

Abstract
The purpose of this study was to investigate labetalol as a potential high permeability reference standard for the application of Biopharmaceutics Classification Systems (BCS). Permeabilities of labetalol and metoprolol were investigated in animal intestinal perfusion models and Caco-2 cell monolayers. After isolating specific intestinal segments, in situ single-pass intestinal perfusions (SPIP) were performed in rats and mice. The effective permeabilities (Peff) of labetalol and metoprolol, an FDA standard for the low/high Peff class boundary, were investigated in two different segments of rat intestine (proximal jejunum and distal ileum), and in the proximal jejunum of mouse. No significant difference was found between Peff of metoprolol and labetalol in the jejunum and ileum of rat (0.33±0.11 ×10−4 vs. 0.38±0.06 ×10−4 and 0.57±0.17 ×10−4 vs. 0.64±0.30 ×10−4 cm/s, respectively) and in the jejunum of mouse (0.55±0.05 ×10−4 vs. 0.59±0.13 ×10−4 cm/s). However, Peff of metoprolol and labetalol were 1.7 and 1.6 times higher in the jejunum of mouse, compared to the jejunum of rat, respectively. Metoprolol and labetalol showed segmental dependent permeability through the rat intestine, with increased Peff in the distal ileum in comparison to the proximal jejunum. Most significantly, Peff of labetalol was found to be concentration dependent. Decreasing concentrations of labetalol in the perfusate resulted in decreased Peff compared to Peff of metoprolol. The intestinal epithelial permeability of labetalol was lower than that of metoprolol in Caco-2 cells at both apical pH 6.5 and 7.5 (5.96±1.96 ×10−6 vs. 9.44±3.44 ×10−6 and 15.9±2.2 ×10−6 vs. 23.2±7.1 ×10−6 cm/s, respectively). Labetalol exhibited higher permeability in basolateral to apical (BL-AP) compared to AP-BL direction in Caco-2 cells at 0.1 times the highest dose strength (HDS) (46.7±6.5 ×10−6 vs. 14.2±1.5 ×10−6 cm/s). The P-gp inhibitor, verapamil significantly increased AP-BL and decreased BL-AP direction transport of labetalol. Overall, labetalol showed high Peff in rat and mouse intestinal perfusion models similar to metoprolol at a concentration based on HDS. However, the concentration dependent permeability of labetalol in mice due to P-gp and the inhibition study with verapamil in Caco-2 cells indicated that labetalol is not an ideal reference standard for BCS classification.
Keywords: Biopharmaceutics Classification Systems (BCS), Caco-2, labetalol, metoprolol, permeability, reference standard, single-pass intestinal perfusion
1. Introduction
Worldwide, numerous regulatory bodies are responsible for ensuring safety, quality and efficacy of drug products essential for public health. Therefore, the marketing authorization process for ensuring the therapeutic performance of drug products following manufacturing changes and for approval of generic drug product efficacy claims relies on the demonstration of bioequivalence (BE) based on comparative in vivo bioavailability (BA) using pharmacokinetic (PK) endpoints.1, 2 BE is defined as the absence of a significant difference in the rate and extent to which the active ingredient or active moiety in pharmaceutical equivalents or pharmaceutical alternatives becomes available at the site of drug action when administered at the same molar dose under similar conditions in an appropriately designed study.1
Since the Biopharmaceutics Classification System (BCS) was first introduced in 1995, it has had an increasing impact on regulatory practice for oral BE.3, 4 BCS is a scientific framework for classifying drugs into four groups according to their aqueous solubility and their intestinal permeability: class I (high solubility-high permeability), class II (low solubility-high permeability), class III (high solubility-low permeability) and class IV (low solubility-low permeability).3 Recently, BCS has been implemented for waiving BE studies on the basis of the solubility and gastrointestinal (GI) permeability of drug substance.2, 5, 6
The US Food and Drug Asministration (FDA) and the European Medicine Agency (EMA) have issued guidelines related to the BCS biowaivers.2, 6 Current FDA has promoted the BCS as a scientific approach to permit waiver of in vivo BE testing for immediate release solid dosage forms for Class I drugs (high solubility and high permeability), when such drug products also exhibit rapid dissolution.6 The 2010 EMA BE Guideline further extends its discussion of biowaivers to Class III drugs (high solubility and low permeability) with very rapid dissolution2. Thus, a BCS-based biowaiver has become an important and cost saving tool for the pharmaceutical industry throughout drug discovery and development.7–9
A drug substance is considered highly permeable when the extent of absorption in humans is determined to be more than 90% (or 85% according to EMA) of an administered dose based on a mass balance determination or in comparison to an intravenous (I.V.) reference dose in the absence of evidence suggesting instability in the GI tract.2, 6
Various methods can be used for the determination of the permeability of a drug from the GI tract. These methods include in vivo intestinal perfusion studies in humans, in vivo or in situ intestinal perfusion studies in animals, in vitro permeation studies using excised human or animal intestinal tissues and in vitro permeation studies across a monolayer of cultured epithelial cells.6
Among the nonclinical methods, in situ intestinal perfusion in a suitable animal model and/or in vitro permeability methods using monolayers of suitable epithelial cells have been widely utilized to predict the extent of intestinal absorption of drugs in humans.10–14 Using permeability methods, a test drug substance is classified as highly permeable when its permeability is equal to or greater than that of the selected internal standard (IS) with high permeability.6 The FDA BCS Guidance includes a list of model drugs and/or chemicals suggested for use in establishing suitability of a permeability method.6 On the other hand, other drugs for which there is sufficient information available on the mechanism of absorption and reliable estimates of the extent of drug absorption in humans may also be used for this purpose. Metoprolol, an FDA reference drug for the low/high permeability class boundry, has been widely used as a reference to categorize the drugs into the proper permeability class in permeability studies.11, 15, 16 However, metoprolol is a conservative reference standard, as it is absorbed completely in human based on the urinary excretion of the unchanged compound and its total radioactive metabolites after oral and I.V. administration in healthy volunteers.17
The aim of the present study was to assess labetalol as a potential high permeability reference standard for the application of BCS. Labetalol has been used as a reference for classifying drugs according to their permeabilities in some in vitro permeability studies.18–20 This drug has been selected as a potential high permeability internal standard (HP-IS), with permeability in close proximity to the low/high class boundary, from the model drugs used in these studies.
In the present study, in situ intestinal perfusion and in vitro cell culture methods were performed for the determination of permeability of labetalol. The intestinal epithelial permeability of metoprolol and labetalol was investigated across Caco-2 cell monolayers in both apical (AP)-basolateral (BL) and BL-AP directions. The effective permeability (Peff) was investigated in the in situ single-pass intestinal perfusion (SPIP) in rats and mice. Metoprolol, an FDA reference drug was coperfused with labetalol for the direct comparison of permeability.
