

Abstract
The activity of a key mitochondrial tricarboxylic acid cycle enzyme, α-ketoglutarate dehydrogenase complex (KGDHC), declines in many neurodegenerative diseases. KGDHC consists of three subunits. The dihydrolipoyl succinyltransferase (DLST) component is unique to KGDHC. DLST+/- mice showed reduced mRNA and protein levels and decreased brain mitochondrial KGDHC activity. Neurotoxic effects of mitochondrial toxins were exacerbated in DLST+/- mice. MPTP produced a significantly greater reduction of striatal dopamine and tyrosine hydroxylase-positive neurons in the substantia nigra pars compacta of DLST+/- mice. DLST deficiency enhanced the severity of lipid peroxidation in the substantia nigra after MPTP treatment. Striatal lesions induced by either malonate or 3-nitropropionic acid (3-NP) were significantly larger in DLST+/- mice than in wildtype controls. DLST deficiency enhanced the 3-NP inhibition of mitochondria enzymes, and 3-NP induced protein and DNA oxidations. These observations support the hypothesis that reductions in KGDHC may impair the adaptability of the brain and contribute to the pathogenesis of neurodegenerative diseases.
Keywords: Mitochondria, neurodegenerative diseases, Parkinson, Huntington, oxidative damage, α-ketoglutarate dehydrogenase complex
INTRODUCTION
The α-ketoglutarate dehydrogenase complex (KGDHC) is a mitochondrial enzyme that plays an important role in brain metabolism. KGDHC is crucial in the cellular production of reducing equivalents (NADH) and in the maintenance of the mitochondrial redox state of the brain (Gibson et al. 2000a). There is burgeoning evidence that KGDHC may be involved in the pathophysiology of multiple neurodegenerative diseases. Reductions in KGDHC activity have been reported in Alzheimer’s disease (AD) (Gibson et al. 1988), Huntington’s disease (HD) (Klivenyi et al. 2004), Parkinson’s disease (PD) (Mizuno et al. 1990; Mizuno et al. 1994; Mizuno et al. 1995; Gibson et al. 2003), Progressive Supranuclear Palsy (PSP) (Park et al. 2001) and spinocerebellar ataxia (Mastrogiacomo and Kish 1994). Diminished KGDHC activity occurs in both normal and pathologically involved brain areas of patients with a number of neurodegenerative diseases including AD (Gibson et al. 1988; Gibson et al. 2003). The reduction in KGDHC activity is highly correlated to the clinical dementia rating of AD patients (Gibson et al. 2000b; Bubber et al. 2005). These results suggest that KGDHC may play a central role in the pathophysiology of neurodegenerative diseases. Oxidative stress, a common feature of these neurodegenerative diseases, is known to inactivate KGDHC (Park et al. 1999).
KGDHC consists of 3 protein subunits, namely α-ketoglutarate dehydrogenase (E1k; E.C. 1.2.4.2 encoded by the ogdh gene), dihydrolipoyl succinyl transferase (DLST; E.C. 2.3.1.61 encoded by the dlst gene) and dihydrolipoamide dehydrogenase (DLD; E.C. 1.8.1.4 encoded by the dld gene). KGDHC is a member of the KGDHC complex family. This family also includes the pyruvate dehydrogenase complex (PDHC) and the branched chain α-ketoacid dehydrogenase complex. A subunit contained in all of these dehydrogenases is dihydrolipoamide dehydrogenase (DLD), which is also known as E3, a critical subunit shared by all three dehydrogenases. It is a flavin-containing protein that transfers reducing equivalents from a dihydrolyl moiety to NAD+ to form NADH and complete the catalytic process of the complex. OGDH and DLST are unique to KGDHC. Encoded by the dlst gene, DLST catalyzes an important reaction sequence in α-ketoacid oxidation. It contains covalently bound lipoyl moieties and catalyzes the transfer of succinyl moiety to CoA substrate to form succinyl-CoA (Reed and Hackert 1990).
Mice deficient in DLST have been developed. Although these mice have reduced KGDHC activity, they have no overt phenotype. If KGDHC deficiency plays a role in neurodegeneration, we hypothesized that mice lacking KGDHC subunits would be more susceptible to mitochondrial toxins. Prior work showed that cells deficient in DLST are more susceptible to H2O2 (Shi et al. 2005). In the current investigation, we carried out studies of neurotoxicity induced by 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP), which has been used to model PD, and with malonate and 3-nitropropionic acid (3-NP) which have been used to mimic HD neuropathology. Mice deficient in DLST were tested for altered susceptibility to these mitochondrial toxins. We previously demonstrated that DLD deficiency exacerbated the neuropathological effects of these toxins (Klivenyi et al. 2004). Given the role of KGDHC in mitochondrial bioenergetics, we hypothesized that DLST+/- mice, heterozygous for the disruption of the DLST gene, would be more vulnerable to mitochondrial neurotoxins mimicking PD and HD.
MATERIALS AND METHODS
Animals
Mice deficient in the DLST subunit (DLST+/-; C57BL/6 and 129SV/EV hybrid) were obtained from Lexicon Pharmaceuticals (The Woodlands, TX). The founder mice were bred with B6129F/mice (Taconia Germantown, NY). They have no overt phenotype. Mice were housed in a room maintained at 20-22°C on a 12-hr light-dark cycle with food and water available ad libitum. All experiments were conducted in accordance with guidelines approved by the Institution for the Care and Use of Animals at Weill Medical College of Cornell University.
For genotyping heterozygous DLST+/- mice, genomic DNA was isolated from the tails by phenol:chloroform:isoamyl alcohol (25:24:1) extraction and ethanol precipitation. Polymerase chain reaction (PCR) amplification of genomic DNA was performed using primers (Invitrogen) listed in Table 1. The cycle conditions were: initial denaturation at 94°C for 2 min followed by 94°C for 30 sec, 53°C for 30 sec and 72°C for 40 sec (last 3 steps 29x), and 72°C for 10 min.
Table 1.
Primers used for genotyping.
| Primer | Sequence | PCR product (bp) | DLST+/+ | DLST+/- |
|---|---|---|---|---|
| primer 5 | 5’-TAGGTTCCTAGGTAGGGATACAGC-3’ | 186 | + | + |
| primer 3 | 5’-CTACTCTCTAACCTACCAAGCTGG-3’ | |||
|
| ||||
| primer 5 | 5’-TAGGTTCCTAGGTAGGGATACAGC-3’ | 210 | - | + |
| LTR-rev | 5’-ATAAACCCTCTTGCAGTTGCATC-3’ | |||
Note: Two sets of primers were used for PCR to genotype the DLST+/+ and DLST+/- mice. Genomic DNA from each pup will be subjected to two PCR. PCR using primer 5/primer 3 will only amplify the wild-type gene and generate product of 186 bp. PCR with primer 5/LTR-rev will only amplify the mutated gene and produce a fragment of 210 bp. Thus, DLST+/+ mice will only give rise to one PCR product of 186 bp. While, DLST+/- mice will have both of 186 and 210 bp fragments from PCR.
Collection of brain samples for biochemical measurements
Wildtype (DLST+/+) and heterozygous (DLST+/-) mice at E15.5, P10, P30, P42, P105 and P196 (n = 3- 9) were used for biochemical analyses. For embryonic tissue, pregnant female mice were sacrificed by CO2 and the embryos were removed. Brains from embryos and adult mice were quickly removed from the skull and pulverized in liquid nitrogen into fine powders. The powders are divided and transferred into 1.5 ml Eppendorf tubes and stored at -80°C.
Quantitative real-time PCR, SDS-PAGE and Western blotting
Total RNA was extracted from brain samples using an RNeasy Mini kit (Qiagen, Valencia, CA). This was followed by first strand cDNA synthesis using the First Strand cDNA synthesis kit for RT-PCR (AMV) (Roche Applied Science, Indianapolis, IN) with oligo-p(dT)15 as primer. Real-time PCR of OGDH, DLST and DLD was performed using an Applied Biosystems 7500 Real-Time PCR system with pre-designed Taqman® gene expression assays (Applied Biosystems, Foster City, CA) as described previously (Shi et al. 2007). In brief, each amplification mixture (50 μl) contained 22.5 μl of cDNA template, 25 μl of TaqMan® Universal PCR Master Mix, 2.5 μl of a FAM™ dye labeled TaqMan® MGB probe and two PCR primers. Thermal cycler conditions were 95°C for 10 min, and 40 cycles of 95°C for 15 sec and 60°C for 1 min. All samples were normalized for beta-2-microglobulin (b2m) expression in parallel in the same run. A comparative Ct (the threshold cycle of PCR at which amplified product was first detected) method was used to compare the mRNA expression in samples from DLST+/- to that of the DLST+/+ mice.
For protein analysis, brain samples were homogenized in ice-cold cell lysate buffer containing Tris-HCl (50 mM, pH 7.2), dithiothreitol (1 mM), EGTA (0.2 mM), Triton X-100 (0.4%), and the protease inhibitor leupeptin (50 μM) (Shi et al. 2007). About 10 μg of total protein was mixed with SDS-loading buffer and denatured further at 100°C for 5 min. SDS-PAGE and Western blotting were performed as described previously (Shi et al. 2007).
