

Background: DPP stimulates differentiation of mesenchymal cells to osteogenic lineage.
Results: DPP promotes differentiation by activating the intracellular Ca2+ influx and CaMKII-Smad1 signaling.
Conclusion: DPP activates Ca2+-mediated signaling events, thereby playing a crucial role in osteoblast differentiation.
Significance: DPP stimulates osteogenic phenotypic alterations in pluripotent stem cells by activating CaMKII-Smad1 signaling.
Keywords: Bone, Calcium, Calcium-Calmodulin-dependent Protein Kinase (CaMK), Differentiation, Signal Transduction
Abstract
Dentin phosphophoryn (DPP) is a major noncollagenous protein in the dentin matrix. In this study, we demonstrate that pluripotent stem cells such as C3H10T1/2 and human bone marrow cells can be committed to the osteogenic lineage by DPP. Treatment with DPP can stimulate the release of intracellular Ca2+. This calcium flux triggered the activation of Ca2+-calmodulin-dependent protein kinase II (CaMKII). Activated CaMKII induced the phosphorylation of Smad1 and promoted nuclear translocation of p-Smad1. Inhibition of store Ca2+ depletion by 1,2-bis(2-aminophenoxy)ethane-N,N,N′,N′-tetraacetic acid tetrakis(acetoxymethyl ester) or down-regulation of CaMKII by KN-62, a selective cell-permeable pharmacological inhibitor or a dominant negative plasmid of CaMKII, blocked DPP-mediated Smad1 phosphorylation. Activation of Smad1 resulted in the expression of osteogenic markers such as Runx2, Osterix, DMP1, Bone sialoprotein, Osteocalcin, NFATc1, and Schnurri-2, which have been implicated in osteoblast differentiation. These findings suggest that DPP is capable of triggering commitment of pluripotent stem cells to the osteogenic lineage.
Introduction
Differentiation of mesenchymal stem cells toward the osteogenic lineage is a collaborative/synergistic process involving a combination of several intracellular signaling pathways triggered by the extracellular matrix (ECM)3 proteins and growth factors. Increased evidence indicates that during osteogenesis cells employ specific mechanisms to decipher signals that facilitate interaction with the components of the dynamic three-dimensional ECM to initiate osteoblast differentiation. One such ECM protein in the dentin and bone matrix is dentin phosphophoryn (DPP).
DPP is a highly acidic, noncollagenous phosphoprotein localized predominantly in the dentin matrix (1). It is highly negatively charged with aspartyl and seryl residues making up about 75% of the total amino acids of which 85–90% of the seryl residues are phosphorylated (2–4). It has extensive sequences of (DSS)n repeats with n as large as 24 (5, 6), and these domains are responsible for matrix mineralization. Published reports show that DPP is synthesized from a larger compound protein called dentin sialophosphoprotein, which appears to be cleaved immediately in vivo into N-terminal dentin sialoprotein and C-terminal DPP (7). Increasing evidence indicates that DPP has a high affinity for calcium (8, 9) and can regulate matrix mineralization (1, 7, 10, 11). This is clearly evident in dentinogenesis imperfecta type II disorders, where a decrease in DPP content leads to impaired dentin mineralization (7).
DPP not only enhances matrix mineralization, it also stimulates gene expression responsible for osteogenic differentiation in a variety of cell types, including mesenchymal stem cells, pre-osteoblasts, and nonosseous fibroblasts (12). Several reports suggest that DPP is present in other tissues like lungs, kidneys, and salivary glands (2, 13). The signaling function of DPP was demonstrated during embryonic development of the kidneys, particularly facilitating epithelial-mesenchymal interactions in meristic tissues (14). Recently, we have shown that the RGD domain in DPP is functional and signals through cell surface integrins (15). DPP signaling via the MAPK and Smad pathways independent of bone morphogenetic protein has been demonstrated previously (12). However, little is known regarding the transcription factors and signaling pathways by which DPP mediates the commitment of pluripotent stem cells to the osteogenic lineage.
In this study, we report that stimulation of undifferentiated mesenchymal C3H10T1/2 cells by DPP results in a transient increase in the intracellular Ca2+ flux. As DPP is known to be a calcium-binding protein, we postulated that DPP could sequester the store-released Ca2+. Sequestered Ca2+ then binds to calmodulin (CaM) and activates CaM kinase II (CaMKII) in C3H10T1/2 and HMSCs. Activated CaMKII can then phosphorylate Smad1, inducing its translocation to the nucleus where it activates osteogenic target genes.
MATERIALS AND METHODS
Expression and Purification of DPP
The recombinant DPP protein was expressed and purified as published earlier (16). Briefly, bovine DPP cDNA was cloned into pGEX-4T-3 (Invitrogen) and expressed as glutathione S-transferase (GST) fusion protein in BL21-DE3 cells. A glutathione-Sepharose column was used to purify the GST-DPP protein, and the recombinant protein DPP (rDPP) was released using thrombin.
Cell Culture
Pluripotent mouse mesenchymal stem cells (C3H10T1/2) were cultured in Eagle's basal medium (Cellgro) supplemented with 10% FBS and 1% penicillin/streptomycin at 37 °C and 5% CO2. Human bone marrow cells (HMSCs) (Tulane Cancer Centre) were grown in cultured in α-minimum essential medium (Invitrogen) supplemented with 20% fetal bovine serum (FBS, Invitrogen), 1% l-glutamine, and 1% antibiotic and antimycotic solution (Invitrogen). Cells were allowed to proliferate on 6-well plates until attaining 70% confluency. Media were changed to BME medium supplemented with 1% FBS (basal medium) 12–16 h before the start of the experiment.
Total RNA Isolation and Quantitative Real Time PCR
The cells were pretreated with BAPTA-AM (50 μm) or KN-62 (10 μm) for 30 min followed by stimulation with 500 ng of rDPP. Total RNA was extracted at 4 and 24 h for quantitative real time PCR using an RNeasy kit (Qiagen). A total of 2.5 μg of total RNA was reverse-transcribed for 90 min at 50 °C with Superscript III (Invitrogen). Quantitative real time PCR analysis was then carried out using ABI Step-One-Plus instrument. Gene expressions for Runx2, DMP1, BSP, OCN, Osterix, SHN-2, NFATc1, Smad1, Smad5, and GAPDH transcripts were analyzed by quantitative PCR during its linear phase. The relative gene expression level was estimated by using the comparative threshold cycle (CT) method, where the CT value = log linear plot of PCR signal versus the cycle number. The amount of the target, normalized to GAPDH, is given by 2−ΔΔCT, where ΔCT = ΔCT value of target gene − ΔCT value of GAPDH. Primers were obtained from Qiagen.
