

Background: Toxoplasma gondii requires a perforin-like protein (PLP1) for rapid host cell egress.
Results: Loss of the PLP1 N-terminal domain reduced parasite egress and lytic activity, but not virulence.
Conclusion: PLP1 has a unique accessory N-terminal domain, which binds membranes and promotes rapid egress.
Significance: Understanding the role of accessory domains is critical for defining the activity of pore-forming proteins.
Keywords: Cell Permeabilization, Gene Knock-out, Host-Pathogen Interactions, Microbial Pathogenesis, Mutant, Parasite, Protein Secretion, Protein Self-assembly, Egress, Toxoplasma gondii
Abstract
The recently discovered role of a perforin-like protein (PLP1) for rapid host cell egress by the protozoan parasite Toxoplasma gondii expanded the functional diversity of pore-forming proteins. Whereas PLP1 was found to be necessary for rapid egress and pathogenesis, the sufficiency for and mechanism of membrane attack were yet unknown. Here we further dissected the PLP1 knock-out phenotype, the mechanism of PLP1 pore formation, and the role of each domain by genetic complementation. We found that PLP1 is sufficient for membrane disruption and has a conserved mechanism of pore formation through target membrane binding and oligomerization to form large, multimeric membrane-embedded complexes. The highly conserved, central MACPF domain and the β-sheet-rich C-terminal domain were required for activity. Loss of the unique N-terminal extension reduced lytic activity and led to a delay in rapid egress, but did not significantly decrease virulence, suggesting that small amounts of lytic activity are sufficient for pathogenesis. We found that both N- and C-terminal domains have membrane binding activity, with the C-terminal domain being critical for function. This dual mode of membrane association may promote PLP1 activity and parasite egress in the diverse cell types in which this parasite replicates.
Introduction
Pore-forming proteins involved in infection have been classically viewed as aiding pathogen invasion into host cells and tissues or promoting pathogen clearance by the host immune system. For example, the Gram-positive bacterium, Listeria monocytogenes, utilizes listeriolysin O (LLO)2 to escape its initial endosomal vacuole to access its replication niche in the host cytosol (1). Also, Plasmodium sporozoites utilize pore-forming proteins to promote tissue migration during multiple stages of their life cycle (2, 3). To combat infection, mammalian hosts circulate components of the complement cascade, which upon activation culminates in the assembly of the membrane attack complex on target microbes, such as the Gram-negative bacterium Neisseria meningitidis (4). Additionally, perforin, a pore-forming protein secreted by cytotoxic T lymphocytes, delivers proteases into virus-infected or transformed cells to initiate programmed cell death (5). Related pore-forming proteins are found in diverse organisms throughout the tree of life (6).
Our group recently identified a perforin-like protein (PLP1) that is crucial for rapid host cell exit of the protozoan parasite, Toxoplasma gondii (7). T. gondii is the etiological agent of toxoplasmosis, which causes severe disease when acquired congenitally or reactivated in immune-compromised individuals, manifesting as tissue destruction from unchecked lytic growth (8–10). T. gondii belongs to the phylum Apicomplexa, which includes other pathogens of human and veterinary significance such as Plasmodium, Cryptosporidium, and Eimeria (11–14). These microorganisms have a unique set of apical secretory organelles termed micronemes and rhoptries. Proteins secreted from these organelles are involved in parasite gliding motility, cell invasion, and manipulation of the host cell to protect the intracellular replication compartment, the parasitophorous vacuole (15–18). Thus, the majority of characterized secretory proteins function at the point of cell entry.
The Toxoplasma lytic cycle occurs through multiple rounds of cell entry, replication, and egress. Whereas molecular mechanisms of cell entry are well characterized, those governing cell exit are largely unknown (19, 20). The discovery that a micronemal protein, PLP1, is required for rapid egress revealed that the parasite produces specific factors for cell exit. PLP1 is a lytic protein with a unique postreplicative function in niche escape rather than a prereplicative function to access a replication site as seen for the pore-forming proteins of the aforementioned pathogens. This and other studies (21–23) support an emerging hypothesis of egress as an event the parasite actively regulates instead of a strictly passive process occurring upon exceeding a critical capacity. PLP1-deficient parasites failed to egress rapidly from the parasitophorous vacuole after calcium ionophore treatment, which induces microneme secretion and motility (24, 25). Additionally, whereas wild-type parasites effectively permeabilized the parasitophorous vacuole membrane (PVM), PLP1-deficient parasites retained an intact surrounding membrane (7). These results suggested that PLP1 functions during egress by disrupting the PVM, which allows parasites to cross this physical barrier rapidly. PLP1-knock-out parasites were also noted to have an invasion defect; however, the contribution of PLP1 to invasion remained unknown.
Recent work on pore-forming proteins has demonstrated conservation of structure between the cholesterol-dependent cytolysins (CDCs) and membrane attack complex/perforin family (MACPF) proteins (26–28). The conservation of structure leads to a proposed conservation in mechanism of membrane permeabilization (6, 29, 30). The pore-forming protein is secreted as a soluble monomer and binds a receptor on the target cell membrane. Following membrane binding, monomers oligomerize into ring-like structures and undergo a structural rearrangement to create a lesion in the target membrane (31, 32). Whereas many pore-forming proteins, such as perforin and LLO, are sufficient for membrane damage, others, such as the membrane attack complex, require assembly of multiple proteins in a specific order (33). Thus, it is important to identify whether PLP1 is sufficient for membrane damage or requires other parasite or host factors.
Predicted MACPF domains are found in genomes of diverse organisms and are associated with a wide variety of adjacent domains. Only a few MACPF-associated domains, however, have been characterized. PLP1 has a highly conserved central MACPF domain, responsible for oligomerization and membrane permeabilization in other MACPF domain-containing proteins. PLP1 also has unique N- and C-terminal extensions. The C-terminal domain is predicted to be β-sheet-rich, a general feature that is conserved among C-terminal domains of other pore-forming proteins (6). The position and structural features of the PLP1 C-terminal domain implicate it in membrane binding, but its role has not been addressed previously. The PLP1 N-terminal domain sequence contains no conserved domains and is found only in Toxoplasma and the related Neospora sequences. It may be more distantly related to the N-terminal extensions seen on some of the Plasmodium MACPF proteins (34). N-terminal domains in other MACPF proteins include a low density lipoprotein receptor A domain and thrombospondin type I domains in several complement proteins (35, 36). Examples of N-terminal domains in CDCs include the lectin binding domain of lectinolysin and an N-terminal PEST-like sequence in LLO (37–41). Because of its unique characteristics, no obvious function for the PLP1 N-terminal domain has been proposed.
Here we investigated the contribution of PLP1 to cell invasion, PLP1 sufficiency for membrane damage, the general mechanism of membrane attack and the role of each domain of PLP1 in membrane damage.
MATERIALS AND METHODS
Ethics Statement
This study was carried out in strict accordance with the Public Health Service Policy on Humane Care and Use of Laboratory Animals and Association for the Assessment and Accreditation of Laboratory Animal Care guidelines. The animal protocol was approved by the University of Michigan Committee on the Use and Care of Animals (Animal Welfare Assurance A3114–01, protocol 09482). All efforts were made to minimize pain and suffering.
Parasite Culture
Parasites were maintained in human foreskin fibroblasts (HFFs) in Dulbecco's modified Eagle's medium supplemented with 10% calf serum, 10 mm HEPES, 2 mm l-glutamine, and 50 μg/ml penicillin/streptomycin. The parent strain RHΔku80Δhxg (42) is derived from the commonly used, virulent RH strain (43). The genetically modified strains used in this study are described in other sections of the text.
