

Background: Endoplasmic reticulum (ER) stress-induced apoptosis is mediated by IRE1α-JNK pathway.
Results: Knockdown or inactivation of RNF13 results in resistance to ER stress-induced apoptosis, whereas RNF13 overexpression induces JNK activation and apoptosis. RNF13 interacts with IRE1α to promote JNK activation and apoptosis.
Conclusion: RNF13 mediates ER stress-induced apoptosis through IRE1α/JNK pathway.
Significance: RNF13 is a novel regulator of ER stress-induced apoptosis.
Keywords: Apoptosis, Cell Biology, Cell Signaling, Endoplasmic Reticulum Stress, Jun N-terminal Kinase (JNK)
Abstract
Disturbance of homeostasis at endoplasmic reticulum (ER) causes stress to cells that in turn triggers an adaptive signaling pathway termed unfolded protein response for the purpose of restoring normal cellular physiology or initiating signaling events leading to apoptosis. Identification of those genes that are involved in the unfolded protein response-mediated apoptotic signaling pathway would be valuable toward elucidating the molecular mechanism underlying the relationship between ER stress and apoptosis. We initiated a genetic screen by using the retroviral insertion mutation system to search for genes whose inactivation confers resistance to apoptosis induction by staurosporine. Using this approach, RING finger protein 13 (RNF13) was identified. Interestingly, RNF13 is highly enriched in ER. RNF13 knockdown cells are resistant to apoptosis and JNK activation triggered by ER stress. Conversely, overexpression of RNF13 induces JNK activation and caspase-dependent apoptosis. The RING and transmembrane domains of RNF13 are both required for its effects on JNK activation and apoptosis. Moreover, systematic analysis of the involvement of individual signaling components in the ER stress pathway using knockdown approach reveals that RNF13 acts upstream of the IRE1α-TRAF2 signaling axis for JNK activation and apoptosis. Finally, RNF13 co-immunoprecipitates with IRE1α, and the intact RING domain is also required for mediating its interaction. Together, our data support a model that RNF13 is a critical mediator for facilitating ER stress-induced apoptosis through the activation of the IRE1α-TRAF2-JNK signaling pathway.
Introduction
Multiple factors including accumulation of misfolded proteins, glucose deprivation, lipid overload, change in the redox state of the ER3 lumen, alterations of Ca2+ concentrations in the ER, and viral infection can all induce ER stress in mammalian cells (1, 2). Abnormality in ER stress response has been implicated as a contributing factor in a variety of diseases such as neurodegenerative diseases, cancer, diabetes, obesity, and viral infections (1, 3, 4). Under ER stress conditions, an intracellular unfolded protein response (UPR) is activated. Mammalian UPR is initiated by the activation of three ER stress sensors: PKR-like ER kinase, activating transcription factor 6 (ATF6), and inositol- requiring transmembrane kinase/endonuclease (IRE1α) (5). These proteins trigger the cascade of signaling events leading to the attenuation of protein synthesis and transcriptional up-regulation of genes encoding ER chaperones, folding enzymes, and ER-associated degradation (ERAD) components. Collectively, these events can cause a reduction of ER protein load by increasing ER-mediated protein folding as well as accelerating degradation of unfolded proteins.
Activated PKR-like ER kinase inhibits protein translation by inactivating the translation initiation factor eIF2α to reduce the load of newly synthesized proteins, thereby also misfolded proteins (6). ATF6 is initially synthesized as an ER-resident protein (7). In response to the accumulation of unfolded proteins, ATF6 is transported to Golgi apparatus and subsequently cleaved to become an active transcription factor (2, 7). Signaling emulating from IRE1α is the most evolutionarily conserved branch of the UPR. It can be found from yeast to mammals (8). IRE1α is localized to ER and possesses both kinase and endoribonuclease activities that can be regulated by oligomerization. Oligomeric IRE1α juxtaposes their kinase domains, which would facilitate trans-autophosphorylation resulting in autoactivation (9). The activated IRE1α cleaves the mRNA of X-box-binding protein 1 (XBP1) (10). In addition, the activated IRE1α also interacts with tumor necrosis factor receptor-associated factor-2 (TRAF2), leading to the activation of apoptosis signal-regulating kinase 1 (ASK1) and c-Jun NH2-terminal kinase (JNK) (6, 11). Phosphorylated JNK has been reported to activate cytochrome c-mediated apoptotic pathway by phosphorylating specific members of BCL-2 family of proteins (12, 13). Recently, the ER-localized BH3 domain-only proteins, BIM and PUMA, are shown to selectively activate IRE1α-TRAF2 pathway to promote apoptosis (14–16). A recent high-throughput chemical screen for inhibitors of ER stress-induced cell death reveals a crucial role of ASK1 in the ER stress-induced apoptosis pathway, as thapsigargin (Tg)-induced apoptosis is drastically inhibited when the level of ASK1 was reduced (17). Thus, mounting evidences suggest that the IRE1α-ASK1-JNK pathway plays a critical role in ER stress-induced apoptosis. However, the upstream signaling components involved in the activation of the IRE1α-ASK1-JNK pathway leading to ER stress-mediated apoptosis remain unclear. Here, we report identification of RNF13 as a critical mediator required for executing the ER stress-mediated apoptosis via activation of the IRE1α-JNK signaling pathway.
MATERIALS AND METHODS
Chemical Reagents
Tunicamycin (Tn), Tg, staurosporine (STS), caspase-3 specific inhibitor (AC-DEVD-CHO), pan caspase inhibitor (benzyloxycarbonyl-VAD-fluoromethyl ketone), and JNK inhibitor (SP600125) were obtained from Sigma. Hoechst 33342, Alexa Fluor 488-conjugated anti-mouse secondary antibody, and Alexa Fluor 568-conjuncted anti-rabbit secondary antibody were purchased from Molecular Probes.
Cell Culture
Mammalian cell lines (SHSY-5Y, COS-7, 293T) were cultured in DMEM supplemented with 10% heat-inactivated fetal bovine serum, 100 units/ml penicillin, and 100 μg/ml streptomycin. SHSY-5Y insertion mutant cells (STS-108) were maintained in 300 μg/ml G418 (Invitrogen). Apoptosis was induced either by 1 μm STS, 10 mJ/cm2 UV radiation, 1.25 μg/ml Tn, or 1 μm Tg. An equal volume of dimethyl sulfoxide was used in controls. Caspase inhibitors (50 μm benzyloxycarbonyl-VAD-fluoromethyl ketone and AC-DEVD-CHO) and JNK inhibitor (10 μm SP600125) were added into cells 30 min before drug treatment.