Protein Isolation and Western Blotting
Total proteins were extracted from either rDPP-treated C3H10T1/2 and HMSCs or cells treated with KN-62 (10 μm) or BAPTA-AM (50 μm) followed by stimulation with rDPP, using M-per reagent (Pierce) at 30 min, 60 min, and 2 h, respectively. Nuclear and cytoplasmic proteins were extracted from C3H10T1/2 cells using NE-PER reagent (Pierce). Total proteins were also isolated from C3H10T1/2 cells infected with adCaMKII-WT (WT), adCaMKII-CAδ, or adCaMKII-DNδ for 6 h at a multiplicity of infection of 100 (kindly provided by Dr. Joan Heller Brown, University of California at San Diego). Western blotting was performed as described earlier (15). Specifically, 35 μg of the total proteins were resolved on a 10% SDS-polyacrylamide gel under reducing conditions. The proteins were then electrotransferred onto nitrocellulose membrane (Bio-Rad). After blocking, antigen detection was performed using anti-CaMKII (1:500) (Santa Cruz Biotechnology), anti-phospho-CaMKII (1:500) (Santa Cruz Biotechnology), anti-phospho-Smad1/5/8 (1:500) (Santa Cruz Biotechnology) and anti-Smad1/5/8 (1:500) (Santa Cruz Biotechnology) antibodies for 16 h at 4 °C. Blots were then incubated with HRP-conjugated goat anti-rabbit IgG secondary antibody (Chemicon International). After several washes with PBS, the bands were visualized by the ECL-Western blot reagent (PerkinElmer Life Sciences). The blots were then stripped and reprobed with anti-tubulin antibody (1:10,000) (Sigma) and then incubated with HRP-conjugated goat anti-mouse IgG secondary antibody and developed as above. Equal loading of proteins in the nuclear extract was confirmed by reprobing the blot with lamin A/C (1:500) (Santa Cruz Biotechnology).
Cytosolic Ca2+ Measurements in C3H10T1/2 Cells
C3H10T1/2 cells were grown to confluence on tissue culture glass coverslips. Before the start of the experiment, the cells were incubated for 2 h at 37 °C in basal medium. Ca2+-sensitive fluorescent dye Fura-2 AM was used to measure the changes in [Ca2+]i (17). Cells were washed with PBS without Ca2+ and loaded with 3 μm Fura-2 AM for 30 min. Changes in [Ca2+]i were measured as published earlier (18). To study the role of released Ca2+ in activating downstream signaling, cells were pretreated with various blocking agents like BAPTA-AM (50 μm), U73122 (10 μm) (Sigma), or KN-62 (10 μm) (Sigma) for 30 min. The cells were then loaded with Fura-2 AM for 30 min. rDPP (500 ng/ml) was then added and cytoplasmic Ca2+ concentration measured. Cells triggered with DMSO (0.01%) served as negative control.
von Kossa Staining
Differentiation of C3H10T1/2 cells was performed using osteogenic media containing Eagle's basal medium (Cellgro) supplemented with 10% FBS, 10 mm β-glycerophosphate, 100 μg/ml ascorbic acid, and10 nm dexamethasone. The cells were then stimulated with either 500 ng of rDPP or 10 μm KN-62 for 7, 14, and 21 days to promote differentiation. The presence of phosphate was determined using von Kossa staining. The cells were fixed in formalin for 1 h, washed twice with distilled water, and then stained with 5% silver nitrate solution.
Alizarin Red S Staining
DPP-stimulated C3H10T1/2 cells were grown in osteogenic medium for 7, 14, and 21 days. The cells were fixed in formalin for 1 h at room temperature and washed with distilled water. 2% Alizarin Red solution (Sigma) was added to fixed cells and incubated for 10–20 min. The cells were then rinsed with distilled water and imaged.
Alkaline Phosphatase Staining
Undifferentiated mesenchymal C3H10T1/2 cells were grown in the presence of DPP for 7, 14, and 21 days in osteogenic medium The cells were then fixed in ice-cold methanol for 1 h and then incubated with the alkaline phosphatase substrate reagent (Bio Rad) at room temperature for 30 min in dark. Samples were then washed with deionized water and imaged.
Alkaline Phosphatase Activity
Total proteins were extracted from DPP-treated C3H10T1/2 cells grown in mineralization medium for 7, 14, and 21 days. 200 μl of reagent from SIGMAFASTTM alkaline phosphatase kit (Sigma) was added to a 20-μl aliquot of cell lysate and incubated for 30 min in the dark at 37 °C. Absorbance at 405 nm was measured using a BioTek Synergy2 plate reader. Experiments were performed in triplicate.
Immunofluorescence
Intracellular localization of Smad1 was analyzed by immunofluorescence staining. C3H10T1/2 cells in basal media were stimulated with either rDPP or treated with CaMKII inhibitor KN-62 or BAPTA-AM a calcium chelator for 1 h. After treatment, the cells were fixed using 10% formalin, and immunofluorescence was performed as per the published protocol (15). Nuclear localization of Smad1 was detected using anti-phospho-Smad1 (1:100) (Cell Signaling Technologies) antibody. All images were acquired using a Zeiss LSM 510 or a 510 META confocal microscope equipped with a ×63 water immersion objective.
Statistical Analyses
Comparisons between the two groups were done using the Student's t test. The error bars represent mean ± S.E. All experiments were performed in triplicate.
