

Background: IL-1β plays a major role in the pathogenesis of Lyme disease.
Results: The induction of autophagy by B. burgdorferi reduces the production of IL-1β.
Conclusion: Autophagy acts as an important regulatory mechanism of inflammation in Lyme disease.
Significance: The modulation of autophagy might lead to novel therapeutic targets for the treatment of Lyme disease.
Keywords: Autophagy, Bacteria, Cytokine, Interleukin, Tumor Necrosis Factor (TNF), Borrelia
Abstract
Borrelia burgdorferi sensu lato is the causative agent of Lyme disease. Recent studies have shown that recognition of the spirochete is mediated by TLR2 and NOD2. The latter receptor has been associated with the induction of the intracellular degradation process called autophagy. The present study demonstrated for the first time the induction of autophagy by exposure to B. burgdorferi and that autophagy modulates the B. burgdorferi-dependent cytokine production. Human peripheral blood mononuclear cells treated with autophagy inhibitors showed an increased IL-1β and IL-6 production in response to the exposure of the spirochete, whereas TNFα production was unchanged. Autophagy induction against B. burgdorferi was dependent on reactive oxygen species (ROS) because cells from patients with chronic granulomatous disease, which are defective in ROS production, also produced elevated IL-1β. Further, the enhanced production of the proinflammatory cytokines was because of the elevated mRNA expression in the absence of autophagy. Our results thus demonstrate the induction of autophagy, which, in turn, modulates cytokine production by B. burgdorferi for the first time.
Introduction
Lyme disease, the most common vector-borne disease in the United States and Western Europe, is caused by bacteria of the species Borrelia burgdorferi sensu lato (1). Localized infection is typically manifested by an erythema migrans skin lesion. Depending on the subspecies, disseminated disease is associated mainly with arthritis (B. burgdorferi), skin disorders (Borrelia afzelii), or neurological alterations (B. garinii) (2). Despite great advances in our understanding of the immunologic pathways triggered by the Lyme disease spirochete in humans (3–5), much remains to be learned regarding the immunopathogenesis of the disease. Several in vitro studies indicate the importance of lipid-modified proteins in the pathogenesis of the infection (6–8). The immune system recognizes these lipoproteins by different pattern recognition receptors, of which Toll-like receptor 2 (TLR2)5 and nucleotide-binding oligomerization domain-containing protein 2 (NOD2) are suggested to be the most important (4, 9). The activation of pattern recognition receptors after recognition of Borrelia results in the secretion of the proinflammatory cytokine IL-1β which is known to be involved in the pathogenesis of Lyme disease (10–12). IL-1β production requires two important steps: transcription of mRNA resulting in the production of proIL-1β protein, and cleavage of the immature precursor into mature bioactive IL-1β by the inflammasome-enzyme caspase-1 (13).
In addition to the induction of intracellular signals leading to the production of cytokines, engagement of pattern recognition receptors such as NOD2 activates autophagy, a process in which damaged organelles or long lived proteins are degraded (14–17). Autophagy involves the sequestration of dysfunctional proteins in a double-layered membrane called autophagosome, which is formed by the elongation of small membrane structures. The formation of this isolation membrane is initiated by autophagy-related gene (Atg) 16 and type III phosphatidylinositol 3-kinase (PI3K) (18). The delivery of dysfunctional proteins to the autophagosomes is regulated by autophagic adaptors such as p62. This latter protein can bind to the intracellular target as well as to the microtubule-associated protein 1 light chain 3 (LC3), which associates with the autophagosome after being processed (19). Autophagosomes mature through fusion with lysosomes, leading to the breakdown of the protein content (20).
The link between autophagy and the innate defense mechanism has been made in several studies describing the connection between dysfunctional autophagy and autoinflammatory diseases (21–24). It has been shown that the inhibition of autophagy by chemical inhibitors of PI3 kinases leads to an enhancement of extracellular IL-1β after stimulation with bacterial wall components such as LPS (25). This observation, next to the fact that B. burgdorferi is thought to be recognized by the autophagy-inducing receptor NOD2, prompted us to investigate the role of autophagy in host defense during infection with B. burgdorferi. By stimulating human peripheral blood mononuclear cells (PBMCs), we demonstrate that inhibition of autophagy increases IL-1β and IL-6 production after stimulation with Borrelia bacteria. The enhanced production was specific for IL-1β and IL-6, whereas TNFα production was unchanged. The robust increase in mRNA synthesis of the proinflammatory cytokines IL-1β and IL-6 indicated that autophagy regulates Borrelia-induced IL-1β production at the transcriptional level.
EXPERIMENTAL PROCEDURES
Borrelia burgdorferi Cultures
B. burgdorferi, ATCC strain 35210, was cultured at 33 °C in Barbour-Stoenner-Kelley (BSK)-H medium (Sigma-Aldrich) supplemented with 6% rabbit serum. Spirochetes were grown to late logarithmic phase and examined for motility by dark-field microscopy. Organisms were quantitated by fluorescence microscopy after mixing 10-μl aliquots of culture material with 10 μl of an acridine orange solution and counted using a Petroff-Hauser counting chamber. Bacteria were harvested by centrifugation of the culture at 7000 × g for 15 min, washed twice with sterile PBS (pH 7.4), and diluted in the specified medium to required concentrations.
