

Background: Two splice variants of caspase-9 can be generated by the inclusion/exclusion of the exon 3,4,5,6 cassette.
Results: hnRNP U is an enhancer of this exonic cassette and is opposed by phosphorylation of hnRNP L via the AKT pathway.
Conclusion: hnRNP U promotes the exon cassette inclusion to form caspase-9a.
Significance: Understanding the regulation of caspase-9 alternative splicing is important for the treatment of lung cancer.
Keywords: Akt, Alternative Splicing, Cancer Biology, RNA Splicing, RNA-binding Proteins, RNA-Protein Interaction, Signal Transduction, Caspase 9b, hnRNP L, hnRNP U
Abstract
Caspase-9 has two splice variants, pro-apoptotic caspase-9a and anti-apoptotic caspase-9b, which are regulated by RNA trans-factors associated with exon 3 of caspase-9 pre-mRNA (C9/E3). In this study, we identified hnRNP U as an RNA trans-factor associated with C9/E3. Down-regulation of hnRNP U led to a decrease in the caspase-9a/9b mRNA ratio, demonstrating a novel enhancing function. Importantly, hnRNP U bound specifically to C9/E3 at an RNA cis-element previously reported as the binding site for the splicing repressor, hnRNP L. Phosphorylated hnRNP L interfered with hnRNP U binding to C9/E3, and our results demonstrate the importance of the phosphoinositide 3-kinase/AKT pathway in modulating the association of hnRNP U to C9/E3. Taken together, these findings show that hnRNP U competes with hnRNP L for binding to C9/E3 to enhance the inclusion of the four-exon cassette, and this splice-enhancing function is blocked by the AKT pathway via phosphorylation of hnRNP L.
Introduction
Apoptosis is a distinctive mode of “programmed cell death” and plays critical roles in the regulation of tissue homeostasis (1). Initiation of apoptosis is highly coordinated and often energy-dependent via activation of a group of cysteine proteases called caspases. Among the 14 caspases identified to date, caspase-9 is one of the most important initiators of the intrinsic apoptotic pathway. For example, the activation of caspase-9 via interaction with Apaf-1 and cytochrome c to form the apoptosome activates downstream caspases to amplify apoptotic signaling. There are two splice variants of caspase-9: caspase-9a and caspase-9b, that can be generated by the inclusion or exclusion of the exon 3,4,5,6 cassette in the mature caspase-9 mRNA. Different from caspase-9a (e.g., procaspase), caspase-9b lacks the catalytic domain but retains the caspase recruitment/Apaf-1 association domain. Thus, caspase-9b functions as an apoptotic inhibitor by competing with caspase-9a for binding to Apaf-1 and consequently suppressing caspase-cascade activation (2, 3).
The ratio of caspase-9a/9b isoform is dysregulated in non-small cell lung cancer (NSCLC)2 cells (4, 5), and the factors implicated in regulating the alternative splicing of caspase-9 include endogenous ceramides (6), SRSF1 (SRp30a or ASF/SF2) (5, 7, 8), and heterogeneous nuclear ribonucleoprotein L (hnRNP L) (4). De novo ceramide synthesis and SRSF1 have been documented to down-regulate the level of caspase-9b mRNA in contrast to hnRNP L, which represses the inclusion of four-exon cassette to favor the formation of caspase-9b. Importantly, phosphorylation of SRSF1 or hnRNP L and subsequent effects on the caspase-9 splicing process have been shown to be important in sensitizing NSCLC cells to chemotherapeutics and promoting the tumorigenic capacity of these cells (4, 8).
hnRNP U, also known as SP120 or scaffold attachment factor A, is capable of binding to both DNA and RNA through the acidic region in the N terminus or the RGG box in the C terminus (9, 10). hnRNP U has been reported to have various biological functions from regulation of gene transcription (11–14) to controlling RNA stability (15) and even participating in the X inactivation process (16, 17) and telomere length control (18). The splicing regulatory function of hnRNP U has just emerged recently (19) with hnRNP U shown to modulate SMN2 splicing via controlling U2 snRNP maturation. Here, we reveal its involvement in regulation of caspase-9 splicing through interacting with target mRNA. In particular, hnRNP U was demonstrated to bind to an RNA cis-element in the exon 3 of caspase-9 (C9/E3) and consequently promoted the four-exon cassette inclusion in the mature caspase-9 mRNA. Our studies also elucidate the mechanism that regulates the interaction between hnRNP U and C9/E3, thereby determining the preferential expression of caspase-9a or caspase-9b via the AKT pathway.
EXPERIMENTAL PROCEDURES
Cell Culture
A549 lung adenocarcinoma cells were cultured in DMEM with 10% (v/v) FBS (Sigma-Aldrich), 100 units/ml penicillin, and 100 μg/ml streptomycin sulfate (Invitrogen). Human bronchial epithelial primary cells (HBEpCs) were cultured in bronchial/tracheal epithelial cell growth medium (Cell Applications). All cells were maintained at less than 80% confluency under standard incubator conditions. For the comparison studies between A549 versus HBEpC cell lines, the cells were plated in tissue culture plates (100 mm), which gave 40% confluency. The following day, the medium was changed to serum-free DMEM, and the cell plates were incubated overnight before total protein or cell lysates were isolated for analysis.
Electrophoretic Mobility Shift Assay
RNA-binding reaction mixtures (20 μl) containing 10 μg of A549 or HBEpC cell lysates, 40 units of RNasin, 11.3 μg of tRNAs, 10 mm HEPES, 5 mm DTT, 120 mm KCl, 3 mm MgCl, 5% glycerol, and 10 μm of FITC-tagged RNA oligonucleotides (C9/E3 WT, C9/E3 Mut1, C9/E3 Mut2, C9/E3 Mut3, or C9/E3 Mut4) were incubated on ice for 20 min. Then 2 μg of hnRNP U antibody or BSA (control) was added to each binding mixture, and the mixtures were incubated for additional 30 min. The samples were loaded on a 5% TBE-polyacrylamide gel for electrophoresis separation. RNA-protein complexes were then visualized by using Molecular Imager FX (Bio-Rad) with a 488-nm EX (530-nm BYPASS) laser.