2. Materials and Methods
2.1. Materials
Labetalol, metoprolol, verapamil, phenol red, calcium chloride, magnesium chloride, sodium dihydrogen phosphate, D-glucose, 2-morpholinoethanesulfonic acid (MES), 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES) and trifluoroacetic acid (TFA) were purchased from Sigma-Aldrich Chemical Co. (St. Louis, MO). Potassium chloride, sodium chloride and HPLC grade acetonitrile were obtained from Fisher Scientific Inc. (Pittsburgh, PA). Physiological saline solution was purchased from Hospira Inc. (Lake Forest, IL). Cell culture reagents were obtained from Gibco® Life Technologies Inc. (Carlsbad, CA) and cell culture supplies were from Corning Costar Co. (Corning, NY). All other chemicals were of analytical grade.
2.2. Methods
2.2.1. Single-Pass Intestinal Perfusion (SPIP) Studies in Rats and Mice
All animal experiments were conducted using protocols approved by the University of Michigan Committee of Use and Care of Animals (UCUCA). The animals were housed and handled according to the University of Michigan Unit for Laboratory Animal Medicine guidelines. Male Wistar rats (Charles River, IN) weighing 250–310 g and male BALB/c mice (Charles River, IN) weighing 20–25 g were used for all perfusion studies. Prior to each experiment, the rats and mice were fasted over night (12–18 h) with free access to water. Animals were randomly assigned to the different experimental groups.
The procedures for the in situ SPIP were performed on Wistar rats and BALB/c mice according to previously published reports.11, 21 Briefly, rats and mice were anesthetized with an intramuscular injection of 5 mg/kg xylazine and 80 mg/kg ketamine. After they were placed on a heated surface maintained at 37°C (Harvard Apparatus Inc., Holliston, MA), the abdomen was opened by a midline incision of 3–4 and 1.5 cm for rats and mice, respectively. For in situ SPIP in rats, the proximal jejunal segment (~3 cm average distance of the inlet from the ligament of Treitz) or the distal ileal segment (~3 cm average distance of the outlet from the ligament of cecum) of approximately 10 cm was carefully exposed and cannulated on two ends with flexible PVC tubing (2.29 mm i.d., Fisher Scientific Inc., Pittsburgh, PA). For in situ SPIP in mice, the proximal jejunal segment of approximately 8 cm was exposed and glass cannulas (2.0 mm o.d.) were inserted at each end of the jejunal segment and cannulated with flexible PVC tubing (2.06 mm i.d., Fisher Scientific Inc., Pittsburgh, PA). Care was taken to avoid disturbing the circulatory system and the exposed segment was kept moist with 37°C normal saline solution. All solutions were incubated in a 37°C water bath. The isolated segment was rinsed with normal saline solution in order to clean out any residual debris.
Two perfusion solutions, containing 5 mM MES, pH 6.5 and 5 mM HEPES, pH 7.5 were used for the corresponding segments, jejunum and ileum, respectively. Perfusion buffers were prepared with 1 mM calcium chloride, 0.5 mM magnesium chloride, 145 mM sodium chloride, 1 mM sodium dihydrogen phosphate, 3 mM potasium chloride and 5 mM D-glucose. Drug concentrations in the perfusion solution were based on the highest dose strength (HDS) of the drug product dissolved in 250 ml water. At the start of the study, the perfusion solution containing labetalol (1200 μg/ml), metoprolol (400 μg/ml) and phenol red (0.17 mM) was perfused through the intestinal segment (Watson Marlow Pumps 323S, Watson-Marlow Bredel Inc., Wilmington, MA), at a flow rate of 0.2 and 0.1 ml/min for rats and mice, respectively. Metoprolol and labetalol were also tested at 0.1 and 0.01 times the HDS dissolved in 250 ml water in a separate intestinal perfusion study in mice.
Phenol red was added to the perfusion buffer as a nonabsorbable marker for water flux measurements. Metoprolol was coperfused with labetalol as a reference standard for permeability in close proximity to the low/high permeability class boundry. The perfusion buffer was first perfused for 1 h in order to ensure steady state conditions. After reaching steady state, perfusion samples were taken in 10 min intervals for 1 h. All samples were frozen immediately after collection and stored at −20°C until analyzed. Drug concentrations in all samples and perfusion solutions were analyzed by high performance liquid chromatography (HPLC). The length of each perfused intestinal segment was accurately measured at the end of the experiment.
2.2.2. Net Water Flux Measurement
The net water flux in the SPIP studies was determined by measuring phenol red, a nonabsorbed and nonmetabolized marker. The measured Cout/Cin ratio was corrected for water transport according to Eq. 1:
| (1) |
where Cin phenol red and Cout phenol red are equal to the concentrations of phenol red in the inlet and outlet samples, respectively.
2.2.3. Caco-2 Cell Culture and Permeability Studies
Caco-2 cells (passage 33–38) from American Type Culture Collection (Rockville, MD) were routinely maintained in Dulbecco’s modified Eagle’s medium (DMEM, Gibco® Life Technologies, Carlsbad, CA) containing 10% fetal bovine serum, 1% nonessential amino acids, 1 mM sodium pyruvate and 1% L-glutamine. Cells were grown in an atmosphere of 5% CO2 and 90% relative humidity at 37°C.
Caco-2 cells were seeded on collagen treated PTFE membrane inserts with 0.4 μm pore size and 24 mm diameter (6-well Transwell plate, Corning Costar® Co., Corning, NY). The cells on the inserts were cultured for 21 days at 37°C in a humidified incubator containing 5% CO2 in air. The differentiation status of the formed monolayer was evaluated by measuring the transepithelial electrical resistance (TEER) (Millicell-ERS epithelial Voltohmmeter, Millipore Co., Bedford, MA). Permeability studies were conducted with the monolayers that developed TEER values >300 Ω cm2 following 21 days in cell culture.
The transport buffers (pH 6.5 and 7.5) were identical to the buffers used in the in situ SPIP studies. HEPES buffer (pH 7.5) was used in the basolateral (BL) side in Caco-2 permeability studies. The effect of pH conditions on the permeability across the cells in both AP-BL and BL-AP directions was investigated using MES buffer, pH 6.5 and HEPES buffer, pH 7.5 in the AP side. Drug concentrations used in the permeability studies were based on the HDS of the drug product dissolved in 250 ml water. The effect of verapamil, a known P-gp inhibitor on the bidirectional transport of labetalol across Caco-2 cell monolayers was investigated at 0.1 times the HDS dissolved in 250 ml water. The AP pH 7.5 was used in the inhibition study. 0.1 mM verapamil was added to the drug solution in the buffer.