Mitochondria isolation
Non-synaptic mouse brain mitochondria were isolated by a modified isopycnic centrifugation procedure employing Percoll density gradient (Sims 1990). Briefly, cerebral cortex was homogenized in the MSEGTA buffer comprising 225 mM mannitol, 75 mM sucrose, 20 mM HEPES (pH 7.4), 1 mM EGTA, 0.2% (w/v) fatty acid free bovine serum albumin (BSA) and centrifuged at 2,000 g × 4 min. The supernatant was collected and centrifuged at 12,000 g × 10 min. The pellet was resuspended in MSEGTA and layered over 25% (v/v) Percoll prepared in MSEGTA mixture and centrifuged at 30,000 g × 10 min. Purified mitochondria fraction was collected at the bottom of the tube, resuspended in MSEGTA without added BSA and washed 2 times by centrifuging at 12,000g × 10 min. Final mitochondrial pellet was resuspended in MS buffer comprising 225 mM mannitol, 75 mM sucrose, 20 mM HEPES (pH 7.4) and stored on ice. Protein content was estimated by a commercial BCA assay (“Pierce Biotechnology” “Thermo Scientific”, USA). Mitochondrial enzymes activities were measured by conventional procedures (Leong et al. 1984; Klivenyi et al. 2004; Starkov et al. 2004).
KGDHC activity measurement
KGDHC catalyzes the following reaction: α-ketoglutarate + NAD+ + CoA → succinyl CoA + CO2 + NADH. In brief, brain samples were homogenized with 250 μl of KGDHC lysis buffer containing Tris-HCl (50 mM, pH 7.2), dithiothreitol (1 mM), EGTA (0.2 mM), Triton X-100 (0.4%), and the protease inhibitor leupeptin (50 μM) and KGDHC activities were assayed immediately as described previously (Gibson et al. 1988). In brief, KGDHC standard (Sigma) or brain samples (25 μl) were added into wells (3 wells/sample) of a 96-well plate. Reaction mix (100 μl containing 50 mM Tris-HCl (pH 7.8), 1 mM MgCl2, 1 mM CaCl2, 0.5 mM EDTA, 0.3 mM TPP, 0.1% Triton X-100 and 1 mM DTT) was added into each well followed by 50 μl of assay mix (3 mM NAD and 0.75 mM CoA). Immediately after addition of 25 μl of α-ketoglutarate (3 mM), the plate was read at 340 nm using a SPECTRA MAX 250 microplate reader (Molecular Devices) for 30 min. Values were corrected for the presence of NADH oxidase.
In situ KGDHC activity staining
Mice were administrated a lethal dose of sodium pentobarbital (2 mg/10 g body weight; Abbott Laboratories, North Chicago, IL) intraperitoneally, and were perfused via the ascending aorta with approximately 50 ml of saline to wash away the blood. The brains were removed immediately and stored at -80°C. The brain was dissected on a Rodent Brain Matrix (ASI Instruments, Warren, MI), and 20 μm sagittal sections were cut using a cryostat (Hacker-Bright OTF microtome cryostat, Fairfield, NJ) and used for in situ KGDHC activity stain according to previously described methods (Gibson et al. 2000a; Park et al. 2000; Shi et al. 2007). In brief, sections were incubated with reaction mixture (50 mM Tris-HCl, 1 mM MgCl2, 0.1 mM CaCl2, 50 μM EDTA, 0.2%Triton X-100, 0.3 mM thiamine pyrophosphate (TPP), 5 μg/ml rotenone, 35 mg/ml polyvinyalcohol, 3 mM β-NAD, 0.75 mM Co-A, 3 mM α-ketoglutarate (α-KG), 0.75 mM nitroblue tetrazolium (NBT) and 0.05 mM phenazine methosulfate (PMS)) for 20 or 30 min. A reaction mixture without Co-A and α-KG was used for blank. NBT and PMS were added immediately before the reaction was initiated. The reaction was stopped by removing the sections from the reaction mixture and rinsing the sections twice with distilled water for 10 seconds each time. The sections were air-dried overnight before quantification. Images were acquired (4x magnification) with Nikon eclipse 80i microscope using ACT-1 software. The MetaMorph program (Molecular Devices, Downingtown, PA) was used to draw the region of interest and to quantify staining density of each region. The density of each region after normalization to the blank was compared between DLST+/- mice and control groups. The percentage change of the regional density in the DLST+/- mice compared to the DLST+/+ mice represented the change of the in situ KGDHC activity.
MPTP lesion
MPTP, prepared at 2 mg/ml in 0.1M sodium phosphate buffered saline (PBS, pH 7.4), was administered intraperitoneally to seven month-old male wildtype (n=12) and DLST+/- mice (n=10) at a dose of 20 mg/kg three times at 2-hr intervals. PBS was administered to a separate group of wildtype (n=4) and DLST+/- mice (n=4). Mice were sacrificed 7 days after MPTP treatment. The striata were dissected from the brain for biochemical analysis while the midbrains were fixed by immersion in 4% paraformaldehyde for 48 hours for histological studies.
High-Performance Liquid Chromatography (HPLC) assay for catecholamines
Striatal tissues were sonicated and centrifuged in cold 0.1 M perchloric acid (100 μl/mg tissue). The supernatant was used for measurements of dopamine, 3,4-dihydroxyphenylacetic acid (DOPAC) and homovanillic acid (HVA) by HPLC. Briefly, 10 μl supernatant was isocratically eluted through an 80 × 4.6 mm C18 column (ESA Inc., Chelmsford, MA) with a mobile phase containing Li3PO4, 0.85 mM 1-octanesulfonic acid and 10% (v/v) methanol, and detected by a 2-channel Coulochem II electrochemical detector (ESA, Inc.). Protein concentrations of tissue homogenates were determined using the Bio-Rad protein assay kit (Bio-Rad Laboratories, Hercules, CA) and the Perkin Elmer Bio Assay Reader (Norwalk, CT). Dopamine, DOPAC and HVA concentrations are expressed as nanograms per milligram protein.
Malonate lesion
The selective neuropathology in human Huntington’s disease brain can be mimicked in laboratory animals by direct intrastriatal infusion of the mitochondrial complex II inhibitor, malonate. Thus, malonate lesioning in rodents has been widely used as a model of Huntington’s disease (Beal et al. 1993a). Four month-old wildtype (n=11) and DLST+/- (n=10) mice were anesthetized with isoflurane inhalation (1-3%). Mice received a unilateral stereotaxically-guided injection of malonate (1.5 μmol in 1.0 μl saline) into the left striatum using coordinates (anterior to bregma +0.5 mm; lateral +1.5 mm; ventral from dura –4 mm) ascertained from the mouse brain atlas of Paxinos and Franklin (2003). The injection needle was left in place for 2-5 min to prevent backflow of infusate up the needle tract. Mice were allowed to recover, and on day 7 post-surgery, mice were sacrificed under deep anesthesia with sodium pentobarbital (120 mg/kg i.p.) by transcardial perfusion with 0.9% saline, followed by 4% paraformaldehyde. The brains were removed and post-fixed in the same fixative overnight.
3-nitropropionic (3-NP) acid lesion
3-NP, a plant toxin that inactivates mitochondrial succinate dehydrogenase, causes striatal neuropathy similar to that seen in clinical HD. We treated eight month-old male DLST+/- (n=8) and wildtype controls (n=8) with 3-NP delivered by subcutaneously implanted miniature osmotic pumps. Mice were anesthetized by isoflurane inhalation and were prepared for surgery. A small incision was made through the skin at the nape of the neck, and the Alzet pump was inserted into the subcutaneous space. The dose of 3NP was 60 mg/kg/day. The incision site was then sutured and animals were allowed to recover from the anesthesia. Five days after surgery, mice were sacrificed and the brains were harvested. Half of the brain was frozen in isopentane for biochemical studies and the other half was fixed by immersion in 4% paraformaldehyde for 48 hours for histological analysis.
Histological analysis
After fixation, the tissues were cryoprotected in 30% sucrose and sectioned coronally (50 μm) using a cryostat. Immunohistochemistry was performed using a modified avidin-biotin-peroxidase technique. Free-floating sections were pretreated with 3% H2O2 in 0.1 M PBS for 30 min. The sections were incubated sequentially in (a) 1% bovine serum albumin (BSA) and 0.2% Triton X-100 in PBS for 30 min, (b) primary antibody diluted in PBS/0.5% BSA, (c) appropriate secondary antibody, either biotinylated anti-rabbit or anti mouse IgG (Vector Laboratories, Burlingame, CA) diluted at 1:200 in PBS/0.5% BSA for 18 hrs, and (d) avidin-biotin-peroxidase complex (Vector) with both reagents A and B diluted at 1:200 in PBS for 1 hr. The immunoreaction product was visualized using 3,3’-diaminobenzidine tetrahydrochloride dihydrate (DAB, Vector). The primary antibodies used were: affinity-purified rabbit antibody against tyrosine hydroxylase (TH) from Chemicon (Temecula, CA), polyclonal rabbit anti-malondialdehyde (provided by Dr. Craig E. Thomas from Hoechst-Marion-Roussel; 1:1,000), mouse antibody against neuron-specific nuclear protein (NeuN) (Chemicon), rabbit anti-nitrotyrosine (Upstate, Charlottesville, VA) 1:100, and goat anti-8-hydroxydeoxyguanosine (Millipore, Temecula, CA) 1:100.
Double label immunofluorescence was performed to demonstrate whether increased malondialdehyde staining occurs in dopaminergic neurons. Sections were incubated for 18 hrs in the primary antibody mixture containing anti-tyrosine hydroxylase (1:100) and anti-malondialdehyde (1:100). After rinsing with PBS, sections were incubated for 1 hr in a mixture of Cy2-conjugated anti-mouse IgG (1:200) and Cy3-conjugated anti-rabbit IgG (1:200; Jackson Immuno Research, West Grove, PA). Sections were examined using a confocal microscope.