Isolation of Human Peripheral Blood Mononuclear Cells and in Vitro Cytokine Production
Venous blood was drawn from the cubital vein of healthy volunteers or patients with chronic granulomatous disease (CGD) into 10-ml EDTA tubes (Monoject, Covidien, Mansfield, MA). The mononuclear cell fraction was obtained by density centrifugation of blood diluted 1:1 in pyrogen-free saline over Ficoll-Paque (Pharmacia Biotech). Cells were washed twice in saline and suspended in culture medium (RPMI 1640 medium; Invitrogen) supplemented with 10 mg/ml gentamicin, 10 mm l-glutamine, and 10 mm pyruvate. Cells were counted in a Coulter counter (Coulter Electronics), and the number was adjusted to 5 × 106 cells/ml. A total of 5 × 105 mononuclear cells in a 100-ml volume was added to round-bottom 96-well plates (Greiner, Monroe, NC) and incubated with either 100 ml of culture medium (negative control), or B. burgdorferi (multiplicity of infection (m.o.i.), 0.2). In some experiments, PBMCs were preincubated with culture medium or the autophagy inhibitors 3-methyl adenine (3MA, 10 mm; Sigma), wortmannin (10 μg/ml; BioLegend) or LY294002 (100 μm; Invivogen) for 60 min. After 24 h, supernatants were collected and stored at −20 °C until being assayed.
Real-time PCR
RNA from PBMCs was isolated using TRIzol reagent (Invitrogen) following the manufacturer's instructions. Isolated RNA was reverse-transcribed into complementary DNA using iScript cDNA synthesis kit (Bio-Rad). Quantitative real-time PCR was performed using Power SYBR Green PCR Master Mix (Applied Biosystems) using a 7300 Real-time PCR system (Applied Biosystems). In each PCR a melting curve analysis was included to control for a specific PCR amplification. Primers used for the experiments (final concentration 10 μm) are shown below. Real-time quantitative PCR data were corrected for expression of the housekeeping gene human GAPDH: human IL-1β forward, 5′-GCC-CTA-AAC-AGA-TGA-AGT-GCT-C-3′ and reverse, 5′-GAA-CCA-GCA-TCT-TCC-TCA-G-3′; human IL-6 forward, 5′-AAT-TCG-GTA-CAT-CCT-CGA-CGG-3′ and reverse, 5′-GGT-TGT-TTT-CTG-CCA-GTG-CCT-3′; human TNFα forward, 5′-TGG-CCC-AGG-CAG-TCA-GA-3′ and reverse, 5′-GGT-TTG-CTA-CAA-CAT-GGG-CTA-CA3-′; human GAPDH forward, 5′-AGG-GGA-GAT-TCA-GTG-TGG-TG-3′ and reverse, 5′-CGA-CCA-CTT-TGT-CAA-GCT-CA-3′. Cycling conditions were 2 min at 50 °C and 10 min at 95 °C followed by 40 cycles of 95 °C for 15 s and 1 min at 60 °C.
Cytokine Measurements
Concentrations of human IL-1β, pro-IL-1β IL-6, TNFα, or IL-1 receptor antagonist (IL-1ra) were determined in duplicate using specific commercial ELISA kits (Sanquin, Amsterdam; or R&D Systems, Minneapolis), in accordance with the manufacturers' instructions. Levels of bioactive IL-1 were measured using a murine thyoma cell line EL4/NOB1 that produces IL-2 in response to active IL-1 as described above.
Western Blotting
For Western blotting of LC3 and GAPDH, cells were washed and samples prepared for SDS-PAGE by lysing the cells in radioimmuneprecipitation buffer. After protein electrophoresis in a 15% polyacrylamide gel, proteins were transferred to a nitrocellulose membrane by Western blotting. The membrane was first incubated with rabbit primary antibody specific for LC3-II or GAPDH, followed by a secondary goat anti-rabbit antibody conjugated to horseradish peroxidase. Enhance chemiluminescence was used to detect the proteins. For Western blotting of caspase-1 and actin, cells were washed twice with sterile PBS and lysed in buffer (150 mm NaCl, 10 mm Tris (pH 7.4), 5 mm EDTA, 1 mm EGTA, 0.1% Nonidet P-40) supplemented with a Roche protease inhibitor mixture tablet. Proteins were separated using SDS-PAGE (12% polyacrylamide gel) and subsequently transferred to a PVDF membrane. The membranes were coated with primary antibodies, and active caspase-1 or actin was detected using secondary anti-rabbit antibody conjugated to horseradish peroxidase followed by enhanced chemiluminescence.
Fluorescence Microscopy
HeLa cells were transfected with a plasmid containing GFP-LC3 (kindly provided by Dr. T. Yoshimori, Osaka, Japan) using the transfection medium FuGENE 6 (Roche Applied Science) according to the manufacturer's instructions. GFP-LC3+ HeLa cells were grown and stimulated on coverslips (19-mm diameter) in 12-well plates. Cells were fixed with 2% paraformaldehyde for 15 min at room temperature and permeabilized for 10 min with cold methanol (100%). After washing with PBS (three times), the coverslips were mounted onto glass slides with Vectashield +DAPI and analyzed on a fluorescence microscope.
Confocal Microscopy
HeLa cells were grown and stimulated on coverslips (19-mm diameter) in 12-well plates. Cells were fixed with 2% paraformaldehyde for 15 min at room temperature. After washing with PBS, cells were stained with anti-HLA class I Alexa Fluor 649 + DAPI and analyzed on a confocal microscope.
Reactive Oxygen Species (ROS) Measurement
PBMCs were suspended in Hanks' buffered salt solution and exposed to different concentrations of B. burgdorferi. ROS formation was measured by a chemiluminescence assay using luminol (5 mm, 5-amino-2,3-dihydro-1,4-phthalazinedione; Sigma). The luminometer measured chemiluminescence in the integration mode at 37 °C for 1 h after luminol had been added.
Ethics Statement
All human experiments were conducted according to the principles expressed in the Declaration of Helsinki. Before taking blood, informed written consent of each human subject was provided. The study was approved by the review board of Radboud University Nijmegen Medical Centre.
Statistical Analysis
Data are expressed as mean ± S.E. unless otherwise indicated. Differences between experimental groups were tested using the two-sided Mann-Whitney U-test performed on GraphPad Prism 4.0 software (GraphPad). p values of ≤0.05 were considered significant.