Invasion Assays
Parasites were inoculated at a high (∼300 μl) or low (∼50 μl) passage in T25 flasks of HFF. Following 2-day growth, parasites in high passage flasks had egressed, whereas parasites in low passage flasks remained intracellular. Egressed parasites were filter-purified in Endo buffer (44.7 mm K2SO4, 106 mm sucrose, 10 mm MgSO4, 20 mm Tris-H2SO4 (pH 8.2), 5 mm glucose, 3.5 mg/ml bovine serum albumin) (44), and a red-green invasion assay was performed as previously described (45). Low passage flasks were washed twice with room temperature Endo buffer, scraped, and syringe-passed prior to filter purification in Endo buffer and subsequent invasion assay. Immunofluorescence was performed as previously described (45), and 15 fields of view (total magnification, ×400) per strain were examined on a Zeiss Axio inverted microscope for attached and invaded parasites. Graphs depict the average ± S.D. of three independent experiments.
Egress Assays
Parasites were inoculated into HFFs in an 8-well chamber slide and incubated at 37 °C at 5% CO2 for 30 h. Wells were washed twice with warm phosphate-buffered saline (PBS), and egress was tested by adding 120 μl/well of vehicle dimethyl sulfoxide or 2 μm A23187 in egress assay buffer (Hanks' buffered salt solution containing 1 mm CaCl2, 1 mm MgCl2, and 10 mm HEPES) and incubated for 2 min in a 37 °C water bath. Egress was stopped by addition of 2× fixative (8% formaldehyde in 2× PBS). Immunofluorescence was performed to stain parasites with antibodies to surface antigen 1 and the parasitophorous vacuole membrane with antibodies to dense granule protein 7. At least 10 fields of view (magnification, ×400) per condition were imaged on a Zeiss Axio inverted microscope, and a minimum of 100 vacuoles, with 4 or more parasites per vacuole, was scored as occupied or unoccupied. Graphs depict the average ± S.D. of three independent experiments.
PLP1 Knock-out and Complementation
PLP1 was genetically ablated in the RHΔku80Δhxg strain using the same approach described previously (7). The PLP1 complementation vector was constructed by replacing the 5′ and 3′ flanks of the microneme protein 2 (MIC2)-associated protein (M2AP) complementation vector with PLP1 5′- and 3′-untranslated regions amplified from genomic DNA (45). The PLP1 signal anchor, amplified from cDNA, restriction enzyme cloning sites, and the epitope tag, were generated by fusion PCR and cloned into the vector by restriction enzyme digestion and ligation. PLP1 domain boundaries were based on computational predictions of the conserved MACPF domain, delineating the protein into a central MACPF domain and an N- and C-terminal domain (7). PLP1 domain-deleted constructs were PCR amplified from cDNA and cloned into the complementation vector by restriction enzyme digest and ligation.
The RHΔku80Δhxg, RHΔku80Δplp1 strains were transfected with 50 μg of the dsRed expression plasmid (46), linearized within the tub promoter, and cloned by limiting dilution. DsRed strains were maintained with chloramphenicol selection. The PLP1 complementation vector was digested within the 5′ and 3′ flanks and transfected into the RHΔku80Δplp1dsRed strain and maintained under 340 μg/ml 6-thioxanthine in Dulbecco's modified Eagle's medium/10 mm HEPES/1% dialyzed FBS and cloned by limiting dilution. Clones were confirmed by PCR of genomic DNA and by immunoblotting.
Recombinant Protein Cloning, Expression, and Purification
PLP1 sequences were PCR amplified from cDNA and cloned into the pET15b expression vector by restriction enzyme digestion and ligation. The expression vector was transformed into BL21 codon + Escherichia coli. Expression was induced with 1 mm isopropyl 1-thio-β-d-galactopyranoside at OD 0.4–0.6 at room temperature for 4–10 h and confirmed by Coomassie Blue staining and immunoblotting. Soluble constructs (PLP1 Nterm and Cterm) were purified by nickel affinity column purification. Insoluble constructs (mature recombinant PLP1, recombinant MACPF domain) were isolated by extracting inclusion bodies as described by the manufacturer (Novagen). Briefly, inclusion bodies were denatured with 6 m guanidine hydrochloride, allowed to bind to the column for 1 h at room temperature or 4 °C overnight, washed, and eluted with nondenaturing buffers. All recombinant protein samples were buffer-exchanged into 50 mm NaCl, 10 mm Tris-HCl (pH 8.0).
Recombinant M2AP was generated as described previously (47). The recombinant LLO expression vector was a kind gift from Mary O'Riordan (48). LLO was expressed and purified in the same manner as mature recombinant PLP1.
Antibody Production
Recombinant PLP1 was used to generate antibodies in mice as described previously with 50 μg of recombinant protein per injection (49) and in rabbits by a commercial service (Cocalico). Recombinant N- and C-terminal domains were used to affinity-purify domain-specific antibodies from rabbit sera using an amino-link kit according to the manufacturer's instructions (Pierce). Specificity and approximately optimal dilutions were determined by immunoblotting and immunofluorescence.
Lytic Activity
Lytic activity on host cells was tested by incubating HFFs in an 8-well chamber slide with 100 nm recombinant protein (M2AP, PLP1, or LLO) in egress assay buffer/12.5 μg/ml propidium iodide/10 mm dithiothreitol. Cells were incubated for 10 min at 37 °C, washed twice with warm PBS, and fixed with formaldehyde. Immunofluorescence was performed for PLP1, and nuclei were stained with 4′,6-diamidino-2-phenylindole.
Hemolysis was tested by incubating washed sheep red blood cells at 2% final hematocrit in PBS with varying concentrations of recombinant protein. Red blood cells were incubated for 1 h at 37 °C and pelleted by centrifugation at 500 × g for 5 min. Supernatant was collected and absorbance at 540 nm read in a 96-well plate reader. The assay was performed with triplicate wells, normalized to 1% Triton X-100 lysis.
Membrane Flotation
Membrane flotation was performed on host cell debris isolated from fully egressed cultures. Parasites were separated from host cell debris by centrifugation at 1000 × g for 10 min. Supernatant was collected and centrifuged again. The second supernatant was subjected to ultracentrifugation at 25,000 rpm for 1 h in a Thermo Scientific Sorvall WX Ultra Series centrifuge. The high speed pellet (HSP) was washed once by resuspension in PBS and centrifugation as in the previous step. A sucrose density gradient was applied to the pellet. The pellet was resuspended in 750 μl of 85% sucrose and overlaid with 3 ml of 65% sucrose and 1 ml of 10% sucrose. Sucrose solutions were prepared in 100 mm NaCl, 10 mm Tris-HCl (pH 7.4). Sucrose gradient samples were centrifuged at 35,000 rpm for 16 h at 4 °C in a Thermo Scientific Sorvall WX Ultra Series centrifuge. Fractions (0.5 ml each) were collected from the top of the gradient and applied to SDS-PAGE. Blots were probed for PLP1, dense granule protein 4, or microneme protein 5.
Membrane flotation of recombinant N- and C-terminal domains was accomplished by preparing red blood cell ghosts as previously described (50). 20 μm (final concentration) recombinant protein was mixed with or without 2 × 107 red blood cell ghosts in 100 μl of PBS and incubated at 37 °C for 15 min. A sucrose density gradient was applied by mixing the protein sample with 400 μl of 85% sucrose and then overlaying with 900 μl of 65% sucrose and 200 μl of 10% sucrose. Samples were centrifuged in a Thermo Scientific Sorvall MTX 150 microultracentrifuge at 90,000 rpm for 3 h. Fractions (200 μl each) were collected and examined by SDS-PAGE and immunoblotting for the His6 epitope tag.
SDS-Agarose Gel Electrophoresis
SDS-agarose gels were prepared by boiling 1% (w/v) agarose in SDS-running buffer (25 mm Tris (pH 8), 250 mm glycine, 0.1% (w/v) SDS), poured into 15 × 17-cm vertical glass plates with a 1.5-mm gap, and cooled to room temperature. Parasite lysate was obtained by filter purification and lysis with hot sample buffer. Lysate and HSP were electrophoresed at 5–8 mA and transferred to PVDF membrane in a semidry transfer machine at 20 V for 70 min. Titin from mouse muscle lysate was used as a size standard, and PVDF membranes were briefly stained with Coomassie Blue prior to immunoblotting for PLP1.