Densitometric Analysis of the EMSA Supershift
The fold differences in hnRNP U-E3/C9 binding between A549 and HBEpC cells were determined by densitometric measurement of the hnRNP U supershift in the scanned EMSA gels from three independent experiments (n = 4) using ImageJ densitometry software (National Institutes of Health).
Mass Spectrometry Analysis
Analysis of RNA-protein complexes was performed at the Emory University Mass Spectrometry Center (Atlanta, GA) as previously described (4). Briefly, RNA-protein complexes retrieved from EMSA were excised from the gel following by in-gel trysin digestion. The tryptic mixtures were then analyzed by nano-LC-MS/MS. Nano-LC-MS/MS results were obtained by searching through Mascot database (Matrix Science).
siRNA Transfection
A549 or HBEpC cells were transfected with negative control siRNA or hnRNP U SMARTpool siRNA or hnRNP R siRNA (Dharmacon) using Dharmafect 1 transfection reagent (Dharmacon). The cells (3–4 × 105) were plated in each well of 6-well tissue culture dishes in regular growth medium. The following day, the cells were plated in Opti-MEM I medium without antibiotics/FBS and transfected with 100 nm of siRNA (diluted in 1× siRNA buffer). After 4 h of incubation, 0.5 ml of Opti-MEM I medium containing 3-fold the normal concentration of antibiotics/FBS was added to the 1 ml of transfection mixture. The cells in transfection mixture were incubated for an additional 4 h before the medium was changed to normal growth medium. After 48 h, total RNA or protein was collected from the cell lysates.
Quantitative/Competitive RT-PCR
Total RNA was extracted from the cells using the RNeasy mini kit (Invitrogen) and then reverse transcribed to cDNA using SuperScript III reverse transcriptase kit (Invitrogen). The reverse transcription reaction products were utilized in PCR for the endogenous caspase-9 primers (sense primer, 5′-CATGCTGGCTTCGTTTCTG-3′, and antisense primer, 5′-AGGGGCAAACAACAGATGG-3′). PCR was performed in 25 cycles of 94 °C for 30 s, 58 °C for 30 s, and 72 °C for 1 min. The final PCR products were resolved on 5% TBE polyacrylamide gel, stained with SYBR Gold (Invitrogen), and visualized by using Molecular Imager FX (Bio-Rad) with a 488-nm EX (530-nm BYPASS) laser.
Quantitative RT-PCR
Total RNA was extracted from the cells using the RNeasy mini kit (Invitrogen) and then reverse transcribed to cDNA using SuperScript III reverse transcriptase kit (Invitrogen). The reverse transcription reaction products were utilized for real time PCR for Casp9a, Casp9b, and 18 S using TaqMan PCR master mix and the Applied Biosystems 7500 real time PCR system. Casp9a and 18 S quantitative PCR primers were identification numbers Hs00154261_m1 and Hs99999901_s1. The quantitative PCR primers for Casp9b were 5′-GGATTTGGTGATGTCGAGCAG-3′ (forward), 5′-CATCTGGCTCGGGGTTACT-3′ (reverse) and 5′-TTCCCCTGAAGACGAGTCCCCTGG-3′ (probe). The relative amount of Casp9a or Casp9b was normalized to 18 S.
Cell Lysate Preparation
Cell lysates, except for the calf intestinal phosphatase (CIP) experiments, were prepared by lysing the cells in Nonidet P-40 lysis buffer (50 mm Tris-HCl, pH 8.0, 150 mm NaCl, 1% Nonidet P-40, and 1× protease/protein phosphatase inhibitor mixture). For cell lysates utilized for the CIP treatment experiments, the cells were lysed in the same Nonidet P-40 buffer as detailed above, but without protein phosphatase inhibitors. Lysates were also utilized “fresh” for these experiments (e.g., same day) and not stored.
Streptavidin-Biotin Affinity Purification
Reaction mixtures (20 μl) containing 10 μg of cell lysates or 4 μg of IgG (control), 40 units of RNasin, 11.3 of μg tRNAs, 10 mm HEPES, 5 mm DTT, 120 mm KCl, 3 mm MgCl, 5% glycerol, and 10 μm biotinylated RNA oligonucleotide (C9/E3 WT, C9/E3 Mut1, C9/E3 Mut2, C9/E3 Mut3, or C9/E3 Mut4) were incubated on ice for 30 min. Prewashed streptavidin-agarose beads were then added to the reactions following by 2 h of incubation with gentle agitation (4 °C). After incubation, the reactions were washed three times with preblock buffer (100 mm KCl, 20 mm Tris-HCl, pH 7.5, and 0.2 mm EDTA) and centrifuged to collect the pellet. The pellet was resuspended in Laemmli buffer, dry-boiled for 10 min, and subjected to western immunoblotting.
In Vitro Phosphatase Treatment
Phosphatase reaction mixtures containing 10 μg of cell lysate and 10 units of CIP or heat-inactivated CIP (denatured CIP, control) in 1× NEBuffer 3 (New England BioLabs) were incubated at 37 °C. After 1 h of incubation, 1× phosphatase inhibitor mixture was added to the mixtures. The reaction mixtures were then utilized in EMSA or SBAP for RNA-protein binding analysis.
Immunoprecipitation
The cells were lysed in 1× IP buffer containing 1% Triton X-100, 150 mm NaCl, 10 mm Tris, pH 7.4, 1 mm EDTA, pH 8.0, 0.5% Nonidet P-40, and 1× phosphatase/protease inhibitor mixture. Sample lysates (200 μg) were precleared with prewashed protein G-Sepharose beads for 1 h at 4 °C in gentle agitating condition. After preclearing, the samples were centrifuged and collected from the supernatants. 4 μg of hnRNP U or hnRNP L monoclonal antibody was placed in each sample and in 4 °C for 2 h with gentle agitation before prewashed protein G-Sepharose beads were added. The following day, the bead complexes were centrifuged, washed extensively with 1× IP buffer, pelleted by centrifugation, resuspended in Laemmli buffer, dry-boiled for 10 min, and subjected to SDS-PAGE/immunoblotting. For IP experiments coupled with CIP treatment, the cells were lysed in the same IP buffer, but without protein phosphatase inhibitors. The IPs were then subjected to CIP treatments as detailed above.