Quantification
A stereological technique (optical fractionator) based upon unbiased principles of systematic uniformly random sampling was used to estimate the number of TH-immunoreactive, Nissl-positive cells, as well as malondialdehyde-immunoreactive neurons in substantia nigra pars compacta (SNpc). The system consisted of a Nikon Eclipse E600 photomicroscope equipped with LEP motorized X-Y mechanical stage and StereoInvestigator software (v3.45; Microbrightfield, Burlington, VT). Three series of 50-μm thick cryosections were cut. Each series contained ~7 sections containing the SNpc. The final mounted section thickness averaged ~20 μm. The size of the x-y sampling grid was 140 μm × 140 μm. The counting frame thickness was 14 μm with 3-μm guard zones. The stereological cell counts represent the estimated total number of cells in the SNpc.
For estimating 3-NP and malonate lesion volume, 50 μm serial cryosections (250 μm apart) per animal were analyzed. The cavalieri method (StereoInvestigator software, v3.45; Microbrightfield, Burlington, VT) was used. The sections were analyzed under 4x objective with 100 μm grid spacing.
A qualitative examination was performed to analyze immunoreactivities of 8-hydroxy-2-deoxyguanosine (8OH2dG) and nitrated tyrosine. The analysis was done using 7 sections per animal (n=5 per group).
Statistics
Data are expressed as mean ± standard error of the mean (SEM). Statistical analysis of the data was performed using student t test or one way analysis of variance followed by the Student-Newman-Keuls post test or unpaired Student’s t test, when appropriate, using the GraphPad Instat software (San Diego, CA). A p value of < 0.05 was considered statistically significant.
RESULTS
Levels of mRNA and protein of KGDHC subunits in DLST+/- and DLST+/+ mice
Brain samples from the wildtype and DLST+/- littermates were assayed for the levels of mRNA and protein of the three subunits of KGDHC by real-time PCR. In DLST+/- mice, the DLST mRNA and protein contents were 53% and 54%, respectively, compared to that of DLST+/+ mice (Fig 1). In contrast, the mRNA and protein levels of the other 2 subunits of KGDHC in DLST+/- mice did not differ from the DLST+/+ mice (Fig 1).
Figure 1.
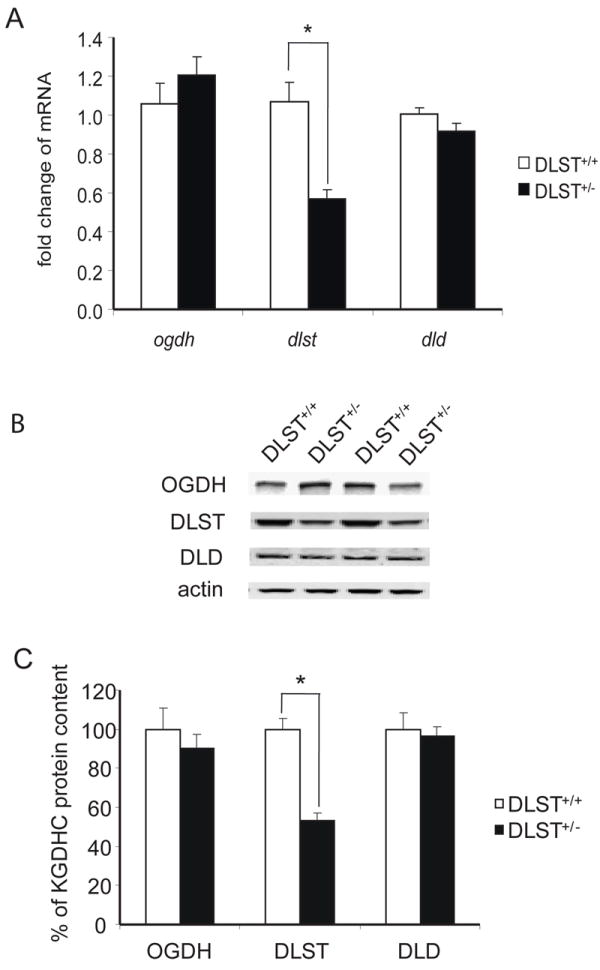
Gene expression and protein levels of KGDHC subunits in DLST+/- mice. A. Brain homogenates were subjected to quantitative real-time PCR to assess changes in the mRNA level of ogdh , dlst and dld subunits of KGDHC. mRNA for β-2-microglobulin (β2m) was measured in the same sample as an internal control. Values represent means ± SEM of fold changes over DLST+/+ mice from at least two independent experiments done in triplicate after normalization to β2m. * P < 0.05. B. Total protein was isolated from brains of mice and subjected to SDS-PAGE followed by Western blotting probed with antibodies against OGDH, DLST or DLD. β-actin immunoreactivity was used as an internal control. Representative blots are shown for three subunits and β-actin. C. Quantitation of protein levels of OGDH, DLST and DLD. Values represent means ± SEM of relative densities of the subunit from three independent experiments after normalization to β-actin. * P < 0.05.
KGDHC activity in brains of DLST+/- mice
KGDHC activity reaches adult level by postnatal day 30 (Buerstatte et al. 2000). Thus, 6-week-old age was chosen for the characterization of DLST+/- mice. The consequences of a reduction in the DLST protein on the overall KGDHC activity in brains of DLST+/+ and DLST+/- mice (6 weeks old) were examined by two methods. KGDHC activity in whole brain homogenates of DLST+/- mice was reduced by ~40% compared to that of DLST+/+ mice (Fig. 2A). KGDHC activities in different regions of brains from DLST+/+ and DLST+/- mice were assessed by an in situ histochemistry activity stain method. Significant reduction in the KGDHC activity of DLST+/- compared to the DLST+/+ mice was observed in olfactory blub (73%), hippocampus (70%), neocortex (79%), striatum (74%), thalamus (72%), hypothalamus (78%), midbrain (73%), cerebellum (86%) and hindbrain (71%) (Fig. 2B).
Figure 2.
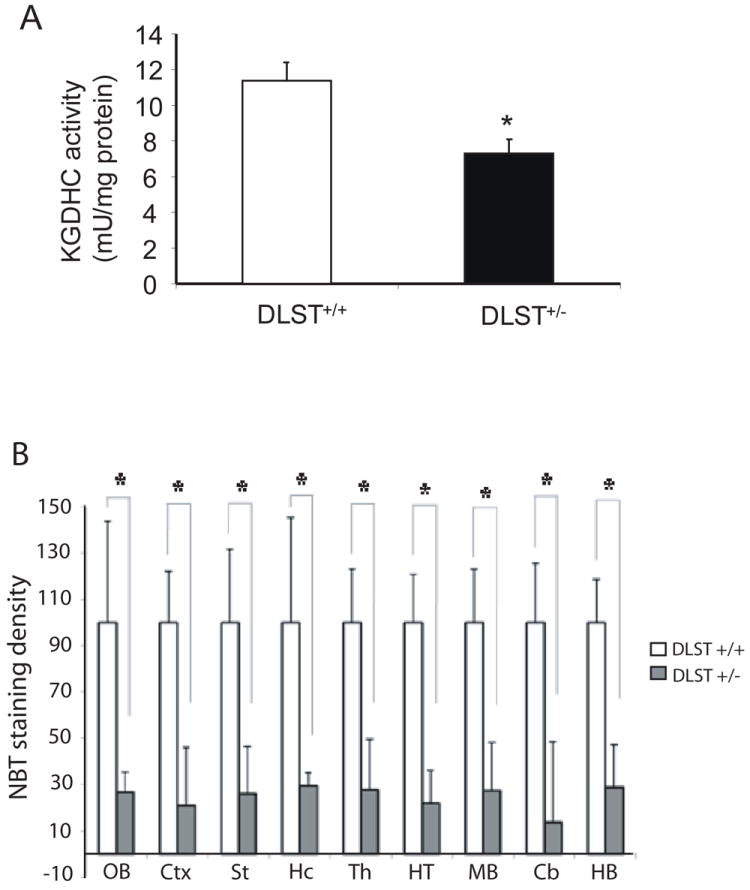
KGDHC activity was reduced in brains of DLST+/- mice. A. Enzymatic activity was measured in whole brain homogenates (6 week old mice). DLST+/- brains showed significantly lower activity. B. Nitroblue tetrazolium (NBT) staining density, measured in brain sections by in situ activity staining, was lower in each region of DLST+/- brains. Abbreviations; Cb, cerebellum; Ctx, neocortex; HB, hindbrain; Hc, hippocampus; HT, hypothalamus; MB, midbrain; OB, olfactory bulb; St, striatum; Th, thalamus. Data for each group were done in at least three brains, in triplicate. * P < 0.05. All values represent means ± SEM.
KGDHC activity during development
KGDHC activity was measured in brain homogenates obtained from mice at ages that ranged from embryonic day 15.5 (E15.5) to postnatal day 10 (P10), P30, P42, P105 and P196. KGDHC activity was detectable at E15.5, reached 12.9 mU/mg at P30 and did not increase further (Fig. 3). Further, KGDHC specific activity at each time point was diminished in the DLST+/- mice.
Figure 3.