On the day of the experiment, DMEM was removed and the monolayers were rinsed and incubated with a blank transport buffer for 15 min. Following 15 min incubation, blank transport buffer was removed from the AP side and replaced by 1.5 ml of the drug solution in the buffer (pH 6.5 or 7.5) and 2.5 ml of uptake buffer (pH 7.5) was added to the receiver compartment on the BL side in the AP-BL direction studies. In the BL-AP direction studies, 2.5 ml of drug solution in HEPES buffer was added to the BL side and 1.5 ml of blank buffer was added to the receiver compartment on the AP side. Throughout the experiment, the transport plates were kept in an incubator at 37°C. Samples (100 μl) were taken from the receiver side at 15., 30., 45., 60., 75., 90., and 120. min and similar volumes of blank buffer were added following each sample withdrawal. Samples were taken from the donor side at 60. and 120. min. The appearance rates of the drugs on the receiver side were obtained under sink conditions. (i.e., the overall transported amount does not exceed 10% of the amount in the donor solution). Drug concentrations in the samples were immediately analyzed by HPLC.
2.2.4. Data Analysis
The effective permeability (Peff, cm/s) through the rat and mouse gut wall in the SPIP studies was determined assuming the “plug flow” model expressed in Eq. 222:
| (2) |
where Q is the perfusion buffer flow rate (0.2 and 0.1 ml/min for rats and mice, respectively), C′out/C′in is the ratio of the outlet and inlet concentration of the tested drug that has been adjusted for water transport, R is the radius of the intestinal segment (set to 0.2 and 0.1 cm for rats and mice, respectively) and L is the length of the intestinal segment.
The apparent permeability (Papp, cm/s) across Caco-2 cell monolayers was calculated from the linear plot of drug accumulated in the receiver side versus time using Eq. 3:
| (3) |
where dQ/dt is the steady-state appearance rate of the drug on the receiver (serosal in the case of AP-BL studies or mucosal in the case of BL-AP studies) side, C0 is the initial concentration of the drug in the donor side and A is the monolayer growth surface area (4.67 cm2). Linear regression was carried out to obtain the steady-state appearance rate of the drug on the receiver side.
The efflux ratio, ER was determined by calculating the ratio of Papp in the secretory (BL-AP) to the absorptive (AP-BL) direction according to the Eq. 4:
| (4) |
2.2.5. Analytical Methods
The simultaneous analysis of labetalol and metoprolol in Caco-2 medium and in the mouse/rat perfusion buffer with phenol red was performed using a HPLC system (Waters 2695 Separation Module) with a photodiode array UV dedector (Waters 996). Separations were carried out using XTerra, RP18, 5 μm, 4.6 × 250 mm column (Waters Co., Milford, MA). Perfusion aliquots of 20 μl or Caco-2 medium aliquots of 100 μl were injected into the HPLC system. The mobile phase consisted of 0.03 or 0.1% TFA in water and 0.1% TFA in acetonitrile with the organic phase gradient changing from 23 to 30% over 9 min and 10 to 50% over 6 min for the analysis of the perfusion and Caco-2 samples, respectively. The flow rate was 1 ml/min at room temperature. The detection wavelengths were 275, 224 and 265 nm for metoprolol, labetalol and phenol red, respectively. The retention times of metoprolol, labetalol and phenol red were 5.0, 6.4 and 7.2 min for the perfusion samples; 8.7 and 10.0 min for Caco-2 samples, respectively. Separate standards curves were used for each experiment. Between-day and within-day precisions expressed as coefficient of variation were less than 1.0%.
2.2.6. Statistical Analysis
All animal experiments were performed in either quadruplicate or sextuplicate and all Caco-2 experiments were performed in triplicate. The data are presented as mean ± SD. The independent t-test and one-way Anova were used to assess differences for two groups and multiple comparisons, respectively. Differences were considered statistically significant at p<0.05.
3. Results
3.1. In Situ Permeability in the Single-Pass Intestinal Perfusion (SPIP) Model in Rats and Mice
Peff values obtained for labetalol and metoprolol in small intestinal segments at physiological pH of each region in rats and mice are presented in Fig. 1 and summarized in Table 1. The permeabilities of labetalol and metoprolol are approximately two times greater in the ileum than in the jejunum, suggesting an intestinal site dependency for the absorption of these drugs. The in situ permeabilities of metoprolol and labetalol obtained from SPIP models were comparable. No significant difference was found between Peff of metoprolol and labetalol in the jejunum and ileum of rat and in the jejunum of mouse at concentrations based on HDS. However, Peff of metoprolol and labetalol were 1.7 and 1.6 times higher in the jejunum of mouse than in the jejunum of rat, respectively.
Figure 1.

Effective permeability (Peff, cm/s) values obtained for labetalol and metoprolol following in situ single-pass intestinal perfusion (SPIP) to the rat proximal jejunum at pH 6.5 and distal ileum at pH 7.5 and to the mouse jejunum at pH 6.5. Drug concentrations in the perfusate are 400 and 1200 μg/ml for metoprolol and labetalol, respectively. Data are presented as the mean ± SD; n = 6 and 4 in each experimental group for rat and mouse, respectively.
Table 1.
Effective permeability (Peff, cm/s) values obtained for labetalol and metoprolol following in situ single-pass intestinal perfusion (SPIP) in rat and mouse and the fraction of dose absorbed (Fabs) in humans
| Human permeability (10−4 cm/s) | Fb abs | Drug concentration in perfusate (μg/ml) | Pa eff (10−4 cm/s)
|
|||
|---|---|---|---|---|---|---|
| Rat jejunum (10−4 cm/s) | Rat ileum (10−4 cm/s) | Mouse jejunum (10−4 cm/s) | ||||
| Metoprolol | 1.5 ± 0.915 | 9615 | 400 | 0.33 ± 0.11 (p = 0.439) | 0.57 ± 0.17 (p = 0.666) | 0.55 ± 0.05 (p = 0.589) |
| Labetalol | NA | >9023 | 1200 | 0.38 ± 0.06 | 0.64 ± 0.30 | 0.59 ± 0.13 |
Effective permeability (cm/s). Data are presented as the mean ± SD; n = 6 and 4 in each experimental group for rat and mouse, respectively;
Fraction of dose absorbed; p values indicate the significance of difference between the effective permeability values of metoprolol and labetalol.
To determine the pH effect on the permeability of metoprolol and labetalol in the jejunum and ileum, Peff values were measured in two additional pH conditions. Fig.2 compares the drugs’ jejunal and ileal permeabilities at the physiological pH of the segments (pH 6.5 and 7.5, respectively) vs. the alternate pH 7.5 and 6.5, respectively. This experiment showed that the change of pH did not significantly affect the permeabilities of metoprolol and labetalol in the jejunum and ileum of rat. Labetalol and metoprolol exhibited segmental dependent permeability through the rat intestine, with increased Peff in the distal ileum in comparison to the proximal region of the intestine at the HDS of these drugs.
Figure 2.