RESULTS
DPP Induces Release of Intracellular Ca2+ in Pluripotent Mesenchymal Cells
We first tested the possibility whether stimulation by DPP induced intracellular Ca2 + fluxes. Pluripotent C3H10T1/2 stem cells were used as a model cell culture system to study intracellular signaling events in response to DPP. Cells grown on coverslips were loaded with Fura-2AM and fluorescence-monitored in the presence of rDPP. When compared with unstimulated cells, the addition of rDPP triggered a 3-fold increase in fluorescence that corresponded to the release of intracellular Ca2 + fluxes (Fig. 1A). It has to be noted that these events occurred in the absence of extracellular Ca2+. To investigate the specificity of DPP stimulus, we suppressed the rise in free [Ca2+]i by introducing the Ca2+ chelator BAPTA-AM for 30 min followed by stimulation with rDPP. The release of intracellular Ca2 + was completely abrogated in the presence of BAPTA-AM (Fig. 1B), thus supporting our hypothesis that DPP stimulus is responsible for the transient increase in free Ca2+. As DMSO was used as a vehicle to dissolve BAPTA-AM, U73122, and KN-62, we therefore investigated if DMSO played a role in the release of intracellular calcium. Cells treated with DMSO did not influence intracellular calcium release (Fig. 1C). To prove that DPP-mediated intracellular Ca2+ release was from the endoplasmic reticulum, we measured Ca2+ release in the presence of the phospholipase C inhibitor U73122 that can potentially inhibit endoplasmic reticulum Ca2+ release. Treatment of cells with U73122 significantly inhibited the intracellular Ca2 + fluxes that were observed by rDPP stimulation (Fig. 1D). Thus, DPP-mediated activation of phospholipase C could play a pivotal role in mediating transient increase in intracellular Ca2+.
FIGURE 1.

A, effect of rDPP on [Ca2+]i in mesenchymal stem cells. DPP-induced [Ca2+]i was measured in C3H10T1/2 cells as described under “Materials and Methods.” Fura-2AM-loaded cells were washed three times, placed in Ca2+-free and Mg2+-free HBSS, and then stimulated with rDPP (500 ng/ml). Experiments were repeated three times, and a representative plot is shown. Picture shots display DPP-induced [Ca2+]i release in C3H10T1/2 cells at 250 s. Yellow color within the cells indicates the release of calcium from the intracellular stores when C3H10T1/2 cells were stimulated with DPP or the intake of calcium upon depletion of the stores when extracellular calcium was added. B, BAPTA-AM is a calcium chelator and can block DPP-stimulated increase in [Ca2+]i in C3H10T1/2 cells. Cells pretreated with BAPTA-AM (50 μm) were loaded with Fura-2AM and placed in Ca2+- and Mg2+-free HBSS. The cells were then stimulated with rDPP (500 ng/ml) and then observed for Ca2+ store release. Cells with no BAPTA-AM treatment served as control. Experiments were performed in triplicate. A representative plot is shown in this panel. Picture shots display DPP-induced release in BAPTA-AM-pretreated C3H10T1/2 cells at 250 s. C, DMSO treatment does not inhibit DPP-induced [Ca2+]i release in C3H10T1/2 cells. Cells pretreated with DMSO (0.01%) were loaded with Fura-2AM and placed in Ca2+- and Mg2+-free HBSS. The cells were then stimulated with rDPP (500 ng/ml) and then observed for Ca2+ store release. As DMSO was used as a solvent vehicle to dissolve all inhibitors, it was therefore necessary to identify if the vehicle had an effect on [Ca2+]i release in C3H10T1/2 cells. Screen shots of the live image of DPP induced [Ca2+]i release in DMSO-pretreated C3H10T1/2 cells. D, U-73122 inhibitor of the phospholipase C pathway inhibits DPP-induced [Ca2+]i release in C3H10T1/2 cells. DPP-induced [Ca2+]i was measured in confluent mesenchymal stem cells upon stimulation with 500 ng/ml rDPP after 30 min of pretreatment with U-73122 (phospholipase C inhibitor). Cells were loaded with Fura-2AM and placed in Ca2+- and Mg2+-free HBSS and stimulated with rDPP (500 ng/ml) to measure store-Ca2+ release. Arrow indicates the time at which cells were stimulated with rDPP. Experiments were repeated a minimum of four times. A representative plot is shown in this panel.
DPP Induces Commitment of Pluripotent Stem Cells to the Osteogenic Lineage
We next sought to examine if DPP influenced the osteogenic differentiation of C3H10T1/2 and HMSCs by analyzing for osteogenic markers. Real time PCR analysis showed that DPP stimulation resulted in an increase in the relative expression of osteoblastic markers like Runx2, DMP1, BSP, OCN, and Osterix (Fig. 2, A and B). However, these markers were significantly suppressed in the presence of BAPTA-AM. These studies show that Ca2+ signaling is required for DPP-mediated osteogenic differentiation.
FIGURE 2.

DPP enhances osteoblastic gene expression in mesenchymal stem cells. C3H10T1/2 cells and HMSCs in basal media for 24 h were stimulated with 500 ng/ml rDPP with or without BAPTA-AM treatment for 4 and 24 h. Total RNA was isolated and subjected to real time PCR and analyzed for gene expression of Runx2, BSP, DMP1, Osterix, and OCN. Untreated C3H10T1/2 cells served as control. These results were normalized with the loading control GAPDH. Experiments were done in triplicate. *, #, (), and [caret], p < 0.05. C, control.
CaMKII-mediated Osteogenic Differentiation
We next explored the idea that activation of CaMKII promotes osteogenic differentiation. The involvement of CaMKII was sought after stimulation of C3H10T1/2 cells and HMSCs with rDPP for 30 min, 60 min and 2 h followed by Western blot analysis. Results in Fig. 3A show increased phosphorylation of CaMKII with time in C3H10T12 cells, although in HMSCs maximum phosphorylation was observed at 60 min when compared with the 2 h unstimulated control cells (Fig. 3B). Treatment of cells with BAPTA-AM reduced the phosphorylation levels in C3H10T1/2 cells (Fig. 3C). We next examined if KN-62, a selective cell-permeable pharmacological inhibitor of calcium/calmodulin-dependent kinase II, had an effect on DPP-mediated intracellular Ca2+ release. Results from this study showed that the rise in intracellular Ca2+ induced by DPP was unaffected by KN-62 (Fig. 3D). However, lower levels of phosphorylated CaMKII were observed in the presence of KN-62 in C3H10T1/2 (Fig. 3E) and HMSCs (Fig. 3F).
FIGURE 3.