Molecular Cloning of RNF13 and Deletion Mutants
Full-length human RNF13 was generated by PCR from cDNA isolated from SHSY-5Y cells with following pair of primers: 5′-CCCAGGCCTCTGCTCTCCATAGGGATG-3′ (forward) and 5′-CCGCTCGAGTCAAACAGTATTTGCTATG-TTG-3′ (reverse) in which Xho1 and HindIII restriction sites were added to forward and reverse primers, respectively. The 1143-bp full-length gene was cloned in-frame into mammalian expression vector pXJ40 with 3′-Myc tag. Using pXY-Myc-RNF13 construct as the template, a series of N- and C-terminal deletion mutants of RNF13 (aa 1–183, aa 165–381), ΔPA (aa Δ52–162), ΔRING (aa Δ240–296), ΔTM (aa Δ184–206), and C243W were generated by PCR. For generating siRNA-resistant full-length RNF13, nucleotide sequences between 292 and 297 (5′-AGAAGA-3′) in the coding sequence of RNF13 cDNA were changed to 5′-CGGCGG-3′ without causing any change in amino acid (silent mutation). All constructs were confirmed by sequencing.
Transfection
Transient transfection experiments were performed with LipofectamineTM2000 (Invitrogen) according to manufacturer's recommendation. Briefly, 1 × 105 SHSY-5Y, COS-7, or 293T cells were seeded in 35-mm dishes overnight before transfection with 1 μg of WT RNF13 and the different mutants. For co-transfections, an additional 0.2 μg of pEGFP-N1, IRE1α, TRAF2, and/or ASK1 were used.
Identification of STS-resistant Cells (STS-108) from SHSY-5Y
An SHSY-5Y clone that exhibited a survival rate of less than 1 in 105 after 12 h of exposure to 0.5 μm STS was used in the experiments. 5 × 106 cells were infected with pDisrup virus, and a yield of ∼103 G418-resistant clones was obtained (18). The G418 resistant clones and the wild-type SHSY-5Y cells were treated with 0.5 μm STS for 10 h and kept in the incubator for 10 h. The surviving cells were cloned and cultured, and the disrupted genes were identified by 3′-rapid amplification of cDNA ends as described. Two sets of primers were designed to check the RNF13 expression level in STS-108 cell line. RNA was extracted using the TRIzol reagent (Invitrogen). Primers used to detect the full-length RNF13 are RNF13F (5′-ATGCTGCTCTCCATAGGGATG-3′) and RNF13R (5′-TCAAACAGTATTTGCTATGTTG-3′), whereas primers used to detect truncated RNF13 were RNF13F (5′-ATGCTGCTCTCCATAGGGATG-3′) and NeoR (5′-TCTTGAATTTATGTACAGGAAG-3′). Actin was used as a loading control.
RNA Interference
Various siRNAs against RNF13 were designed according to siDirect 2.0 and tested for their Knockdown efficiency by RT-PCR and semiquantitative PCR analysis. Two siRNA sequences (5′-TTAGAAGACTTGATTGTAA-3′ and 5′-GAAACTTCCTGTACATAAA-3′) that correspond to codons 290–308 and 672–690 relative to the start codon of RNF13 cDNA were identified to be effective in knocking down RNF13. shRNA corresponding to 5′-TTAGAAGACTTGATTGTAA-3′ was generated and subcloned into lentiviral vector. As a control, two point single nucleotide mutations (G293C, A297T) were introduced into the shRNA sequence used for RNF13 silencing. Lentivirus containing shRNA for RNF13 or its control were generated and used to infect SHSY-5Y cells for the generation of stable cells in 400 μg/ml hygromycin (Sigma). The siRNAs used for knockdown IRE1α, ASK1, and TRAF2 were 5′-AGACAGAGGCCAAGAGCAA-3′, 5′-GGUAUACAUGAGUGGAAUUTT-3′, and 5′-AGGGCAUAUAUGAAGAAGGCA-3′, respectively according to previous publications (17, 19).
Apoptosis Assay
Apoptosis was assessed by examining the nuclear morphology of cells. Cells were incubated with 0.1 μg/ml H-33342 (a cell permeable, blue fluorescent chromatin dye) for 30 min. Apoptosis was characterized by counting condensed and fragmented fluorescent nuclei. Each set of experiments was repeated at least 3 times, with at least 300 cells in each assay.
Subcellular Fractionation of RNF13
RNF13-transfected SHSY-5Y cells or COS-7 cells on 10-cm dish were washed twice with PBS, harvested, and suspended in buffer A (10 mm HEPES, pH 7.8, 250 mm sucrose, 25 mm potassium chloride, and 1 mm EGTA). Extraction buffer was kept at 4 °C and supplemented with protease inhibitor mixture (Roche Applied Science). Cells were homogenized by gentle Dounce (50 strokes) followed by centrifugation at 1300 × g for 10 min at 4 °C to pellet the nuclei and unbroken cells. The supernatant was collected and centrifuged at 12,000 × g for 30 min to yield a mitochondrial enriched pellet.
Mitochondria were purified by step sucrose gradient centrifugation as previously described (20). The supernatant collected was the starting material for preparation of ER microsomes. After centrifugation at 30,000 × g for 20 min, the supernatant was precipitated with 7 mm CaCl2 for 15 min at 4 °C. Last, the mixture was centrifuged at 8000 × g for 10 min at 4 °C to yield the enriched ER microsomal pellet, which was then purified by sucrose gradient centrifugation (20). The components obtained were subjected to Western blotting and detected with corresponding antibodies.
Immunofluorescent Analysis
Cos-7 cells were seeded on 12-mm coverslips and transfected with Myc-tagged RNF13 or the different mutants for 20 h. Cells were loaded with 20 nm MitoTracker Red CMXRos or 0.1 μg/ml H-33342 for 2 h (Molecular Probes) and washed twice with DMEM before treatment. After treatment, cells were washed once with PBS and fixed in 4% paraformaldehyde before permeabilization with 0.1% Trion X-100. Immunostaining was performed using the following primary antibodies; monoclonal mouse anti-Myc (1:200; Santa Cruz Biotechnology, Santa Cruz, CA), polyclonal rabbit anti-PDI (1:1200; Sigma) for 1 h followed by incubation with the appropriate secondary antibodies conjugated with either Alexa Fluor 488 or Alexa Fluor 568. Fluorescence images were collected and analyzed with microscopy equipment with Colored CCD camera (Zeiss AxioCam).
Co-immunoprecipitation Assays
Cells were transfected with indicated plasmids. At 24 h after transfection cells were harvested with lysis buffer (20 mm Tris/HCl, pH 7.4, 150 mm NaCl, 1 mm EGTA, 1 mm EDTA, 1 mm sodium orthovanadate, 1% Triton X-100, 1 mm glycerol phosphate, 2.5 mm sodium pyrophosphate, 1 μg/ml leupeptin, and 1 mm PMSF). Cell extracts were sonicated 6 times (1-s burst each time) and centrifuged at 14,000 × g for 15 min at 4 °C. Supernatants were subjected to immune-precipitations with anti-FLAG M2-agarose beads or A/G Plus beads with indicated antibodies at 4 °C for 3–5 h. Beads were then centrifuged at 3000 × g for 3 min and washed 3 times with 1 ml of lysis buffer. The immune-precipitated proteins were dissolved in 2× SDS sample buffer (20% (v/v) glycerol, 0.48% SDS, 10% (v/v) 2-mercaptoethanol and 0.1 m Tris, pH 6.8) and were analyzed by Western blots.