Comparison of the effective permeability (Peff, cm/s) values obtained for labetalol and metoprolol in the rat jejunum and ileum at the physiological pH of the segments (pH 6.5 and 7.5, respectively; n = 6) vs. the alternate pH of 7.5 and 6.5, respectively (n = 6). Drug concentrations in the perfusate are 400 and 1200 μg/ml for metoprolol and labetalol, respectively. Data are presented as the mean ± SD. (p = 0.583 and p = 0.385 for metoprolol, p = 0.898 and p = 0.372 for labetalol at the physiological pH vs. the alternate pH in the jejunum and ileum, respectively.)
Peff values of labetalol and metoprolol obtained following in situ SPIP in the mouse jejunum, at different initial drug concentrations (0.1 and 0.01 times the HDS dissolved in 250 ml water) are presented in Fig. 3. No significant difference was found between the Peff values of metoprolol in all experimental groups (p>0.05). However, labetalol displayed a two fold decreased permeability which was lower than that of metoprolol following the perfusion of low drug concentrations compared to the concentration based on HDS.
Figure 3.

Effective permeability (Peff, cm/s) values obtained for different perfusate concentrations of labetalol and metoprolol following in situ single-pass intestinal perfusion (SPIP) to the mouse proximal jejunum at pH 6.5. Data are presented as the mean ± SD, n = 4 in each experimental group.
3.2. Transport Studies Across Caco-2 Monolayers
The result of the transport studies for labetalol and metoprolol across Caco-2 cell monolayers in the absorptive (AP-BL) and the secretory (BL-AP) directions is presented in Fig.4. The effect of efflux transporter inhibitor, verapamil on the permeability of labetalol and metoprolol across Caco-2 cell monolayers is presented in Fig. 5. Labetalol exhibited significantly higher permeability in the BL-AP in comparison to the AP-BL direction at 0.1 times the HDS (46.7±6.5 ×10−6 vs. 14.2±1.5 ×10−6 cm/s, p=0.001). Efflux ratio (ER) was 3.3. The P-gp inhibitor, verapamil caused a 1.5 times increase and a 2.3 times decrease in the AP-BL and BL-AP direction transports of labetalol, respectively. ER approached 1 in the presence of 0.1 mM verapamil. Verapamil showed no effect on the permeability of metoprolol across Caco-2 cell monolayers in the AP-BL and BL-AP directions.
Figure 4.

The apparent permeability (Papp, cm/s) of labetalol (1200 μg/ml) and metoprolol (400 μg/ml) in the absorptive (AP-BL) and the secretory (BL-AP) directions. Data are presented as the mean ± SD, n = 3 in each experimental group.
Figure 5.

The apparent permeability (Papp, cm/s) of labetalol (x 0.1 Highest Dose Strength, 120 μg/ml) and metoprolol (x 0.1 Highest Dose Strength, 40 μg/ml) in the absorptive (AP-BL) and the secretory (BL-AP) directions in the presence of 0.1 mM verapamil. Data are presented as the mean ± SD, n = 3 in each experimental group.
4. Discussion
It has been known that to demonstrate the suitability of a permeability method intended for the application of the BCS, a rank order relationship between test permeability values and the extent of drug absorption data in human subjects should be established using a sufficient number of model drugs. After demonstrating the suitability of a method, a low and a high permeability model drug should be used as an IS and then the method can be used to classify a drug substance’s permeability. It is apparent that careful selection of a HP-IS may simplify the permeability classification of a test drug. Therefore, the purpose of this study was to investigate the intestinal permeability of labetalol and compare the drug with metoprolol, an FDA high permeability reference drug to assess the suitability of labetalol as a reference for permeability methods.
Metoprolol is a well absorbed drug, which was demonstrated by means of an intestinal intubation technique in humans. It was reported that approximately 60% of the drug is absorbed from the duodenum and about 50% of that leaving the duodenum is absorbed from the first part of the jejunum.24 It was shown that the absorption rates of metoprolol are similar in jejunum and ileum.25 About 95% of the oral dose of metoprolol is excreted in the urine within 72 h, mainly in the metabolized form. The fraction of dose absorbed (Fabs) for metoprolol is well documented based on the fractions of the radioactive dose excreted as the parent compound and metabolites after oral and I.V. administration.17
On the other hand, from the radiochemical analysis of urine and faeces for labetalol, it is known that 55 and 60% of the orally administered dose of radioactivity was excreted by urine within 24 h in two subjects who took an 200 mg oral dose of labetalol. The remainder of the dose radioactivity (12.4 and 27.4%) was excreted by faeces for 48 and 97 h after the administration of dose in these subjects, respectively. The percentage of the dose excreted in the urine as unchanged drug is up to 5% in man. The plasma levels of radioactivity and high urinary excretion show that labetalol is well absorbed by man but extensively metabolized during its passage through the gut and liver.26 In this respect, it resembles highly metabolized metoprolol. When given orally both drugs undergo significant first pass metabolism.17, 26 Systemic BA of the labetalol varies from 11 to 86% (mean 33%) in hypertensive subjects.27 Labetalol is also absorbed rapidly with a time to reach maximum plasma concentration (tmax) of 0.8 h after the administration of 100 mg oral dose in healthy volunteers.28 According to the PK data compilation in literature, it is stated that labetalol is a well absorbed drug with Fabs >90 reported elsewhere.23, 26, 29–31 Surprisingly, the mass balance or PK data supporting Fabs >90 are not publicly available, though stated as such in references.20, 23 Labetalol has been categorized as Class I (high solubility-extensive metabolism) drug according to the Biopharmaceutics Drug Disposition Classification System (BDDCS) proposed by Wu and Benet. 32 However, data concerning the extent of metabolism for labetalol following I.V. dose in humans are not available.
It is also stated that the drug is completely absorbed from the GI tract in the FDA product information of Trandate® (labetalol hydrochloride).33 On the other hand, the FDA draft guidance on labetalol hydrochloride recommends single-dose, two-way crossover in vivo study.34 In contrast to the FDA current approach, it has been categorized as BCS Class I drug on the basis of the in vitro permeability measured using the Caco-2 cell line and solubility data.35
In the FDA laboratory, Volpe et al. validated a traditional Caco-2 cell permeability assay with over 20 model drugs with an initial drug concentration based on the HDS in 250 ml of water.19 Labetalol, near the low/high permeability class boundary, was determined to be the HP-IS within their laboratory. The assay was then employed to correctly classify four fluoroquinolone drugs (ciprofloxacin, levofloxacin, lomefloxacin and ofloxacin) as high or low permeability.18 Based on comparison to labetalol, ciprofloxacin was classified as a low permeability drug, whereas lemofloxacin, levofloxacin and ofloxacin were classified as high permeability drugs by the investigator although Caco-2 permeabilities of these drugs were lower than metoprolol’s. However, it was stated that the data showed active transport and/or efflux for these fluoroquinole drugs in the Caco-2 cells. Therefore, the use of a cellular model would not be acceptable for a biowaiver application with these drugs according to the BCS Guidance.