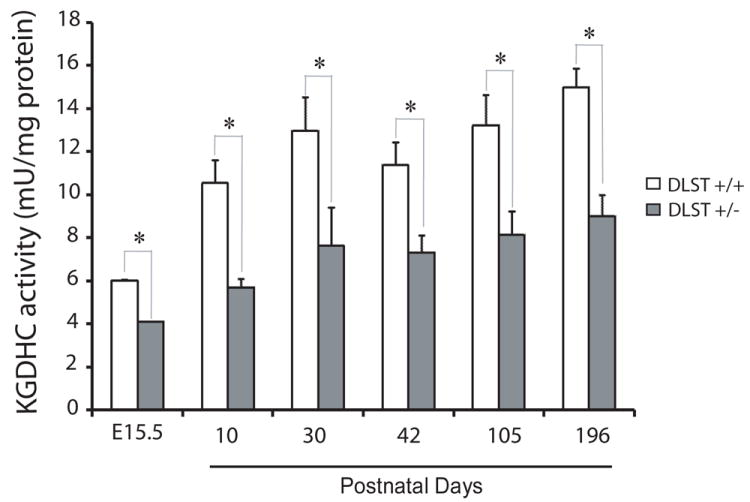
KGDHC activity during development. Specific enzymatic activity was measured in triplicate. Values represent means ± SEM. * P < 0.05. n = 3-9. No significant increase in the activity after P30.
Mitochondrial enzymes activities
We compared the activities of several key tricarboxylic acid cycle (TCA) enzymes (KGDHC, PDHC, MDH), respiratory chain complexes (Complex I and IV) and antioxidant enzymes (GR, GPx1, ME, MnSOD) in brain mitochondria isolated from adult DLST+/+ and DLST+/- mice (see the abbreviation section for enzymes’ full names). Fig. 4A shows that except KGDHC, the activity of which in DLST+/- mice mitochondria was reduced by about 40% compared to that in DLST+/+ mice mitochondria, other activities of evaluated mitochondrial enzymes were similar in DLST+/- and DLST+/+ mitochondria. There is also no difference in protein levels of mitochondria antioxidant enzymes (GR, GPx1, MnSOD) (Fig. 4B). Therefore, it may be concluded that DLST+/- brain mitochondria exhibit no overt bioenergetics abnormality except the loss of DLST.
Figure 4.
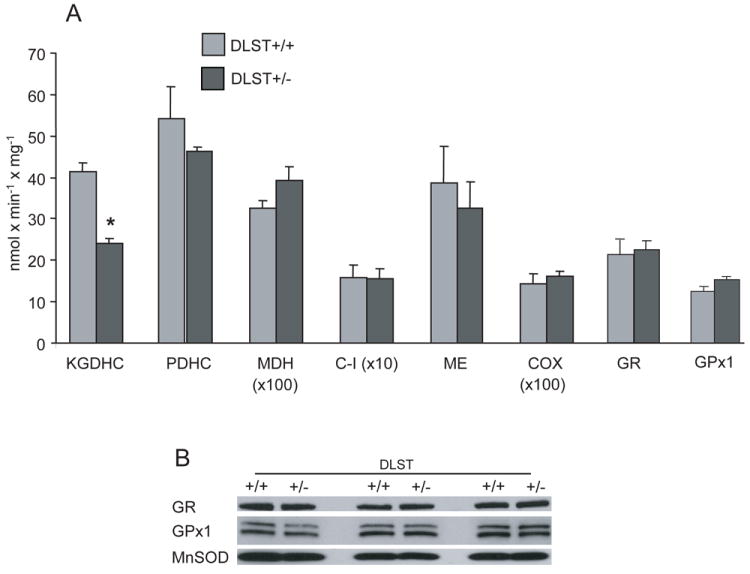
Enzyme activities in mitochondria of DLST deficient mice. A. The activity of mitochondria KGDHC isolated from DLST+/- brains was half of that from DLST+/+ brains. Other evaluated mitochondrial enzymes activities were similar in DLST+/- and DLST+/+ mitochondria. Values are means ± SEM. * p < 0.05. n = 12. B. Immunoblotting of MnSOD, GR, and GPx1 proteins in isolated mitochondria shows no difference between DLST+/- and DLST+/+. C-I, respiratory chain Complex I; COX, cytochrome oxidase; MDH, malate dehydrogenase; PDHC, pyruvate dehydrogenase complex; KGDHC, α-ketoglutarate dehydrogenase complex; ME, malic enzyme; GR, glutathione reductase; GPx1, mitochondrial glutathione peroxidase 1; MnSOD, manganese superoxide dismutase.
Body weight, sex and genotype ratios
No significant difference in body weight was observed between the DLST+/+ (20.61 ± 0.69 g) and DLST+/- (20.86 ± 0.96 g) mice at the age of 6 weeks. The sex ratio for DLST+/+ pups was 1: 0.91 (male: female) whereas the ratio for DLST+/- pups was 1:1.16. The genotype ratio of DLST+/+ to DLST+/- pups from breeding of DLST+/- mice was nearly 1:2, as expected of Mendelian inheritance.
MPTP Studies
Administration of MPTP produced a significantly greater reduction of striatal dopamine as well as tyrosine hydroxylase-positive neurons in the SNpc of DLST+/- mice than in wildtype controls. MPTP induced dopamine depletion in the striatum by 59% in the wildtype control mice and 73% in the DLST+/- mice (Fig. 5A). Stereological analysis of TH-immunostained nigral sections revealed that MPTP reduced the number of dopaminergic neurons in the SNpc by 25% in wildtype control mice and 42% in the DLST+/- mice (Fig. 5B and C).
Figure 5.
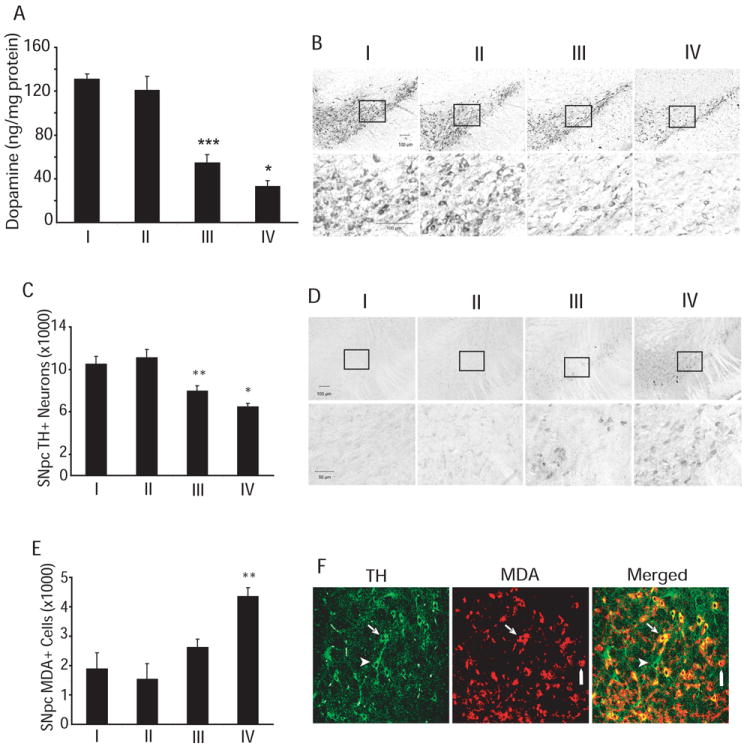
DLST deficiency enhances MPTP toxicity. A. DLST deficiency enhances MPTP-induced dopamine depletion in the striatum. *** p < 0.001, versus wildtype/PBS; * p < 0.05, versus wildtype/MPTP. B. Tyrosine hydroxylase (TH) immunoreactivity in the substantia nigra pars compacta (SNpc). Boxed areas on top are magnified in lower panel of photomicrographs. C. Stereological analysis of TH-immunoreactive (TH+) neurons in SNpc showing exacerbation of MPTP-induced TH+ neuron loss in the SNpc of DLST+/- mice. Values are means ± SEM. * p < 0.05, versus wildtype/MPTP; ** p < 0.01 versus wildtype/PBS. D. Malondialdehyde (MDA) immunoreactivity in the SNpc. Boxed areas on top are magnified in lower panel of photomicrographs. E. Stereological analysis of MDA-immunoreactive neurons in SNpc showing increased number of intensely stained neurons in MPTP-treated DLST-deficient mice compared to MPTP-treated wildtype control. Values are means ± SEM. ** p < 0.01 versus wildtype/MPTP. n = 10-12. I, Wildtype/PBS; II, DLST+/-/PBS; III, Wildtype/MPTP; IV, DLST+/-/MPTP. F. Confocal images of combined immunofluorescence of TH (green) and MDA (red) antisera showing co-existence (yellow) in neurons in the SNpc of a DLST+/- mouse treated with MPTP. Arrows show a TH-positive neuron with increased MDA immunoreactivity. Point bars show MDA immunostaining in a TH-negative cell. Arrowheads show a TH-immunoreactive neuron without MDA-immunoreactivity.
MPTP treatment increases malondialdehyde, a product of lipid peroxidation, in mouse brain (Sriram et al. 1997). Using an antibody against malondialdehyde-modified protein, levels of lipid peroxidation were examined in the SNpc. MPTP induced increases in the number of neurons intensely stained with malondialdehyde in wildtype control mice (Fig. 5D). DLST deficiency enhanced the severity of MPTP-induced lipid peroxidation in the SNpc as evidenced by a significantly higher number of malondialdehyde-labeled neurons in the MPTP-treated DLST+/- mice compared to MPTP-treated wildtype controls (p<0.01; Fig. 5E). Double label immunofluorescence sections analyzed with confocal image show that increased malondialdehyde staining occurs in TH positive and TH negative neurons in SNpc (Fig. 5F).