RESULTS
B. burgdorferi Induces Autophagy
Previous studies demonstrated that autophagy inhibits LPS-induced IL-1β in human PBMCs (25). Because IL-1β is associated with the pathogenesis of Lyme disease (12), we assessed the role of autophagy during the exposure to B. burgdorferi based on LC3. During autophagy, the cytoplasmic form (LC3-I) is processed into the phosphatidylethanolamine-conjugated form (LC3-II), the latter indicating activation of autophagy inside the cells. GFP-LC3-expressing HeLa cells were exposed to B. burgdorferi alone or in combination with the autophagy inhibitors 3MA or wortmannin (Fig. 1A). An increase in autophagosome formation can be seen after exposure to B. burgdorferi demonstrated by a high number of GFP-LC3+ punctae. 3MA and wortmannin inhibited the formation of those vesicles attesting the validity of this model. Previously, it has been demonstrated that primary cells as PBMCs but not all cell lines are able to phagocytose the spirochetes (26). Therefore, the ability of the used model (HeLa cells) to internalize B. burgdorferi has been investigated. Internalized spirochetes can be seen indicating the legality of the model (Fig. 1B). Western blot analysis of LC3 in murine bone marrow-derived macrophages (BMDMs) showed an increased amount of LC3-II after exposure to B. burgdorferi (Fig. 1C).
FIGURE 1.

Activation of autophagy by B. burgdorferi. A, GFP-LC3-expressing HeLa cells were preincubated for 1 h at 37 °C in RPMI 1640 medium alone, containing 10 mm 3MA or 100 nm wortmannin in which inhibitors of lysosomal fusion such as ammonium chloride (20 mm) and leupeptine (100 mm) have been added. After 2 h of stimulation with culture medium or B. burgdorferi (m.o.i., 0.4), cells were fixed, nuclei stained with DAPI (blue), and slides were analyzed by fluorescence microscopy. Data are representative of at least four experiments. B, HeLa cells were exposed to GFP-labeled B. burgdorferi (m.o.i., 1) for 2 h, cells were fixed, membrane stained with anti HLA class I Alexa Fluor 649 (red), and nuclei stained with DAPI (blue). Slides were analyzed by confocal microscopy. Data are representative of at least four experiments. C, Western blot analysis of LC3-II in lysates of mouse BMDMs stimulated with B. burgdorferi (m.o.i., 5) was performed for the indicated periods of time.
Inhibition of Autophagy Enhances IL-1β and IL-6 Production but Not TNFα
To determine the role of autophagy in the induction of proinflammatory cytokines by B. burgdorferi, PBMCs obtained from healthy volunteers were stimulated for 24 h with the spirochete alone or in combination with different autophagy inhibitors. 3MA blocks the activity of PI3-kinases, which are mandatory for the initiation of autophagy. PBMCs treated with autophagy inhibitors showed significantly higher IL-1β and IL-6 production after exposure to B. burgdorferi (Fig. 2, A and B). To corroborate the findings of 3MA, other inhibitors were used (wortmannin, LY294002). In contrast, no increase in TNFα production could be observed by blocking autophagy with 3MA, wortmannin, or LY294003 (Fig. 2C). In line with the results of the secreted IL-1β protein, intracellular concentrations of total IL-1β were also enhanced (Fig. 2G). To examine whether inhibition of autophagy during exposure to Borrelia results in altered mRNA levels, we measured transcription of several cytokines. IL-1β and IL-6 mRNA levels were strongly increased in human PBMCs after Borrelia recognition when autophagy was inhibited (Fig. 2, D and E). In contrast, the level of TNFα mRNA was not influenced (Fig. 2F). To exclude any alteration of cytokine production due to the autophagy inhibitors alone, we blocked autophagy in nonstimulated PBMCs, and no increase of cytokine production was observed (data not shown). Using murine ATG7-deficient BMDMs, an enhanced production of IL-1β could be seen after the exposure to B. burgdorferi (data not shown). These findings underscore our results that inhibition of autophagy leads to enhanced IL-1β production after Borrelia exposure.
FIGURE 2.

Modulation of B. burgdorferi-induced inflammatory cytokine production by the inhibition of autophagy. Freshly isolated human PBMCs were preincubated for 1 h at 37 °C in culture medium in the presence or absence of autophagy inhibitors such as 3MA (10 mm), wortmannin (100 nm), and LY294002 (100 μm) and afterward stimulated with B. burgdorferi (m.o.i. 0.2) for 24 h. IL-1β (A), IL-6 (B), and TNFα (C) in the supernatants as well as intracellular IL-1β (G) in cell lysates were measured by specific ELISA. IL-1β (D), IL-6 (E), and TNFα (F) expression levels have been determined by RT-PCR. Bars represent mean ± S.E. (error bars) of cells harvested from eight volunteers. *, p < 0.05; **, p < 0.01; ***, p < 0.0001, Mann-Whitney U-test, two-sided.
Increased Extracellular IL-1β Is Not Based on the Secretion of Immature IL-1β Protein
To exclude the possibility of an increased secretion of immature (and inactive) IL-1β by autophagy inhibitors, the bioactivity of extracellular IL-1β was measured using a murine thyoma cell line EL4/NOB1, which produces IL-2 in response to active IL-1. The amount of bioactive IL-1 increased in the presence of autophagy inhibitors (Fig. 3A). To support these data, the amount of pro-IL-1β was measured in the supernatants of PBMCs incubated with B. burgdorferi alone or in combination with autophagy inhibitors. No difference in the amount of proIL-1β could be found in the supernatant of the various samples (Fig. 3B). An increase in proinflammatory cytokines due to cell death was also excluded because the level of lactate dehydrogenase, a marker for cell death, stayed below the detection level in all samples (data not shown).