In another study by Thiel-Demby et al., labetalol was used as the reference drug defining the low/high permeability class boundary for the validated in vitro permeability assay using the MDCKII-MDR1 cell line to classify drugs according to the FDA Guidance on BCS.20 The investigators demonstrated a rank order relationship for BCS permeability classification and standardized the method according to the cell type, pH conditions, transport direction, incubation time, drug concentration and reference standards. Metoprolol, pindolol, labetalol and ranitidine were used as reference standards to show assay reproducibility in that study.20
In the present study, the intestinal permeability of labetalol compared to metoprolol was determined using both mouse and rat SPIP models. It was previously demonstrated that rat permeability data can be used to predict Fabs in human based on the correlation between the human and rat intestinal permeability of drugs with both carrier-mediated absorption and passive diffusion mechanism.10 On the other hand, in situ SPIP technique has also been applied in mouse model to determine the intestinal permeability of drugs in literature.36–38 Mouse perfusions generally require less active pharmaceutical ingredient (API) or drug candidate and may be a more useful preclinical model for predicting human absorption and BCS classification. More recently, Escribano et al. have assessed the intestinal perfusion technique in mouse using five model drugs proposed by FDA for establishing the suitability of a permeability method.39 The investigators showed a high correlation between Peff values of the model drugs in mouse and human. They also found that drug permeabilities in mouse were more similar to the values in human than those obtained in rat, suggesting the use of the mouse perfusion model to classify drugs into their appropriate BCS permeability class.39 In the present study, the jejunal Peff values obtained for metoprolol and labetalol were 1.7 and 1.6 times higher in mouse compared to rat, respectively. Thus, the in situ mouse intestinal perfusion is a suitable model to determine the permeability of drugs and to predict Fabs in human.
The present study showed that metoprolol and labetalol have similar permeabilities in the jejunal segments of rat and mouse, suggesting a rapid absorption of labetalol from the proximal jejunum like metoprolol. Therefore, the upper intestinal site in the GI tract seems to be the main absorption site for labetalol.
The physicochemical properties of drugs are of great impact on the GI absorption. In this respect, the chemical structure and physicochemical properties of metoprolol and labetalol can explain the high permeabilities of both drugs through the GI tract. The lipophilicities of metoprolol and labetalol are similar as the compounds have similar values of Log P and S+Log P (2.2 vs. 3.09 and 1.96 vs. 2.30, respectively) (Table 2). While metoprolol possesses a basic secondary amine functionality (pKa: 9.5140), labetalol is a diprotic molecule and has two acidity constants, which strengthen its amphiprotic behaviour. The first pKa value (7.4) can be attributed to the ionisation of the phenolic group, taking into account this group has more acid character than the secondary amine group.41 The unionized fraction (fu) of metoprolol is zero at low pH (up to pH 5.5) due to the protonation of the secondary amine and as pH rises, the fu increases to give the sigmoidal profile (~pH 11). The fu of labetalol is also zero at low pH (up to pH 4.5) due to protonation of the secondary amine. As pH rises, the fu of labetalol gradually increases, reaching a maximum at the isoelectric point of labetalol (pH=8.4). However, the isoelectric point of labetalol is beyond the physiologically relevant pH range. As pH is increased further above the isoelectric point, fu gradually decreases again to zero (~pH 11) as labetalol is ionized due to the presence of a phenolic group. Therefore, the high log P (3.09) and fu of labetalol can explain its high permeability at pH 6.5 and 7.5 observed in the present in situ study.
Table 2.
Physicochemical properties of metoprolol and labetalol
| Compound | Chemical Structure | pKa | Log P | S+Log Pa |
|---|---|---|---|---|
| Metoprolol |
|
9.5140 | 2.242 | 1.96 |
| Labetalol |
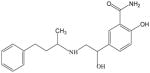
|
7.35 (OH), 9.42 (NH)41 | 3.0943 | 2.30 |
Calculated values using MedChem Designer™ Version 1.0.1.15, Simulations Plus, Inc.
The present study also showed that while labetalol and metoprolol exhibited similar in situ permeabilities, the permeability of labetalol was lower than that of metoprolol in Caco-2 cells at both AP pH 6.5 and 7.5. This was due to the P-gp-mediated efflux mechanism in the absorption of labetalol, since labetalol is known to be a substrate of P-gp.20, 44 It was also confirmed by the inhibition of P-gp-mediated transport of labetalol using verapamil in Caco-2 cells in the present study. The involvement of a carrier-mediated process in the absorption of labetalol was suggested in another study in which labetalol absorption kinetics was studied in rat small intestine and colon.44 The investigators found that labetalol is well absorbed from the intestine by means of a combined process of saturable absorption and passive diffusion. The inhibition studies showed that it is secreted from the enterocyte by the P-gp. They showed that verapamil produces a significant increase in the apparent absorption rate constant of labetalol according to the concentration of drug and the intestinal site. At the lowest concentration (10 mM), an increase of about 84% was observed, while at the highest concentration (100 mM) of labetalol, the increase was about 53% at the small intestine. It was also shown that the secretion process is saturated before the active one (around 500 mM), as explained by the corresponding Michaelis-Menten constants.44
It is well known that the carrier-mediated intestinal absorption of drugs is poorly predicted using Caco-2 cell model. It primarily measures the passive drug transport (both transcellular and paracellular), which is one of the limitations of Caco-2 cell model.
In the present study, Caco-2 permeabilities of metoprolol and labetalol decreased significantly as the pH of the AP side buffer was decreased (p=0.039 and p=0.004 for metoprolol and labetalol, respectively). The ER of metoprolol and labetalol increased remarkably (3.6 and 6.2 times, respectively) under AP pH 6.5. This can be easily misinterpreted as an indicator of active efflux.45 Since metoprolol is an apparently passively transported drug46 and it does not interact with P-gp20 while labetalol is a substrate of P-gp.20, 44 Thus, low AP pH may not be appropriate to study active efflux mechanism for basic drugs.45
The effect of AP pH on the absorptive permeability of 24 test compounds in rabbit intestinal tissue and Caco-2 monolayers were investigated in another study.47 The investigators found that Caco-2 permeability of acidic and basic compounds changed significantly upon the AP pH changed from 7.4 to 6.5. However, the rabbit intestinal permeability did not change, which was attributed to the presence of mucous layer that retains its microclimate pH regardless of the luminal pH. The investigators suggested that Caco-2 permeability at pH 7.4 is a good qualitative predictor for the physiological intestinal permeability from duodenum to colon.47
The result of the present SPIP study conducted in mice clearly showed that the in situ permeability of labetalol was dependent on the initial drug concentration. Decreasing concentrations of labetalol resulted in decreased permeability compared to the permeability of metoprolol, which may also be attributed to the P-gp effect on the drug absorption. However, it is known that the transporter effects may not be always clinically relevant. Since it is believed that almost all drugs are substrates for some transporters.48 Moreover, it has been shown that the P-gp plays a minimal role in the intestinal absorption process of the high permeability drugs like verapamil, although P-gp affects the BA of those drugs due to the drug efflux function in the liver and other organs.49
Investigations showed that the P-gp protein expression follows a gradient pattern, increasing from the proximal to the distal intestinal segments of intestine.50, 51 Therefore, it is also possible that the regional differences in the enzyme activity of the P-gp efflux function might contribute to the regional dependency for the absorption of labetalol at low concentrations. However, this process appears not to have a significant impact on the rate and extent of in vivo intestinal absorption of labetalol at therapeutic doses, as it is highly permeabl and the secretion process will be saturated at the high drug concentrations. The present study clearly showed that the in situ intestinal permeability of labetalol was similar to that of metoprolol at HDS. It is very likely that the passive diffusion dominates its intestinal absorption. In addition, there is no publicly available in vivo human data confirming the extent of absorption for labetalol.