Malonate lesions
We also carried out studies to test the impact of DLST deficiency on vulnerability of mice to malonate neurotoxicity. Areas of striatal neuron loss were measured in tissue sections that were stained with an antibody against the neuronal marker NeuN (Fig. 6A). Malonate lesion volumes in the DLST+/- mice were twice as large as the wildtype controls (2.6 ± 0.5 versus 1.3 ± 0.4 mm3 respectively; p< 0.05; Fig. 6B). Immunostaining with an antibody against the inflammatory mediator, CD40 ligand (CD40L) revealed intensely stained astrocytes at the site of malonate lesion in wildtype controls (Fig. 6C). In DLST+/- mice, the CD40L-labeled astrocytes were hypertrophied and astrogliosis was more robust than in controls following malonate (Fig. 6C). Furthermore, the striatum in the lesioned hemisphere was significantly smaller in the DLST+/- mice (95.8 ± 0.5% of contralateral hemisphere) than in wildtype controls (98.8 ± 0.3; p<0.001).
Figure 6.
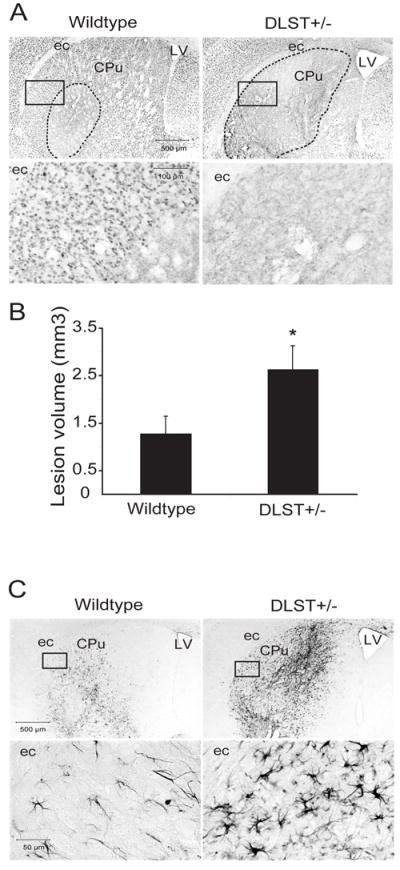
DLST-deficiency enhances malonate toxicity. A. NeuN-stained sections through the caudate putamen lesioned with malonate show an exacerbation of the lesion in DLST+/- mice. Broken lines show demarcation of the areas of NeuN-stained neuron loss. Boxed areas in the photomicrographs are magnified in the lower panel showing the severity of loss of NeuN-labeled neurons. B. Lesion volume analysis using stereological cavalieri method shows an increase of lesion volumes in DLST+/- mice. Values are means ± SEM. * p < 0.05. n = 10-11. C. CD40L immunoreactivity in the caudate putamen lesioned with malonate. There is marked proliferation and hypertrophy of CD40L-labeled astrocytes in DLST+/- mice. Boxed areas in the photomicrographs are magnified in the lower panel. ec= external capsule, LV= lateral ventricle, CPu= caudate putamen.
3-NP lesions
The 3-NP lesioned mice were assessed qualitatively for motor function. After five days of 3-NP intoxication, when suspended by the tail, the DLST+/- mice displayed a sustained flexure of both forelimbs and hindlimbs, while the wildtype mice showed less severe symptoms (Fig. 7A).
Figure 7.
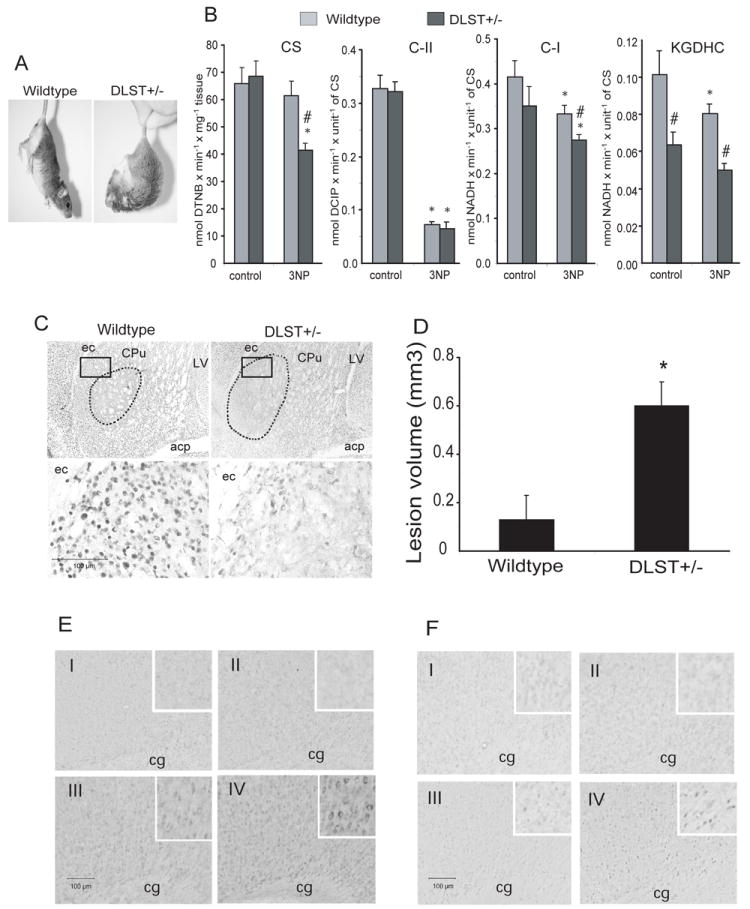
DLST-deficiency enhances 3-NP toxicity. A. Motor function in 3NP lesioned mice when suspended by the tail. After 5 days of 3-NP intoxication, the DLST+/- mouse exhibited sustained flexure of both forelimbs and hindlimbs, while the wildtype mouse showed less severe symptoms. B. Effect of 3-NP treatment on the activity of mitochondrial enzymes in striata of DLST+/- mice and their wild type littermates (see results for interpretation). * p < 0.05 compared with its respective control; # p < 0.05 compared to wildtype/3-NP. n = 3. C. NeuN-stained sections through the caudate putamen lesioned with 3-NP show an exacerbation of lesion in DLST+/- mice. Broken lines show demarcation of the areas of NeuN-stained neuron loss. Boxed areas in the photomicrographs are magnified in the lower panel to show the extent of loss of NeuN-immunoreactive neurons. ec=external capsule, LV=lateral ventricle, CPu=caudate putamen; acp=anterior commissure, posterior part. D. Lesion volumes obtained by stereological cavalieri method show an increase of lesion volumes in DLST+/- mice. Values are means ± SEM, * p < 0.05. n = 8. E. Representative photomicrographs of nitrotyrosine immunoreactivity in the cortex. Insets show the staining in neurons. F. Representative photomicrographs of 8-hydroxy-2-deoxyguanosine immunoreactivity in the cortex. Insets show the staining in neuronal nuclei. n=5 in each group. cg=cingulum. I, Wildtype/PBS; II, DLST+/-/PBS; III, Wildtype/3-NP; IV, DLST+/-/3-NP. C-I, respiratory chain Complex I; C-II, Complex II; CS, citrate synthase; KGDHC, α-ketoglutarate dehydrogenase complex.
For assessment of the extent of 3-NP induced damage to striatal mitochondria, a separate cohort of mice were treated with 3-NP for 3 days to avoid developing massive striatal lesions. We measured the activity of selected key mitochondrial enzymes in striata (Fig. 7B). After 3-NP treatment, the activity of citrate synthase (CS), a classical biochemical marker of tissue mitochondria content, was ~35% lower in the striatum of DLST+/- mice as compared to wildtype mice, in which CS has not been shown to be directly inhibited by 3NP. The activity of complex II (succinate dehydrogenase, SDH), which is a biochemical marker of 3-NP toxicity, was inhibited by ~85% in both wildtype and DLST+/- mice. Note that Complex I and II activities as well as the activity of KGDHC are normalized by the activity of CS (mitochondria content). Surprisingly, we also found ~20% decrease in activities of Complex I and the KGDHC in both wildtype and DLST+/- mice treated with 3-NP. These may not be directly inhibited by 3-NP since in our in vitro control experiments 3-NP (up to 2 mM concentration) did not inhibit the activity of these enzymes (data not presented). Severe inhibition of complex II (SDH) diminishes the activity of Complex I and KGDHC, and to a greater extent in DLST+/- mice than in their wildtype littermates.
The 3-NP-induced striatal damage was also examined in NeuN-stained sections from DLST+/- and control mice after five days of 3-NP intoxication (Fig. 7C). Well-demarcated areas of NeuN-labeled neuronal loss occurred in the striatum of both groups of mice. The 3-NP lesion volumes were almost five-fold larger in the DLST+/- mice than controls (0.6 ± 0.1 vs. 0.13 ± 0.1 mm3 respectively; p< 0.05; Fig. 7D).
DLST deficient mice themselves did not show obvious changes in immunohistochemical staining for nitrotyrosine, a marker for protein oxidation (Fig. 7E), and 8-hydroxy-2-deoxyguanosine (8OH2dG), a marker for DNA oxidation (Fig. 7F), compared to the wildtype controls. However, DLST deficiency enhanced the severity of protein and DNA oxidations in the cortex of 3-NP treated DLST+/- mice as compared to 3-NP treated wildtype controls (Fig. 7E and 7F).
DISCUSSION
There is substantial evidence implicating KGDHC deficiency in the pathogenesis of several neurodegenerative diseases including AD (Gibson et al. 1988), HD (Klivenyi et al. 2004), PD (Mizuno et al. 1990; Mizuno et al. 1994; Mizuno et al. 1995), and spinocerebellar ataxia (Mastrogiacomo and Kish 1994). We previously demonstrated that mice with a deficiency of murine dihydrolipoamide dehydrogenase subunit (DLD+/-) have a 50% decrease in KGDHC activity (Klivenyi et al. 2004). DLD however participates in other enzyme complexes like pyruvate dehydrogenase complex (PDHC) and branched chain α-ketoacid dehydrogenase complex, and we found that DLD+/- mice have a significant reduction in PDHC activity in cerebral cortex (Klivenyi et al. 2004).