Hexafluoroisopropanol (HFIP) Treatment
HSP was TCA-precipitated and resuspended in 500 μl of buffer (50 mm NaCl, 10 mm Tris (pH 7.4)) or HFIP. Samples were allowed to dry in a Speed-Vac, and pellets were resuspended in sample buffer prior to SDS-PAGE and immunoblotting for PLP1.
Domain Deletion Construct Expression and Secretion
Parasites were filter-purified and attached to a glass slide coated with BD-CellTak (BD Biosciences) prior to fixation with ethanol. Immunofluorescence was performed for PLP1 and MIC2, and nuclei were stained with 4′,6-diamidino-2-phenylindole. Intracellular parasites inoculated for 24 h were also fixed with ethanol prior to staining for PLP1 and MIC2. Results are representative of two or more independent experiments.
For immunoblot detection, parasites were filter-purified in PBS, counted, and lysed in hot sample buffer. Parasites (1.5 × 107) were loaded per lane and probed for PLP1 and MIC2. Microneme secretion with ethanol induction was performed as previously described (51), and samples were probed for PLP1 and MIC2.
Plaque Assay
Parasites were filter-purified and counted, and 50 parasites were inoculated into each well of a 6-well plate. Plates were incubated for 7 days and stained with crystal violet. Plaque number was determined by visual examination. Plaque area was determined by measuring plaque diameter in two dimensions in bright field on a Zeiss Axio inverted microscope, averaging the diameters for 10 plaques/strain, and approximating a circle in area. Graphs represent the average ± S.D. of three independent experiments.
PVM Permeabilization and Lytic Activity
PVM permeabilization was tested by inoculating parasites in HFF as described for the egress assay. Following a 30-h incubation, wells were washed twice with warm PBS, and parasites were immobilized by treating with 1 μm cytochalasin D in egress assay buffer (120 μl/well) at 37 °C for 3 min. Microneme secretion was induced by addition of 120 μl of egress assay buffer/1 μm cytochalasin D with dimethyl sulfoxide or 4 μm A23187 for 3 min. Secretion was halted by addition of 240 μl of 2× fixative. Cells were stained with 4′,6-diamidino-2-phenylindole and imaged as above. Infected cells were determined by bright field, whereas release of dsRed from the parasitophorous vacuole to the host cytosol indicated PVM permeabilization. Lytic activity was determined by normalizing PVM permeabilization to wild-type (WT) parasites. Graphs represent the average ± S.D. of three independent experiments.
Mouse Virulence
Ten 6-week-old female Swiss-Webster mice were injected intraperitoneally with 10, 100, or 1000 tachyzoites and observed for morbidity. Mice that became moribund were humanely euthanized.
RESULTS
Loss of PLP1 Leads to a Secondary Invasion Defect
PLP1 has been previously identified as an important egress factor (7). In addition to the striking defect in rapid egress observed on the level of individual infected cells, we show here that this defect is observed on a population level (Fig. 1A). The majority of PLP1-deficient parasites remain trapped in the parasitophorous vacuole 2 min after ionophore addition whereas other strains such as RH (the original parent for all of the strains used in this study), Δhxg, and m2apko efficiently exit from cells. Whereas PLP1 had been noted to have an invasion defect (7), the contribution of PLP1 to parasite invasion was unclear. Video microscopy revealed that plp1ko parasites that were able to egress after a delay appeared to utilize persistent gliding motility to eventually break through the PVM and escape from the cell (7). Accordingly, we reasoned that plp1ko parasites might exhaust limited resources such as the microneme contents in their prolonged attempts to breach the PVM, thus leaving them less competent for cell invasion. To this end, we compared cell invasion of naturally egressed (Fig. 1B) and mechanically liberated parasites (Fig. 1C). The ∼40% decrease in invasion of naturally egressed plp1ko is similar to that observed for m2apko, a parasite strain with a previously reported invasion defect (45). However, mechanically liberating the parasites restored plp1ko invasion to that of the parental strain (Δhxg) but did not rescue m2apko invasion, consistent with the plp1ko invasion defect being secondary to the egress defect. We also noted a 5–7-fold increase in cell invasion for mechanically liberated parasites over naturally egressed parasites. The increased invasiveness of mechanically liberated parasites could be due to preserving limiting components that would normally be depleted during egress and extracellular existence. The findings support a model in which economical egress promotes efficient cell invasion.
FIGURE 1.
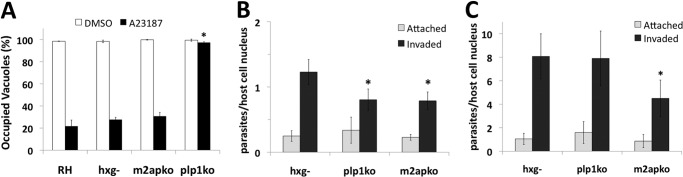
Loss of PLP1 leads to a primary egress defect and a secondary invasion defect. A, 30-h vacuoles were treated with vehicle (dimethyl sulfoxide (DMSO), white bars) or ionophore (A23187, black bars) for 2 min and >100 vacuoles in >10 fields of view scored by immunofluorescence as occupied or unoccupied vacuoles. B and C, naturally egressed (B) or mechanically liberated (C) parasites were tested for invasion defects by red/green invasion assay; light gray bars = attached parasites, dark gray bars = invaded parasites. Fifteen fields of view were counted per strain per experiment. Graphs display the mean ± S.D. (error bars) of three independent experiments (*, p < 0.05 by one-tailed Student's t test compared with RH).
PLP1 Is Sufficient for Membrane Permeabilization, Likely via a Canonical Mechanism of Membrane Attack
Kafsack et al. (7) showed that PLP1 is necessary for ionophore-induced PVM permeabilization, so we next asked whether PLP1 activity was sufficient for membrane damage. We produced recombinant and refolded PLP1 in E. coli and tested it for lytic activity on host cells and erythrocytes. Recombinant protein was incubated with host cells in the presence of propidium iodide, which stains nuclei of cells with membrane damage. Host cells incubated with PLP1 or LLO exhibited propidium iodide-stained nuclei, whereas those incubated with buffer or recombinant M2AP excluded propidium iodide (Fig. 2A). PLP1 and LLO were both hemolytic, whereas M2AP failed to lyse red blood cells (Fig. 2B). Recombinant PLP1 and LLO were of similar purity (supplemental Fig. S1). These assays confirm that PLP1 is sufficient for membrane damage.
FIGURE 2.

PLP1 is sufficient for lytic activity and binds and oligomerizes on host membranes. A, host cells were incubated for 10 min at 37 °C with buffer alone, 100 nm recombinant M2AP, PLP1, or LLO and propidium iodide, which stains nuclei of cells with membrane damage. Immunofluorescence was performed for PLP1. The PLP1 antibody cross-reacted with LLO, but not M2AP. B, lytic activity was observed by hemolysis assay with varying concentrations of recombinant protein. Graph displays the mean ± S.D. (error bars) of triplicate wells and is representative of three independent experiments. C, sucrose gradient membrane flotation of cell debris after parasite egress revealed PLP1 and dense granule protein 4 (GRA4) signal associated with lighter density (membrane) fractions. Endogenous microneme protein 5 (MIC5) was not detected due to lack of membrane binding activity. Recombinant MIC5 added to the cell debris sample was associated with the higher density fractions, also indicative of no membrane binding. Low density, high molecular mass PLP1 signal is absent in the PLP1 knock-out sample. D, membrane-associated PLP1 is resistant to alkaline treatment, indicative of membrane integration. E, in parasite lysate (L), PLP1 migrated rapidly on SDS-agarose, whereas in the HSP (host cell debris), PLP1 signal occurred in multiple, slowly migrating complexes (arrowheads). F, treatment of HSP with HFIP, which disrupts hydrophobic interactions, led to PLP1 migrating at the same molecular mass as in parasite lysate (L), along with some smaller processed forms (arrowheads). Molecular mass markers are indicated in kDa.