At this time, there is no acceptance criterion for the degree of intestinal efflux or active transport that may be present in a permeability test system. It is clear that the variability of P-gp expression in various laboratories makes labetalol more risky to use as a reference compound for BCS in Caco-2 cell models.
It should be noted that metoprolol is a reliable high permeability reference standard based on explicit in vivo human data on the extent of absorption compared to labetalol. However, metoprolol is known to be a conservative reference for the low/high permeability class boundry, as it is absorbed completely. This is of great importance especially for the borderline drugs, which have permeabilities close to that of the internal reference standard. The permeability classification of these drugs is not easy because of the variability of the method used to determine permeability. The choice of internal reference standard can significantly impact the borderline between high and low permeability. To overcome this, statistical analysis employing the “0.8–1.25” confidence interval rule (in situ BE) was suggested to compare a drug’s in situ permeability with that of the IS.15 FDA 90% Fabs criterion is also considered to be conservative. EMA states that complete absorption is considered to be established where measured extent of absorption is ≥85%. An ≥85% cut-off is recommended and it is still under discussion.
In conclusion, the present study clearly demonstrated that labetalol exhibits high in situ intestinal permeability at the highest clinical dose, ensuring its high permeability classification. However, the IS intended to be used for BCS classification of drugs should not be subject to active or efflux transport processes. Thus, labetalol is not an ideal candidate for a high permeability IS for BCS classification, due to the concentration dependent permeability indicating the P-gp efflux process involved in its intestinal absorption.
Acknowledgments
This work was supported by NIH Grant NIGMD-2R01GM037188. T. Incecayir was supported by International Postdoctoral Research Scholarship Program (2219) from TUBITAK (The Scientific and Technological Research Council of Turkey) and Gazi University in Ankara, Turkey.
The authors acknowledge the discussion with Dr. Donna A. Volpe (Food & Drug Administration, Division of Drug Safety Research, Silver Spring, MD) and thank her for her constructive comments on this manuscript. Opinions expressed in this research are those of the authors and do not necessarily reflect the views or policies of the FDA.
References
- 1.U.S. Food and Drug Administration, Center for Drug Evaluation and Research (CDER) Guidance for Industry: Bioavailability and Bioequivalence Studies for Orally Administered Drug Products– General Considerations. Division of Drug Information Branch, HFD-240, Center for Drug Evaluation and Research, FDA; 5600 Fishers Lane, Rockville, MD 20857: Mar, 2003. < http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM070124.pdf. [Google Scholar]
- 2.European Medicines Agency (EMEA) Committee for Medicinal Products for Human Use. Guideline on the Investigation of Bioequivalence, Doc.Ref.: CPMP/EWP/QWP/1401/98Rev.1/Corr. EMEA; London: Jan 20, 2010. < http://www.emea.europa.eu/docs/en_GB/document_library/Scientific_guideline/2010/01/WC500070039.pdf>. [Google Scholar]
- 3.Amidon GL, Lennernäs H, Shah VP, Crison JR. A Theoretical Basis for a Biopharmaceutic Drug Classification: The Correlation of in Vitro Drug Product Dissolution and in Vivo Bioavailability. Pharm Res. 1995;12:413–420. doi: 10.1023/a:1016212804288. [DOI] [PubMed] [Google Scholar]
- 4.Polli JE, Abrahamsson BSI, Yu LX, Amidon GL, Baldoni JM, Cook JA, Fackler P, Hartauer K, Johnston G, Krill SL, Lipper RA, Malick WA, Shah VP, Sun D, Winkle HN, Wu Y, Zhang H. Summary Workshop Report: Bioequivalence, Biopharmaceutics Classification System, and Beyond. AAPS J. 2008;10:373–379. doi: 10.1208/s12248-008-9040-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chen ML, Amidon GL, Benet LZ, Lennernas H, Yu LX. The BCS, BDDCS, and Regulatory Guidances. Pharm Res. 2011;28:1774–1778. doi: 10.1007/s11095-011-0438-1. [DOI] [PubMed] [Google Scholar]
- 6.U.S. Food and Drug Administration, Center for Drug Evaluation and Research (CDER) Guidance for Industry: Waiver of In Vivo Bioavailability and Bioequivalence Studies for Immediate-Release Solid Oral Dosage Forms Based on a Biopharmaceutics Classification System. 2000 Aug; < http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/ucm070246.pdf>.