Encoded by the dlst gene, DLST is a component unique to KGDHC. DLST contains covalently bound lipoyl moieties and catalyzes the important intermediate step in the KGDHC reaction sequence of α-ketoacid oxidation, transferring the succinyl moiety to the CoA substrate to form succinyl-CoA (Reed and Hackert 1990). Several studies reported that there is an association between polymorphisms (SNPs) in DLST and AD or PD (Nakano et al. 1997; Kobayashi et al. 1998; Sheu et al. 1999b; Sheu et al. 1999a). However, other reports from later studies concluded that there is no association (Kunugi et al. 1998; Matsushita et al. 2001; Prince et al. 2001; Brown et al. 2004). These controversial results leave an uncertainty as to the possible role of polymorphisms in DLST in relation to these neurodegenerative diseases.
The present study tested whether a reduction in the DLST subunit of KGDHC alter the overall KGDHC activity and susceptibility to mitochondrial toxins. The DLST+/- mice were characterized at levels of mRNA, protein and activity of KGDHC, as well as phenotypes including body weight, ratio of sex and ratio of genotype. Characterization of the DLST+/- mice at the levels of mRNA and protein showed that the DLST mRNA and protein were both reduced by about 50% in DLST+/- mice as compared to DLST+/+ mice, while the mRNA and protein levels of the other two subunits (OGDH and DLD) were not altered. In comparisons of the spectrum of key mitochondrial enzymes in isolated mitochondria from the brain tissue we found that except for the reduction in KGDHC activity in DLST+/- mice, the other evaluated mitochondrial enzyme activities were similar in DLST+/- and DLST+/+ mitochondria. These findings demonstrate a specific disruption of the DLST mRNA, and indicate that no compensation occurs in the other two subunits (OGDH and DLD) in KGDHC as well as in other key mitochondria enzymes when the DLST subunit is reduced by 50%.
Furthermore, the DLST+/+ and DLST+/- mice showed no significant differences in body weight. The sex ratio for either DLST+/+ or DLST+/- mice is about 1 to 1. The ratio of DLST+/+ to DLST+/- pups from breeding of DLST+/- mice is close to 1:2, suggesting that DLST+/- mice have a similar survival rate as the DLST+/+. No DLST-/- mice were born, which agrees with the previous study on DLD mice in which DLD-/- mice die prenatally with apparent development delay (Johnson et al. 1997). Thus, a complete loss of DLST is fatal to embryo development, and about half of KGDHC activity is enough for normal development, as shown by the developmental study of KGDHC activity. The KGDHC activity is low in prenatal E15.5 and increases in P10 and reaches adult levels in P30. This agrees with a previous study showing that KGDHC activity rises steeply from between P10 and attains adult levels by P30 (Buerstatte et al. 2000). A neurohistological survey of cresyl violet-stained brain sections of DLST+/- mice did not reveal any apparent structural abnormalities (Calingasan et al. 2008). The size and morphology of different brain regions, ventricles and the neurons appeared normal. Using an antibody against glial fibrillary acidic protein (GFAP) as a marker of astrocytes, no astrogliosis was observed. Immunostaining with an antibody against the lipid peroxidation marker, malondialdehyde, did not show any alteration in the DLST+/- mice. Furthermore, silver staining did not show any neurodegenerative changes in the DLST+/- brain sections. The DLST+/- mice therefore appear to have normal development and brain morphology.
Previous studies show that MPTP and isoquinoline derivative neurotoxicity are associated with reduced activity of KGDHC (McNaught et al. 1995; Joffe et al. 1998). The MPTP metabolite MPP+ inhibits KGDHC, these effects may be mediated by oxidative stress. KGDHC is sensitive to hyperoxia and a number of oxidants. Exposure of Chinese hampster ovary cells to hyperoxia results in cell death and complete inactivation of KGDHC, whereas KGDHC activity is preserved in cells that are resistant to hyperoxia (Schoonen et al. 1990, 1991). KGDHC is also inhibited by an oxidized derivative of dopamine (Shen et al. 2000) and other oxidants including hydroxynonenal, H2O2 and peroxynitrite (Chinopoulos et al. 1999; Park et al. 1999; Gibson and Zhang 2002; Kumar et al. 2003). The inactivation of KGDHC by peroxynitrite is particularly interesting since peroxynitrite mediated neurotoxicity is strongly implicated in MPTP neurotoxicity (Schulz et al. 1995a; Hantraye et al. 1996; Przedborski et al. 1996). Peroxynitrite is also implicated in PD since increases in 3-nitrotyrosine staining of Lewy bodies and increased staining with antibodies to nitrated α-synuclein are seen in Lewy bodies in the substantia nigra of patients with PD (Good et al. 1998; Giasson et al. 2000).
We previously investigated mice with a deficiency of DLD (Klivenyi et al. 2004). We examined heterozygous DLD+/- mice since the homozygous DLD-/- mice die in utero at a very early gastrulation stage (Johnson et al. 1997). Similarly, the DLST-/- homozygous mice are never born. We demonstrated that mice which were deficient in DLD showed increased vulnerability to MPTP, malonate and 3-nitropropionic acid (3-NP), which have been proposed as models for PD and HD. Administration of MPTP resulted in a significantly greater depletion of tyrosine hydroxylase positive neurons in the substantia nigra of DLD+/- mice than that seen in wildtype littermate controls. Striatal lesion volumes produced by malonate and 3-NP were significantly increased in DLD+/- mice. Studies of isolated brain mitochondria treated with 3-NP showed that both succinate supported respiration and membrane potential were suppressed to a greater extent in the DLD+/- mice.
In the current experiments, we tested whether a deficiency of DLST, a subunit that is unique to KGDHC, also alters the vulnerability of mice to MPTP neurotoxicity. If a deficiency in KGDHC contributes to PD pathogenesis, one might expect that these mice would show increased vulnerability to MPTP, which is known to impair mitochondrial function. We found that following MPTP administration the DLST+/- mice showed a greater loss of TH stained neurons in the SNpc as compared to wildtype mice. There was also a significantly greater reduction in dopamine levels in the striatum. The number of positive immunoreactive neurons of malondialdehyde, a biomarker for lipid peroxidation, in the SNpc was significantly increased following MPTP administration in the DLST+/- mice as compared to wildtype mice. This shows that the DLST+/- mice are vulnerable to oxidative damage produced by mitochondrial toxins.
We examined the effects of malonate lesions in the DLST+/- mice. Malonate is a reversible inhibitor of mitochondria complex II (SDH). We and others have previously demonstrated that intrastriatal injections of malonate produced lesions that in many respects resemble the neuropathology of HD (Beal et al. 1993a; Greene et al. 1993). The lesions are blocked by excitatory amino acid antagonists, and they are accompanied by increases in markers of free radical damage and are attenuated by free radical scavengers (Beal et al. 1993a; Greene et al. 1993; Schulz et al. 1995b). In the present experiments, we found that striatal lesions produced by malonate were markedly exacerbated in the DLST+/- mice. Lesion volumes were approximately two-fold larger in the DLST+/- mice. Furthermore, the proliferation of CD40L-labeled astrocytes was much greater in the DLST+/- mice as compared to wild-type controls. This is consistent with the possibility that inhibition of the TCA cycle at both the SDH as well as the KGDHC site may greatly exacerbate bioenergetic defects, and enhance the production of free radicals.
We also examined the effects of 3-NP in DLST+/- mice. 3-NP is a suicide inhibitor of the mitochondrial TCA cycle enzyme, SDH (Alston et al. 1977; Coles et al. 1979). It is also a reversible inhibitor in vitro of fumerase, another TCA cycle enzyme. Systemic administration of 3-NP inhibits SDH in the whole brain, as well as in the liver and heart. It results in abnormal succinate build up and inhibition of the mitochondrial TCA cycle. Systemic administration of 3-NP produces selective lesions in the striatum which closely mimic the neuropathologic features of HD, since it produces relative sparing of NADPH diaphorase neurons, a characteristic feature of HD neuropathology (Beal et al. 1993b). In baboons, it also produces a choreiform of movement disorder as well as dystonia (Brouillet et al. 1995). Baboons show frontal type learning deficits, which are also characteristic of HD deficits (Palfi et al. 1996). 3-NP induced lesions are attenuated in mice overexpressing copper zinc superoxide dismutase (Beal et al. 1995). In current study mice deficient with DLST were markedly more vulnerable to the neurotoxic effects of 3-NP. They showed marked clasping of the fore- and hind-limbs, which was absent in wildtype mice. 3-NP inhibited SDH activity by ~85% and also caused a ~20% decreases in the activities of Complex I and KGDHC, which was greater in DLST+/- mice. 3-NP treatment significantly reduced citrate synthase, consistent with a reduction in mitochondria content, in DLST+/- mice. The lesions in these mice were more than four-fold larger than those seen in controls. 3-NP induced protein and DNA oxidations in the cerebral cortex of DLST+/- mice were much more severe than those in wildtype controls. These results show that a selective deficiency in KGDHC increases the vulnerability of mice to neurotoxic effects of 3-NP.
In summary, we found that DLST+/- mice show increased vulnerability to MPTP, malonate and 3-NP. These deficits appear to be due to a specific decrease in KGDHC, since the DLST subunit is confined to this enzyme complex. These findings, therefore, provide further evidence that reductions in KGDHC may impair the ability of the brain to adapt and contribute to the pathogenesis of neurodegenerative diseases.
