

Abstract
The zebrafish has emerged as a powerful model organism for studying intestinal development1-5, physiology6-11, disease12-16, and host-microbe interactions17-25. Experimental approaches for studying intestinal biology often require the in vivo introduction of selected materials into the lumen of the intestine. In the larval zebrafish model, this is typically accomplished by immersing fish in a solution of the selected material, or by injection through the abdominal wall. Using the immersion method, it is difficult to accurately monitor or control the route or timing of material delivery to the intestine. For this reason, immersion exposure can cause unintended toxicity and other effects on extraintestinal tissues, limiting the potential range of material amounts that can be delivered into the intestine. Also, the amount of material ingested during immersion exposure can vary significantly between individual larvae26. Although these problems are not encountered during direct injection through the abdominal wall, proper injection is difficult and causes tissue damage which could influence experimental results.We introduce a method for microgavage of zebrafish larvae. The goal of this method is to provide a safe, effective, and consistent way to deliver material directly to the lumen of the anterior intestine in larval zebrafish with controlled timing. Microgavage utilizes standard embryo microinjection and stereomicroscopy equipment common to most laboratories that perform zebrafish research. Once fish are properly positioned in methylcellulose, gavage can be performed quickly at a rate of approximately 7-10 fish/ min, and post-gavage survival approaches 100% depending on the gavaged material. We also show that microgavage can permit loading of the intestinal lumen with high concentrations of materials that are lethal to fish when exposed by immersion. To demonstrate the utility of this method, we present a fluorescent dextran microgavage assay that can be used to quantify transit from the intestinal lumen to extraintestinal spaces. This test can be used to verify proper execution of the microgavage procedure, and also provides a novel zebrafish assay to examine intestinal epithelial barrier integrity under different experimental conditions (e.g. genetic manipulation, drug treatment, or exposure to environmental factors). Furthermore, we show how gavage can be used to evaluate intestinal motility by gavaging fluorescent microspheres and monitoring their subsequent transit. Microgavage can be applied to deliver diverse materials such as live microorganisms, secreted microbial factors/toxins, pharmacological agents, and physiological probes. With these capabilities, the larval zebrafish microgavage method has the potential to enhance a broad range of research fields using the zebrafish model system.
Keywords: Developmental Biology, Issue 72, Molecular Biology, Anatomy, Physiology, Basic Protocols, Surgery, Zebrafish, Danio rerio, intestine, lumen, larvae, gavage, microgavage, epithelium, barrier function, gut motility, microsurgery, microscopy, animal model
Protocol
We use published protocols for standard zebrafish husbandry and maintenance to obtain larvae for microgavage27. We primarily use natural breeding to propagate fish, and they are kept on a 14 hr light/10 hr dark cycle. This protocol has been optimized for microgavage of zebrafish larvae at 6-7 days post-fertilization (dpf). We anticipate that these methods will be generally applicable to younger and older developmental stages with minor modifications. Unless otherwise noted, we used the wild-type TL zebrafish strain at 6 dpf, and the fish were not fed. All experiments were performed in system water acquired from our conventional zebrafish aquaculture facility; however, any appropriate zebrafish media (e.g. embryo medium or gnotobiotic zebrafish medium (GZM)28) can be used. In this protocol, we refer to 'zebrafish media' to represent the user's media of choice.
1. Preparation of Methylcellulose, Gavage Mold, and Microgavage Needle Fabrication
The day before or early the day of the experiment, prepare the methylcellulose, agarose mold for larvae stabilization, and the microgavage needles.
Dissolve 3 g of methylcellulose in 100 ml of sterile, deionized water or zebrafish media (depending on your application) to make a 3% solution. Add a magnetic stir bar to the bottle, heat water to close to boiling, and place on a stir plate at high heat. While stirring, slowly sprinkle in methylcellulose to ensure it does not clump. Slowly decrease the speed of stirring and temperature of the stir plate throughout the day until it is room temperature.
Continue stirring slowly overnight until the methylcellulose is completely dissolved. Solution can be stored at room temperature for several months in a sealed container.