In Vitro Kinase Assay
Kinase reaction mixtures (20 μl) containing 80 ng of recombinant hnRNP L, 10 mm HEPES, 5 mm DTT, 120 mm KCl, 3 mm MgCl, 5% glycerol, 0.5 mm ATP, 1.25 mm glycerol-2-phosphate, and 200 ng of AKT2 (Sigma) were incubated at 30 °C for 30 min. The reaction mixtures were then utilized in EMSAs for RNA-protein binding analysis or SBAP-based competitive binding assays.
Competitive Binding Assay
Recombinant hnRNP L was subjected to in vitro kinase assay. Kinase reaction mixtures (20 μl) were then incubated with 40 units of RNasin, 11.3 μg of tRNAs, 10 mm HEPES, 5 mm DTT, 120 mm KCl, 3 mm MgCl2, 5% glycerol, and 10 μm biotinylated C9/E3 RNA oligonucleotides (ROs) on ice for 30 min. Prewashed streptavidin-agarose beads were then added to the reactions following by 2 h of incubation with gentle agitation (4 °C). After incubation, the reactions were washed three times with preblock buffer (100 mm KCl, 20 mm Tris-HCl, pH 7.5, and 0.2 mm EDTA) and centrifuged to pellet the bead complexes. The bead complexes were then incubated with binding mixtures (20 μl) containing 10 μg of HBEpC cell lysates, 40 units of RNasin, 11.3 μg of tRNAs, 10 mm HEPES, 5 mm DTT, 120 mm KCl, 3 mm MgCl, and 5% glycerol for additional 2 h. After incubation, the bead complexes were washed three times with preblock buffer (100 mm KCl, 20 mm Tris-HCl, pH 7.5, and 0.2 mm EDTA), pelleted by centrifugation, resuspended in Laemmli buffer, dry-boiled for 10 min, and subjected to SDS-PAGE/immunoblotting.
Statistics
Statistical differences in the mRNA level between the control and hnRNP U siRNA-treated cells were determined by a two-tailed, unpaired Student's t test. Statistical differences in the association of hnRNP U with E3/C9 between A549 and HBEpC cells were determined by one-sample t test. p values of less than or equal to 0.05 were considered significant.
RESULTS
Identification of hnRNP U Associated with Exon 3 of Caspase-9 and Suppressing the Formation of the Caspase-9b Isoform
Using EMSA coupled to nanospray LC-MS/MS analysis, two RNA trans-factors, hnRNP L and hnRNP A2/B1, were previously identified to associate with the purine-rich sequences in C9/E3 (Table 1) (4). hnRNP L was then further characterized to modulate the alternative splicing of caspase-9 pre-mRNA as a repressor of the inclusion of the exon 3,4,5,6 cassette. Here, we found two additional RNA trans-factors interacting with C9/E3: hnRNP U and hnRNP R. The association between hnRNP U/R and C9/E3 suggested a possible biological function in regulating caspase-9 mRNA processing, which was further examined by employing siRNA technology. Whereas down-regulation of hnRNP U reduced the endogenous caspase-9a/9b mRNA ratio in both transformed A549 cells and nontransformed HBEpCs (Fig. 1, A–D), hnRNP R depletion yielded no change in the ratio of caspase-9a/9b mRNA (Fig. 1, A and B). Importantly, the effect of hnRNP U on the mRNA ratio translated to the protein level (Fig. 1E). These data indicate that hnRNP U inhibits the formation of caspase-9b via suppressing the exclusion of four-exon cassette in the mature caspase-9 mRNA.
TABLE 1.
RNA trans-factors associated with exon 3 of caspase-9 in NSCLC cells
| RNA trans-factors |
|---|
| hnRNP A2/B1 |
| hnRNP L |
| hnRNP R |
| hnRNP U |
FIGURE 1.

hnRNP U, but not hnRNP R, represses the formation of caspase-9b via RNA splicing. A549 or HBEpC cells were transfected with control siRNA (100 nm), hnRNP U SMARTpool siRNA (100 nm), or hnRNP R siRNA (100 nm) for 48 h. A, total RNA was isolated and analyzed by competitive/quantitative RT-PCR for caspase-9 splice variant ratio using ratio-based quantitation. Ctr, control. B, total protein extracts were subjected to SDS/PAGE/immunoblotting for hnRNP U, hnRNP R, or β-actin. C, graphs were created from the results in A. D, total RNA isolated from A549 cells transfected with control or hnRNP U siRNA was analyzed for relative amount of caspase-9a or 9b mRNA by quantitative RT-PCR using the standard curve method and 18 S rRNA as a normalizing control. E, total protein extracts were subjected to SDS/PAGE/immunoblotting for caspase-9 or β-actin. The data are shown as the means ± S.D. (n ≥ 4 on at least two independent occasions). F, A549 cells were transfected with siRNA as in A. After 48 h, the cells were treated with actinomycin D (5 μg/ml) for 0, 2, 4, or 8 h. RNA was isolated, and the relative amount of caspase-9a or 9b mRNA (fold change compared with the si-control or si-hnRNP U samples at 0 h) was individually determined by quantitative RT-PCR. G, half-life of caspase-9a or caspase-9b mRNA was calculated based on the data in F. H, total protein from siRNA transfected cells (48 h) was subjected to SDS/PAGE/immunoblotting for hnRNP U or β-actin. Statistical significance was evaluated by Student's t test (p < 0.0001, n = 5 on two independent occasions).
