrinology 142, 4272–4281 [DOI] [PubMed] [Google Scholar]
- 47. Bartke A., Musto N., Caldwell B. V., Behrman H. R. (1973) Effects of a cholesterol esterase inhibitor and of prostaglandin F2α on testis cholesterol and on plasma testosterone in mice. Prostaglandins 3, 97–104 [DOI] [PubMed] [Google Scholar]
- 48. Osuga J., Ishibashi S., Shimano H., Inaba T., Kawamura M., Yazaki Y., Yamada N. (1997) Suppression of neutral cholesterol ester hydrolase activity by antisense DNA of hormone-sensitive lipase. Biochem. Biophys. Res. Commun. 233, 655–657 [DOI] [PubMed] [Google Scholar]
- 49. Ström K., Gundersen T. E., Hansson O., Lucas S., Fernandez C., Blomhoff R., Holm C. (2009) Hormone-sensitive lipase (HSL) is also a retinyl ester hydrolase. Evidence from mice lacking HSL. FASEB J. 23, 2307–2316 [DOI] [PubMed] [Google Scholar]
- 50. Azhar S., Reaven E. (2002) Scavenger receptor class BI and selective cholesteryl ester uptake. Partners in the regulation of steroidogenesis. Mol. Cell Endocrinol. 195, 1–26 [DOI] [PubMed] [Google Scholar]
- 51. Rao R. M., Jo Y., Leers-Sucheta S., Bose H. S., Miller W. L., Azhar S., Stocco D. M. (2003) Differential regulation of steroid hormone biosynthesis in R2C and MA-10 Leydig tumor cells. Role of SR-B1-mediated selective cholesteryl ester transport. Biol. Reprod. 68, 114–121 [DOI] [PubMed] [Google Scholar]
- 52. Kolmakova A., Wang J., Brogan R., Chaffin C., Rodriguez A. (2010) Deficiency of scavenger receptor class B type I negatively affects progesterone secretion in human granulosa cells. Endocrinology 151, 5519–5527 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Servetnick D. A., Brasaemle D. L., Gruia-Gray J., Kimmel A. R., Wolff J., Londos C. (1995) Perilipins are associated with cholesteryl ester droplets in steroidogenic adrenal cortical and Leydig cells. J. Biol. Chem. 270, 16970–16973 [DOI] [PubMed] [Google Scholar]
- 54. Clifford G. M., Londos C., Kraemer F. B., Vernon R. G., Yeaman S. J. (2000) Translocation of hormone-sensitive lipase and perilipin upon lipolytic stimulation of rat adipocytes. J. Biol. Chem. 275, 5011–5015 [DOI] [PubMed] [Google Scholar]
- 55. Rigamonti E., Helin L., Lestavel S., Mutka A. L., Lepore M., Fontaine C., Bouhlel M. A., Bultel S., Fruchart J. C., Ikonen E., Clavey V., Staels B., Chinetti-Gbaguidi G. (2005) Liver X receptor activation controls intracellular cholesterol trafficking and esterification in human macrophages. Circ. Res. 97, 682–689 [DOI] [PubMed] [Google Scholar]
- 56. Willy P. J., Umesono K., Ong E. S., Evans R. M., Heyman R. A., Mangelsdorf D. J. (1995) LXR, a nuclear receptor that defines a distinct retinoid response pathway. Genes Dev. 9, 1033–1045 [DOI] [PubMed] [Google Scholar]
- 57. Tazoe F., Yagyu H., Okazaki H., Igarashi M., Eto K., Nagashima S., Inaba T., Shimano H., Osuga J., Ishibashi S. (2008) Induction of ABCA1 by overexpression of hormone-sensitive lipase in macrophages. Biochem. Biophys. Res. Commun. 376, 111–115 [DOI] [PubMed] [Google Scholar]
- 58. Wu Q., Sucheta S., Azhar S., Menon K. M. (2003) Lipoprotein enhancement of ovarian theca-interstitial cell steroidogenesis. Relative contribution of scavenger receptor class B (type I) and adenosine 5′-triphosphate-binding cassette (type A1) transporter in high-density lipoprotein-cholesterol transport and androgen synthesis. Endocrinology 144, 2437–2445 [DOI] [PubMed] [Google Scholar]
- 59. Robertson K. M., Schuster G. U., Steffensen K. R., Hovatta O., Meaney S., Hultenby K., Johansson L. C., Svechnikov K., Söder O., Gustafsson J. A. (2005) The liver X receptor-β is essential for maintaining cholesterol homeostasis in the testis. Endocrinology 146, 2519–2530 [DOI] [PubMed] [Google Scholar]
- 60. Volle D. H., Mouzat K., Duggavathi R., Siddeek B., Déchelotte P., Sion B., Veyssière G., Benahmed M., Lobaccaro J. M. (2007) Multiple roles of the nuclear receptors for oxysterols liver X receptor to maintain male fertility. Mol. Endocrinol. 21, 1014–1027 [DOI] [PubMed] [Google Scholar]
- 61. Tamura K., Chen Y. E., Horiuchi M., Chen Q., Daviet L., Yang Z., Lopez-Ilasaca M., Mu H., Pratt R. E., Dzau V. J. (2000) LXRα functions as a cAMP-responsive transcriptional regulator of gene expression. Proc. Natl. Acad. Sci. U.S.A. 97, 8513–8518 [DOI] [PMC free article] [PubMed] [Google Scholar]