A, DPP activates CaMKII in C3H10T1/2 cells. C3H10T1/2 cells were stimulated with rDPP (500 ng/ml), and Western blotting was performed. Experiments were done in triplicate. *, #, and [caret], p < 0.05 as compared with untreated cells. B, DPP activates CaMKII in HMSCs. C3H10T1/2 cells were stimulated with rDPP (500 ng/ml), and Western blotting was performed with Abs against CaMKII. Experiments were done in triplicate. *, #, and [caret], p < 0.05 as compared with untreated cells. C, DPP-mediated activation of CaMKII can be inhibited by the treatment with BAPTA-AM. C3H10T1/2 cells were pretreated with BAPTA-AM (50 μm) followed by stimulation with rDPP for the indicated time points, and Western blotting was performed with Abs against CaMKII. Experiments were done in triplicate. *, #, and [caret], p < 0.05 as compared with untreated cells. D, CaMKII inhibitor KN-62 inhibits DPP-induced [Ca2+]i release in C3H10T1/2 cells. DPP-induced [Ca2+]i was measured in confluent C3H10T1/2 cells loaded with 10 μm KN-62 for 30 min prior to the addition of Fura-2AM. Fura-2AM-loaded cells were washed three times, placed in Ca2+- and Mg2+-free HBSS, and then stimulated with rDPP (500 ng/ml). The experiment was repeated four times, and a representative plot is shown. Picture images display DPP-induced [Ca2+]i release in KN-62-pretreated C3H10T1/2 cells at 250 s. E, KN-62 inhibits CaMKII activation in C3H10T1/2 cells. Treatment of C3H10T1/2 cells with KN-62 (10 μm) inhibits CaMKII activation. Total proteins were isolated, and Western blots were developed with CaMKII antibody and anti-phospho-CaMKII antibody. Equal protein loading was confirmed by stripping the blot followed by probing it with tubulin antibody. Experiments were done in triplicate. *, #, and [caret], p < 0.05 as compared with untreated cells. F, KN-62 inhibits DPP-mediated phosphorylation of CaMKII in HMSCs. Total proteins were isolated from DPP-stimulated HMSCs, and immunoblots were performed with CaMKII antibody and anti-phospho-CaMKII antibody. Experiments were done in triplicate. *, #, and [caret], p < 0.05 as compared with untreated cells. C, control.
CaMKII-mediated Smad1 Phosphorylation in C3H10T1/2 and HMSCs
It is known that activated CaMKII can phosphorylate multiple downstream targets. Therefore, we examined if Smad1 is phosphorylated by CaMKII. Western blot analysis in Fig. 4, A and B, shows that DPP stimulation can activate the phosphorylation of Smad1 as early as 30 min. Maximum level of activation was observed at 2 h when compared with the 2 h of unstimulated control C3H10T1/2 and HMSCs. Equal loading of proteins was confirmed by stripping the blot followed by re-probing with tubulin.
FIGURE 4.

Activation of Smad1/5/8 in C3H10T1/2 and HMSCs by DPP can be inhibited by the treatment with BAPTA-AM and KN-62. A, C3H10T1/2 cells were stimulated with rDPP (500 ng/ml), and Western blotting was performed with Abs against Smad1/5/8. *, #, and (), p < 0.05 as compared with control cells. B, HMSCs were stimulated with rDPP (500 ng/ml), and Western blotting was performed with Abs against Smad1/5/8. *, #, and (), p < 0.05 as compared with control cells. C, C3H10T1/2 cells were pretreated with BAPTA-AM (50 μm) followed by stimulation with rDPP for the indicated time points, and Western blotting was performed with Abs against Smad1/5/8. *, #, and (), p < 0.05 as compared with control cells. D, HMSCs were treated with BAPTA-AM (50 μm) followed by stimulation with rDPP, and Western blotting was performed with Abs against Smad1/5/8. *, #, and (), p < 0.05 as compared with control cells. E, treatment of C3H10T1/2 cells with KN-62 (10 μm) inhibits Smad1/5/8 activation. Total proteins were isolated, and Western blots were developed with anti-Smad1/5/8 antibody and anti-phospho-Smad1/5/8 antibody. Equal loading of the proteins was confirmed by stripping the blot followed by probing it with tubulin. *, #, and (), p < 0.05 as compared with control cells. F, inhibition of Smad1/5/8 in KN-62-treated HMSCs, and Western blotting was performed with Abs against Smad1/5/8. *, #, and (), p < 0.05 as compared with control cells. G, C3H10T1/2 cells were transduced with wild type (WT), constitutively active (CA), and dominant negative (DN) CaMKII and then stimulated with rDPP. Western blotting was performed with Abs against Smad1/5/8. C, control.
To determine whether intracellular Ca2+ was necessary for activation of Smad1, cells were pre-treated with BAPTA-AM and then stimulated with DPP for the specified time points. Western blot analysis in Fig. 4, C and D, showed lower phosphorylation levels of Smad1 indicating a role for intracellular Ca2+ in phosphorylation of Smad1. To assess the role of activated CaMKII in phosphorylating Smad1, cells were then treated with KN-62 followed by stimulation with DPP. Interestingly, KN-62 like BAPTA-AM had the same inhibition effect on Smad1 phosphorylation (Fig. 4, E and F). Strong inhibition of Smad1 phosphorylation was seen from 30 min to 2 h. To further confirm the specificity of the results obtained with KN-62 and BAPTA-AM, C3H10T1/2 cells were transfected with adenovirus wild type (WT) and constitutively active and dominant negative CaMKII. Dominant negative adenovirus CaMKII inhibited the phosphorylation of Smad1 when compared with the cells transduced with the viral particles carrying the WT and constitutively active forms of CaMKII (Fig. 4G). All Western blots were re-probed for tubulin that served as loading control.
Calcium and CaMKII-dependent Nuclear Translocation of Phosphorylated Smad1
A hallmark of the noncanonical BMP signaling is stabilization and nuclear accumulation of Smad1. Phosphorylated Smad1 translocates from the cytoplasm to the nucleus in DPP-stimulated C3H10T1/2 cells (Fig. 5B). In marked contrast, untreated cells displayed predominant cytoplasmic localization of p-Smad1 (Fig. 5A). Interestingly, predominant cytoplasmic localization and reduced nuclear accumulation of phosphorylated Smad1 were observed in cells treated with DPP and BAPTA-AM (Fig. 5D). Cells treated with BAPTA-AM by itself served as a control and did not have an effect on the translocation of p-Smad1 (Fig. 5C). These data support our hypothesis that release of intracellular calcium by DPP is required for the activation of Smad1 in pluripotent stem cells.
FIGURE 5.