Because the two splice variants of caspase-9, caspase-9a and caspase-9b, are generated from the same gene and have the same 5′-UTR and 3′-UTR sequences, the effect of hnRNP U depletion on caspase-9a/9b ratio is likely attributable to the regulation of the alternative splicing of caspase-9 rather than mRNA stability. Indeed, no significant difference in the decay rate (specifically, RNA half-life t½) of caspase-9a or caspase-9b mRNA was observed after treatment of the control versus hnRNP U-depleted cells with actinomycin D (Fig. 1, F–H). These data “rule out” the possibility that hnRNP U represses the production of caspase-9b by stabilizing caspase-9a mRNA or destabilizing caspase-9b mRNA. Hence, regulation of the alternative splicing of caspase-9 is most likely the mechanism contributing to the observed enhancing function of hnRNP U in the inclusion of the four-exon cassette.
hnRNP U Binds Specifically to Exon 3 of Caspase-9 Pre-mRNA
The LC-MS/MS results strongly suggested that hnRNP U interacted with C9/E3. To determine whether hnRNP U interacts specifically with C9/E3, EMSA coupled with an hnRNP U “supershift” was performed (Fig. 2A and Table 2 for sequences utilized). Indeed, a supershift was observed after the addition of an anti-hnRNP U antibody, indicating that hnRNP U is present in the C9/E3-protein complex and thereby verifying the LC-MS/MS results (Fig. 2B). Furthermore, this supershift was specific for hnRNP U because reduction in levels of hnRNP U via siRNA resulted in the loss of the supershift (Fig. 2, C and D).
FIGURE 2.

hnRNP U binds to exon 3 of caspase-9. A, schematic illustration of caspase-9 pre-mRNA structure. 18-mer C9/E3 ROs used in RNA binding assays are corresponding to the purine-rich sequence in C9/E3. oligos, oligonucleotides. B and C, EMSAs were performed in the absence of protein phosphatase inhibitors using 5′-FITC-tagged C9/E3 ROs and in the presence of IgG (control), A549 cell lysates (B), or lysates from A549 cells transfected with control siRNA or hnRNP U SMARTpool siRNA (C). Ab, antibody. D, cell lysate input in C was subjected to SDS-PAGE and immunoblotted for hnRNP U and β-actin. E, SBAP was performed using 5′-biotinylated nonspecific control (NSC), 5′-biotinylated wild-type C9/E3 ROs (E3/C9 WT), or 5′-biotinylated mutant C9/E3 ROs (Mut1–Mut4) in the presence of IgG (control) or A549 cell lysates. F, EMSAs were performed as in C, but using additional ROs (5′-FITC-tagged C9/E3 Mut1–Mut4). Arrows indicate the supershifts obtained by the addition of the hnRNP U antibody. The graphs shown in B, C, E, and F are representative of n = 4 from three independent experiments.
TABLE 2.
RNA oligonucleotides utilized in the RNA binding assays (EMSA or SBAP)
| RNA oligonucleotide code | Sequence |
|---|---|
| NSC | CAGAAUUCGCACGUUACU |
| C9/E3 WT | GAGAGUUUGAGGGGAAAU |
| C9/E3 Mut1 | CAGAAUUUGAGGGGAAAU |
| C9/E3 Mut2 | GAGAGCCCGAGGGGAAAU |
| C9/E3 Mut3 | GAGAGUUUCUACUGAAAU |
| C9/E3 Mut4 | GAGAGUUUGAGGGUUACU |
Mutagenesis analysis was employed to further determine/validate the binding site for hnRNP U in C9/E3. As shown in Fig. 2 (E and F), the interaction between hnRNP U and C9/E3 was completely abolished in one of four C9/E3 mutations (Mut3). Interestingly, this interaction site has been previously identified as the binding site for hnRNP L, a repressor that regulates the alternative splicing of caspase-9 (4). Two additional mutations (Mut2 and Mut4) flanking this site also significantly diminished the association of hnRNP U to C9/E3 (Fig. 2, E and F). These findings reveal that hnRNP U specifically interacts with the hnRNP L binding site in C9/E3. Also of note, an almost complete loss of protein-RNA complexes was observed with Mut3 in accord with our previous findings of hnRNP L binding to this region. These data also suggest that hnRNP L, hnRNP U, or both may also target additional RNA trans-factors to exon 3 of caspase-9.
Phosphorylation Regulates the Interaction between hnRNP U and Exon 3 of Caspase-9 Pre-mRNA
Although hnRNP U was expressed at the same level in nontransformed (HBEpC) versus transformed (A549) cells (Fig. 3A), the association of this RNA trans-factor to C9/E3 in HBEpC cells was significantly higher than in A549 cells (Fig. 3, B and C). Specifically, the fold difference as assayed by EMSA analysis was 1.70 ± 0.18 (mean ± S.D.) (p < 0.01).
FIGURE 3.

Phosphorylation regulates the association of hnRNP U with exon 3 of caspase-9. A, hnRNP U is expressed at the same level in both transformed and nontransformed cells. A549 (transformed) and HBEpC (nontransformed) cells were cultured under the same condition (DMEM with/without FBS) and confluency. Total protein extracts were subjected to SDS-PAGE/immunoblotting for hnRNP U and β-actin. B and C, hnRNP U binds to C9/E3 in higher amounts in HBEpCs than in A549 cells. SBAP assays (B) or EMSAs in the presence of protein phosphatase inhibitors (C) were performed with either IgG (control) or lysates from A549 or HBEpC cells using a 5′-biotinylated or 5-FITC-tagged wild-type C9/E3 ROs. Ab, antibody. D and E, dephosphorylation increases the binding affinity of hnRNP U to C9/E3. A549 cell lysates (fresh) were preincubated with denatured calf intestinal alkaline phosphatase (De-CIP) or active protein phosphatase (CIP) followed by EMSA (D) in the absence of protein phosphatase inhibitors or SBAP (E) to assay the binding with 5′-FITC tagged (D) or 5′-biotinylated (E) C9/E3 ROs. IgG was used as a control. F, dephosphorylation abolishes the difference between A549 versus HBEpC regarding the binding of hnRNP U to E3/C9. A549 (transformed) and HBEpC (nontransformed) cells were cultured under the same conditions and confluency, and cell lysates were produced. Lysates (fresh) were again treated and assayed as in E. G, hypothetical model for regulation of hnRNP U/L-C9/E3 interaction by phosphorylation. Whereas the phosphorylation event augments the association between hnRNP L and C9/E3, phosphorylation of hnRNP U attenuates its binding to C9/E3. The graphs shown in A–F are representative of n = 4 from three independent experiments.