Previous work on CDCs, complement, and perforin suggest a common mechanism for pore formation. These proteins are generally secreted as monomers that bind to a receptor(s) on the target cell, oligomerize, and undergo a structural rearrangement to cross the target cell membrane, creating a large transmembrane lesion (32). To examine PLP1 membrane binding and oligomerization in vivo, we isolated host cell debris in an HSP from naturally egressed cultures of RH and plp1ko and tested for PLP1 membrane association via flotation in a sucrose density gradient. In an immunoblot of gradient fractions, PLP1 was observed in the lighter density fractions along with another PVM protein, dense granule protein 4 (Fig. 2C). Microneme protein 5 is a secreted protein with no known membrane binding capacity; accordingly, no microneme protein 5 was observed in the HSP (data not shown) or in the membrane-associated fractions. Recombinant microneme protein 5 added to the bottom of the gradient with the HSP remained associated with the higher density fractions, confirming a lack of membrane association. Treatment of the HSP with sodium bicarbonate, pH 11.5, did not extract PLP1 from the membrane fractions, suggesting it is embedded in membranes (Fig. 2D).
Interestingly, whereas monomeric PLP1 in parasite lysate migrates at 130 kDa, we observe membrane-associated PLP1 in the well and at the interface between the stacking gel and separating gel (Fig. 2C). Additional PLP1 was observed between these landmarks. This severely retarded migration suggests that PLP1 forms large, highly stable oligomeric complexes that are resistant to boiling in SDS-PAGE sample buffer. To resolve these putative oligomers further, we performed SDS-agarose gel electrophoresis (SDS-AGE). PLP1 in parasite lysate migrated as a single band of ∼130 kDa whereas PLP1 associated with host cell debris in the HSP migrated as multiple, high molecular mass complexes (Fig. 2E, arrowheads). These complexes were not observed in samples from plp1ko parasites, confirming the specificity of the antibody. Because PLP1 is partially processed upon secretion in extracellular parasites (52), we next asked whether PLP1 is proteolytically processed in these large molecular mass complexes. We treated the HSP with HFIP to disrupt protein-protein interactions and analyzed the samples by SDS-PAGE (Fig. 2F). HFIP treatment resulted in the loss of PLP1 from the well and stacking gel along with the concurrent appearance of a major band at 130 kDa, similar to that observed in parasite lysate. We also observed multiple minor, processed forms of PLP1 upon HFIP treatment, which suggests some processed PLP1 is incorporated into the high molecular mass complexes, although the majority of the complex is composed of mature PLP1 (Fig. 2F). We conclude that PLP1 forms large, membrane-embedded oligomers, consistent with the established mechanism of oligomerization and membrane insertion by other members of the MACPF and CDC protein families.
Precise Generation of plp1ko Domain Deletion Complementation Strains
Whereas PLP1 has a highly conserved MACPF domain, the N- and C-terminal domains flanking it are unique (7, 34). We hypothesized the MACPF domain to be required for oligomerization and membrane permeabilization. The PLP1 C-terminal domain is predicted to be β-sheet rich, a feature of other pore-forming protein C-terminal domains involved in target cell membrane binding. The PLP1 N-terminal domain has no conserved sequence, and most pore-forming proteins lack N-terminal domains. To identify functional domains of PLP1, we devised a novel complementation strategy involving regenerating the PLP1 knock-out in the genetically tractable RHΔku80 background and complementing the knock-out at the PLP1 locus by double homologous recombination and negative selection against the hypoxanthine xanthine guanine phosphoribosyltransferase (HXG) drug selection marker (Fig. 3A). The recreated knock-out was confirmed by PCR and immunofluorescence (Fig. 3, B and D). A panel of domain deletion mutants was designed based on the computational prediction of the MACPF domain (Fig. 3C). Replacement of HXG with PLP1 cDNA at the PLP1 locus was confirmed by PCR (Fig. 3B). Fig. 3E indicates the genotypes and hereafter used colloquial names for these complementation strains. Fig. 3E also lists the presence or absence of spheres in egressed cultures, a characteristic of PLP1 deficiency and egress failure (see also supplemental Fig. S2 for images of egressed cultures). The absence of spheres in the WT comp and MACPF+Cterm strains preliminarily suggests normal or near normal egress whereas the presence of spheres in plp1ko and all of the other domain deletion strains implies defective egress.
FIGURE 3.

Generation of plp1 knock-out and complementation strains for domain deletion analysis and immunofluorescence of complemented strains. A, the plp1 knock-out was generated in the RHΔku80Δhxg strain by replacing the PLP1 locus with the HXG drug selection marker. The plp1ko was complemented at the endogenous locus by double homologous recombination and negative selection. B, PCR on parasite genomic DNA demonstrated plp1 locus replacement with HXG in the knock-out and HXG replacement with PLP1 cDNAs in complemented strains. C, PLP1 domain structure and domain deletions tested in this study are shown. D, immunofluorescence with a polyclonal PLP1 antibody showed some PLP1 complementation constructs co-localized with MIC2, whereas other constructs were poorly detected. Scale bars, 2 μm. E, parasite strains investigated in this study are listed, including the genotype, common name used throughout this paper, and the presence or absence of spheres in postegressed cultures. Spheres were observed by bright field examination of egressed cultures, and images were acquired on bright field at a magnification of ×200 on a Zeiss Axio inverted microscope.
Expression and localization of the domain deletions were tested by immunofluorescence, immunoblotting, and a microneme secretion assay. Although the complementation plasmid included a C-terminal epitope tag, tagged protein was not detected by immunofluorescence, and only a faint proform of PLP1 was observed by immunoblotting (data not shown). These findings suggest that the epitope tag is removed after the protein is synthesized, possibly by proteolysis whereas the protein traffics to the micronemes. Immunofluorescence with an antibody to the entire mature PLP1 (αPLP1) showed absence of PLP1 in plp1ko and the presence of apical fluorescence in the complemented strains, which co-localized with microneme protein 2 in most extracellular parasites (Fig. 3D) and intracellular parasites (supplemental Fig. S3). Immunoblotting parasite lysate with αPLP1 showed that the WT comp is expressed at a slightly lower level than the WT parental PLP1, despite complementation at the endogenous locus (Fig. 4A). This lower expression could be due to the absence of introns and mRNA splicing or the transient presence of the epitope tag. As confirmed by affinity-purified domain-specific antibodies (αNterm, αCterm), the domain deletions were expressed at levels similar to the WT comp except for the N-terminal domain and the MACPF domain, which were poorly detected (middle and lower panels of Fig. 4A). Low detection may be due to poor antigenicity or from protein instability. Ethanol-induced secretion was observed for all of the domain deletions, further supporting the localization of these constructs to the micronemes (Fig. 4B). Because the PLP1 signal anchor is the only translated element common to all of the constructs, our findings are consistent with it being sufficient to target the constructs to the micronemes. In secreted samples, we detected proteolytic processing of constructs containing the N-terminal domain, which mirrors the previously reported processing of PLP1 by subtilisin 1 (SUB1), a micronemal subtilisin-like protease (Fig. 4, B and C) (52).
FIGURE 4.