FIGURE 3.
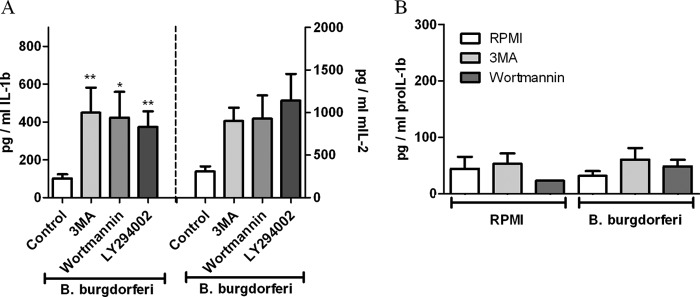
Bioactivity of extracellular IL-1β. Freshly isolated human PBMCs were preincubated for 1 h at 37 °C in culture medium in the presence or absence of autophagy inhibitors such as 3MA (10 mm), wortmannin (100 nm), LY294002 (100 μm) and afterward stimulated with B. burgdorferi (m.o.i., 0.2) for 24 h. A, supernatant was added to murine NOB-1 cells which react to functional human IL-1 with the secretion of IL-2. Murine IL-2 was measured in the supernatant of NOB-1 cells after 24 h. B, pro-IL-1β was measured in supernatants using a specific ELISA. Data from four volunteers are shown as mean ± S.E. (error bars). *, p < 0.05; **,p < 0.01, Mann-Whitney U-test, two-sided.
Inhibition of Autophagy Does Not Increase Caspase-1 Activity
IL-1β is synthesized as an immature pro-form (pro-IL-1β) which requires activated caspase-1, a component of the inflammasome, for cleavage into mature IL-1β secreted from the cells. To determine the role of autophagy in the activation of caspase-1 leading to processing of immature IL-1β, PBMCs obtained from healthy volunteers were stimulated with B. burgdorferi alone or in combination with several autophagy inhibitors for 3 h. The amount of active caspase-1 stayed the same independent of the inhibition of autophagy (Fig. 4). Furthermore, the influence of caspase-1 on the induction of autophagy by B. burgdorferi has been analyzed using GFP-LC3 expressing HeLa cells incubated with the caspase-1 inhibitor YVAD and Borrelia. No effect on autophagosome formation by caspase-1 inhibition was seen after exposure of the GFP-LC3 cells to B. burgdorferi (data not shown).
FIGURE 4.
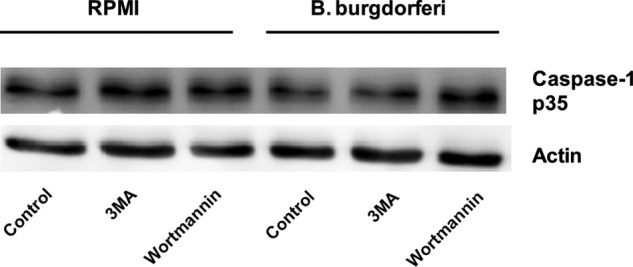
Autophagy does not influence the activation of caspase-1. Western blot analysis of caspase-1 in lysates of autophagy-impaired human PBMCs stimulated with B. burgdorferi (m.o.i., 2) was performed. PBMCs were preincubated for 1 h at 37 °C in RPMI 1640 medium alone, containing 10 mm 3MA or 100 nm wortmannin. After 3 h of stimulation with culture medium or B. burgdorferi, cells were lysed and further analyzed with Western blotting. The picture is representative for results obtained from four volunteers.
B. burgdorferi-induced ROS Reduce the Production of IL-1β
It has been proposed that the formation of ROS forms a necessary step in the induction of autophagy (27, 28). We therefore hypothesized that defects in the production of ROS would result in a defective autophagic process, followed by an increase in IL-1β production. The exposure of B. burgdorferi to PBMCs resulted in a significant increase in ROS compared with unstimulated samples (Fig. 5A). To further assess the production of cytokines in response to ROS, PBMCs of patients with CGD, who have a defective production of ROS, were exposed to B. burgdorferi for 24 h (Fig. 5B). Cells of CGD patients showed higher production of IL-1β compared with healthy controls, supporting our previous data of increased B. burgdorferi-induced cytokines during impaired autophagy (Fig. 2A).
FIGURE 5.
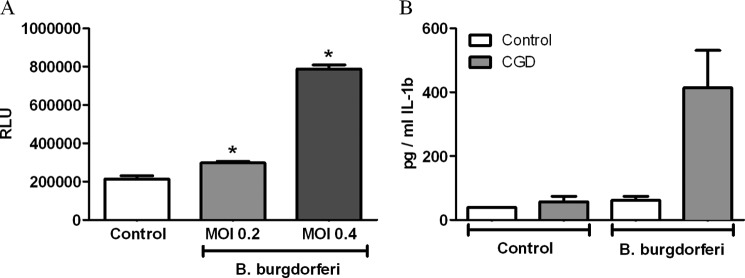
Production of IL-1β after B. burgdorferi stimulation is controlled by ROS. A, freshly isolated human PBMCs were stimulated with B. burgdorferi for 1 h. The production of ROS was measured by chemiluminescence using luminol. B, freshly isolated human PBMCs of healthy volunteers and CGD patients were stimulated with B. burgdorferi for 24 h. IL-1β was measured in the supernatant by specific ELISA. Bars represent mean ± S.E. (error bars). *, p < 0.05, Mann-Whitney U-test, two-sided. n = 4 controls, patients.