Western Blot Analysis
Cells were lysed in lysis buffer (20 mm Tris-HCl, pH 7.5, 120 mm NaCl, 1 mm Na3VO4, 2 mm EDTA, 1 mm phenyl methanesulfonyl fluoride, 40 mm β-glycerophosphate) containing 1%Triton X-100. 50 μg of cell lysate was loaded on a 12% SDS gel, protein (30 μg) from gradient-purified mitochondria was separated on 15% SDS gel, or ER fractionation was separated on 10% SDS gel for Western blotting. The following antibodies were used for Western blot analysis; polyclonal rabbit anti-actin (1:2000), polyclonal rabbit anti-caspase-3 (1:1,000), polyclonal rabbit anti-JNK (1:1,000), polyclonal rabbit anti-phospho-JNK (1:1,000), monoclonal rabbit anti-c-Jun (1:1000), polyclonal rabbit anti-phospho-c-Jun (1:1,000), monoclonal rabbit anti-IRE1α (1:1,000) and polyclonal rabbit anti-ASK1 antibody (1:1,000, Cell Signaling, Beverly, MA), polyclonal rabbit anti-FLAG (1:2,000), polyclonal rabbit anti-calnexin (1:10,000, Sigma); polyclonal rabbit anti-IRE1α (1:1,000), polyclonal rabbit anti-phospho-IRE1α (1:1,000, Novus Biological); monoclonal mouse anti-Myc (1:1,000), monoclonal mouse anti-tubulin (1:2,000), monoclonal mouse anti-COX4 (1:1,000, Molecular Probes, Eugene, OR), and polyclonal rabbit anti-TRAF2 (1:1000, Bethyl Laboratories). Appropriate horseradish peroxidase-linked secondary antibodies (Amersham Biosciences) were detected by enhanced chemiluminescence (Pierce). Membranes probed with more than one antibody were stripped before re-probing.
3′-Rapid Amplification of cDNA Ends
Total RNA was isolated using TRIzol reagent (Invitrogen), and reverse transcription was performed with RT primers (5′-CCA GTG AGC AGA GTG ACG AGG ACT CGA GCT CAA GC(T)17-3′. A nested PCR was performed using primers P1/Q1(5′-ATG GGC TGA CCG CTT CCT-3′/5′-CCA GTG AGC AGA GTG ACG-3′) and P2/Q2(5′-GAC GAG TTCTTC TGA CTA GCA GCT AG-3′/5′-GAG GAC TCG AGC TCA AGC-3′), respectively. P1 and P2 are located on the neo-resistant gene, whereas Q1 and Q2 are on the anchor sequence of QT. The PCR fragments were subcloned into the TA-cloning vector and confirmed by sequencing.
Statistical Analysis
Statistical significance (p < 0.05) was assessed using Student's t test or one-way analysis of variance.
RESULTS
Identification of RNF13 as a Novel Apoptotic Regulator
To identify novel genes involved in apoptosis regulation, we initiated a screen using retroviral random insertion approach (Gene trap) (18). This screen allows the identification of genes whose disruption would render SHSY-5Y cells resistant to apoptosis triggered by the kinase inhibitor STS (Fig. 1A). Approximately 30 positive clones that showed resistance to STS were selected, and the genes containing viral vector insertion were cloned using 3′-rapid amplification of the cDNA ends analysis. Three clones (STS-108) that showed strong resistance to STS-induced apoptosis contained a retroviral vector insertion at exon 8 of RING finger protein 13 (RNF13), corresponding to nucleotide 501 bp of the coding sequence of RNF13 cDNA (supplemental Fig. S1, A and B). Insertion of Neo vector at this position would predict the generation of truncated RNF13 protein containing only aa 1–167. Consistent with the prediction, a short coding transcript of RNF13 encodes the N-terminal portion of RNF13 (aa 1–183) was readily detected in STS-108 cells (supplemental Fig. S1C). Primary amino acid sequences of RNF13s from various species are evolutionary conserved (supplemental Fig. S1D), and full-length RNF13 protein contains an N-terminal protease-associated (PA) domain, a potential transmembrane (TM) domain, and a C-terminal RING domain (Fig. 1B) (21). To further confirm that RNF13 is indeed involved in STS-induced apoptosis, we treated wild-type SHSY-5Y and STS-108 cells with STS. STS-108 cells were highly resistant to STS-induced apoptosis with only 20% of apoptotic cells detected even after 48 h of STS treatment, whereas more than 80% of the wild-type cells underwent apoptosis by 48 h (Fig. 1B). Furthermore, the STS-108 cells were also found to be substantially more resistant than the WT SHSY-5Y cells to UV-induced apoptosis (Fig. 1C). To further test the role of RNF13 in mediating apoptosis, we tested several siRNAs against RNF13 and determined their efficiency in knocking down RNF13 and found two of them to be effective in knocking down endogenous RNF13 evaluated by semi-quantitative RT-PCR or RT-PCR analysis (supplemental Fig. S2, A and B). Furthermore, these two siRNAs were effective in reducing the expression of ectopically expressed Myc-tagged RNF13 (supplemental Fig. S2C). We then generated stable RNF13 knockdown SHSY-5Y cells using lentivirus containing shRNA derived from siRNF13–1 of RNF13. In agreement with the idea that inactivation of RNF13 would cause blockade of STS-mediated apoptosis, RNF13 knockdown SHSY-5Y cells indeed exhibited a similar degree of resistance to STS as the STS-108 cells (Fig. 1D). When RNF13 cDNA containing two silent mutations at the region targeted by the siRNF13–1 was introduced back into the RNF13 knockdown cells through transient transfection, sensitivity to STS-induced apoptosis was largely restored to level similar to that of the WT SH5Y-5Y cells (Fig. 1D).
FIGURE 1.

Identification of RNF13 as a novel apoptotic regulator. A, a schematic diagram depicts procedures for conducting the retroviral random insertion screen. B, top, a schematic illustrates domain organization in RNF13. The predicted full-length RNF13 protein consists of a protease-associated domain (PA) at the N terminus, a transmembrane domain (TM) in the middle, and a really interesting new gene containing domain (RING) at the C terminus as indicated. Bottom, STS-108 is a SHSY-5Y cell clone identified to be STS-resistant during the screening. Wild-type and STS-108 SHSY-5Y cells were treated with 1 μm STS for the indicated period of time. The nucleus was visualized by staining with 100 ng/ml Hoechst 33342, and cells containing the condensed or broken nuclei were defined as apoptotic cells under fluorescence microscope. 300 cells were chosen randomly for scoring, and the percentage of apoptotic cells was calculated by the number of apoptotic cells over total number of cells. C, STS-108 is also resistant to UV-induced apoptosis. Wild-type and STS-108 SHSY-5Y cells were exposed to 10 mJ/cm2 of UV radiation and incubated for the indicated periods of time before they were examined for apoptosis as described in B. D, reintroduction of siRNA-resistant Myc-tagged RNF13 into RNF13 knockdown (shRNF13) cells restores their sensitivity to STS-induced apoptosis. Wild-type and RNF13 knockdown SHSY-5Y cells were co-transfected with 0.2 μg of GFP and 3 μg of CMV5 or Myc-RNF13 plasmid for 36 h. Cells were then treated with 1 μm STS for the indicated amounts of time and subsequently evaluated for apoptosis. The inset panel is the result of RT-PCR for RNF13 mRNA levels. E, overexpression of RNF13 induces caspase-3 activation. COS-7 cells were treated with 1 μm STS for 3 h or transfected with 3 μg of indicated plasmids for 30 h. Cell lysates were blotted with anti-caspase-3 antibody. M-cofilin is the mitochondrial-targeted cofilin that induces caspase activation and apoptosis as shown previously (20). F, RNF13 induces caspase-dependent apoptosis. COS-7 cells transfected with 3 μg of plasmid encoding M-cofilin or RNF13 in the presence or absence of indicated caspase inhibitors were analyzed. At 36 h of post-transfection, cells were evaluated for apoptosis as in B. Data are representative of three independent experiments. ZVAD-fmk, benzyloxycarbonyl-VAD-fluoromethyl ketone. ***, p < 0.001.