PLP1 domain deletion constructs are expressed by the parasites and secreted from the micronemes. A, parasite lysate immunoblotted with a polyclonal (αPLP1) or affinity-purified (αNterm, αCterm) PLP1 antibodies. Arrowheads indicate faint bands specifically detected by the antibodies. The schematic below indicates domains recognized by the antibodies. B, microneme secretion induced with 1% ethanol and the secreted fraction immunoblotted for PLP1 and MIC2. C, immunoblot of secreted fractions demonstrating that PLP1 is processed in the N-terminal domain, indicated by the presence of multiple bands detected by the PLP1 antibody in constructs with the N-terminal domain and absent in the plp1ko. Arrowheads indicate specific bands for single domain constructs. Molecular mass markers are indicated in kDa.
PLP1 Domain Deletion Leads to Intermediate or Complete Loss of Function
Having established expression and secretion of the mutant constructs, we next tested for defects in the lytic cycle by plaque assay. Equal numbers of parasites for each strain were allowed to form plaques in HFF cell monolayers for 1 week. We observed no significant differences in plaque numbers among WT, plp1ko, and complemented strains (Fig. 5A), suggesting equal viability and that none of the deleted PLP1 domains is required for suppressing PLP1 lytic activity during membrane transport or storage in the micronemes. Accordingly, we experienced no obvious problems in complementing the plp1ko with any of the domain deletions as might be expected for expression of a toxic mutant.
FIGURE 5.

Phenotypic analysis of PLP1 domain deletion strains reveals intermediate or nonfunctional complementation. A, 50 parasites per well were allowed to form plaques for 7 days. B, plaque area was measured by determining the average diameter in two dimensions and approximating a circle. C, egress was tested following ionophore (A23187) treatment by immunostaining vacuoles for dense granule protein 7 (GRA7) and parasites for surface antigen 1 (SAG1) and quantifying vacuoles containing parasites (occupied) and parasite-free vacuoles (unoccupied). D, time course of parasite egress is shown as in C with indicated times for each strain. E, PVM permeabilization was determined by treating infected cells with cytochalasin D prior to ionophore (A23187) treatment and fixation; infected cells were identified by bright field, and PVM permeabilization was indicated by release of the PV-targeted fluorescent protein dsRed into the host cytosol. F, lytic activity determined from PVM permeabilization was normalized to WT parasites. Graphs depict the average ± S.D. (error bars) of three independent experiments. *, p < 0.05 by one-tailed Student's t test. G, mouse survival after infection. Ten Swiss-Webster mice per strain were injected intraperitoneally with the indicated numbers of parasites and monitored for morbidity and mortality.
Although plaque numbers were similar between strains, we observed a conspicuous difference in plaque size between WT and plp1ko parasites. WT parasites formed large plaques, and plp1ko parasites formed much smaller plaques. This decrease in plaque size is likely due to both the pronounced egress defect and the moderate invasion defect. Interestingly, plaque size of the complemented strains mirrored WT or the plp1ko for the majority of the strains; however, the MACPF+Cterm (i.e. deletion of the N-terminal domain) strain had an intermediate plaque size (Fig. 5B).
In an induced egress assay, the majority of WT parasites rapidly leave the vacuole whereas plp1ko parasites remain associated with the vacuole over the course of several minutes. In the complemented strains, the WT comp is similar to WT in egress and most other strains resemble the knock-out, indicating nonfunctional complementation. Again, the MACPF+Cterm strain showed an intermediate phenotype with the majority of parasites still in vacuoles, although more parasites egress than the plp1ko (Fig. 5C). We then compared the kinetics of egress for the WT comp, MACPF+Cterm, and plp1ko strains. The egress time course showed that the MACPF+Cterm parasites have a notable delay in egress but do not remain trapped as long as the plp1ko parasites (Fig. 5D).
To determine whether the differences in egress were due to differences in PLP1 lytic activity, we tested the complemented strains for their ability to permeabilize the PVM by immobilizing intracellular parasites with cytochalasin D prior to ionophore treatment. PVM permeabilization was observed by release of the fluorescent vacuolar marker dsRed into the host cytosol. We found that most WT parasites exhibited a damaged PVM, whereas plp1ko parasites showed an intact PVM (Fig. 5E). The complemented WT strain showed somewhat less PVM damage, likely due to the lower level of PLP1 expression. Most other strains had no significant PVM damage; however, the MACPF+Cterm strain showed more PVM damage than the plp1ko. By normalizing PVM permeabilization to WT, we observed 80% of WT activity in the WT comp and 20% of WT activity in the MACPF+Cterm (Fig. 5F). Loss of the N-terminal domain does not abolish PLP1 lysis activity, but reduces it substantially. The absence of spheres from naturally egressed cultures of MACPF+Cterm parasites is consistent with lytic egress occurring in this strain without the failure events seen in nonfunctional strains. Together, these results support a crucial role for the MACPF and C-terminal domains for PLP1 activity because complemented strains lacking one of these domains had no PLP1 activity.
Although plp1ko parasites can be propagated normally in vitro, previous work demonstrated a substantial virulence defect in the mouse infection model (7), implying that rapid parasite egress is crucial for pathogenesis. The MACPF+Cterm complemented strain provided another tool with which to investigate the role of rapid egress in pathogenesis because this strain had an egress defect that was intermediate between WT and plp1ko. To examine this further, we infected mice with 10 or 100 WT comp or MACPF+Cterm parasites, or 1000 plp1ko parasites and observed the outcome of infection with respect to mouse survival. As expected, plp1ko showed an attenuation of virulence, which was reversed in the WT comp. Notably, MACPF+Cterm parasites were also highly virulent and not significantly different (p = 0.09 for 10 tachyzoites/mouse) from the WT comp (Fig. 5G), despite the survival of one mouse infected with MACPF+Cterm parasites, confirmed by serology. These results suggest that even low levels of lytic activity are sufficient for virulence, and the moderate egress delay does not have a significant impact on infection.
To identify the basis for nonfunctionality of the domain deletions, we assessed PLP1 membrane binding and oligomerization in the complemented strains. Membrane flotation of host cell debris from naturally egressed, complemented cultures showed membrane association of high molecular mass complexes for both WT comp and MACPF+Cterm. This suggests that the Nterm is not required for membrane binding or oligomerization. We also observed membrane binding for the Nterm+Cterm and Nterm+MACPF constructs, but no significant antibody reactivity was observed in the well or stacking gel for these two constructs (Fig. 6A), indicating the absence of oligomerization. We were unable to observe any specific antibody reactivity upon membrane flotation for other constructs due to low expression levels, minimal membrane binding activity, poor detection, or a combination of these possible situations.
FIGURE 6.

Functional analysis of PLP1 domain deletions reveals differences in membrane binding and oligomerization. A, membrane flotation of host cell debris was acquired from complemented strains immunoblotted for PLP1. Arrowheads indicate specific bands. B, membrane flotation of recombinant N- and C-terminal domains used red blood cell ghosts (RBCg) and was immunoblotted for the His6 epitope tag. C, host cell debris from complemented strains was TCA-precipitated, treated with HFIP or buffer, and immunoblotted for PLP1. D, SDS-AGE and PLP1 immunoblotting of WT comp and MACPF+Cterm lysate (L) and HSP were performed. Marker is Coomassie Blue-stained mouse muscle lysate; the indicated sizes are in kDa. Arrows indicate monomers in lysate.
To determine whether the N- or C-terminal domains were sufficient for membrane binding, we produced the corresponding recombinant, His-tagged proteins in E. coli. The N-terminal domain was expressed as a soluble product and the C-terminal domain was partially soluble. Recombinant Nterm was processed by bacterial proteases in a pattern similar to endogenous, secreted Nterm, and the Cterm was unprocessed by parasitic or bacterial proteases (Figs. 4C and 6B). This suggests that similar protease-sensitive sites are exposed on recombinant and endogenous Nterm and that recombinant Nterm is structurally similar to the endogenous construct. We attempted to generate recombinant MACPF domain, but this construct was poorly expressed, largely insoluble and unstable in solution upon refolding (data not shown). Recombinant Nterm and Cterm were incubated with red blood cell ghosts and subjected to membrane flotation. Both processed and unprocessed Nterm as well as the Cterm constructs were detected in lighter density sucrose fractions in the presence of membranes, consistent with these domains each being sufficient for membrane association (Fig. 6B).