Enhanced Production of IL-1β and IL-6 during Inhibition of Autophagy Is Not Based on IL-1β Feedback Loop
Previous studies showed that exogenous IL-1β induces endogenous IL-1β and IL-6 mRNA expression and protein production. To assess the influence of extracellular bioactive IL-1β on the production of cytokines by PBMCs, which have been treated with autophagy inhibitors, we blocked the IL-1 receptor using IL-1ra (Anakinra) and analyzed the production of cytokines at mRNA level as well as secreted protein. PBMCs treated with IL-1ra in combination with 3MA or wortmannin produce a larger amount of IL-1β and IL-6 in response to B. burgdorferi at both mRNA expression and protein levels (Fig. 6, A, B, D, E, G). In contrast, TNFα mRNA expression, as well as the secreted TNFα protein, was not affected by autophagy inhibitors even in the presence of IL-1ra (Fig. 6, C and F).
FIGURE 6.

Effect of IL-1ra on the autophagy-related increase in inflammatory cytokine production. Human PBMCs were preincubated for 1 h at 37 °C in culture medium in the presence or absence of IL-1ra and autophagy inhibitors such as 3MA (10 mm), wortmannin (100 nm), and LY294002 (100 μm). Afterward, cells were stimulated with B. burgdorferi (m.o.i., 0.2) for 24 h. IL-1β (A), IL-6 (B), and TNFα (C) in the supernatants as well as intracellular IL-1β (G) in cell lysates were measured by specific ELISA. IL-1β (D), IL-6 (E), and TNFα (F) expression levels were determined by RT-PCR. Bars represent mean ± S.E. (error bars) of data obtained in six volunteers. *, p < 0.05, Mann-Whitney U-test, two-sided.
DISCUSSION
The link between autophagy and innate immunity, and its role as a defense mechanism against invading pathogens, has been made in several studies (29–31), suggesting a regulatory role of autophagy on inflammasome activation and production of cytokines upon stimulation with microbial ligands (32). Because IL-1β has been demonstrated to contribute to the pathogenesis of Lyme disease (12), we examined the role of autophagy for the production of this pivotal proinflammatory cytokine in response to B. burgdorferi. Although both mRNA expression and production of TNFα were not enhanced in autophagy-blocked PBMCs stimulated with the Borrelia spirochete, secretion of IL-1β and IL-6 protein was highly elevated. In addition, the mRNA synthesis of IL-1 β and IL-6 was strongly increased as well, indicating that the inhibition of autophagy modulates IL-1β and IL-6 production at the transcriptional level.
IL-1β is synthesized as an inactive procytokine (pro-IL-1β) and requires activated caspase-1, a component of the inflammasomes, for cleavage into mature bioactive IL-1β. Because recent studies revealed an association between genetic disorders involving the activation of the inflammasome and autoinflammatory disorders (33–35), we investigated the effect of autophagy on the activation of inflammasome/caspase-1. In contrast to murine studies (36), autophagy did not inhibit inflammasome activation in human PBMCs, supporting our previous studies that demonstrated that autophagy inhibition affected the transcription, rather than processing, of IL-1β. The constitutive active caspase-1 in primary human monocytes, in contrast to the inactive caspase-1 of mouse macrophages, may explain the different results (37). An increased release of pro-IL-1β secretion due to cell death could be excluded by evaluation of the bioactivity of IL-1 that was significantly increased by autophagy inhibitors (Fig. 3).
The increased production of IL-6 after inhibition of autophagy is most probably a consequence of increased bioactive IL-1β, because it has been shown that IL-1 receptor signaling is a major inducer of IL-6 (38). Interestingly, production of TNFα was not affected, which can be explained by different induction pathways. It has been shown that p38-MAPK and NF-κB are main inducers of TNFα transcription, whereas additional pathways such as extracellular signal-related kinase/nuclear factor κB (ERK/NF-κB) are needed for the transcription of IL-1β (39, 40).
Recent studies have revealed that formation of ROS forms a necessary step in the initiation of autophagy (27, 28). Several TLRs are able to activate Nox2, leading to the generation of ROS. These oxidative conditions are essential for autophagy induction because treatment with antioxidative agents abolishes the formation of autophagosomes (27). We therefore hypothesized that defects in the production of ROS would result in a defective autophagic process, followed by an increase in IL-1β production. Consistent with this hypothesis, cells isolated from patients with CGD, who have a defective production of ROS due to genetic defects in the genes forming the NADPH complex, had an enhanced production of IL-1β compared with healthy controls (Fig. 5). These additional data support our results of increased production of B. burgdorferi-induced cytokines in autophagy-impaired cells (Fig. 2, A and B).
Extracellular IL-1β performs its proinflammatory effect via an autocrine mechanism followed by binding to its cognate receptor IL-1 receptor type I (41). In the present study, we used recombinant IL-1ra to determine the contribution of an IL-1-positive feedback loop to the total amount of B. burgdorferi-induced cytokine production. IL-1r blockade in autophagy capable cells resulted in reduced cytokine concentrations after Borrelia exposure. However, in cells in which autophagy was impaired, we noted an increased induction of cytokines by B. burgdorferi even if IL-1ra was added to the medium to block the IL-1-induced loop, suggesting an IL-1r- independent mechanism.
In previous reports it was described that autophagy plays a role in the cytokine response to several TLR-ligands (32, 42). However, this is the first study to show an influence of autophagy on the Borrelia-induced cytokine response. The precise mechanism underlying the effect of autophagy on IL-1β production induced by B. burgdorferi remains to be elucidated in further detail. Lee et al. (43) proposed a model in which autophagy suppresses the IL-1β signaling by down-regulating p62 levels (25, 43). p62 acts a selective autophagy receptor for ubiquitinated protein aggregates (44) and as an important scaffold in the IL-1β signaling pathway by promoting oligomerization and activation of TRAF6 resulting in the transcription of NF-κB (45) and consequently increased IL-1β production. The expression level of p62 is important for these functions, and it is thought to be regulated by autophagy. In the absence of this degradation process, increased p62 levels promote oligomerization and activation of TRAF6 resulting in overactivation of NF-κB. Therefore, we propose that the recognition of B. burgdorferi by TLR2 and NOD2 induces the production of IL-1β. In the absence of autophagy, p62 levels will be elevated, leading to even more TRAF6 aggregation and consequently more IL-1β production.