Next we evaluated intrinsic proapoptotic activity of RNF13 by conducting transient transfection analysis in COS-7 cells. Overexpression of RNF13 induced apoptosis in a time- and dose-dependent manner (supplemental Fig. S3, A and B). Furthermore, similar to apoptosis triggered by overexpression of M-cofilin (20), cleaved caspase-3 was also readily observed in COS-7 cells overexpressing RNF13 (Fig. 1E). The caspase-3 inhibitor DEVD-CHO or a broad spectrum inhibitor of caspases, benzyloxycarbonyl-VAD-fluoromethyl ketone, was equally effective in blocking apoptosis induced by M-cofilin and RNF13 in these cells (Fig. 1F). Together, these data suggest that apoptosis induced by RNF13 is mediated through a caspase-dependent pathway.
The RING and Transmembrane Domains Are Both Required for RNF13-induced Apoptosis
To gain insights into the molecular mechanism of action of RNF13, we next analyzed the subcellular localization of RNF13. Myc-tagged RNF13 was overexpressed in 293T cells, and the cells were subjected to fractionation and immunofluorescent staining. RNF13 protein was found to be highly enriched in the microsomal fraction of ER but was absent in cytosolic or mitochondrial fractions (Fig. 2A). The purity of the mitochondrial and ER microsomal fractions was confirmed by probing the fractions with antibody for COX4 and calnexin, respectively (Fig. 2A). Immunofluorescent staining in COS-7 cells overexpressing Myc-RNF13 showed an intense signal with distinct network patterns near the nuclei region that overlapped with that of protein-disulfide isomerase, an ER-localized protein (Fig. 2B). Disruption of the RING domain structure by a single point mutation at the RING finger domain (C243W) of RNF13 did not affect its ER localization, whereas deletion of the putative transmembrane domain (ΔTM/Δ184–207) disrupted its localization to ER (Fig. 2B). Therefore, RNF13 is highly enriched in ER, and the putative transmembrane domain is required for its ER localization.
FIGURE 2.

RNF13 is highly enriched in the microsomal ER fraction, and both the ER localization and functional RING domain are required for RNF13-induced apoptosis. A, mitochondrial, microsomal ER, and cytosolic fractions of 293T cells transfected with Myc-RNF13 were prepared as described under “Materials and Methods.” Different components were subjected to SDS-PAGE and blotted with the corresponding antibodies as indicated. B, the putative TM of RNF13 is required for mediating its localization to ER. Myc-tagged RNF13 and its mutations were expressed in COS-7 cells. Immunostaining was performed as described under “Materials and Methods”; ER was stained with anti-protein-disulfide isomerase (PDI) antibody. Insets show enlargements of the boxed regions. C, the TM and the intact RING domains of RNF13 are both required for mediating its proapoptotic activity. Left, shown is a schematic representation of RNF13 and mutants used in experiments. Right, COS-7 cells were transfected with either 3 μg of plasmid encoding wild-type RNF13 or the indicated mutants for 36 h before the evaluation of apoptosis. Data are representative of three independent experiments. ***, p < 0.001.
To evaluate the involvement of distinct domains of RNF13 in apoptosis induction, a series of deletion mutants of RNF13 (Fig. 2C) was evaluated in COS-7 cells by transfection analysis. Deletion of the C-terminal region (aa 165–381), transmembrane domain (ΔTM), or disruption of the RING finger structure with a single point mutation in RING finger domain (C243W) of RNF13 completely abolished its apoptotic activity (Fig. 2C). In contrast, deletion of the N-terminal region (aa 1–183) and the domain in RNF13 did not affect its proapoptotic activity (Fig. 2C). Therefore, the RING and TM domains are both required for mediating apoptosis induced by RNF13.
RNF13 Mediates ER Stress-induced Apoptosis and Splicing of XBP1 mRNA
We next checked the sensitivity of wild-type and RNF13 knockdown SHSY-5Y cells to the apoptotic response triggered by tunicamycin (Tn), which is a known inducer of ER stress. Although wild-type SHSY-5Y cells were sensitive to Tn-induced apoptosis, RNF13 knockdown SHSY-5Y cells were resistant to the apoptotic effect of Tn (Fig. 3A). Most importantly, introduction of siRNA-resistant RNF13 back into the knockdown cells restored their ability to respond to Tn-induced apoptosis (Fig. 3A). We further studied the molecular pathway by which RNF13 mediates ER stress response. ER stress induces activation of IRE1α, which in turn causes splicing of XBP1 mRNA. Similar to treatment with Tn or overexpression of IRE1α, overexpression of RNF13 in SHSY-5Y cells dramatically promoted the splicing of XBP1 mRNA (Fig. 3B). We then checked the splicing of XBP1 in response to ER stress in RNF13 knockdown SHSY-5Y cells. In wild-type cells, Tn induced a time-dependent increase in splicing of XBP1 mRNA, with the appearance of spliced product at 3 h and reaching highest levels at 6 h (Fig. 3C). Interestingly, splicing of XBP1 mRNA induced by Tn was significantly reduced in the RNF13 knockdown cells (Fig. 3C). Expression of RNF13 in SHSY-5Y cells was not altered upon STS exposure (supplemental Fig. S4A). Similarly, overexpression or knockdown of RNF13 did not increase the expression levels of CHOP, a downstream target of PKR-like ER kinase or ATF6 (supplemental Fig. S4, B and C). These data clearly suggest that RNF13 is specifically required for mediating ER stress-induced splicing of XBP1 mRNA via activation of IRE1α (23, 24).
FIGURE 3.