Nuclear localization of p-Smad1 in C3H10T1/2 cells stimulated by DPP and abrogated by BAPTA-AM. C3H10T1/2 cells were stimulated with rDPP (500 ng/ml) or BAPTA-AM (50 μm) for 1 h. The cells were fixed and immunostained for p-Smad1 (red). Control cells exhibited diffused staining throughout the cells for p-Smad1 (A). Upon stimulation with DPP for 1 h, nuclear translocation of p-Smad1 was observed. Arrows indicate nuclear localization of p-Smad1 (B). Cells treated with BAPTA-AM alone displayed cytoplasmic staining of p-Smad1 (C). However, DPP-stimulated and BAPTA-AM-pretreated cells failed to show nuclear translocation of p-Smad1 (D). DAPI (blue) was used to stain the nucleus. Bar, 10 μm.
We next tested if Smad1 phosphorylated by activated CaMKII translocates to the nucleus. For this, C3H10T1/2 cells were treated with rDPP or KN-62 or both for 1 h, respectively. Confocal images clearly show unstimulated cells having diffused cytoplasmic staining for phosphorylated Smad1 (Fig. 6A). Cells stimulated with rDPP for 60 min showed distinct nuclear translocation of p-Smad1 (Fig. 6B). However, cells pretreated with KN-62 inhibitor and upon DPP stimulation abolished nuclear translocation of p-Smad1 (Fig. 6D). Cells pretreated with KN-62 inhibitor alone, which served as control, showed similar cytoplasmic localization as in untreated cells (Fig. 6C). Nuclear and cytoplasmic proteins isolated from DPP-stimulated C3H10T1/2 cells demonstrate greater amounts of p-Smad1 in the nuclear fraction at 60 min when compared with the cytoplasmic fraction (Fig. 6E). Results from the immunoblot clearly show that KN-62 inhibited nuclear localization of p-Smad1. These results suggest that Smad1 is phosphorylated by CaMKII, as the inhibitor KN-62 abolished CaMKII activation and Smad1 phosphorylation.
FIGURE 6.

DPP-mediated nuclear localization of p-Smad1 in C3H10T1/2 is abrogated in the presence of KN-62. C3H10T1/2 cells were stimulated with rDPP (500 ng/ml) for 1 h. Cells were fixed and immunostained for p-Smad1. Immunofluorescence shows p-Smad1 predominantly located in the cytoplasm of untreated cells (A). With DPP stimulation, the predominant nuclear localization of p-Smad1 is observed. Arrows indicate nuclear localization of p-Smad1 (B). Diffused cytoplasmic staining of p-Smad1 was observed in cells pretreated with KN-62 alone (C). However, C3H10T1/2 cells stimulated with rDPP and KN-62 prevented nuclear localization of p-Smad1 (D). Bar, 10 μm. Nuclear and cytoplasmic proteins were extracted from C3H10T1/2 cells stimulated with DPP, and Western blotting was performed with anti-p-Smad1/5/8 and Smad1/5/8 antibody. The blots were stripped and reprobed with lamin A/C and tubulin (E). C, control.
Phosphorylated Smad1 Activates Shnurri-2 and Nfatc1
We then examined whether activated Smad1 was responsible for inducing the expression of downstream Smad targets. Real time PCR results showed an enhanced expression of Smad1, Smad5 (supplemental Fig. S1), Shnurri-2, and NFATc1 (Fig. 7) mRNA levels at 4 and 24 h in C3H10T1/2 and HMSCs. Expression levels were reduced when cells were pretreated with either BAPTA-AM or KN-62 followed by DPP stimulation for 4 and 24 h (Fig. 7 and supplemental Fig. S1). These results suggest that DPP could play a pivotal role in potentially mediating a cross-talk between CaMKII and the Smad signaling pathway to initiate osteoblast differentiation.
FIGURE 7.

DPP activates Smad pathway-related gene expression. C3H10T1/2 and HMSCs were stimulated with rDPP (500 ng/ml) or KN-62 (10 μm) or BAPTA-AM (50 μm) for 4 and 24 h, respectively. Total RNA was extracted, and expression levels of NFATc1 and SHN2 were estimated based on real time PCR. Upon stimulation with DPP, expression levels of these genes increased from 4 to 24 h. Lower expression levels were observed with KN-62 and BAPTA-AM pretreatment. Experiments were performed in triplicate. * and #, p < 0.05. C, control.
Functional Analysis to Demonstrate Terminal Differentiation of Osteoblasts
To monitor terminal differentiation of the pluripotent stem cells to functional osteoblasts, an in vitro nodule formation assay was performed. Nodule formation due to secretion of extracellular matrix proteins in the presence of phosphate ions and ascorbic acid has been considered to be an important feature for mineralization and precedes mineralization. Therefore, nodule formation assays were performed in the presence of DPP. Osteogenic differentiation was observed at 7, 14, and 21 days. The presence of phosphate in the calcified nodules, seen as dark deposits in the extracellular matrix, was high at 14 and 21 days of DPP-stimulated culture (Fig. 8, A and B). In contrast, cells grown in mineralization media for 21 days in the absence of DPP showed no staining (Fig. 8A). No mineralized nodules were observed in cells treated with KN-62 alone (Fig. 8B) or in cells treated with KN-62 and DPP (Fig. 8B). Terminal differentiation of C3H10T1/2 cells into osteogenic lineage was further confirmed by Alizarin Red S and alkaline phosphatase staining. Alizarin Red staining showed the presence of calcium deposits in the mineralized matrix at 21 days (Fig. 8A). Alkaline phosphatase staining was observed at 14 and 21 days in DPP-treated cells grown under osteogenic conditions (Fig. 8A). Interestingly, no staining was detected in control cells grown in the absence of DPP for 21 days. In addition, quantitative analysis of the alkaline phosphatase assay showed an increase in the alkaline phosphatase activity at 14 and 21 days in DPP-treated cells when compared with the control cells (Fig. 8C). Together, these results suggest that DPP can stimulate the terminal differentiation of C3H10T1/2 cells into fully functional osteoblasts.
FIGURE 8.