Selective phosphorylation of hnRNP U has been reported in a study by Berglund and Clarke (20). Thus, we hypothesized that the enhanced binding of hnRNP U to C9/E3 in nontransformed cells compared with transformed cells was due to the differences in post-translational modification, particularly the phosphorylation state. To address this possibility, protein phosphatase treatments coupled with RNA binding assays were undertaken. Congruent with our hypothesis, the dephosphorylation of NSCLC (A549) cell lysates resulted in significant enhancement of the association of hnRNP U to C9/E3 (Fig. 3, C–E). These findings implicate phosphorylation as a regulatory mechanism for hnRNP U-C9/E3 interaction, which may explain the differences observed in nontransformed versus transformed cells regarding hnRNP U-C9/E3 binding.
To determine whether the phosphorylation of RNA trans-factors was the mitigating factor in modulating the differential association of hnRNP U to C9/E3 in transformed cells versus nontransformed cells, both A549 and HBEpC cell lysates were subjected to phosphatase (CIP) treatment (denatured versus active) in the absence of protein phosphatase inhibitors. Dephosphorylation of A549 cell lysates increased the association of hnRNP U to C9/E3 to comparable levels of the binding observed in HBEpC cell lysates, whereas the association of hnRNP U in the nontransformed cells was unaffected (Fig. 3F). These data demonstrated that the disparity in hnRNP U binding between transformed and nontransformed cells is due to the phosphorylation state of RNA trans-factors (Fig. 3G).
Inhibition of the AKT Pathway Promotes the Association of hnRNP U with C9/E3 and Alters the Ratio of Caspase-9 Splice Variants
The phosphoinositide 3-kinase/AKT pathway has been reported to modulate the alternative splicing of caspase-9 (5), and we hypothesized that this signaling pathway modulates the ability of hnRNP U to bind C9/E3. Indeed, treatment of the NSCLC cells with AKT1/2 inhibitor increased the binding of hnRNP U to C9/E3 in a dose-responsive manner but did not significantly affect its expression level (Fig. 4A). Importantly, enhanced association of hnRNP U to C9/E3 by AKT inhibitor treatment correlated with the increasing caspase-9a/9b ratio in NSCLC cells (Fig. 4A). In contrast, ectopic expression of constitutively active AKT2 (caAKT2) in nontransformed HBEpC cells attenuated the binding of hnRNP U to C9/E3, which correlated with a decrease in the caspase-9a/9b ratio (Fig. 4B). These data demonstrate that AKT signaling regulates the interaction of hnRNP U with caspase-9 pre-mRNA.
FIGURE 4.

AKT pathway regulates the association of hnRNP U to exon 3 of caspase-9. A, inhibition of AKT1/2 activity increases the caspase-9a/9b ratio and enhances the binding of hnRNP U to C9/E3 in a dose-response manner. A549 cells were treated with 0.1% DMSO control or increasing concentrations of the AKT1/2 inhibitor (AKT VIII). Total RNA was isolated and analyzed for caspase-9 splice variants by competitive/quantitative RT-PCR. Cell lysates from a concomitant experiment were utilized in SBAP to assay the binding of hnRNP U to 5′-biotinylated C9/E3 ROs. Cell lysate input for the SBAP was subjected to SDS-PAGE and immunoblotted for hnRNP U and β-actin. B, overexpression of AKT decreases both caspase-9a/9b ratio and hnRNP U-E3/C9 association. HBEpC cells were transfected with the control adenovirus or the adenovirus expressing constitutive active AKT2 (caAKT2; multiplicity of infection of 25) for 48 h. Total protein or RNA extracts from the transfected cells were then subjected to SDS-PAGE/immunoblotting or competitive/quantitative RT-PCR. Cell lysates were also generated for SBAP to assay the binding of hnRNP U to E3/C9 ROs. The graphs shown in A and B are representative of n = 4 from three independent experiments.
The Phospho-state of hnRNP U Is Not Affected by AKT Inhibition
The culmination of our data coupled to reports in the literature of hnRNP U being selectively phosphorylated led to the initial hypothesis that the phospho-state of hnRNP U regulates the association of the RNA trans-factor to C9/E3 (Fig. 3G). In direct opposition to this hypothesis, treatment of A549 cells with an AKT inhibitor had no significant effect on the phosphorylation state of hnRNP U (Fig. 5A), although in vitro phosphatase treatment showed a minor effect on serine phosphorylation (Fig. 5B). These data demonstrate that phospho-state of hnRNP U does not play an important role in regulating the association of the RNA trans-factor with C9/E3.
FIGURE 5.
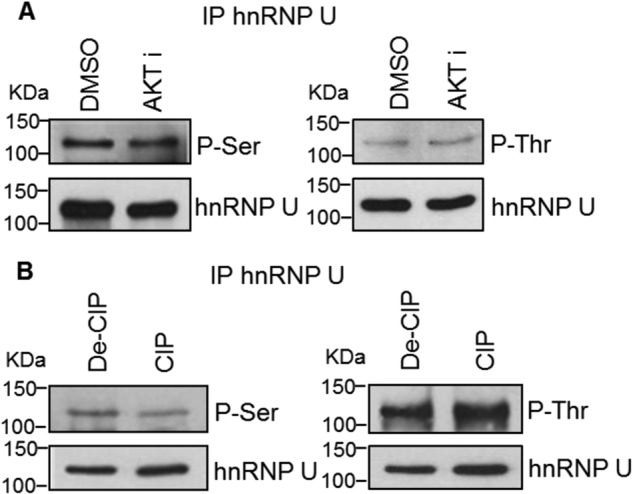
AKT inhibition does not alter the phospho-state of hnRNP U. A, A549 cells were treated with 0.1% dimethyl sulfoxide (DMSO) control or AKT1/2 inhibitor (20 μm). Endogenous hnRNP U was then immunoprecipitated (IP) and resolved by SDS-PAGE/immunoblotting for phospho-serine/threonine or hnRNP U. B, A549 protein extracts were incubated with denatured or active CIP. Endogenous hnRNP U in the resulted protein extracts was immunoprecipitated and resolved by SDS-PAGE/immunoblotting for phosphoserine/phosphothreonine or hnRNP U. The graphs shown in A and B are representative of n = 3 from two independent experiments.