RNF13 is required for ER stress-induced apoptosis. A, RNF13 knockdown SHSY-5Y cells are resistant to Tn-induced apoptosis, and introduction of the expression plasmid encoding the siRNA-resistant RNF13 cDNA into the knockdown cells restores its sensitivities to Tn. Wild-type and RNF13 knockdown SHSY-5Y cells were co-transfected with 0.2 μg of GFP plasmid and 3 μg of CMV5 or siRNF13–1-resistant Myc-RNF13 plasmid for 36 h. Cells were then treated with Tn (1.25 μg/ml) for the indicated amount of time before apoptosis assay was performed. B, overexpression of RNF13 induces XBP1 splicing. SHSY-5Y cells were treated with 1.25 μg/ml Tn for 6 h or transfected with 3 μg of either IRE1α or RNF13 plasmid for 24 h. Cells were harvested, RNA was extracted, and the levels of unspliced and spliced XBP1 were evaluated by RT-PCR analysis. β-Actin was used as a loading control. C, Tn-induced splicing of XBP1 mRNA is attenuated in the RNF13 knockdown SHSY-5Y cells. Control and RNF13 knockdown SHSY-5Y cells were treated with 1.25 μg/ml Tn for indicated periods of time before harvesting for RNA extraction. RT-PCR analysis was then performed to observe subsequent XBP1 splicing. β-Actin was served as a loading control.
RNF13 Is Required for ER stress-mediated JNK Activation
Next, we checked whether RNF13-mediated ER stress could induce JNK activation by measuring levels of phosphorylated JNK and its target gene c-Jun in the wild-type and RNF13 SHSY-5Y knockdown cells treated with ER stress stimuli Tn and Tg. Levels of JNK phosphorylation were quickly elevated in the wild-type cells treated with either Tn (Fig. 4, A and B) or Tg (supplemental Fig. S5, A and B). Consistent with the idea that RNF13 plays a role in promoting activation of JNK-Jun pathway in the ER stress response, levels of phosphorylated JNK and c-Jun were induced at significantly lower levels by Tn or Tg in the RNF13 knockdown cells (Fig. 4, A and B; (supplemental Fig. S5, A and B).
FIGURE 4.
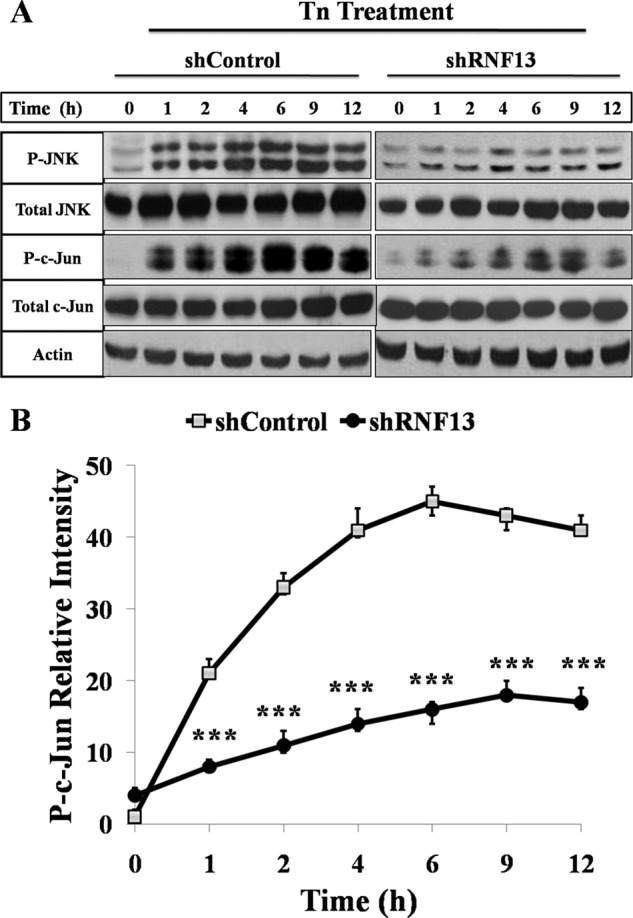
Activation of JNK and phosphorylation of c-Jun by Tn is suppressed in the RNF13 knockdown cells. A, SHSY-5Y cells were treated with 1.25 μg/ml Tn for indicated amounts of time before harvesting for Western blot analysis. Western blot analysis of the levels of phosphorylated endogenous JNK and c-Jun in wild-type and RNF13 knockdown cells is shown. B, shown is quantitative analysis of the relative levels of phosphorylated c-Jun in A using Quantity One software. β-Actin was used as a loading control. Data presented are the mean ± S.D. from three independent experiments. ***, p < 0.001.
The RING and TM Domains Are Both Required for RNF13-mediated JNK Activation
To define the region on RNF13 that mediates ER stress-induced JNK activation, we expressed a series of truncated or mutated RNF13 proteins in COS-7 cells and determined the levels of phosphorylated JNK and c-Jun in the transfected cells. Expression of wild-type RNF13 (aa 1–381) resulted in a significantly increased JNK and c-Jun phosphorylation (Fig. 5A). Deletion of the N-terminal domain (1–183) or the PA domain (ΔPA) of RNF13 did not affect its activity in activating JNK. On the contrary, deletion of the C-terminal region (165–381) or the transmembrane domain (ΔTM) or disruption of the RING domain (C243W) resulted in a complete loss of activity in promoting JNK activation (Fig. 5A). These data indicate that RNF13-induced JNK activation, similar to its proapoptotic activity, is dependent on functional RING structure and the TM domain. To test whether RNF13-induced apoptosis is dependent on JNK activity, we preincubated cells with 10 μm SP600125, a specific JNK inhibitor, before transfection with Myc-RNF13. Interestingly, cells pretreated with JNK inhibitor showed marked reduction in RNF13-induced apoptosis (Fig. 5B). Taken together, these data strongly suggest that RNF13-induced apoptosis requires the activation of JNK.
FIGURE 5.