We next examined potential oligomers of strains with detectable PLP1 construct membrane association by HFIP treatment of the HSP. We observed PLP1 in the well and stacking gel for both WT comp and MACPF+Cterm as previously noted, and no significant high molecular mass bands for the Nterm+Cterm and Nterm+MACPF (Fig. 6C). Treatment of the HSP with HFIP resulted in mature and processed PLP1 bands observed in the WT comp and a single band in the MACPF+Cterm sample. Bands equivalent to the lysate form are observed in both buffer and HFIP-treated samples of the Nterm+Cterm and Nterm+MACPF samples. Together, these results indicate an essential role for the MACPF and Cterm in PLP1 oligomerization. Although membrane binding was observed for the Nterm+Cterm, loss of the MACPF domain precluded oligomerization. For the Nterm+MACPF domain, the membrane binding observed did not lead to detectable amounts of oligomerization, suggesting that the protein is not positioned to oligomerize when membrane-bound by only the N-terminal domain.
To determine whether WT comp and MACPF+Cterm oligomers differed in their high molecular mass complexes, we applied lysate and HSP from both samples to SDS-AGE and immunoblotted for PLP1. We observed similar high molecular mass smears for both WT comp and MACPF+Cterm samples, confirming that the N-terminal domain is not required for formation of large molecular mass complexes (Fig. 6D).
DISCUSSION
Here we show that PLP1 is sufficient for membrane permeabilization and appears to function as a bona fide pore-forming protein. Loss of PLP1 activity led to a marked egress defect and a secondary invasion defect. PLP1 has unique functional domains associated with its conserved MACPF domains including a membrane binding C-terminal domain that is required for functionality. Interestingly, the N-terminal domain also contains membrane binding activity, which promotes PLP1-mediated membrane damage but is not required for pore formation or pathogenesis.
Our findings indicate that loss of PLP1 leads to a secondary invasion defect based on showing that mechanical release of plp1ko parasites restored invasion to parental levels. Thus, PLP1 is likely dedicated to parasite egress and does not play a direct role in invasion. Because PLP1 is probably secreted from micronemes during both egress and invasion, how its activity is differentially regulated during these events remains unknown. PLP1 is expected to be active during egress to permeabilize the PVM but inactive during invasion to maintain membrane integrity for the formation of the nascent PV. Although PLP1 is processed in its N-terminal domain, this proteolysis may play an inhibitory role rather than an activating one. In support of this notion, loss of the N-terminal domain reduced PLP1 activity and the majority of PLP1 observed in native oligomers is unprocessed. Additionally, genetic ablation of SUB1, the protease responsible for processing PLP1 in the secreted fraction, does not affect egress (52), indicating that processing is not required for PLP1 activity. On the other hand, if SUB1 processing of PLP1 suppresses lytic activity, proteolysis may be down-regulated during egress to promote PVM lysis and enhanced when parasites are extracellular to protect surrounding host cells or the parasite itself. SUB1-deficient parasites have an invasion defect due to the lack of processing several invasion-related proteins. Whether the absence of PLP1 proteolysis in SUB1-deficient parasites and the proposed derepression of lytic activity play a role in this invasion defect is unknown. If SUB1 activity is suppressed during egress there may be functional differences in other SUB1 substrates during egress and invasion.
It is also interesting to note the substantial increase in parasite attachment and invasion for mechanically lysed versus naturally egressed parasites. The enhanced invasion of mechanically liberated parasites suggests that such parasites are more fit for entry. Although parasite inoculation and harvest occurred at the same time for both conditions, natural egress occurs somewhat asynchronously, and this is likely a source of variation in invasion assays because parasites continuously secrete micronemes while extracellular.
Previous work demonstrated that PLP1 is necessary for PVM permeabilization (7). Here we have shown that PLP1 is sufficient for membrane disruption. Although it is possible that other parasite or host factors regulate PLP1 activity, the results with recombinant protein PLP1 indicate that they are not necessary for its function. The Toxoplasma genome contains another putative pore-forming protein termed perforin-like protein 2 (PLP2). Although PLP2 expression has not been carefully examined, the lack of lytic activity in the absence of PLP1 suggests that PLP2 does not have lytic activity independent of PLP1. Determining the extent to which PLP2 contributes to egress or another function awaits the creation of a PLP2-deficient strain.
To examine the role of each PLP1 domain in lytic activity, we complemented the knock-out parasites with domain deletion constructs. The enhanced homologous recombination of the RHΔku80 strain permitted targeting the constructs to the endogenous locus, resulting in expression under the native promoter. We found that the PLP1 signal anchor is sufficient for microneme targeting of all tested PLP1 domains as we observed apical localization by immunofluorescence and ethanol-induced secretion. Similar to the previously generated randomly integrated PLP1-complemented strain (7), we observed less PLP1 expression in the WT-complemented parasites than the parental strain. This suggests that intronic sequences promote expression or that the epitope tag interferes with expression, or a combination of the two scenarios. Importantly, we observe similar levels of expression for most of the domain deletion constructs using affinity-purified domain-specific antibodies. The lower levels of expression observed for the Nterm and MACPF domains expressed individually may be due to proteolytic processing of the Nterm and a lack of epitopes in the MACPF domain, and/or construct instability and degradation. Regardless, the domain deletion complementation strategy was useful for determining the functional role for each domain in pore formation in vivo, a first of its kind for a MACPF protein.
The finding of predicted functions for the MACPF and β-sheet-rich C-terminal domain highlights the conservation of this domain family. The MACPF domain is necessary for oligomerization and membrane permeabilization whereas the C-terminal domain provides membrane binding activity necessary for pore formation. We also identified a novel activity for a MACPF-associated N-terminal domain in membrane binding. We speculate that binding through the N-terminal domain provides another means of enhancing lytic activity by allowing recognition of more than one membrane receptor. An N-terminal lectin-binding domain in the CDC lectinolysin binds to Lewis blood group antigen on the surface of erythrocytes and is thought to enhance pore formation by concentrating the toxin at fucose-rich sites on target membranes (37, 39). Although C8γ has lectin binding activity, it is a lipocalin protein associated with C8α and C8β and does not contain a MACPF domain (53). Whereas the protein sequence of the Nterm is not highly conserved, other MACPF proteins in apicomplexan genomes also contain N-terminal domains of varying lengths. Apicomplexan parasites complete their life cycle in a wide variety of hosts and tissues. These domains may provide unique regulatory elements necessary for augmenting lytic activity at each stage of the life cycle.
Although the loss of PLP1 results in an egress delay and profound virulence attenuation, we did not detect a significant virulence defect for the MACPF+Cterm strain, which also has a marked delay in egress. This suggests that the presence of lytic activity, rather than the absolute rate of parasite egress, is the deciding factor in pathogenesis. The basis of virulence attenuation for the plp1ko parasites is still an open question. What factors lead to host control of plp1ko infection yet susceptibility to WT parasites? One deciding factor may be indicated in the different plaque sizes of WT and plp1ko parasites. WT parasites form large plaques in a monolayer, and plp1ko parasites form much smaller plaques despite similar plating efficiency. Plaque size is dependent on multiple factors including invasion, egress, and motility, and is an indication of parasite viability and capacity to complete the lytic cycle. The small plaque size of PLP1-deficient parasites is likely due to the combined egress and secondary invasion defects. plp1ko parasites might expend more resources for egress, thus compromising motility during intercellular migration. The combined invasion and egress defects may render extracellular plp1ko parasites more susceptible to phagocytosis than WT parasites, which are able to rapidly egress and invade a neighboring cell, spending a limited time in the extracellular environment. Additionally, the small plaques in the plp1ko may indicate a limited amount of lytic damage occurring in plp1ko infections. High levels of tissue damage from multiple rounds of lytic replication in WT parasite infections may exacerbate the immune response whereas reduced tissue damage by plp1ko parasites may be more efficiently repaired by the host, resulting in a dampened immune response and recovery from infection. Verification of these hypotheses will require more careful examination of the differences in host immune responses and the lytic damage caused by WT and plp1ko parasites in the mouse infection model.