hnRNP L Is Phosphorylated in an AKT-dependent Manner Inducing Competition with hnRNP U for C9/E3 Binding
Because the phospho-state of hnRNP U is not critical to hnRNP U-C9/E3 interaction, we hypothesized that the phosphorylation of other regulatory RNA trans-factors modulated the ability of hnRNP U to associate with C9/E3. In this regard, hnRNP U shares the same binding site as hnRNP L, which has the opposite function in regards to regulating the alternative splicing of caspase-9 pre-mRNA. Therefore, we examined the direct binding of recombinant hnRNP L purified from nontransformed HEK293 cells to the C9/E3. Interestingly, direct binding of recombinant hnRNP L to this purine-rich sequence was not observed by EMSA (Fig. 6A, first lane), congruent with a report showing the interaction of this recombinant hnRNP L to be specific for CA repeats (21). Because caspase-9 RNA splicing is regulated by the phosphorylation of hnRNP L (4), we examined whether AKT directly phosphorylates hnRNP L and induces the association of the RNA trans-factor with C9/E3. Intriguingly, AKT efficiently phosphorylated hnRNP L, which induced its binding to C9/E3 (Fig. 6A). Moreover, inhibition of AKT signaling in NSCLC cells directly by AKT inhibitor treatment or indirectly by blocking EGFR activity with erlotinib treatment significantly reduced the phospho-state of hnRNP L (Fig. 6B). Lastly, hnRNP L phosphorylated by AKT interfered with hnRNP U for binding to C9/E3 (Fig. 6C) in contrast to nonphosphorylated hnRNP L. Taken together, these data demonstrate that the phospho-status of hnRNP L is regulated by AKT signaling in NSCLC, which induces the association of this RNA trans-factor and restricts the access of the enhancing factor, hnRNP U, to E3/C9 (Fig. 6D).
FIGURE 6.

AKT-dependent phosphorylation of hnRNP L leads to the competition with hnRNP U for C9/E3 binding. A, phosphorylation of hnRNP L by AKT enhances binding to E3/C9. Recombinant FLAG-tagged hnRNP L was phosphorylated in vitro using active AKT2 (denatured AKT2 was utilized as a negative control). Products from the kinase assay were then subjected to SDS-PAGE/immunoblotting for phosphoserine/phosphothreonine, or the FLAG tag and also utilized in the EMSA to examine the interaction with E3/C9 ROs. B, AKT or EGFR inhibition in NSCLC cells induces a reduction in the phospho-status of hnRNP L. A549 cells were treated with either 0.1% dimethyl sulfoxide (DMSO) control, AKT inhibitor (20 μm), or erlotinib (EGFR inhibitor, 1 μm). Endogenous hnRNP L was immunoprecipitated (IP) from the protein extracts. Immunoprecipitated hnRNP L was resolved by SDS-PAGE and immunoblotted with Ser(P)52-hnRNP L or hnRNP L antibodies. C, exogenous hnRNP L phosphorylated by AKT competes with endogenous hnRNP U for binding to E3/C9. Recombinant hnRNP L was incubated with denatured or active AKT2 in the in vitro kinase assay and then utilized in the SBAP-based competitive binding assay using HBEpC cell lysates and E3/C9 ROs. D, model for controlling hnRNPU-hnRNP L-E3/C9 interaction by AKT pathway. In the nontransformed cells, the recruitment of hnRNP L to E3/C9 is interfered with by hnRNP U binding to the same position. Through activation of AKT pathway in transformed cells, hnRNP L is phosphorylated, which results in the enhanced binding of this splicing repressor to E3/C9. Consequently, the elevated levels of hnRNP L associated with E3/C9 prevent the access of hnRNP U to E3/C9 for splice-enhancing function. The graphs shown in A–C are representative of n = 4 from two independent experiments.
DISCUSSION
In this study, two additional C9/E3-associated proteins were identified by LC-MS/MS: hnRNP R and hnRNP U. Whereas hnRNP R had no effect on caspase-9 splicing, hnRNP U was revealed as a splicing enhancer and validated for interaction with C9/E3. To date, little is known about the regulatory function of hnRNP U in RNA splicing, although it is regarded as an RNA-binding protein. A recent study suggested that hnRNP U might be a global regulator of alternative splicing; however, >90% of reported cases about an RNA splicing change induced by hnRNP U down-regulation did not accompany binding to respective mRNA (19). Our findings expand the situations where hnRNP U-regulated splicing events are associated with target mRNA interactions.
The precise site for hnRNP U binding on C9/E3 was also examined. Previously, hnRNP U has been reported to preferentially bind G/U-rich motifs in RNA (9, 10). In line with these reports, hnRNP U was demonstrated to specifically interact with the G-rich sequence, GAGGG, in C9/E3. Furthermore, mutation of the upstream U motif (UUUGAGGG) also significantly reduced the association of hnRNP U with the C9/E3. Importantly, this sequence has been reported as a binding site for hnRNP L. The opposing functions of hnRNP U versus hnRNP L and the same binding site on C9/E3 suggest regulatory competition between hnRNP U and hnRNP L, and our data further suggest that the presence of hnRNP U likely restricts the exon exclusion effect of nonphosphorylated hnRNP L. Furthermore, the dramatic loss of the protein-RNA complex by mutation of this RNA cis-element suggests that hnRNP L and U may target additional RNA trans-factors to exon 3. Current experimental directions are focused on determining whether the phosphorylation of hnRNP L leads to loss or gain of binding partners, which aid in regulating the inclusion/exclusion of the exon 3,4,5,6 cassette.