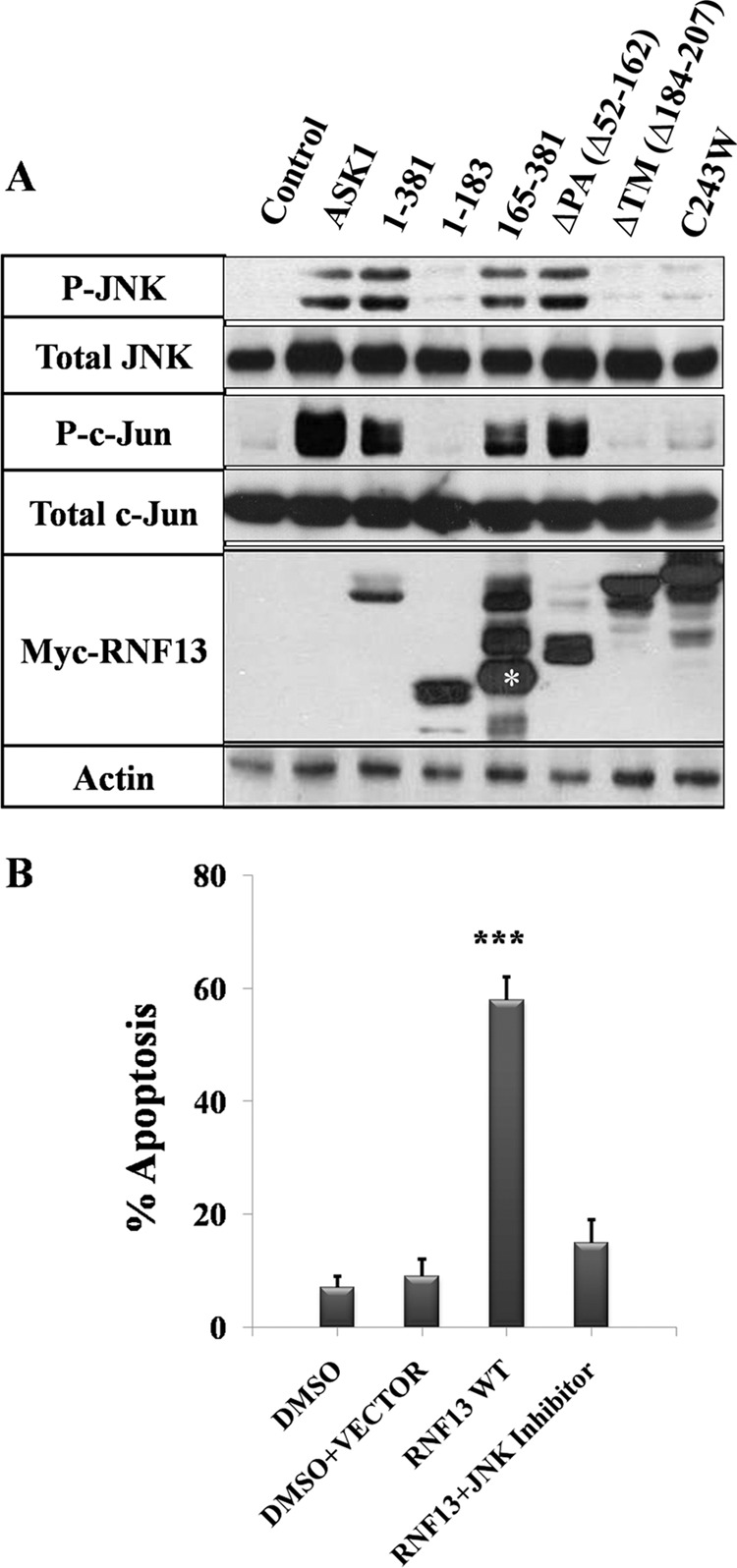
The RING and TM domains are both required for RNF13-induced JNK activation. A, COS-7 cells were transfected with 3 μg of plasmid encoding the Myc-tagged RNF13 or the indicated mutant for 30 h. Cells were harvested, and a Western blot was performed using antibodies against phospho-JNK and phospho-c-Jun. β-Actin was used as a loading control. The asterisk represents the correct sized protein band for RNF13 truncation (aa 165–381). B, inhibition of JNK activity blocks RNF13-induced apoptosis. COS-7 cells preincubated with 10 μm JNK inhibitor (SP600125) were transfected with 3 μg of Myc-tagged RNF13 for 30 h before apoptosis of was analyzed. Data presented are the mean ± S.D. values from three independent experiments. ***, p < 0.001.
RNF13 Acts Upstream of IRE1α-TRAF2 Pathway to Activate JNK
Because ER stress-induced JNK activation is mediated by IRE1α signaling pathway, we decided to investigate the relative position where RNF13 acts in the IRE1α-JNK signaling axis by systematically reducing the activity of individual components in the pathway by siRNA knockdown or by expression of dominant negative mutants. Overexpression of RNF13, IRE1α, TRAF2, or ASK1 all resulted in the activation of JNK as the phosphorylation levels of c-Jun were significantly increased (Fig. 6, A and B; supplemental Fig. S6, A and B). Knockdown of IRE1α, TRAF2, ASK1, or MKK4/7 in cells expressing RNF13 all markedly reduced the levels of JNK activation (Fig. 6, A and B; supplemental Fig. S6, A and B, lane 3 and 4). When dominant negative forms of TRAF2 or ASK1 (25) were introduced into cells expressing TRAF2 or ASK1, levels of phosphorylated c-Jun were significantly decreased (Fig. 6C, lane 6 and 8). Interestingly, when these dominant negative proteins were introduced into RNF13 expressing cells, they were able to markedly reduce JNK activity (Fig. 6C, lane 3 and 4). Together, these data suggest that RNF13 is at the ER to act upstream of the IRE1α pathway to mediate ER stress-induced JNK activation and apoptosis.
FIGURE 6.

RNF13 induces JNK activation via the IRE1α-TRAF2-ASK1-MKK4/7-JNK signaling axis. A, knockdown of IRE1α or TRAF2 blocks RNF13-induced c-Jun phosphorylation. SHSY-5Y cells were co-transfected with 3 μg of plasmid of RNF13 and 3 μg of indicated siRNAs corresponding to IRE1α and TRAF2 for 30 h before harvesting for Western blot analysis. Western blotting was performed using antibodies against c-Jun, phosphorylated c-Jun, Myc, IRE1α, and TRAF2 as indicated on the left. β-Actin served as a loading control. B, activation of c-Jun by RNF13 is dependent on ASK1 and MKK4/7. Knockdown of ASK1 or MKK4/7 inhibits RNF13-mediated c-Jun phosphorylation in SHSY-5Y cells. Cells were co-transfected with 3 μg of RNF13 and the indicated siRNAs for 30 h. Cells were harvested, and Western blot analysis was performed using antibodies against c-Jun, phospho-c-Jun, Myc, and ASK1. C, expression of the dominant negative (DN) form of TRAF2 or ASK1 inhibits RNF13-mediated c-Jun activation. SHSY-5Y cells were co-transfected with 3 μg of the indicated plasmids and plasmid encoding the indicated dominant negative mutant for 30 h before harvesting for Western blots. Antibodies against c-Jun, phospho-c-Jun, Myc, HA, and FLAG were used to measure the amounts of c-Jun, phosphorylated c-Jun, RNF13, TRAF2, and ASK1. β-Actin was used as a loading control.
RNF13 Associates with IRE1α and Promotes Its Activation
To investigate the relationship between RNF13 and IRE1α, we next evaluated whether RNF13 can interact with IRE1α by co-expressing Myc-tagged RNF13 with FLAG-tagged IRE1α in 293T cells. FLAG-tagged IRE1α were immunoprecipitated with anti-FLAG antibody, and full-length RNF13 was co-precipitated by IRE1α (Fig. 7A). Furthermore, endogenous IRE1α was also shown to interact with the overexpressed RNF13 (Fig. 7B). In addition, RNF13 and IRE1α interaction was not affected by ER stress inducer (Tn) (supplemental Fig. S7). Next, we evaluated the domains in RNF13 required for mediating its interaction with IRE1α. RNF13 mutants containing the C-terminal region (aa 165–381) or with a deletion of the PA domain (ΔPA, Δ52–162) were able to co-precipitated with IRE1α. Interestingly, RNF13 with a single point mutation in RING domain (C243W) failed to associate with IRE1α (Fig. 7A). These data suggest that RNF13 interaction with IRE1α requires a functional RING domain that is also shown to mediate JNK activation and apoptosis. We next checked whether RNF13 is involved in IRE1α phosphorylation by co-expressing RNF13 (or its mutants) with IRE1α. As demonstrated previously (2, 17), treatment of ER stress inducer Tn resulted in an significantly increased IRE1α phosphorylation (Fig. 7C). Interestingly, co-expression of RNF13 with IRE1α resulted in significantly higher levels of IRE1α phosphorylation (Fig. 7C). In contrast, co-transfecting the cells with the RING mutant (C243W) failed to elevate the level of p-IRE1α in comparison with that of control cells (Fig. 7C), raising the possibility that RNF13 activates IRE1α via steps involving protein-protein interaction and IRE1α phosphorylation (Fig. 7D).