The field of pore-forming protein biology has been stimulated with breakthroughs in structural and biochemical techniques. Here we have demonstrated the utility of investigating the function of a pore-forming protein in a parasite system. Future studies will contribute knowledge into the diverse ways pore-forming proteins function and to the molecular mechanisms of virulence in these important pathogens.
Acknowledgments
We thank all members of the Carruthers laboratory for helpful discussions; Tracey Schultz for technical support; Matthew Chapman for advice on disassociating oligomers; and Andrew Perry for input on PLP1 domain boundaries; Rodney Tweten, Mary O'Riordan, and their laboratory members for advice regarding SDS-AGE; and Akira Ono and members of his laboratory for advice on membrane flotation.
This work was supported, in whole or in part, by National Institutes of Health Operating Grant R01AI046675 (to V. B. C.) and Cellular and Molecular Biology Training Grant T32 GM007315. This work was also supported by a University of Michigan Rackham merit fellowship and Rackham student research grants (to M. S. R.).

This article contains supplemental Figs. S1–S3.
- LLO
- listeriolysin O
- CDC
- cholesterol-dependent cytolysin
- HFF
- human foreskin fibroblast
- HFIP
- hexafluoroisopropanol
- HSP
- high speed pellet
- HXG
- hypoxanthine xanthine guanine phosphoribosyltransferase
- MACPF
- membrane attack complex/perforin
- M2AP
- MIC2-associated protein
- MIC2
- microneme protein 2
- PLP1
- perforin-like protein 1
- PLP2
- perforin-like protein 2
- PVM
- parasitophosphorous vacuole membrane
- SDS-AGE
- SDS-agarose gel electrophoresis
- SUB1
- subtilisin 1
- WT comp
- complemented WT.
REFERENCES
- 1. Schnupf P., Portnoy D. A. (2007) Listeriolysin O: a phagosome-specific lysin. Microbes Infect. 9, 1176–1187 [DOI] [PubMed] [Google Scholar]
- 2. Kadota K., Ishino T., Matsuyama T., Chinzei Y., Yuda M. (2004) Essential role of membrane-attack protein in malarial transmission to mosquito host. Proc. Natl. Acad. Sci. U.S.A. 101, 16310–16315 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Ishino T., Chinzei Y., Yuda M. (2005) A Plasmodium sporozoite protein with a membrane attack complex domain is required for breaching the liver sinusoidal cell layer prior to hepatocyte infection. Cell. Microbiol. 7, 199–208 [DOI] [PubMed] [Google Scholar]
- 4. Schneider M. C., Exley R. M., Ram S., Sim R. B., Tang C. M. (2007) Interactions between Neisseria meningitidis and the complement system. Trends Microbiol. 15, 233–240 [DOI] [PubMed] [Google Scholar]
- 5. Voskoboinik I., Smyth M. J., Trapani J. A. (2006) Perforin-mediated target-cell death and immune homeostasis. Nat. Rev. Immunol. 6, 940–952 [DOI] [PubMed] [Google Scholar]
- 6. Rosado C. J., Kondos S., Bull T. E., Kuiper M. J., Law R. H., Buckle A. M., Voskoboinik I., Bird P. I., Trapani J. A., Whisstock J. C., Dunstone M. A. (2008) The MACPF/CDC family of pore-forming toxins. Cell. Microbiol. 10, 1765–1774 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Kafsack B. F., Pena J. D., Coppens I., Ravindran S., Boothroyd J. C., Carruthers V. B. (2009) Rapid membrane disruption by a perforin-like protein facilitates parasite exit from host cells. Science 323, 530–533 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Sibley L. D., Mordue D., Howe D. K. (1999) Experimental approaches to understanding virulence in toxoplasmosis. Immunobiology 201, 210–224 [DOI] [PubMed] [Google Scholar]
- 9. McLeod R., Kieffer F., Sautter M., Hosten T., Pelloux H. (2009) Why prevent, diagnose and treat congenital toxoplasmosis? Mem. Inst. Oswaldo Cruz 104, 320–344 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Pereira-Chioccola V. L., Vidal J. E., Su C. (2009) Toxoplasma gondii infection and cerebral toxoplasmosis in HIV-infected patients. Future Microbiol. 4, 1363–1379 [DOI] [PubMed] [Google Scholar]
- 11. Boothroyd J. C. (2009) Toxoplasma gondii: 25 years and 25 major advances for the field. Int. J. Parasitol. 39, 935–946 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Van Den Steen P. E., Deroost K., Geurts N., Heremans H., Van Damme J., Opdenakker G. (2011) Malaria: Host-pathogen interactions, immunopathological complications and therapy. Verh. K. Acad. Geneeskd. Belg. 73, 123–151 [PubMed] [Google Scholar]
- 13. McDonald V. (2011) Cryptosporidiosis: host immune responses and the prospects for effective immunotherapies. Expert Rev. Anti Infect. Ther. 9, 1077–1086 [DOI] [PubMed] [Google Scholar]
- 14. Shirley M. W., Smith A. L., Blake D. P. (2007) Challenges in the successful control of the avian coccidia. Vaccine 25, 5540–5547 [DOI] [PubMed] [Google Scholar]
- 15. Singh S., Chitnis C. E. (2012) Signalling mechanisms involved in apical organelle discharge during host cell invasion by apicomplexan parasites. Microbes Infect. 14, 820–824 [DOI] [PubMed] [Google Scholar]
- 16. Keeley A., Soldati D. (2004) The glideosome: a molecular machine powering motility and host-cell invasion by Apicomplexa. Trends Cell Biol. 14, 528–532 [DOI] [PubMed] [Google Scholar]
- 17. Besteiro S., Dubremetz J. F., Lebrun M. (2011) The moving junction of apicomplexan parasites: a key structure for invasion. Cell. Microbiol. 13, 797–805 [DOI] [PubMed] [Google Scholar]
- 18. Howard J. C., Hunn J. P., Steinfeldt T. (2011) The IRG protein-based resistance mechanism in mice and its relation to virulence in Toxoplasma gondii. Curr. Opin. Microbiol. 14, 414–421 [DOI] [PubMed] [Google Scholar]
- 19. Carruthers V., Boothroyd J. C. (2007) Pulling together: an integrated model of Toxoplasma cell invasion. Curr. Opin. Microbiol. 10, 83–89 [DOI] [PubMed] [Google Scholar]
- 20. Lavine M. D., Arrizabalaga G. (2007) Invasion and egress by the obligate intracellular parasite Toxoplasma gondii: potential targets for the development of new antiparasitic drugs. Curr. Pharm. Des. 13, 641–651 [DOI] [PubMed] [Google Scholar]
- 21. Lourido S., Tang K., Sibley L. D. (2012) Distinct signalling pathways control Toxoplasma egress and host-cell invasion. EMBO J. 31, 4524–4534 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Nagamune K., Hicks L. M., Fux B., Brossier F., Chini E. N., Sibley L. D. (2008) Abscisic acid controls calcium-dependent egress and development in Toxoplasma gondii. Nature 451, 207–210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Nagamune K., Moreno S. N., Chini E. N., Sibley L. D. (2008) Calcium regulation and signaling in apicomplexan parasites. Subcell. Biochem. 47, 70–81 [DOI] [PubMed] [Google Scholar]
- 24. Carruthers V. B., Sibley L. D. (1999) Mobilization of intracellular calcium stimulates microneme discharge in Toxoplasma gondii. Mol. Microbiol. 31, 421–428 [DOI] [PubMed] [Google Scholar]