The autophagy-dependent increase of IL-1β in PBMCs seems to be Borrelia-specific, because the exposure of different pathogens such as Candida albicans to autophagy-impaired cells resulted in a decreased IL-1β response (data not shown). Therefore, a better understanding of the precise mechanisms underlying the altered IL-1β production upon B. burgdorferi stimulation in autophagy-impaired cells may lead to the identification of novel therapeutic targets that may be important in the pathogenesis of Lyme disease in which IL-1-dependent inflammation is central.
It is tempting to speculate that inhibition of autophagy could be beneficial during the early phase of Lyme disease leading to higher concentrations of IL-1β and therefore a possibly improved clearance of the pathogen. However, because Lyme disease occurs in different stadia, enhanced IL-1β production by autophagy modulation in disseminated Lyme disease might be detrimental for the disease activity. Several studies indicated a link between a high concentrations of IL-1β and pathogenic Th17 cells l leading to increased joint damage in rheumatoid arthritis or psoriasis patients (46, 47). Therefore, the induction of autophagy in the disseminated state of Lyme disease could be favorable for Lyme patients through a decreased IL-1β production and inhibition of pathogenic Th17 cells.
In summary, we have elucidated the link between B. burgdorferi recognition and an important regulatory mechanism of inflammation by autophagy in Lyme disease. These findings may reveal novel therapeutic targets to treat Lyme disease patients in the future.
Acknowledgments
We thank Carla Bartels (Department of Medical Microbiology, Radboud University Nijmegen Medical Centre) for culturing and counting Borrelia spirochetes, Dr. T. Yoshimori (Osaka, Japan) for providing the plasmid containing GFP-LC3, Huib Croes for help using the confocal microscope, and Prof. J. D. Radolf (University of Connecticut) for providing GFP-labeled Borrelia.
This work was supported in part by Dutch Arthritis Association Grant NR 10-1-303.
- TLR
- Toll-like receptor
- Atg
- autophagy-related gene
- BMDM
- bone marrow-derived macrophage
- CGD
- chronic granulomatous disease
- IL-1ra
- IL-1 receptor antagonist
- LC3
- microtubule-associated protein 1 light chain 3
- 3MA
- 3-methyl adenine
- m.o.i.
- multiplicity of infection
- NOD2
- nucleotide-binding oligomerization domain-containing protein 2
- PBMC
- peripheral blood mononuclear cell
- ROS
- reactive oxygen species.
REFERENCES
- 1. Stanek G., Wormser G. P., Gray J., Strle F. (2012) Lyme borreliosis. Lancet 379, 461–473 [DOI] [PubMed] [Google Scholar]
- 2. Derdáková M., Lencáková D. (2005) Association of genetic variability within the Borrelia burgdorferi sensu lato with the ecology, epidemiology of Lyme borreliosis in Europe. Ann. Agric. Environ. Med. 12, 165–172 [PubMed] [Google Scholar]
- 3. Cruz A. R., Moore M. W., La Vake C. J., Eggers C. H., Salazar J. C., Radolf J. D. (2008) Phagocytosis of Borrelia burgdorferi, the Lyme disease spirochete, potentiates innate immune activation and induces apoptosis in human monocytes. Infect. Immun. 76, 56–70 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Oosting M., Ter Hofstede H., Sturm P., Adema G. J., Kullberg B. J., van der Meer J. W., Netea M. G., Joosten L. A. (2011) TLR1/TLR2 heterodimers play an important role in the recognition of Borrelia spirochetes. PLoS One 6, e25998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Cervantes J. L., Dunham-Ems S. M., La Vake C. J., Petzke M. M., Sahay B., Sellati T. J., Radolf J. D., Salazar J. C. (2011) Phagosomal signaling by Borrelia burgdorferi in human monocytes involves Toll-like receptor (TLR) 2 and TLR8 cooperativity and TLR8-mediated induction of IFN-β. Proc. Natl. Acad. Sci. U.S.A. 108, 3683–3688 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Vincent M. S., Roessner K., Sellati T., Huston C. D., Sigal L. H., Behar S. M., Radolf J. D., Budd R. C. (1998) Lyme arthritis synovial γδT cells respond to Borrelia burgdorferi lipoproteins and lipidated hexapeptides. J. Immunol. 161, 5762–5771 [PubMed] [Google Scholar]
- 7. Hefty P. S., Jolliff S. E., Caimano M. J., Wikel S. K., Radolf J. D., Akins D. R. (2001) Regulation of OspE-related, OspF-related, and Elp lipoproteins of Borrelia burgdorferi strain 297 by mammalian host-specific signals. Infect. Immun. 69, 3618–3627 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Norgard M. V., Arndt L. L., Akins D. R., Curetty L. L., Harrich D. A., Radolf J. D. (1996) Activation of human monocytic cells by Treponema pallidum and Borrelia burgdorferi lipoproteins and synthetic lipopeptides proceeds via a pathway distinct from that of lipopolysaccharide but involves the transcriptional activator NF-κB. Infect. Immun. 64, 3845–3852 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Oosting M., Berende A., Sturm P., Ter Hofstede H. J., de Jong D. J., Kanneganti T. D., van der Meer J. W., Kullberg B. J., Netea M. G., Joosten L. A. (2010) Recognition of Borrelia burgdorferi by NOD2 is central for the induction of an inflammatory reaction. J. Infect. Dis. 201, 1849–1858 [DOI] [PubMed] [Google Scholar]