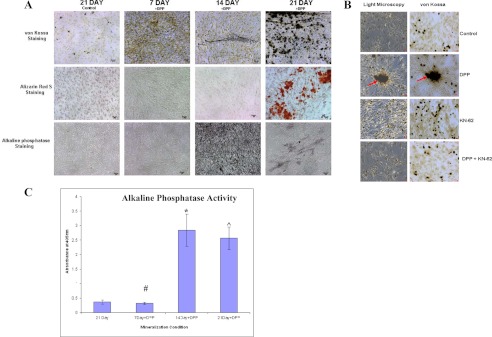
DPP stimulates mineralized nodule formation. C3H10T1/2 cells were cultured in mineralization media for 30 days either with rDPP (500 ng/ml) or KN-62 (10 μm) or both. von Kossa, Alizarin Red S, and alkaline phosphatase staining were performed (A). Terminal differentiation and mineralized nodule formation were observed when cells were stimulated with rDPP. However, untreated control cells, cells treated with inhibitor alone, and cells treated with the inhibitor in the presence of DPP did not show evidence of mineralized nodule formation (B). Arrows point to the mineralized nodule formed. Alkaline phosphatase activity was highly expressed at 14 and 21 days in DPP-treated cells (C). *, #, and [caret], p < 0.05 as compared with control cells. Bar, 20 μm.
DISCUSSION
Pluripotent stem cells such as C3H10T1/2 and HMSCs have the capacity to undergo commitment to muscle, bone, adipose, or cartilage lineages. Our findings in this study show that treatment of pluripotent stem cells with dentin phosphophoryn gives rise to osteoblasts. Although it is well established that DPP promotes extracellular matrix mineralization, however, its role in signaling has only been recently described (15, 19). The function of DPP as a signaling molecule has been demonstrated in human mesenchymal stem cells via the MAPK pathway and the Smad pathway; however, no mechanisms were reported (12, 20). Understanding the precise mechanism by which DPP initiates signaling from the ECM is unknown. Accordingly, the central goal of this study was to identify specific components of the signaling cascade mediated by DPP during commitment of pluripotent stem cells to the osteogenic lineage.
We first identified that DPP stimulates intracellular calcium release in embryonic mesenchymal cells. This depletion of calcium from the endoplasmic reticulum evokes a number of downstream responses. Furthermore, we demonstrate that stimulation by DPP leads to Smad1 phosphorylation and subsequent translocation to the nucleus. The prevention of Smad1 phosphorylation and nuclear translocation by the CaMKII inhibitor KN-62 and the intracellular calcium chelator BAPTA-AM shows the important role played by both intracellular calcium flux and ubiquitously expressed Ca2+-calmodulin-dependent CaMKII in Smad1 phosphorylation and subsequent nuclear localization. Thus, in this study we have uncovered a novel interaction between the CaMKII and the Smad1 signaling pathway stimulated by DPP in undifferentiated mesenchymal cells.
CaMKII is a serine/threonine multifunctional kinase. It is activated in response to Ca2+ signals and phosphorylates several downstream proteins. The role of CaMKII in osteoblast differentiation has been reported, and α-CaMKII, an isoform of CaMKII, has specifically been shown to be a critical regulator of osteoblast differentiation (24). In osteoblasts, stretch-dependent Ca2+ influx was shown to activate TGF-β-activated kinase 1 via CaMKII to induce IL-6 expression (25). Furthermore, α-CaMKII was shown to control different signaling pathways, namely CREB/ATF, MAPK, as well as AP-1 transcription factor activity, by regulating the expression of c-fos (26).
In this study, we have shown that the Smad1 function can be directly controlled by the cytoplasmic CaMKII in response to DPP stimulus. To elucidate the specificity of this enzyme, we have used KN-62, a specific and selective calcium-CaM-dependent protein kinase inhibitor. KN-62 has been shown to inhibit both the substrate phosphorylation and autophosphorylation of CaMKII, with the calmodulin-independent activity of the enzyme suggesting that KN-62 affects the enzyme and calmodulin interactions directly. This inhibitor functions by binding to the calmodulin binding region of CaMKII (21, 22). The inhibitory role of KN-62 has been established previously. Published results show that KN-62 strongly inhibited the activation of MAPK/c-Myc and autocrine keratinocyte proliferation (23). In our study, use of KN-62 not only abrogated the phosphorylation and nuclear translocation of Smad1 following DPP stimulation but also resulted in reduced osteogenic gene expression levels and abrogated mineralized nodule formation. Furthermore, expression of dominant negative CaMKII significantly reduced the phosphorylation of Smad1. This confirms the role of CaMKII in regulating the Smad1/5/8 pathway following DPP stimulation in mouse pluripotent stem cells.
Smad signaling pathway plays an essential role in transmitting and regulating activin, BMP, and TGF-β signals from the cell surface to the nucleus, thus regulating the expression of target genes during osteogenesis (27–30). Recently, it has become apparent that Smads are also extensively regulated by direct input from a variety of receptor signaling pathways. One example of such a pathway other than BMP/TGF-β is the MAPK pathway (31, 32). In another study, functional interaction of Smad2 with CaMKII inhibited TGF-β signaling (33). In the environment of the complex signaling network, Smads have begun to appear as a central junction that receives input from different signaling pathways to thereby interpret cues from the ECM. Thus, Smads play a crucial role in ECM-mediated cell differentiation and tissue remodeling process.
In this study, we show that DPP activates Smad1, independent of the known ligands via CaMKII. Nuclear translocation of phosphorylated Smad1 results in directly affecting gene responses. It was apparent from our studies that DPP stimulation led to a significant fold increase in the expression of Runx2, Osterix, dentin matrix protein 1 (DMP1), BSP, and OCN. Treatment with KN-62 followed by DPP stimulation significantly reduced the mRNA expression of these genes. Increase in the expression of Runx2 was particularly interesting as it is a “master” transcription factor for osteoblast differentiation, maturation, and bone formation. Furthermore, Runx2 can regulate the expression of several extracellular matrix proteins required for mineralized matrix formation (34). Other genes of interest that were expressed in response to DPP stimulation are Schnurri2 (SHN-2) and NFATc1. SHN-2 is a zinc finger protein, and lack of SHN-2 expression reduced bone remodeling by suppressing both osteoblastic bone formation and osteoclastic bone resorption activities in vivo. Thus, SHN-2 is a novel regulator of osteoblasts and modulates the expression of factors required for osteoblast function like Osterix, NFATc1, and OCN (35). Expression of NFATc1 is particularly interesting as activated NFATc1 was shown to activate Wnt signaling components necessary for directing osteoblast proliferation (36).