The nature of the regulation of the hnRNP U and L competition was also elucidated in this study. For example, simple changes in expression of the RNA trans-factors did not account for the observed loss of hnRNP U binding in transformed cells. Specifically, the expression of hnRNP U and L was not significantly different between nontransformed and transformed cells. Previous studies in our laboratory have pointed to the importance of phosphorylation in regulating hnRNP L-C9/E3 interaction (4), and hnRNP U has been reported to be selectively phosphorylated in response to DNA double-strand breaks (20, 22). Therefore, we hypothesized that phosphorylation events might impact the association between hnRNP U and C9/E3. Nevertheless, our findings in this study demonstrate that phosphorylation of hnRNP U is unlikely to play a role, and the regulatory mechanism is mainly via phosphorylation of hnRNP L. Together with the findings that caspase-9a/9b ratio is higher in nontransformed than in transformed cells (4, 5) and the enhanced association of hnRNP U in nontransformed cells, these data extend the mechanistic insights into the dysregulation of caspase-9 splicing in transformed cells. In this paradigm, certain survival/oncogenic kinases are activated and phosphorylate hnRNP L in transformed cells. As a result, the hnRNP L-C9/E3 interaction and subsequent splice-repressing function is promoted, whereas hnRNP U is prevented from binding and competing with hnRNP L (Fig. 6D). Therefore, the exclusion of the four-exon cassette is favored, shifting the alternative splicing of caspase-9 to the lower caspase-9a/9b ratio observed in NSCLC cells.
Because the enhanced exclusion of exon 3,4,5,6 cassette has been linked to phosphorylation events modulated by phosphoinositide 3-kinase/AKT pathway (5), this cell survival regulatory pathway likely mediates the phosphorylation of hnRNP L and subsequently the competitive interaction between hnRNP L and hnRNP U to C9/E3. Indeed, inhibition of AKT signaling in transformed cells augmented hnRNP U-C9/E3 binding and the enhanced binding to the C9/E3 were correlated perfectly with alteration in caspase-9 splice variant ratio. Hence, the AKT pathway regulates the alternative splicing of caspase-9 via hindering the interaction between hnRNP U and C9/E3 and subsequently suppressing its splice-enhancing function. Our study also demonstrates that hnRNP L is a direct substrate for AKT, and thus, AKT may be directly influencing the alternative splicing of caspase-9 via hnRNP L. Direct phosphorylation of hnRNP L by AKT is not heretical because AKT has been shown to directly phosphorylate SRSF1 (SRp30a or ASF/SF2), another RNA trans-factor known to regulate the alternative splicing of caspase-9 (5).
In regards to SRSF1 and caspase-9 RNA splicing, the presented study fills a missing “gap” in our knowledge of how the exon 3,4,5,6 cassette is regulated. Previously, we reported that the phosphorylation of SRSF1 on novel phosphorylation sites flanking the RS domain were partially required for AKT activation to exclude the exon 3,4,5,6 cassette (5). Specifically, we showed that a phospho-mimic of SRSF1 could only partially block the exclusion of the exonic cassette of caspase-9 pre-mRNA induced by an AKT inhibitor. This study now places hnRNP L phosphorylation and subsequent hnRNP U displacement from exon 3 as the plausible missing mechanism by which AKT activation maximally induces the exclusion of the exon 3,4,5,6 cassette.
One of the more intriguing findings is the observation that phosphorylation of hnRNP L induces the association of the RNA trans-factor with a nonstandard binding site. Numerous reports in the literature have demonstrated that hnRNP L prefers CA repeats. Indeed, we initially found that recombinant hnRNP L did not bind the purine sequence, in line with previous reports; however, hnRNP L can be modulated to associate with different sequence specificity by phosphorylation. Our data therefore suggest that hnRNP L may have both constitutive roles in RNA splicing, as well as activated roles in modulating a subset of splicing events. How phosphorylation would change the sequence specificity of an RNA trans-factor has not been described, but possibly one or more quasi-RNA-recognition motif could be “unmasked” by an intramolecular change in the structure of the RNA trans-factor stimulated by phosphorylation. This mechanism is plausible because hnRNP K, a related RNA trans-factor, has been reported to have a quasi-RNA-recognition motif that does not recognize the standard GC-rich RNA sequence (23). Overall, phosphorylation may be a key regulatory step in activating RNA splicing factors to induce specific RNA splicing events (e.g., caspase-9 RNA splicing) in response to external stimuli outside of their roles in constitutive RNA splicing.
In conclusion, our presented work implicates hnRNP U as a limiting factor for caspase-9b formation, which has been previously demonstrated to enhance the anchorage-dependent growth and tumorigenic capacity of NSCLC cells (4). Moreover, cascape-9b is not significantly expressed in nontransformed cells (4), and the induction of caspase-9b expression is due to activation of hnRNP L via phosphorylation to compete/inhibit hnRNP U association with C9/E3. Therefore, targeting the phosphorylation of hnRNP L by specific kinase inhibitors to further augment the formation of apoptotic caspase-9a over anti-apoptotic caspase-9b would be an attractive cancer-specific approach for treatment of NSCLC, the leading cause of cancer-related death in both men and women worldwide (24).
This work was supported, in whole or in part, by National Institutes of Health Grants CA154314 (to C. E. C.) and R01 GM067719 (to K. W. L.). This work was also supported by a merit review and research career scientist award from the Department of Veterans Affairs (to C. E. C.), a fellowship from the Vietnam Education Foundation (to N. T. V.), and a fellowship from the American Heart Association (to S. A. S.).
- NSCLC
- non-small cell lung cancer
- CIP
- calf intestinal alkaline phosphatase
- C9/E3
- exon 3 of caspase-9
- HBEpC
- human bronchial epithelial primary cells
- IP
- immunoprecipitation
- RO
- RNA oligonucleotide
- SBAP
- streptavidin-biotin affinity purification.