- 25. Endo T., Sethi K. K., Piekarski G. (1982) Toxoplasma gondii: calcium ionophore A23187-mediated exit of trophozoites from infected murine macrophages. Exp. Parasitol. 53, 179–188 [DOI] [PubMed] [Google Scholar]
- 26. Rosado C. J., Buckle A. M., Law R. H., Butcher R. E., Kan W. T., Bird C. H., Ung K., Browne K. A., Baran K., Bashtannyk-Puhalovich T. A., Faux N. G., Wong W., Porter C. J., Pike R. N., Ellisdon A. M., Pearce M. C., Bottomley S. P., Emsley J., Smith A. I., Rossjohn J., Hartland E. L., Voskoboinik I., Trapani J. A., Bird P. I., Dunstone M. A., Whisstock J. C. (2007) A common fold mediates vertebrate defense and bacterial attack. Science 317, 1548–1551 [DOI] [PubMed] [Google Scholar]
- 27. Hadders M. A., Beringer D. X., Gros P. (2007) Structure of C8α-MACPF reveals mechanism of membrane attack in complement immune defense. Science 317, 1552–1554 [DOI] [PubMed] [Google Scholar]
- 28. Law R. H., Lukoyanova N., Voskoboinik I., Caradoc-Davies T. T., Baran K., Dunstone M. A., D'Angelo M. E., Orlova E. V., Coulibaly F., Verschoor S., Browne K. A., Ciccone A., Kuiper M. J., Bird P. I., Trapani J. A., Saibil H. R., Whisstock J. C. (2010) The structural basis for membrane binding and pore formation by lymphocyte perforin. Nature 468, 447–451 [DOI] [PubMed] [Google Scholar]
- 29. Anderluh G., Lakey J. H. (2008) Disparate proteins use similar architectures to damage membranes. Trends Biochem. Sci. 33, 482–490 [DOI] [PubMed] [Google Scholar]
- 30. Lukoyanova N., Saibil H. R. (2008) Friend or foe: the same fold for attack and defense. Trends Immunol. 29, 51–53 [DOI] [PubMed] [Google Scholar]
- 31. Tweten R. K. (2005) Cholesterol-dependent cytolysins, a family of versatile pore-forming toxins. Infect. Immun. 73, 6199–6209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Dunstone M. A., Tweten R. K. (2012) Packing a punch: the mechanism of pore formation by cholesterol-dependent cytolysins and membrane attack complex/perforin-like proteins. Curr. Opin. Struct. Biol. 22, 342–349 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Müller-Eberhard H. J. (1985) Transmembrane channel-formation by five complement proteins. Biochem. Soc. Symp. 50, 235–246 [PubMed] [Google Scholar]
- 34. Kafsack B. F., Carruthers V. B. (2010) Apicomplexan perforin-like proteins. Commun. Integr. Biol. 3, 18–23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Scibek J. J., Plumb M. E., Sodetz J. M. (2002) Binding of human complement C8 to C9: role of the N-terminal modules in the C8α subunit. Biochemistry 41, 14546–14551 [DOI] [PubMed] [Google Scholar]
- 36. Tegla C. A., Cudrici C., Patel S., Trippe R., 3rd, Rus V., Niculescu F., Rus H. (2011) Membrane attack by complement: the assembly and biology of terminal complement complexes. Immunol. Res. 51, 45–60 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Farrand S., Hotze E., Friese P., Hollingshead S. K., Smith D. F., Cummings R. D., Dale G. L., Tweten R. K. (2008) Characterization of a streptococcal cholesterol-dependent cytolysin with a lewis y and b specific lectin domain. Biochemistry 47, 7097–7107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Bouyain S., Geisbrecht B. V. (2012) Host glycan recognition by a pore forming toxin. Structure 20, 197–198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Feil S. C., Lawrence S., Mulhern T. D., Holien J. K., Hotze E. M., Farrand S., Tweten R. K., Parker M. W. (2012) Structure of the lectin regulatory domain of the cholesterol-dependent cytolysin lectinolysin reveals the basis for its lewis antigen specificity. Structure 20, 248–258 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Decatur A. L., Portnoy D. A. (2000) A PEST-like sequence in listeriolysin O essential for Listeria monocytogenes pathogenicity. Science 290, 992–995 [DOI] [PubMed] [Google Scholar]
- 41. Schnupf P., Portnoy D. A., Decatur A. L. (2006) Phosphorylation, ubiquitination and degradation of listeriolysin O in mammalian cells: role of the PEST-like sequence. Cell. Microbiol. 8, 353–364 [DOI] [PubMed] [Google Scholar]
- 42. Huynh M. H., Carruthers V. B. (2009) Tagging of endogenous genes in a Toxoplasma gondii strain lacking Ku80. Eukaryot. Cell 8, 530–539 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Sabin A. (1941) Toxoplasmic encephalitis in children. JAMA 116, 801–807 [Google Scholar]
- 44. Endo T., Tokuda H., Yagita K., Koyama T. (1987) Effects of extracellular potassium on acid release and motility initiation in Toxoplasma gondii. J. Protozool. 34, 291–295 [DOI] [PubMed] [Google Scholar]
- 45. Huynh M. H., Rabenau K. E., Harper J. M., Beatty W. L., Sibley L. D., Carruthers V. B. (2003) Rapid invasion of host cells by Toxoplasma requires secretion of the MIC2-M2AP adhesive protein complex. EMBO J. 22, 2082–2090 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Pepper M., Dzierszinski F., Wilson E., Tait E., Fang Q., Yarovinsky F., Laufer T. M., Roos D., Hunter C. A. (2008) Plasmacytoid dendritic cells are activated by Toxoplasma gondii to present antigen and produce cytokines. J. Immunol. 180, 6229–6236 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Rabenau K. E., Sohrabi A., Tripathy A., Reitter C., Ajioka J. W., Tomley F. M., Carruthers V. B. (2001) TgM2AP participates in Toxoplasma gondii invasion of host cells and is tightly associated with the adhesive protein TgMIC2. Mol. Microbiol. 41, 537–547 [DOI] [PubMed] [Google Scholar]
- 48. Glomski I. J., Gedde M. M., Tsang A. W., Swanson J. A., Portnoy D. A. (2002) The Listeria monocytogenes hemolysin has an acidic pH optimum to compartmentalize activity and prevent damage to infected host cells. J. Cell Biol. 156, 1029–1038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Laliberté J., Carruthers V. B. (2011) Toxoplasma gondii toxolysin 4 is an extensively processed putative metalloproteinase secreted from micronemes. Mol. Biochem. Parasitol. 177, 49–56 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Bjerrum P. J. (1979) Hemoglobin-depleted human erythrocyte ghosts: characterization of morphology and transport functions. J. Membr. Biol. 48, 43–67 [DOI] [PubMed] [Google Scholar]
- 51. Carruthers V. B., Moreno S. N., Sibley L. D. (1999) Ethanol and acetaldehyde elevate intracellular [Ca2+] and stimulate microneme discharge in Toxoplasma gondii. Biochem. J. 342, 379–386 [PMC free article] [PubMed] [Google Scholar]
- 52. Lagal V., Binder E. M., Huynh M. H., Kafsack B. F., Harris P. K., Diez R., Chen D., Cole R. N., Carruthers V. B., Kim K. (2010) Toxoplasma gondii protease TgSUB1 is required for cell surface processing of micronemal adhesive complexes and efficient adhesion of tachyzoites. Cell. Microbiol. 12, 1792–1808 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Schreck S. F., Parker C., Plumb M. E., Sodetz J. M. (2000) Human complement protein C8γ. Biochim. Biophys. Acta 1482, 199–208 [DOI] [PubMed] [Google Scholar]