- 10. Liu N., Belperron A. A., Booth C. J., Bockenstedt L. K. (2009) The caspase 1 inflammasome is not required for control of murine Lyme borreliosis. Infect. Immun. 77, 3320–3327 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Petzke M. M., Brooks A., Krupna M. A., Mordue D., Schwartz I. (2009) Recognition of Borrelia burgdorferi, the Lyme disease spirochete, by TLR7 and TLR9 induces a type I IFN response by human immune cells. J. Immunol. 183, 5279–5292 [DOI] [PubMed] [Google Scholar]
- 12. Pietruczuk A., Swierzbińska R., Pietruczuk M., Hermanowska-Szpakowicz T. (2004) [Role of interleukin-18, interleukin-1β and its soluble receptor (sIL-1RII) in early and late Lyme borreliosis]. Pol. Merkur Lekarski 17, 446–450 [PubMed] [Google Scholar]
- 13. Dinarello C. A. (2009) Immunological and inflammatory functions of the interleukin-1 family. Annu. Rev. Immunol. 27, 519–550 [DOI] [PubMed] [Google Scholar]
- 14. Cooney R., Baker J., Brain O., Danis B., Pichulik T., Allan P., Ferguson D. J., Campbell B. J., Jewell D., Simmons A. (2010) NOD2 stimulation induces autophagy in dendritic cells influencing bacterial handling and antigen presentation. Nat. Med. 16, 90–97 [DOI] [PubMed] [Google Scholar]
- 15. Plantinga T. S., Joosten L. A., Netea M. G. (2011) ATG16L1 polymorphisms are associated with NOD2-induced hyperinflammation. Autophagy 7, 1074–1075 [DOI] [PubMed] [Google Scholar]
- 16. Netea M. G., Joosten L. A. (2010) A NOD for autophagy. Nat. Med. 16, 28–30 [DOI] [PubMed] [Google Scholar]
- 17. Anand P. K., Tait S. W., Lamkanfi M., Amer A. O., Nunez G., Pagès G., Pouysségur J., McGargill M. A., Green D. R., Kanneganti T. D. (2011) TLR2 and RIP2 pathways mediate autophagy of Listeria monocytogenes via extracellular signal-regulated kinase (ERK) activation. J. Biol. Chem. 286, 42981–42991 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Oh J. E., Lee H. K. (2012) Modulation of pathogen recognition by autophagy. Front. Immunol. 3, 44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Ponpuak M., Davis A. S., Roberts E. A., Delgado M. A., Dinkins C., Zhao Z., Virgin H. W., 4th, Kyei G. B., Johansen T., Vergne I., Deretic V. (2010) Delivery of cytosolic components by autophagic adaptor protein p62 endows autophagosomes with unique antimicrobial properties. Immunity 32, 329–341 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Kundu M., Thompson C. B. (2008) Autophagy: basic principles and relevance to disease. Annu. Rev. Pathol. 3, 427–455 [DOI] [PubMed] [Google Scholar]
- 21. Márquez A., Núñez C., Martínez A., Mendoza J. L., Taxonera C., Fernández-Arquero M., Díaz-Rubio M., de la Concha E. G., Urcelay E. (2009) Role of ATG16L1 T300A polymorphism in inflammatory bowel disease: a study in the Spanish population and a meta-analysis. Inflamm. Bowel Dis. 15, 1697–1704 [DOI] [PubMed] [Google Scholar]
- 22. Zhang H. F., Qiu L. X., Chen Y., Zhu W. L., Mao C., Zhu L. G., Zheng M. H., Wang Y., Lei L., Shi J. (2009) ATG16L1 T300A polymorphism and Crohn's disease susceptibility: evidence from 13,022 cases and 17,532 controls. Hum. Genet. 125, 627–631 [DOI] [PubMed] [Google Scholar]
- 23. Plantinga T. S., Joosten L. A., van der Meer J. W., Netea M. G. (2012) Modulation of inflammation by autophagy: consequences for Crohn's disease. Curr. Opin. Pharmacol. 12, 497–502 [DOI] [PubMed] [Google Scholar]
- 24. Plantinga T. S., Crisan T. O., Oosting M., van de Veerdonk F. L., de Jong D. J., Philpott D. J., van der Meer J. W., Girardin S. E., Joosten L. A., Netea M. G. (2011) Crohn's disease-associated ATG16L1 polymorphism modulates pro-inflammatory cytokine responses selectively upon activation of NOD2. Gut 60, 1229–1235 [DOI] [PubMed] [Google Scholar]
- 25. Criŝan T. O., Plantinga T. S., van de Veerdonk F. L., Farcaŝ M. F., Stoffels M., Kullberg B. J., van der Meer J. W., Joosten L. A., Netea M. G. (2011) Inflammasome-independent modulation of cytokine response by autophagy in human cells. PLoS One 6, e18666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Moore M. W., Cruz A. R., LaVake C. J., Marzo A. L., Eggers C. H., Salazar J. C., Radolf J. D. (2007) Phagocytosis of Borrelia burgdorferi and Treponema pallidum potentiates innate immune activation and induces γ-interferon production. Infect. Immun. 75, 2046–2062 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Scherz-Shouval R., Shvets E., Fass E., Shorer H., Gil L., Elazar Z. (2007) Reactive oxygen species are essential for autophagy and specifically regulate the activity of Atg4. EMBO J. 26, 1749–1760 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Huang J., Brumell J. H. (2009) NADPH oxidases contribute to autophagy regulation. Autophagy 5, 887–889 [DOI] [PubMed] [Google Scholar]
- 29. Nakagawa I., Amano A., Mizushima N., Yamamoto A., Yamaguchi H., Kamimoto T., Nara A., Funao J., Nakata M., Tsuda K., Hamada S., Yoshimori T. (2004) Autophagy defends cells against invading group A Streptococcus. Science 306, 1037–1040 [DOI] [PubMed] [Google Scholar]