Mineralized nodule formation confirmed that DPP stimulation favored terminal differentiation of osteoblasts. Interestingly, inhibition of CaMKII by KN-62 abrogated mineralized nodule formation, demonstrating the pivotal role played by CaMKII in the process of osteoblastic differentiation of C3H10T1/2 cells stimulated by DPP.
Previously, DPP has been shown to activate the MAPK pathway, and in this study we show that DPP activates the Smad signaling pathway. Thus, it is possible that DPP activates both the Smad and the MAPK pathway similar to the BMPs. From this, it appears that during bone formation the cells use multiple pathways to decipher signals from the ECM. Signaling by DPP could be one such signaling network that the ECM utilizes to regulate the function during tissue morphogenesis.
In summary, this study identifies a novel mechanism by which DPP plays a functional role in activating Smad signaling through cytoplasmic CaMKII. Translocation of phosphorylated Smad1 then regulates target gene expression and facilitates osteoblastic differentiation of pluripotent stem cells thereby validating our proposed hypothetical model (Fig. 9). Thus, DPP facilitates a cross-talk between calcium-CaMKII signaling and the Smad signaling pathway to initiate osteogenic differentiation in pluripotent stem cells.
FIGURE 9.

Hypothetical model. Model showing DPP-mediated intracellular calcium store flux can trigger the activation of CaMKII, resulting in the activation of Smad1/5/8 signaling cascade. ER, endoplasmic reticulum; SERCA, sarco/endoplasmic reticulum Ca2+-ATPase; IP3R, inositol 1,4,5-trisphosphate receptor.
Acknowledgment
We thank Dr. Joan Heller Brown for kindly providing us with constitutively active and dominant negative CaMKII adenovirus.
This work was supported, in whole or in part, by National Institutes of Health Grant DE 19633. This work was also supported by The Brodie Endowment Fund.

This article contains supplemental Fig. S1.
- ECM
- extracellular matrix
- CaMKII
- Ca2+-calmodulin-dependent protein kinase II
- DPP
- dentin phosphophoryn
- BAPTA-AM
- 1,2-bis(2-aminophenoxy)ethane-N,N,N′,N′-tetraacetic acid tetrakis(acetoxymethyl ester)
- HMSC
- human bone marrow cell
- HBSS
- Hanks' balanced salt solution
- Ab
- antibody
- BSP
- bone sialoprotein
- OCN
- osteocalcin
- rDPP
- recombinant DPP
- BMP
- bone morphogenetic protein.
REFERENCES
- 1. Veis A., Perry A. (1967) The phosphoprotein of the dentin matrix. Biochemistry 6, 2409–2416 [DOI] [PubMed] [Google Scholar]
- 2. Ogbureke K. U., Fisher L. W. (2004) Expression of SIBLINGs and their partner MMPs in salivary glands. J. Dent. Res. 83, 664–670 [DOI] [PubMed] [Google Scholar]
- 3. Linde A., Bhown M., Butler W. T. (1980) Noncollagenous proteins of dentin. A re-examination of proteins from rat incisor dentin utilizing techniques to avoid artifacts. J. Biol. Chem. 255, 5931–5942 [PubMed] [Google Scholar]
- 4. Richardson W. S., Munksgaard E. C., Butler W. T. (1978) Rat incisor phosphoprotein. J. Biol. Chem. 253, 8042–8046 [PubMed] [Google Scholar]
- 5. Feng J. Q., Luan X., Wallace J., Jing D., Ohshima T., Kulkarni A. B., D'Souza R. N., Kozak C. A., MacDougall M. (1998) Genomic organization, chromosomal mapping, and promoter analysis of the mouse dentin sialophosphoprotein (Dspp) gene, which codes for both dentin sialoprotein and dentin phosphoprotein. J. Biol. Chem. 273, 9457–9464 [DOI] [PubMed] [Google Scholar]
- 6. Sfeir C., Butler S., Lin E., George A., Vies A. (2000) in Chemistry and Biology of Mineralized Tissue (Goldberg M., Boskey A., Robinson C., eds) pp. 181–184, American Academy of Orthopaedic Surgeons, Chicago [Google Scholar]
- 7. MacDougall M., Simmons D., Luan X., Nydegger J., Feng J., Gu T. T. (1997) Dentin phosphoprotein and dentin sialoprotein are cleavage products expressed from a single transcript coded by a gene on human chromosome 4. J. Biol. Chem. 272, 835–842 [DOI] [PubMed] [Google Scholar]
- 8. Zanetti M., de Bernard B., Jontell M., Linde A. (1981) Ca2+-binding studies of the phosphoprotein from rat-incisor dentine. Eur. J. Biochem. 113, 541–545 [DOI] [PubMed] [Google Scholar]
- 9. Lee S. L., Veis A., Glonek T. (1977) Dentin phosphoprotein: an extracellular calcium-binding protein. Biochemistry 16, 2971–2979 [DOI] [PubMed] [Google Scholar]
- 10. Boskey A. L., Maresca M., Doty S., Sabsay B., Veis A. (1990) Concentration-dependent effects of dentin phosphophoryn in the regulation of in vitro hydroxyapatite formation and growth. Bone Miner. 11, 55–65 [DOI] [PubMed] [Google Scholar]
- 11. George A., Hao J. (2005) Role of phosphophoryn in dentin mineralization. Cells Tissues Organs 181, 232–240 [DOI] [PubMed] [Google Scholar]
- 12. Jadlowiec J., Koch H., Zhang X., Campbell P. G., Seyedain M., Sfeir C. (2004) Phosphophoryn regulates the gene expression and differentiation of NIH3T3, MC3T3-E1, and human mesenchymal stem cells via the integrin/MAPK signaling pathway. J. Biol. Chem. 279, 53323–53330 [DOI] [PubMed] [Google Scholar]
- 13. Ogbureke K. U., Fisher L. W. (2007) Sibling expression patterns in duct epithelia reflect the degree of metabolic activity. J. Histochem. Cytochem. 55, 403–409 [DOI] [PubMed] [Google Scholar]
- 14. Alvares K., Kanwar Y. S., Veis A. (2006) Expression and potential role of dentin phosphophoryn (DPP) in mouse embryonic tissues involved in epithelial-mesenchymal interactions and branching morphogenesis. Dev. Dyn. 235, 2980–2990 [DOI] [PubMed] [Google Scholar]