- 7.Cook J, Addicks W, Wu YH. Application of the Biopharmaceutical Classification System in Clinical Drug Development-An Industrial View. AAPS J. 2008;10:306–310. doi: 10.1208/s12248-008-9036-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ku MS. Use of the Biopharmaceutical Classification System in Early Drug Development. AAPS J. 2008;10:208–212. doi: 10.1208/s12248-008-9020-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lennernäs H, Abrahamsson B. The Use of Biopharmaceutic Classification of Drugs in Drug Discovery and Development: Current Status and Future Extension. J Pharm Pharmacol. 2005;57:273–285. doi: 10.1211/0022357055263. [DOI] [PubMed] [Google Scholar]
- 10.Cao X, Gibbs ST, Fang L, Miller HA, Landowski CP, Shin HC, Lennernas H, Zhong Y, Amidon GL, Yu LX, Sun D. Why is it Challenging to Predict Intestinal Drug Absorption and Oral Bioavailability in Human Using Rat Model. Pharm Res. 2006;23:1675–1686. doi: 10.1007/s11095-006-9041-2. [DOI] [PubMed] [Google Scholar]
- 11.Dahan A, Miller JM, Hilfinger JM, Yamashita S, Yu LX, Lennernäs H, Amidon GL. High-Permeability Criterion for BCS Classification: Segmental/pH Dependent Permeability Considerations. Mol Pharmaceutics. 2010;7:1827–1834. doi: 10.1021/mp100175a. [DOI] [PubMed] [Google Scholar]
- 12.Dahan A, Amidon GL. Grapefruit Juice and its Constituents Augment Colchicine Intestinal Absorption: Potential Hazardous Interaction and the Role of P-Glycoprotein. Pharm Res. 2009;26:883–892. doi: 10.1007/s11095-008-9789-7. [DOI] [PubMed] [Google Scholar]
- 13.Volpe DA. Drug-Permeability and Transporter Assays in Caco-2 and MDCK Cell Lines. Future Med Chem. 2011;3:2063–2077. doi: 10.4155/fmc.11.149. [DOI] [PubMed] [Google Scholar]
- 14.Hubatsch I, Ragnarsson EG, Artursson P. Determination of Drug Permeability and Prediction of Drug Absorption in Caco-2 Monolayers. Nat Protoc. 2007;2:2111–2119. doi: 10.1038/nprot.2007.303. [DOI] [PubMed] [Google Scholar]
- 15.Kim JS, Mitchell S, Kijek P, Tsume Y, Hilfinger J, Amidon GL. The Suitability of an in Situ Perfusion Model for Permeability Determinations: Utility for BCS Class I Biowaiver Requests. Mol Pharmaceutics. 2006;3:686–694. doi: 10.1021/mp060042f. [DOI] [PubMed] [Google Scholar]
- 16.Dahan A, Amidon GL. Segmental Dependent Transport of Low Permeability Compounds along the Small Intestine Due to P-Glycoprotein: The Role of Efflux Transport in the Oral Absorption of BCS Class III Drugs. Mol Pharmaceutics. 2008;6:19–28. doi: 10.1021/mp800088f. [DOI] [PubMed] [Google Scholar]
- 17.Regårdh CG, Borg KO, Johansson R, Johnsson G, Palmer L. Pharmacokinetic Studies on the Selective β1-Receptor Antagonist Metoprolol in Man. J Pharmacokinet Biopharm. 1974;2:347–364. doi: 10.1007/BF01061407. [DOI] [PubMed] [Google Scholar]
- 18.Volpe DA. Permeability Classification of Representative Fluoroquinolones by a Cell Culture Method. AAPS Pharm Sci. 2004;6:1–6. doi: 10.1208/ps060213. [DOI] [PubMed] [Google Scholar]
- 19.Volpe DA, Faustino PJ, Ciavarella AB, Asafu-Adjaye EB, Ellison CD, Yu LX, Hussain AS. Classification of Drug Permeability with a Caco-2 Cell Monolayer Assay. Clin Res Reg Affairs. 2007;24:39–47. [Google Scholar]
- 20.Thiel-Demby VE, Humphreys JE, Williams LA, St J, Ellens HM, Shah N, Ayrton AD, Polli JW. Biopharmaceutics Classification System: Validation and Learnings of an in Vitro Permeability Assay. Mol Pharmaceutics. 2008;6:11–18. doi: 10.1021/mp800122b. [DOI] [PubMed] [Google Scholar]
- 21.Jappar D, Wu SP, Hu Y, Smith DE. Significance and Regional Dependency of Peptide Transporter (PEPT) 1 in the Intestinal Permeability of Glycylsarcosine: In Situ Single-Pass Perfusion Studies in Wild-Type and Pept1 Knockout Mice. Drug Metab Dispos. 2010;38:1740–1746. doi: 10.1124/dmd.110.034025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Fagerholm U, Johansson M, Lennarnäs H. Comparison Between Permeability Coefficients in Rat and Human Jejunum. Pharm Res. 1996;13:1336–1342. doi: 10.1023/a:1016065715308. [DOI] [PubMed] [Google Scholar]
- 23.Meier J. Pharmacokinetic Comparison of Pindolol with Other Beta-Adrenoceptor-Blocking Agents. Am Heart J. 1982;104:364–373. doi: 10.1016/0002-8703(82)90127-2. [DOI] [PubMed] [Google Scholar]
- 24.Jobin G, Cortot A, Godbillon J, Duval M, Schoeller JP, Hirtz J, Bernier JJ. Investigation of Drug Absorption From the Gastrointestinal Tract of Man. I. Metoprolol in the Stomach, Duodenum and Jejunum. Br J Clin Pharmac. 1985;19:97S–105S. doi: 10.1111/j.1365-2125.1985.tb02749.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Vidon N, Evard D, Godbillon J, Rongier M, Duval M, Schoeller JP, Bernier JJ, Hirtz J. Investigation of Drug Absorption From the Gastrointestinal Tract of Man. II. Metoprolol in the Jejunum and Ileum. Br J Clin Pharmac. 1985;19:107S–112S. doi: 10.1111/j.1365-2125.1985.tb02750.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Martin LE, Hopkins R, Bland R. Metabolism of Labetalol by Animals and Man. Br J Clin Pharmac. 1976;3:695–710. [PubMed] [Google Scholar]
- 27.McNeil JJ, Anderson AE, Louis WJ, Morgan DJ. Pharmacokinetics and Pharmacodynamic Studies of Labetalol in Hypertensive Subjects. Br J Clin Pharmac. 1979;8:157S–161S. [PMC free article] [PubMed] [Google Scholar]
- 28.Fujimura A, Ohashi K, Tsuru M, Ebihara A, Kondo K Clinical Pharmacology of Dilevalol (I) Comparison of the Pharmacokinetic and Pharmacodynamic Properties of Dilevalol Labetalol After a Single Oral Administration in Healthy Subjects. J Clin Pharmacol. 1989;29:635–642. doi: 10.1002/j.1552-4604.1989.tb03392.x. [DOI] [PubMed] [Google Scholar]
- 29.Hoffman BB. Catecholamines, Sympathomimetic Drugs, and Adrenergic Receptor Antagonists. In: Hardman JG, Limbird LE, Gilman AG, editors. The Goodman and Gilman’s The Pharmacological Basis of Therapeutics. 10. McGraw-Hill Medical Publishing Div; New York: 2001. pp. 215–268. [Google Scholar]
- 30.Dollery C, Boobis AR, Burley D, Davies DM, Davies DS, Harrison PI, Orme MLE, Park BK, Goldberg LI, editors. Labetalol (Hydrochloride) Therapeutic Drugs. Vol. 2. Churchill Livingstone; Edinburgh: 1991. pp. L1–L4. [Google Scholar]
- 31.Wagner JG, Ganes DA, Midha KK, Gonzalez-Younes I, Sackellares JC, Olson LD, Affrime MB, Patrick JE. Stepwise Determination of Multicompartment Disposition and Absorption Parameters from Extravascular Concentration-Time Data. Application to Mesoridazine, Flurbiprofen, Flunarizine, Labetalol and Diazepam. J Pharmacokinet Biopharm. 1991;19:413–455. doi: 10.1007/BF01061665. [DOI] [PubMed] [Google Scholar]
- 32.Wu CY, Benet LZ. Predicting Drug Disposition via Application of BCS: Transport/Absorption/Elimination Interplay and Development of a Biopharmaceutics Drug Disposition Classification System. Pharm Res. 2005;22:11–23. doi: 10.1007/s11095-004-9004-4. [DOI] [PubMed] [Google Scholar]
- 33.Product Information Trandate® (labetalol hydrochloride) Tablets, FDA. < http://www.accessdata.fda.gov/drugsatfda_docs/label/2010/018716s026lbl.pdf>.