- 30. Gutierrez M. G., Master S. S., Singh S. B., Taylor G. A., Colombo M. I., Deretic V. (2004) Autophagy is a defense mechanism inhibiting BCG and Mycobacterium tuberculosis survival in infected macrophages. Cell 119, 753–766 [DOI] [PubMed] [Google Scholar]
- 31. Kleinnijenhuis J., Oosting M., Plantinga T. S., van der Meer J. W., Joosten L. A., Crevel R. V., Netea M. G. (2011) Autophagy modulates the Mycobacterium tuberculosis-induced cytokine response. Immunology 134, 341–348 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Saitoh T., Fujita N., Jang M. H., Uematsu S., Yang B. G., Satoh T., Omori H., Noda T., Yamamoto N., Komatsu M., Tanaka K., Kawai T., Tsujimura T., Takeuchi O., Yoshimori T., Akira S. (2008) Loss of the autophagy protein Atg16L1 enhances endotoxin-induced IL-1β production. Nature 456, 264–268 [DOI] [PubMed] [Google Scholar]
- 33. Franchi L., Eigenbrod T., Muñoz-Planillo R., Nuñez G. (2009) The inflammasome: a caspase-1-activation platform that regulates immune responses and disease pathogenesis. Nat. Immunol. 10, 241–247 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Jha S., Ting J. P. (2009) Inflammasome-associated nucleotide-binding domain, leucine-rich repeat proteins and inflammatory diseases. J. Immunol. 183, 7623–7629 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Vandanmagsar B., Youm Y. H., Ravussin A., Galgani J. E., Stadler K., Mynatt R. L., Ravussin E., Stephens J. M., Dixit V. D. (2011) The NLRP3 inflammasome instigates obesity-induced inflammation and insulin resistance. Nat. Med. 17, 179–188 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Shi C. S., Shenderov K., Huang N. N., Kabat J., Abu-Asab M., Fitzgerald K. A., Sher A., Kehrl J. H. (2012) Activation of autophagy by inflammatory signals limits IL-1β production by targeting ubiquitinated inflammasomes for destruction. Nat. Immunol. 13, 255–263 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Netea M. G., Nold-Petry C. A., Nold M. F., Joosten L. A., Opitz B., van der Meer J. H., van de Veerdonk F. L., Ferwerda G., Heinhuis B., Devesa I., Funk C. J., Mason R. J., Kullberg B. J., Rubartelli A., van der Meer J. W., Dinarello C. A. (2009) Differential requirement for the activation of the inflammasome for processing and release of IL-1β in monocytes and macrophages. Blood 113, 2324–2335 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Tosato G., Jones K. D. (1990) Interleukin-1 induces interleukin-6 production in peripheral blood monocytes. Blood 75, 1305–1310 [PubMed] [Google Scholar]
- 39. Campbell J., Ciesielski C. J., Hunt A. E., Horwood N. J., Beech J. T., Hayes L. A., Denys A., Feldmann M., Brennan F. M., Foxwell B. M. (2004) A novel mechanism for TNF-α regulation by p38 MAPK: involvement of NF-κB with implications for therapy in rheumatoid arthritis. J. Immunol. 173, 6928–6937 [DOI] [PubMed] [Google Scholar]
- 40. Hsu H. Y., Wen M. H. (2002) Lipopolysaccharide-mediated reactive oxygen species and signal transduction in the regulation of interleukin-1 gene expression. J. Biol. Chem. 277, 22131–22139 [DOI] [PubMed] [Google Scholar]
- 41. Segovia J., Sabbah A., Mgbemena V., Tsai S. Y., Chang T. H., Berton M. T., Morris I. R., Allen I. C., Ting J. P., Bose S. (2012) TLR2/MyD88/NF-κB pathway, reactive oxygen species, potassium efflux activates NLRP3/ASC inflammasome during respiratory syncytial virus infection. PLoS One 7, e29695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Delgado M. A., Elmaoued R. A., Davis A. S., Kyei G., Deretic V. (2008) Toll-like receptors control autophagy. EMBO J. 27, 1110–1121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Lee J., Kim H. R., Quinley C., Kim J., Gonzalez-Navajas J., Xavier R., Raz E. (2012) Autophagy suppresses interleukin-1β (IL-1β) signaling by activation of p62 degradation via lysosomal and proteasomal pathways. J. Biol. Chem. 287, 4033–4040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Knaevelsrud H., Simonsen A. (2010) Fighting disease by selective autophagy of aggregate-prone proteins. FEBS Lett. 584, 2635–2645 [DOI] [PubMed] [Google Scholar]
- 45. Huang L. Y., Reis e Sousa C., Itoh Y., Inman J., Scott D. E. (2001) IL-12 induction by a TH1-inducing adjuvant in vivo: dendritic cell subsets and regulation by IL-10. J. Immunol. 167, 1423–1430 [DOI] [PubMed] [Google Scholar]
- 46. Geboes L., Dumoutier L., Kelchtermans H., Schurgers E., Mitera T., Renauld J. C., Matthys P. (2009) Proinflammatory role of the Th17 cytokine interleukin-22 in collagen-induced arthritis in C57BL/6 Mice. Arthritis Rheum. 60, 390–395 [DOI] [PubMed] [Google Scholar]
- 47. Ma H. L., Liang S., Li J., Napierata L., Brown T., Benoit S., Senices M., Gill D., Dunussi-Joannopoulos K., Collins M., Nickerson-Nutter C., Fouser L. A., Young D. A. (2008) IL-22 is required for Th17 cell-mediated pathology in a mouse model of psoriasis-like skin inflammation. J. Clin. Invest. 118, 597–607 [DOI] [PMC free article] [PubMed] [Google Scholar]