REFERENCES
- 1. Elmore S. (2007) Apoptosis. A review of programmed cell death. Toxicol. Pathol. 35, 495–516 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Seol D.-W., Billiar T. R. (1999) A caspase-9 variant missing the catalytic site is an endogenous inhibitor of apoptosis. J. Biol. Chem. 274, 2072–2076 [DOI] [PubMed] [Google Scholar]
- 3. Srinivasula S. M., Ahmad M., Guo Y., Zhan Y., Lazebnik Y., Fernandes-Alnemri T., Alnemri E. S. (1999) Identification of an endogenous dominant-negative short isoform of caspase-9 that can regulate apoptosis. Cancer Res. 59, 999–1002 [PubMed] [Google Scholar]
- 4. Goehe R. W., Shultz J. C., Murudkar C., Usanovic S., Lamour N. F., Massey D. H., Zhang L., Camidge D. R., Shay J. W., Minna J. D., Chalfant C. E. (2010) hnRNP L regulates the tumorigenic capacity of lung cancer xenografts in mice via caspase-9 pre-mRNA processing. J. Clin. Invest. 120, 3923–3939 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Shultz J. C., Goehe R. W., Wijesinghe D. S., Murudkar C., Hawkins A. J., Shay J. W., Minna J. D., Chalfant C. E. (2010) Alternative splicing of caspase-9 is modulated by the phosphoinositide 3-kinase/Akt pathway via phosphorylation of SRp30a. Cancer Res. 70, 9185–9196 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Chalfant C. E., Rathman K., Pinkerman R. L., Wood R. E., Obeid L. M., Ogretmen B., Hannun Y. A. (2002) De novo ceramide regulates the alternative splicing of caspase-9 and Bcl-x in A549 lung adenocarcinoma cells. Dependence on protein phosphatase-1. J. Biol. Chem. 277, 12587–12595 [DOI] [PubMed] [Google Scholar]
- 7. Massiello A., Chalfant C. E. (2006) SRp30a (ASF/SF2) regulates the alternative splicing of caspase-9 pre-mRNA and is required for ceramide-responsiveness. J. Lipid Res. 47, 892–897 [DOI] [PubMed] [Google Scholar]
- 8. Shultz J. C., Goehe R. W., Murudkar C. S., Wijesinghe D. S., Mayton E. K., Massiello A., Hawkins A. J., Mukerjee P., Pinkerman R. L., Park M. A., Chalfant C. E. (2011) SRSF1 regulates the alternative splicing of caspase-9 via a novel intronic splicing enhancer affecting the chemotherapeutic sensitivity of non-small cell lung cancer cells. Mol. Cancer Res. 9, 889–900 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Kiledjian M., Dreyfuss G. (1992) Primary structure and binding activity of the hnRNP U protein. Binding RNA through RGG box. EMBO J. 11, 2655–2664 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Fackelmayer F. O., Dahm K., Renz A., Ramsperger U., Richter A. (1994) Nucleic-acid-binding properties of hnRNP-U/SAF-A, a nuclear-matrix protein which binds DNA and RNA in vivo and in vitro. Eur. J. Biochem. 221, 749–757 [DOI] [PubMed] [Google Scholar]
- 11. Eggert M., Michel J., Schneider S., Bornfleth H., Baniahmad A., Fackelmayer F. O., Schmidt S., Renkawitz R. (1997) The glucocorticoid receptor is associated with the RNA-binding nuclear matrix protein hnRNP U. J. Biol. Chem. 272, 28471–28478 [DOI] [PubMed] [Google Scholar]
- 12. Kim M. K., Nikodem V. M. (1999) hnRNP U inhibits carboxy-terminal domain phosphorylation by TFIIH and represses RNA polymerase II elongation. Mol. Cell Biol. 19, 6833–6844 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Kukalev A., Nord Y., Palmberg C., Bergman T., Percipalle P. (2005) Actin and hnRNP U cooperate for productive transcription by RNA polymerase II. Nat. Struct. Mol. Biol. 12, 238–244 [DOI] [PubMed] [Google Scholar]
- 14. Obrdlik A., Kukalev A., Louvet E., Farrants A. K., Caputo L., Percipalle P. (2008) The histone acetyltransferase PCAF associates with actin and hnRNP U for RNA polymerase II transcription. Mol. Cell Biol. 28, 6342–6357 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Yugami M., Kabe Y., Yamaguchi Y., Wada T., Handa H. (2007) hnRNP-U enhances the expression of specific genes by stabilizing mRNA. FEBS Lett. 581, 1–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Hasegawa Y., Brockdorff N., Kawano S., Tsutui K., Tsutui K., Nakagawa S. (2010) The matrix protein hnRNP U is required for chromosomal localization of Xist RNA. Dev. Cell 19, 469–476 [DOI] [PubMed] [Google Scholar]
- 17. Helbig R., Fackelmayer F. O. (2003) Scaffold attachment factor A (SAF-A) is concentrated in inactive X chromosome territories through its RGG domain. Chromosoma 112, 173–182 [DOI] [PubMed] [Google Scholar]
- 18. Fu D., Collins K. (2007) Purification of human telomerase complexes identifies factors involved in telomerase biogenesis and telomere length regulation. Mol. Cell 28, 773–785 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Xiao R., Tang P., Yang B., Huang J., Zhou Y., Shao C., Li H., Sun H., Zhang Y., Fu X. D. (2012) Nuclear matrix factor hnRNP U/SAF-A exerts a global control of alternative splicing by regulating U2 snRNP maturation. Mol. Cell 45, 656–668 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Berglund F. M., Clarke P. R. (2009) hnRNP-U is a specific DNA-dependent protein kinase substrate phosphorylated in response to DNA double-strand breaks. Biochem. Biophys. Res. Commun. 381, 59–64 [DOI] [PubMed] [Google Scholar]
- 21. Motta-Mena L. B., Heyd F., Lynch K. W. (2010) Context-dependent regulatory mechanism of the splicing factor hnRNP L. Mol. Cell 37, 223–234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Britton S., Froment C., Frit P., Monsarrat B., Salles B., Calsou P. (2009) Cell nonhomologous end joining capacity controls SAF-A phosphorylation by DNA-PK in response to DNA double-strand breaks inducers. Cell Cycle 8, 3717–3722 [DOI] [PubMed] [Google Scholar]
- 23. Matunis M. J., Michael W. M., Dreyfuss G. (1992) Characterization and primary structure of the poly(C)-binding heterogeneous nuclear ribonucleoprotein complex K protein. Mol. Cell. Biol. 12, 164–171 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Molina J. R., Yang P., Cassivi S. D., Schild S. E., Adjei A. (2008) Non-small cell lung cancer. Epidemiology, risk factors, treatment and survivorship. Mayo Clin. Proc. 83, 584–594 [DOI] [PMC free article] [PubMed] [Google Scholar]