FIGURE 7.

RNF13 interacts with IRE1α and promotes IRE1α phosphorylation. A, 293T cells were co-transfected with 2 μg of FLAG-IRE1α plasmid and 3 μg of plasmid encoding Myc-RNF13 or its mutants for 24 h. FLAG-IRE1α protein was immune-precipitated (IP) using anti-FLAG antibody, and the presence of RNF13 or its mutant proteins in the immunoprecipitated products was detected by anti-Myc antibody. IB, immunoblot. B, RNF13 interacts with endogenous IRE1α. 293T cells were transfected with 3 μg Myc-RNF13 plasmid for 24 h. Myc-RNF13 protein was immunoprecipitated using anti-Myc antibody, and the presence of endogenous IRE1α in the immunoprecipitated products was detected by anti-IRE1α antibody. C, 293T cells were transfected with 2 μg of IRE1α plasmid alone or co-transfected with 3 μg of plasmid encoding RNF13 or its RING domain mutant (C243W) for 24 h. For cells treated with Tn, 1.25 μg/ml drug was added at 18 h post-transfection. D, a schematic model illustrates the role of RNF13 in mediating apoptosis through the IRE1α-TRAF2-JNK signaling pathway.
DISCUSSION
Using a retroviral-mediated insertional mutagenesis approach, we have identified RNF13 as a critical regulator of STS-mediated apoptosis. Interestingly, RNF13 is highly enriched in ER, which prompted us to focus our investigation in evaluating its role in mediating ER-stress induced apoptosis. Indeed, RNF13 knockdown cells showed markedly reduced response to the apoptotic effect of Tn and Tg, which are potent triggers of ER stress-mediated apoptosis. Consistent with the idea that RNF13 might play a role in the ER stress-mediated apoptotic pathway, overexpression of RNF13 induced caspase-dependent apoptosis, splicing of XBP1 mRNA, and JNK activation, all of which are known to be the downstream events emulating from ER stress response. The intact RING finger structure and the TM domain, which is required for ER localization, are both required for RNF13 to induce apoptosis and JNK activation. Finally, our data showed that RNF13 associates with IRE1α and promotes its activation, a step crucial for downstream JNK activation and apoptosis induced by ER stress. Interestingly, the truncated RNF13 protein identified from the original screening lacks the ER localization signal and the IRE1α interaction domain. This truncated RNF13 protein may act as a dominant negative form of wild-type RNF13 by inhibiting other positive effector molecules by a mechanism that does not require binding to IRE1α. Although our current data clearly link RNF13 to the ER stress-mediated apoptosis signaling pathway, RNF13 knockdown cells were also shown to be resistant to the apoptotic effect triggered by STS and UV, suggesting that RNF13 may have other roles in regulating apoptosis signaling. Together, our data indicate that RNF13 is a crucial mediator of ER stress-induced JNK activation and apoptosis through its interaction and activation of IRE1α.
RNF13 is a member of the ever increasing RING-domain-containing superfamily of proteins. Among some members in the family, the RING domains have been shown to possess ubiquitin E3 ligase activity and are involved in diverse cellular processes including cancer metastasis (22). However, RNF13 has only been studied quite recently, and its role as an E3 ligase to promote the degradation of downstream substrate has not yet been well characterized (22, 26–28). A recent study has shown that mRNA of RNF13 is highly expressed in proliferating myoblasts, and its expression gradually decreases during skeletal myogenesis (26). Moreover, expression of RNF13 mRNA transcripts is observed in embryonic and adult brain tissues, and overexpression of RNF13 induces spontaneous neurite outgrowth (27, 28). Our data provide evidence for the first time that RNF13 plays an important role in mediating ER stress-induced apoptosis by activating the IRE1α-JNK signaling pathway.
The UPR signals are mediated by three distinct stress sensors, IRE1α, ATF6, and PKR-like ER kinase, which are localized on the ER membrane. The IRE1α branch of the UPR has recently emerged as a highly regulatory process, controlled by many adaptor and regulatory proteins. The IRE1α protein complex is a dynamic scaffold onto which many regulatory components assemble (29). The UPR signals activate IRE1α oligomerization and autophosphorylation, resulting in the recruitment of adaptor protein TRAF2 to form a protein complex and the activation of downstream kinases ASK1, MKK, and JNK. Our data clearly indicate that RNF13 facilitates activation of the ER stress-induced IRE1α-JNK branch but not the ATF6 and PKR-like ER kinase pathways. Furthermore, the ER localization and the functional RING domain of RNF13 are required for its activity to induce JNK activation. The specificity of RNF13 in controlling one arm of ER stress signaling and the dependence on the ER localization and intact RING domain structure for its activity is not clear. Mechanistically, we demonstrated that RNF13 interacts with IRE1α, and the RING finger domain is required for mediating its association with IRE1α. Thus far, we did not observe significant up-regulation of the interaction between RNF13 and IRE1α in the presence of ER stress inducer Tn. It remains to be a subject of interest for future investigation to determine whether E3 ligase activity of RNF13 is involved in the activation of any of the steps in the ER stress-mediated IRE1α/JNK activation and apoptosis.
Acknowledgments
We thank members of the Peng Li laboratory in Tsinghua University for helpful discussions.
This work was supported by National Natural Science Foundation of China Grant 30670418 (to P. L.) and National High Technology Research and Development Program Grant 2010AA023002 from the Ministry of Science and Technology of China.

This article contains supplemental Figs. S1—S7.
- ER
- endoplasmic reticulum
- UPR
- unfolded protein response
- ATF6
- activating transcription factor 6
- RNF13
- RING (really interesting new gene) finger protein 13
- TM
- transmembrane
- PA
- protease-associated
- IRE1α
- inositol-requiring enzyme
- XBP1
- X-box-binding protein
- TRAF2
- tumor necrosis factor receptor-associated factor-2
- ASK1
- apoptosis signal-regulating kinase 1
- MKK4/7
- mitogen-activated protein kinase kinase 4/7
- STS
- staurosporine
- Tn
- tunicamycin
- Tg
- thapsigargin
- PKR
- Protein Kinase R.