- 34.Draft Guidance on Labetalol Hydrochloride, FDA. < http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM194633.pdf>.
- 35.Yang Y, Faustino PJ, Volpe DA, Ellison CD, Lyon RC, Yu LX. Biopharmaceutics Classification of Selected β-Blockers: Solubility and Permeability Class Membership. Mol Pharmaceutics. 2007;4:608–614. doi: 10.1021/mp070028i. [DOI] [PubMed] [Google Scholar]
- 36.Moss AM, Endres CJ, Ruiz-Garcia A, Choi DS, Unadkat JD. Role of the Equilibrative and Concentrative Nucleoside Transporters in the Intestinal Absorption of the Nucleoside Drug, Ribavirin, in Wild-Type and Ent1(−/−) Mice. Mol Pharmaceutics. 2012;9:2442–2449. doi: 10.1021/mp200647a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Holmstock N, Annaert P, Augustijns P. Boosting of HIV Protease Inhibitors by Ritonavir in the Intestine: The Relative Role of Cytochrome P450 and P-Glycoprotein Inhibition Based on Caco-2 Monolayers versus In Situ Intestinal Perfusion in Mice. Drug Metab Dispos. 2012;40:1473–1477. doi: 10.1124/dmd.112.044677. [DOI] [PubMed] [Google Scholar]
- 38.Holmstock N, Mols R, Annaert P, Augustijns P. In Situ Intestinal Perfusion in Knockout Mice Demonstrates Inhibition of Intestinal P-Glycoprotein by Ritonavir Causing Increased Darunavir Absorption. Drug Metab Dispos. 2010;38:1407–1410. doi: 10.1124/dmd.110.032771. [DOI] [PubMed] [Google Scholar]
- 39.Escribano E, Sala XG, Salamanca J, Navarro CR, Regué JQ. Single-Pass Intestinal Perfusion to Establish the Intestinal Permeability of Model Drugs in Mouse. Int J Pharm. 2012;436:472–477. doi: 10.1016/j.ijpharm.2012.07.010. [DOI] [PubMed] [Google Scholar]
- 40.Shalaeva M, Kenseth J, Lombardo F, Bastin A. Measurement of Dissociation Constants (pKa values) of Organic Compounds by Multiplexed Capillary Electrophoresis Using Aqueous and Cosolvent Buffers. J Pharm Sci. 2008;97:2581–2606. doi: 10.1002/jps.21287. [DOI] [PubMed] [Google Scholar]
- 41.Martínez V, Maguregui MI, Jiménez RM, Alonso RM. Determination of the pKa Values of β-Blockers by Automated Potentiometric Titrations. J Pharm Biomed Anal. 2000;23:459–468. doi: 10.1016/s0731-7085(00)00324-1. [DOI] [PubMed] [Google Scholar]
- 42.Henchoz Y, Guillarme D, Martel S, Rudaz S, Veuthey JL, Carrupt PA. Fast log P Determination by Ultra-High-Pressure Liquid Chromatography Coupled With UV and Mass Spectrometry Detections. Anal Bioanal Chem. 2009;394:1919–1930. doi: 10.1007/s00216-009-2862-1. [DOI] [PubMed] [Google Scholar]
- 43.Burgot G, Serrand P, Burgot JL. Thermodynamics of Partitioning in the n-Octanol/Water System of Some β-Blockers. Int J Pharm. 1990;63:73–76. [Google Scholar]
- 44.Abushammala I, Garrigues TM, Casabó VG, Nácher A, Martín-Villodre A. Labetalol Absorption Kinetics: Rat Small Intestine and Colon Studies. J Pharm Sci. 2006;95:1733–1741. doi: 10.1002/jps.20639. [DOI] [PubMed] [Google Scholar]
- 45.Neuhoff S, Ungell AL, Zamora I, Artursson P. pH-Dependent Bidirectional Transport of Weakly Basic Drugs Across Caco-2 Monolayers: Implications for Drug-Drug Interactions. Pharm Res. 2003;20:1141–1148. doi: 10.1023/a:1025032511040. [DOI] [PubMed] [Google Scholar]
- 46.Lennernäs H, Palm K, Fagerholm U, Artursson P. Comparison Between Active and Passive Drug Transport in Human Intestinal Epithelial (Caco-2) Cells in Vitro and Human Jejunum in Vivo. Int J Pharm. 1996;127:103–107. [Google Scholar]
- 47.Lee KJ, Johnson N, Castelo J, Sinko PJ, Grass G, Holme K, Lee Y-H. Effect of Experimental pH on the in Vitro Permeability in Intact Rabbit Intestines Caco-2 Monolayer. Eur J Pharm Sci. 2005;25:193–200. doi: 10.1016/j.ejps.2005.02.012. [DOI] [PubMed] [Google Scholar]
- 48.Custodio JM, Wu CY, Benet LZ. Predicting Drug Disposition, Absorption/Elimination/Transporter Interplay and the Role of Food on Drug Absorption. Adv Drug Deliv Rev. 2008;60:717–733. doi: 10.1016/j.addr.2007.08.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Cao X, Yu LX, Barbaciru C, Landowski CP, Shin HC, Gibbs S, Miller HA, Amidon GL, Sun D. Permeability Dominates in Vivo Intestinal Absorption of P-gp Substrate With High Solubility and High Permeability. Mol Pharmaceutics. 2005;2:329–340. doi: 10.1021/mp0499104. [DOI] [PubMed] [Google Scholar]
- 50.Dahan A, Sabit H, Amidon GL. Multiple Efflux Pumps are Involved in the Transepithelial Transport of Colchicine: Combined Effect of P-Glycoprotein and Multidrug Resistance-Associated Protein 2 Leads to Decreased Intestinal Absorption Throughout the Entire Small Intestine. Drug Metab Dispos. 2009;37:2028–2036. doi: 10.1124/dmd.109.028282. [DOI] [PubMed] [Google Scholar]
- 51.Valenzuela B, Nácher A, Ruiz-Carretero P, Martín-Villodre A, López-Carballo G, Barettino D. Profile of P-Glycoprotein Distribution in the Rat and its Possible Influence on the Salbutamol Intestinal Absorption Process. J Pharm Sci. 2004;93:1641–1648. doi: 10.1002/jps.20071. [DOI] [PubMed] [Google Scholar]