- 15. Eapen A., Ramachandran A., George A. (2012) Dentin phosphoprotein (DPP) activates integrin-mediated anchorage-dependent signals in undifferentiated mesenchymal cells. J. Biol. Chem. 287, 5211–5224 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Srinivasan R., Chen B., Gorski J. P., George A. (1999) Recombinant expression and characterization of dentin matrix protein 1. Connect. Tissue Res. 40, 251–258 [DOI] [PubMed] [Google Scholar]
- 17. Sundivakkam P. C., Kwiatek A. M., Sharma T. T., Minshall R. D., Malik A. B., Tiruppathi C. (2009) Caveolin1 scaffold domain interacts with TRPC1 and IP3R3 to regulate Ca2+ store release-induced Ca2+ entry in endothelial cells. Am. J. Physiol. Cell Physiol. 296, C403–C413 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Eapen A., Sundivakkam P., Song Y., Ravindran S., Ramachandran A., Tiruppathi C., George A. (2010) Calcium-mediated stress kinase activation by DMP1 promotes osteoblast differentiation. J. Biol. Chem. 285, 36339–36351 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Eapen A., Ramachandran A., George A. (2012) DPP in the matrix mediates cell adhesion but is not restricted to stickiness: A tale of signaling. Cell Adh. Migr. 6, 307–311 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Jadlowiec J. A., Zhang X., Li J., Campbell P. G., Sfeir C. (2006) Extracellular matrix-mediated signaling by dentin phosphophoryn involves activation of the Smad pathway independent of bone morphogenetic protein. J. Biol. Chem. 281, 5341–5347 [DOI] [PubMed] [Google Scholar]
- 21. Tokumitsu H., Chijiwa T., Hagiwara M., Mizutani A., Terasawa M., Hidaka H. (1990) KN-62, l-[N,O-bis(5-isoquinolinesulfonyl)-N-methyl-l-tyrosyl]-4-phenylpiperazine, a specific inhibitor of Ca2+/calmodulin-dependent protein kinase II. J. Biol. Chem. 265, 4315–4320 [PubMed] [Google Scholar]
- 22. Schweitzer E. S., Sanderson M. J., Wasterlain C. G. (1995) Inhibition of regulated catecholamine secretion from PC12 cells by the Ca2+/calmodulin kinase II inhibitor KN-62. J. Cell Sci. 108, 2619–2628 [DOI] [PubMed] [Google Scholar]
- 23. Praskova M., Kalenderova S., Miteva L., Poumay Y., Mitev V. (2002) Ca2+/calmodulin-dependent protein kinase (CaM-kinase) inhibitor KN-62 suppresses the activity of mitogen-activated protein kinase (MAPK), c-Myc activation, and human keratinocyte proliferation. Arch. Dermatol. Res. 294, 198–202 [DOI] [PubMed] [Google Scholar]
- 24. Zayzafoon M. (2006) Calcium/calmodulin signaling controls osteoblast growth and differentiation. J. Cell. Biochem. 97, 56–70 [DOI] [PubMed] [Google Scholar]
- 25. Fukuno N., Matsui H., Kanda Y., Suzuki O., Matsumoto K., Sasaki K., Kobayashi T., Tamura S. (2011) TGF-β-activated kinase 1 mediates mechanical stress-induced IL-6 expression in osteoblasts. Biochem. Biophys. Res. Commun. 408, 202–207 [DOI] [PubMed] [Google Scholar]
- 26. Zayzafoon M., Fulzele K., McDonald J. M. (2005) Calmodulin and calmodulin-dependent kinase IIα regulate osteoblast differentiation by controlling c-Fos expression. J. Biol. Chem. 280, 7049–7059 [DOI] [PubMed] [Google Scholar]
- 27. Souchelnytskyi S., Moustakas A., Heldin C. H. (2002) TGF-β signaling from a three-dimensional perspective: insight into selection of partners. Trends Cell Biol. 12, 304–307 [DOI] [PubMed] [Google Scholar]
- 28. Moustakas A., Souchelnytskyi S., Heldin C. H. (2001) Smad regulation in TGF-β signal transduction. J. Cell Sci. 114, 4359–4369 [DOI] [PubMed] [Google Scholar]
- 29. Wrana J. L., Attisano L. (2000) The Smad pathway. Cytokine Growth Factor Rev. 11, 5–13 [DOI] [PubMed] [Google Scholar]
- 30. Massagué J., Wotton D. (2000) Transcriptional control by the TGF-β/Smad signaling system. EMBO J. 19, 1745–1754 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Pera E. M., Ikeda A., Eivers E., De Robertis E. M. (2003) Integration of IGF, FGF, and anti-BMP signals via Smad1 phosphorylation in neural induction. Genes Dev. 17, 3023–3028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Kretzschmar M., Doody J., Massagué J. (1997) Opposing BMP and EGF signaling pathways converge on the TGF-β family mediator Smad1. Nature 389, 618–622 [DOI] [PubMed] [Google Scholar]
- 33. Wicks S. J., Lui S., Abdel-Wahab N., Mason R. M., Chantry A. (2000) Inactivation of Smad-transforming growth factor β signaling by Ca2+-calmodulin-dependent protein kinase II. Mol. Cell. Biol. 20, 8103–8111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Byers B. A., Pavlath G. K., Murphy T. J., Karsenty G., García A. J. (2002) Cell type-dependent up-regulation of in vitro mineralization after overexpression of the osteoblast-specific transcription factor Runx2/Cbfal. J. Bone Miner. Res. 17, 1931–1944 [DOI] [PubMed] [Google Scholar]
- 35. Saita Y., Takagi T., Kitahara K., Usui M., Miyazono K., Ezura Y., Nakashima K., Kurosawa H., Ishii S., Noda M. (2007) Lack of Schnurri-2 expression associates with reduced bone remodeling and osteopenia. J. Biol. Chem. 282, 12907–12915 [DOI] [PubMed] [Google Scholar]
- 36. Wang Y., Jarad G., Tripathi P., Pan M., Cunningham J., Martin D. R., Liapis H., Miner J. H., Chen F. (2010) Activation of NFAT signaling in podocytes causes glomerulosclerosis. J. Am. Soc. Nephrol. 21, 1657–1666 [DOI] [PMC free article] [PubMed] [Google Scholar]