Acknowledgments
This work was supported by a grant from the National Institutes of Health/National Institute on Aging (AG 14930) to Dr. M. Flint Beal and Dr. Gary Gibson. We thank Greta Strong for assistance with the preparation of this manuscript.
Abbreviations
- DLST
dihydrolipoyl succinyl transferase
- DLD
dihydrolipoamide dehydrogenase
- OGDH
α-ketoglutarate dehydrogenase
- KGDHC
α-ketoglutarate dehydrogenase complex
- AD
Alzheimer’s disease
- HD
Huntington’s disease
- PD
Parkinson’s disease
- 8OH2dG
8-hydroxy-2-deoxyguanosine
- MPTP
1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine
- PDHC
pyruvate dehydrogenase complex
- C-I
respiratory chain Complex I
- COX
cytochrome oxidase
- MDH
malate dehydrogenase
- CS
citrate synthase
- ME
malic enzyme (NADPH)
- SDH
succinate dehydrogenase
- GR
glutathione reductase
- GPx1
mitochondrial glutathione peroxidase 1
- MnSOD
mitochondrial manganese superoxide dismutase
- TCA
tricarboxlyic acid cycle
References
- Alston TA, Mela L, Bright HJ. 3-Nitropropionate, the toxic substance of Indigofera, is a suicide inactivator of succinate dehydrogenase. Proc Natl Acad Sci U S A. 1977;74:3767–3771. doi: 10.1073/pnas.74.9.3767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beal MF, Brouillet E, Jenkins B, Henshaw R, Rosen B, Hyman BT. Age-dependent striatal excitotoxic lesions produced by the endogenous mitochondrial inhibitor malonate. J Neurochem. 1993a;61:1147–1150. doi: 10.1111/j.1471-4159.1993.tb03633.x. [DOI] [PubMed] [Google Scholar]
- Beal MF, Ferrante RJ, Henshaw R, Matthews RT, Chan PH, Kowall NW, Epstein CJ, Schulz JB. 3-Nitropropionic acid neurotoxicity is attenuated in copper/zinc superoxide dismutase transgenic mice. J Neurochem. 1995;65:919–922. doi: 10.1046/j.1471-4159.1995.65020919.x. [DOI] [PubMed] [Google Scholar]
- Beal MF, Brouillet E, Jenkins BG, Ferrante RJ, Kowall NW, Miller JM, Storey E, Srivastava R, Rosen BR, Hyman BT. Neurochemical and histologic characterization of striatal excitotoxic lesions produced by the mitochondrial toxin 3-nitropropionic acid. J Neurosci. 1993b;13:4181–4192. doi: 10.1523/JNEUROSCI.13-10-04181.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brouillet E, Hantraye P, Ferrante RJ, Dolan R, Leroy-Willig A, Kowall NW, Beal MF. Chronic mitochondrial energy impairment produces selective striatal degeneration and abnormal choreiform movements in primates. Proc Natl Acad Sci U S A. 1995;92:7105–7109. doi: 10.1073/pnas.92.15.7105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown AM, Gordon D, Lee H, Caudy M, Haroutunian V, Blass JP. Substantial linkage disequilibrium across the dihydrolipoyl succinyltransferase gene region without Alzheimer’s disease association. Neurochem Res. 2004;29:629–635. doi: 10.1023/b:nere.0000014833.54481.1d. [DOI] [PubMed] [Google Scholar]
- Bubber P, Haroutunian V, Fisch G, Blass JP, Gibson GE. Mitochondrial abnormalities in Alzheimer brain: mechanistic implications. Ann Neurol. 2005;57:695–703. doi: 10.1002/ana.20474. [DOI] [PubMed] [Google Scholar]
- Buerstatte CR, Behar KL, Novotny EJ, Lai JC. Brain regional development of the activity of alpha-ketoglutarate dehydrogenase complex in the rat. Brain Res Dev Brain Res. 2000;125:139–145. doi: 10.1016/s0165-3806(00)00134-6. [DOI] [PubMed] [Google Scholar]
- Calingasan NY, Ho DJ, Wille EJ, Campagna MV, Ruan J, Dumont M, Yang L, Shi Q, Gibson GE, Beal MF. Influence of mitochondrial enzyme deficiency on adult neurogenesis in mouse models of neurodegenerative diseases. Neuroscience. 2008;153:986–996. doi: 10.1016/j.neuroscience.2008.02.071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chinopoulos C, Tretter L, Adam-Vizi V. Depolarization of in situ mitochondria due to hydrogen peroxide-induced oxidative stress in nerve terminals: inhibition of alpha-ketoglutarate dehydrogenase. J Neurochem. 1999;73:220–228. doi: 10.1046/j.1471-4159.1999.0730220.x. [DOI] [PubMed] [Google Scholar]
- Coles CJ, Edmondson DE, Singer TP. Inactivation of succinate dehydrogenase by 3-nitropropionate. J Biol Chem. 1979;254:5161–5167. [PubMed] [Google Scholar]
- Giasson BI, Duda JE, Murray IV, Chen Q, Souza JM, Hurtig HI, Ischiropoulos H, Trojanowski JQ, Lee VM. Oxidative damage linked to neurodegeneration by selective alpha-synuclein nitration in synucleinopathy lesions. Science. 2000;290:985–989. doi: 10.1126/science.290.5493.985. [DOI] [PubMed] [Google Scholar]
- Gibson GE, Zhang H. Interactions of oxidative stress with thiamine homeostasis promote neurodegeneration. Neurochem Int. 2002;40:493–504. doi: 10.1016/s0197-0186(01)00120-6. [DOI] [PubMed] [Google Scholar]
- Gibson GE, Park LC, Sheu KF, Blass JP, Calingasan NY. The alpha-ketoglutarate dehydrogenase complex in neurodegeneration. Neurochem Int. 2000a;36:97–112. doi: 10.1016/s0197-0186(99)00114-x. [DOI] [PubMed] [Google Scholar]
- Gibson GE, Sheu KF, Blass JP, Baker A, Carlson KC, Harding B, Perrino P. Reduced activities of thiamine-dependent enzymes in the brains and peripheral tissues of patients with Alzheimer’s disease. Arch Neurol. 1988;45:836–840. doi: 10.1001/archneur.1988.00520320022009. [DOI] [PubMed] [Google Scholar]
- Gibson GE, Kingsbury AE, Xu H, Lindsay JG, Daniel S, Foster OJ, Lees AJ, Blass JP. Deficits in a tricarboxylic acid cycle enzyme in brains from patients with Parkinson’s disease. Neurochem Int. 2003;43:129–135. doi: 10.1016/s0197-0186(02)00225-5. [DOI] [PubMed] [Google Scholar]
- Gibson GE, Haroutunian V, Zhang H, Park LC, Shi Q, Lesser M, Mohs RC, Sheu RK, Blass JP. Mitochondrial damage in Alzheimer’s disease varies with apolipoprotein E genotype. Ann Neurol. 2000b;48:297–303. [PubMed] [Google Scholar]
- Good PF, Hsu A, Werner P, Perl DP, Olanow CW. Protein nitration in Parkinson’s disease. J Neuropathol Exp Neurol. 1998;57:338–342. doi: 10.1097/00005072-199804000-00006. [DOI] [PubMed] [Google Scholar]
- Greene JG, Porter RH, Eller RV, Greenamyre JT. Inhibition of succinate dehydrogenase by malonic acid produces an “excitotoxic” lesion in rat striatum. J Neurochem. 1993;61:1151–1154. doi: 10.1111/j.1471-4159.1993.tb03634.x. [DOI] [PubMed] [Google Scholar]
- Hantraye P, Brouillet E, Ferrante R, Palfi S, Dolan R, Matthews RT, Beal MF. Inhibition of neuronal nitric oxide synthase prevents MPTP-induced parkinsonism in baboons. Nat Med. 1996;2:1017–1021. doi: 10.1038/nm0996-1017. [DOI] [PubMed] [Google Scholar]
- Joffe GT, Parks JK, Parker WD., Jr Secondary inhibition of 2-ketoglutarate dehydrogenase complex by MPTP. Neuroreport. 1998;9:2781–2783. doi: 10.1097/00001756-199808240-00018. [DOI] [PubMed] [Google Scholar]
- Johnson MT, Yang HS, Magnuson T, Patel MS. Targeted disruption of the murine dihydrolipoamide dehydrogenase gene (Dld) results in perigastrulation lethality. Proc Natl Acad Sci U S A. 1997;94:14512–14517. doi: 10.1073/pnas.94.26.14512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klivenyi P, Starkov AA, Calingasan NY, Gardian G, Browne SE, Yang L, Bubber P, Gibson GE, Patel MS, Beal MF. Mice deficient in dihydrolipoamide dehydrogenase show increased vulnerability to MPTP, malonate and 3-nitropropionic acid neurotoxicity. J Neurochem. 2004;88:1352–1360. doi: 10.1046/j.1471-4159.2003.02263.x. [DOI] [PubMed] [Google Scholar]
- Kobayashi T, Matsumine H, Matuda S, Mizuno Y. Association between the gene encoding the E2 subunit of the alpha-ketoglutarate dehydrogenase complex and Parkinson’s disease. Ann Neurol. 1998;43:120–123. doi: 10.1002/ana.410430121. [DOI] [PubMed] [Google Scholar]
- Kumar MJ, Nicholls DG, Andersen JK. Oxidative alpha-ketoglutarate dehydrogenase inhibition via subtle elevations in monoamine oxidase B levels results in loss of spare respiratory capacity: implications for Parkinson’s disease. J Biol Chem. 2003;278:46432–46439. doi: 10.1074/jbc.M306378200. [DOI] [PubMed] [Google Scholar]