REFERENCES
- 1. Kim I., Xu W., Reed J. C. (2008) Cell death and endoplasmic reticulum stress. Disease relevance and therapeutic opportunities. Nat. Rev. Drug Discov. 7, 1013–1030 [DOI] [PubMed] [Google Scholar]
- 2. Walter P., Ron D. (2011) The unfolded protein response. From stress pathway to homeostatic regulation. Science 334, 1081–1086 [DOI] [PubMed] [Google Scholar]
- 3. Cnop M., Foufelle F., Velloso L. A. (2012) Endoplasmic reticulum stress, obesity, and diabetes. Trends Mol. Med. 18, 59–68 [DOI] [PubMed] [Google Scholar]
- 4. Merquiol E., Uzi D., Mueller T., Goldenberg D., Nahmias Y., Xavier R. J., Tirosh B., Shibolet O. (2011) HCV causes chronic endoplasmic reticulum stress leading to adaptation and interference with the unfolded protein response. Plos One 6, e24660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Malhotra J. D., Kaufman R. J. (2007) The endoplasmic reticulum and the unfolded protein response. Semin. Cell Dev. Biol. 18, 716–731 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Tabas I., Ron D. (2011) Integrating the mechanisms of apoptosis induced by endoplasmic reticulum stress. Nat. Cell Biol. 13, 184–190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Ye J., Rawson R. B., Komuro R., Chen X., Davé U. P., Prywes R., Brown M. S., Goldstein J. L. (2000) ER stress induces cleavage of membrane-bound ATF6 by the same proteases that process SREBPs. Mol. Cell 6, 1355–1364 [DOI] [PubMed] [Google Scholar]
- 8. Lee K. P., Dey M., Neculai D., Cao C., Dever T. E., Sicheri F. (2008) Structure of the dual enzyme ire1 reveals the basis for catalysis and regulation in nonconventional RNA splicing. Cell 132, 89–100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Ron D., Walter P. (2007) Signal integration in the endoplasmic reticulum unfolded protein response. Nat. Rev. Mol. Cell Biol. 8, 519–529 [DOI] [PubMed] [Google Scholar]
- 10. Hetz C., Martinon F., Rodriguez D., Glimcher L. H. (2011) The unfolded protein response. Integrating stress signals through the stress sensor Ire1α. Physiol. Rev. 91, 1219–1243 [DOI] [PubMed] [Google Scholar]
- 11. Urano F., Wang X., Bertolotti A., Zhang Y., Chung P., Harding H. P., Ron D. (2000) Coupling of stress in the ER to activation of JNK protein kinases by transmembrane protein kinase IRE1. Science 287, 664–666 [DOI] [PubMed] [Google Scholar]
- 12. Lei K., Davis R. J. (2003) JNK phosphorylation of Bim-related members of the Bcl2 family induces Bax-dependent apoptosis. Proc. Natl. Acad. Sci. U. S. A. 100, 2432–2437 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Tournier C., Hess P., Yang D. D., Xu J., Turner T. K., Nimnual A., Bar-Sagi D., Jones S. N., Flavell R. A., Davis R. J. (2000) Requirement of JNK for stress-induced activation of the cytochrome c-mediated death pathway. Science 288, 870–874 [DOI] [PubMed] [Google Scholar]
- 14. Nickson P., Toth A., Erhardt P. (2007) PUMA is critical for neonatal cardiomyocyte apoptosis induced by endoplasmic reticulum stress. Cardiovasc. Res. 73, 48–56 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Morishima N., Nakanishi K., Tsuchiya K., Shibata T., Seiwa E. (2004) Translocation of bim to the endoplasmic reticulum (ER) mediates ER stress signaling for activation of caspase-12 during ER stress-induced apoptosis. J. Biol. Chem. 279, 50375–50381 [DOI] [PubMed] [Google Scholar]
- 16. Puthalakath H., O'Reilly L. A., Gunn P., Lee L., Kelly P. N., Huntington N. D., Hughes P. D., Michalak E. M., McKimm-Breschkin J., Motoyama N., Gotoh T., Akira S., Bouillet P., Strasser A. (2007) ER stress triggers apoptosis by activating BH3-only protein Bim. Cell 129, 1337–1349 [DOI] [PubMed] [Google Scholar]
- 17. Kim I., Shu C. W., Xu W., Shiau C. W., Grant D., Vasile S., Cosford N. D., Reed J. C. (2009) Chemical biology investigation of cell death pathways activated by endoplasmic reticulum stress reveals cytoprotective modulators of ASK1. J. Biol. Chem. 284, 1593–1603 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Li J., Li Q., Xie C., Zhou H., Wang Y., Zhang N., Shao H., Chan S. C., Peng X., Lin S. C., Han J. (2004) β-Actin is required for mitochondria clustering and ROS generation in TNF-induced, caspase-independent cell death. J. Cell Sci. 117, 4673–4680 [DOI] [PubMed] [Google Scholar]
- 19. Ghosh R., Lipson K. L., Sargent K. E., Mercurio A. M., Hunt J. S., Ron D., Urano F. (2010) Transcriptional regulation of VEGF-A by the unfolded protein response pathway. PLoS ONE 5, e9575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Chua B. T., Volbracht C., Tan K. O., Li R., Yu V. C., Li P. (2003) Mitochondrial translocation of cofilin is an early step in apoptosis induction. Nat. Cell Biol. 5, 1083–1089 [DOI] [PubMed] [Google Scholar]
- 21. Plonne D., Cartwright I., Linss W., Dargel R., Graham J. M., Higgins J. A. (1999) Separation of the intracellular secretory compartment of rat liver and isolated rat hepatocytes in a single step using self-generating gradients of iodixanol. Anal. Biochem. 276, 88–96 [DOI] [PubMed] [Google Scholar]
- 22. Zhang Q., Meng Y., Zhang L., Chen J., Zhu D. (2009) RNF13. A novel RING-type ubiquitin ligase over-expressed in pancreatic cancer. Cell Res. 19, 348–357 [DOI] [PubMed] [Google Scholar]
- 23. Samali A., Fitzgerald U., Deegan S., Gupta S. (2010) Methods for monitoring endoplasmic reticulum stress and the unfolded protein response. Int. J. Cell Biol. 2010, 830307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Back S. H., Schröder M., Lee K., Zhang K., Kaufman R. J. (2005) ER stress signaling by regulated splicing. IRE1/HAC1/XBP1. Methods 35, 395–416 [DOI] [PubMed] [Google Scholar]
- 25. Hoeflich K. P., Yeh W. C., Yao Z., Mak T. W., Woodgett J. R. (1999) Mediation of TNF receptor associated factor effector functions by apoptosis signal-regulating kinase-1 (ASK1). Oncogene 18, 5814–5820 [DOI] [PubMed] [Google Scholar]
- 26. Zhang Q., Wang K., Zhang Y., Meng J., Yu F., Chen Y., Zhu D. (2010) The myostatin-induced E3 ubiquitin ligase RNF13 negatively regulates the proliferation of chicken myoblasts. FEBS J. 277, 466–476 [DOI] [PubMed] [Google Scholar]
- 27. Tranque P., Crossin K. L., Cirelli C., Edelman G. M., Mauro V. P. (1996) Identification and characterization of a RING zinc finger gene (C-RZF) expressed in chicken embryo cells. Proc. Natl. Acad. Sci. U. S. A. 93, 3105–3109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Saito S., Honma K., Kita-Matsuo H., Ochiya T., Kato K. (2005) Gene expression profiling of cerebellar development with high-throughput functional analysis. Physiol Genomics 22, 8–13 [DOI] [PubMed] [Google Scholar]
- 29. Sitia R., Braakman I. (2003) Quality control in the endoplasmic reticulum protein factory. Nature 426, 891–894 [DOI] [PubMed] [Google Scholar]