- Kunugi H, Nanko S, Ueki A, Isse K, Hirasawa H. DLST gene and Alzheimer’s disease. Lancet. 1998;351:1584–1585. doi: 10.1016/S0140-6736(05)61151-8. [DOI] [PubMed] [Google Scholar]
- Leong SF, Lai JC, Lim L, Clark JB. The activities of some energy-metabolising enzymes in nonsynaptic (free) and synaptic mitochondria derived from selected brain regions. J Neurochem. 1984;42:1306–1312. doi: 10.1111/j.1471-4159.1984.tb02788.x. [DOI] [PubMed] [Google Scholar]
- Mastrogiacomo F, Kish SJ. Cerebellar alpha-ketoglutarate dehydrogenase activity is reduced in spinocerebellar ataxia type 1. Ann Neurol. 1994;35:624–626. doi: 10.1002/ana.410350519. [DOI] [PubMed] [Google Scholar]
- Matsushita S, Arai H, Yuzuriha T, Kato M, Matsui T, Urakami K, Higuchi S. No association between DLST gene and Alzheimer’s disease or Wernicke-Korsakoff syndrome. Neurobiol Aging. 2001;22:569–574. doi: 10.1016/s0197-4580(01)00225-1. [DOI] [PubMed] [Google Scholar]
- McNaught KS, Altomare C, Cellamare S, Carotti A, Thull U, Carrupt PA, Testa B, Jenner P, Marsden CD. Inhibition of alpha-ketoglutarate dehydrogenase by isoquinoline derivatives structurally related to 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) Neuroreport. 1995;6:1105–1108. doi: 10.1097/00001756-199505300-00008. [DOI] [PubMed] [Google Scholar]
- Mizuno Y, Suzuki K, Ohta S. Postmortem changes in mitochondrial respiratory enzymes in brain and a preliminary observation in Parkinson’s disease. J Neurol Sci. 1990;96:49–57. doi: 10.1016/0022-510x(90)90056-s. [DOI] [PubMed] [Google Scholar]
- Mizuno Y, Matuda S, Yoshino H, Mori H, Hattori N, Ikebe S. An immunohistochemical study on alpha-ketoglutarate dehydrogenase complex in Parkinson’s disease. Ann Neurol. 1994;35:204–210. doi: 10.1002/ana.410350212. [DOI] [PubMed] [Google Scholar]
- Mizuno Y, Ikebe S, Hattori N, Nakagawa-Hattori Y, Mochizuki H, Tanaka M, Ozawa T. Role of mitochondria in the etiology and pathogenesis of Parkinson’s disease. Biochim Biophys Acta. 1995;1271:265–274. doi: 10.1016/0925-4439(95)00038-6. [DOI] [PubMed] [Google Scholar]
- Nakano K, Ohta S, Nishimaki K, Miki T, Matuda S. Alzheimer’s disease and DLST genotype. Lancet. 1997;350:1367–1368. doi: 10.1016/S0140-6736(05)65139-2. [DOI] [PubMed] [Google Scholar]
- Palfi S, Ferrante RJ, Brouillet E, Beal MF, Dolan R, Guyot MC, Peschanski M, Hantraye P. Chronic 3-nitropropionic acid treatment in baboons replicates the cognitive and motor deficits of Huntington’s disease. J Neurosci. 1996;16:3019–3025. doi: 10.1523/JNEUROSCI.16-09-03019.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park LC, Calingasan NY, Sheu KF, Gibson GE. Quantitative alpha-ketoglutarate dehydrogenase activity staining in brain sections and in cultured cells. Anal Biochem. 2000;277:86–93. doi: 10.1006/abio.1999.4359. [DOI] [PubMed] [Google Scholar]
- Park LC, Albers DS, Xu H, Lindsay JG, Beal MF, Gibson GE. Mitochondrial impairment in the cerebellum of the patients with progressive supranuclear palsy. J Neurosci Res. 2001;66:1028–1034. doi: 10.1002/jnr.10062. [DOI] [PubMed] [Google Scholar]
- Park LC, Zhang H, Sheu KF, Calingasan NY, Kristal BS, Lindsay JG, Gibson GE. Metabolic impairment induces oxidative stress, compromises inflammatory responses, and inactivates a key mitochondrial enzyme in microglia. J Neurochem. 1999;72:1948–1958. doi: 10.1046/j.1471-4159.1999.0721948.x. [DOI] [PubMed] [Google Scholar]
- Prince JA, Feuk L, Sawyer SL, Gottfries J, Ricksten A, Nagga K, Bogdanovic N, Blennow K, Brookes AJ. Lack of replication of association findings in complex disease: an analysis of 15 polymorphisms in prior candidate genes for sporadic Alzheimer’s disease. Eur J Hum Genet. 2001;9:437–444. doi: 10.1038/sj.ejhg.5200651. [DOI] [PubMed] [Google Scholar]
- Przedborski S, Jackson-Lewis V, Yokoyama R, Shibata T, Dawson VL, Dawson TM. Role of neuronal nitric oxide in 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP)-induced dopaminergic neurotoxicity. Proc Natl Acad Sci U S A. 1996;93:4565–4571. doi: 10.1073/pnas.93.10.4565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reed LJ, Hackert ML. Structure-function relationships in dihydrolipoamide acyltransferases. J Biol Chem. 1990;265:8971–8974. [PubMed] [Google Scholar]
- Schoonen WG, Wanamarta AH, van der Klei-van Moorsel JM, Jakobs C, Joenje H. Respiratory failure and stimulation of glycolysis in Chinese hamster ovary cells exposed to normobaric hyperoxia. J Biol Chem. 1990;265:1118–1124. [PubMed] [Google Scholar]
- Schoonen WG, Wanamarta AH, van der Klei-van Moorsel JM, Jakobs C, Joenje H. Characterization of oxygen-resistant Chinese hamster ovary cells. III. Relative resistance of succinate and alpha-ketoglutarate dehydrogenases to hyperoxic inactivation. Free Radic Biol Med. 1991;10:111–118. doi: 10.1016/0891-5849(91)90004-m. [DOI] [PubMed] [Google Scholar]
- Schulz JB, Henshaw DR, Matthews RT, Beal MF. Coenzyme Q10 and nicotinamide and a free radical spin trap protect against MPTP neurotoxicity. Exp Neurol. 1995a;132:279–283. doi: 10.1016/0014-4886(95)90033-0. [DOI] [PubMed] [Google Scholar]
- Schulz JB, Henshaw DR, Siwek D, Jenkins BG, Ferrante RJ, Cipolloni PB, Kowall NW, Rosen BR, Beal MF. Involvement of free radicals in excitotoxicity in vivo. J Neurochem. 1995b;64:2239–2247. doi: 10.1046/j.1471-4159.1995.64052239.x. [DOI] [PubMed] [Google Scholar]
- Shen XM, Li H, Dryhurst G. Oxidative metabolites of 5-S-cysteinyldopamine inhibit the alpha-ketoglutarate dehydrogenase complex: possible relevance to the pathogenesis of Parkinson’s disease. J Neural Transm. 2000;107:959–978. doi: 10.1007/s007020070045. [DOI] [PubMed] [Google Scholar]
- Sheu KF, Brown AM, Kristal BS, Kalaria RN, Lilius L, Lannfelt L, Blass JP. A DLST genotype associated with reduced risk for Alzheimer’s disease. Neurology. 1999a;52:1505–1507. doi: 10.1212/wnl.52.7.1505. [DOI] [PubMed] [Google Scholar]
- Sheu KF, Brown AM, Haroutunian V, Kristal BS, Thaler H, Lesser M, Kalaria RN, Relkin NR, Mohs RC, Lilius L, Lannfelt L, Blass JP. Modulation by DLST of the genetic risk of Alzheimer’s disease in a very elderly population. Ann Neurol. 1999b;45:48–53. [PubMed] [Google Scholar]
- Shi Q, Chen HL, Xu H, Gibson GE. Reduction in the E2k subunit of the alpha-ketoglutarate dehydrogenase complex has effects independent of complex activity. J Biol Chem. 2005;280:10888–10896. doi: 10.1074/jbc.M409064200. [DOI] [PubMed] [Google Scholar]
- Shi Q, Karuppagounder SS, Xu H, Pechman D, Chen H, Gibson GE. Responses of the mitochondrial alpha-ketoglutarate dehydrogenase complex to thiamine deficiency may contribute to regional selective vulnerability. Neurochem Int. 2007;50:921–931. doi: 10.1016/j.neuint.2007.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sims NR. Rapid isolation of metabolically active mitochondria from rat brain and subregions using Percoll density gradient centrifugation. J Neurochem. 1990;55:698–707. doi: 10.1111/j.1471-4159.1990.tb04189.x. [DOI] [PubMed] [Google Scholar]
- Sriram K, Pai KS, Boyd MR, Ravindranath V. Evidence for generation of oxidative stress in brain by MPTP: in vitro and in vivo studies in mice. Brain Res. 1997;749:44–52. doi: 10.1016/s0006-8993(96)01271-1. [DOI] [PubMed] [Google Scholar]
- Starkov AA, Fiskum G, Chinopoulos C, Lorenzo BJ, Browne SE, Patel MS, Beal MF. Mitochondrial alpha-ketoglutarate dehydrogenase complex generates reactive oxygen species. J Neurosci. 2004;24:7779–7788. doi: 10.1523/JNEUROSCI.1899-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]