Prepare a 3% agarose gel mold in a 10 cm Petri dish using a plastic cast (Adaptive Science Tools, TU-1) according to the manufacturer's instructions. Seal with parafilm and store at 4 °C until needed to prevent desiccation. Warm up to room temperature or 28 °C prior to experiment. Agarose molds can generally be re-used several times.
Pull several gavage needles using borosilicate glass capillaries, and a Flaming Brown Micropipette Puller fitted with a wide-trough heat filament (FT330B). The programs we use for needle fabrication are as follows:
| Needle type | Pressure | Heat | Pull | Velocity | Time |
| embryo | 500 | 315 | 100 | 50 | 200 |
| gavage | 500 | 315 | 125 | 75 | 200 |
Since filaments can vary between instruments, record the ramp time (active heat time). Our ramp time ranges between 8.57-8.79 ms for this program. This program creates shorter, stockier needles than traditional microinjection needles (Figure 1). These settings may need to be optimized for use in different micropipette pullers.
2. Clipping, Loading, and Calibration of the Microgavage Needles
Note*: The needle clipping is a critical step in this protocol. Also, the methods for needle loading and calibration will vary depending on the type of microinjection unit being used. We have optimized this protocol using Drummond II microinjectors, but we anticipate that this protocol will also be applicable to other microinjection instruments with minor modification. Since injection rigs and needle pullers will vary between labs, it will be important to measure/calibrate the injection volumes (see section 2.8).
The needles are clipped under a stereomicroscope. We use a Leica S6E stereomicroscope set to 2.5x magnification. At this setting, our eyepiece graticle with 100 divisions is 2 mm in length.
Align the tip of the needle with the end of the graticle ruler (division mark 100), and clip the needle using fine-tipped watchmaker forceps at ~1 mm from the tip (division mark 48-50) (Figure 1A).
The needle should be ~27-30 μm in diameter and blunt. Examine the needle tips at 100x magnification. Sharp or jagged needles lead to higher probability of damage during gavage and should be avoided if possible.
Alternatively, when available, a microforge can be used to better control the needle clip point and to fire-polish the needle tips to remove sharp edges (Figure 1B).
Prepare the gavage solution. For most applications, we prepare the solution with addition of a 1:10 dilution of a 0.5% phenol red solution in Dulbecco's phosphate buffered saline (DPBS) (Sigma-Aldrich) to ensure that the solution can be seen under the stereomicroscope to monitor proper functioning of the microinjector and placement of the gavage solution in the anterior bulb. Loading dyes other than phenol red have not been evaluated but could be used.
Prepare the gavage needle by filling with mineral oil using a 1 ml syringe with a 25G 5/8-needle. Mount onto the Nanoject II microinjection unit.
Wrap a plastic 10 cm Petri dish with parafilm to provide a flat, waterproof surface and aliquot ~2 μl of the gavage solution onto the parafilm. Manually backfill the gavage needle being careful to avoid creating air bubbles in the mineral oil.
To test microinjector function and calibrate the ejection volume, fill a 3 cm Petri dish with mineral oil and inject into the oil until the droplets are consistent in size. Measure the diameter of the droplets using the graticle or a measurement function on the stereomicroscope, if available. Calculate the droplet volume (Equation: V=4/3πr3 ). Volume consistency between needles is plotted (Figure 1C).
Rinse the tip of the needle by repeated submersions in clean zebrafish media to remove residual mineral oil before starting gavage. Mineral oil is very dense and may cause damage to epithelial tissues if not washed off of the needle tip.
3. Anesthetizing, Mounting, and Gavage of Zebrafish Larvae
Warm the agarose mold at room temperature or 28 °C prior to experiment.
Prepare the agarose mold for gavage by covering 3 grooves with 3% methylcellulose. Use an amount of methylcellulose sufficient to cover the grooves and hold the fish, but not so scant that it dries out quickly.
In a clean 6-well plate, aliquot zebrafish larvae for each experimental group into separate wells, each containing 3.5-4.0 ml of zebrafish media.
Prepare 3x tricaine solution (0.05% w/v) in the appropriate zebrafish medium. Anesthetize fish one well at a time by mixing an equal volume of 3x tricaine into the well containing larvae for a final concentration of 1.5x (0.025%), and swirl gently.
As soon as the zebrafish stop moving, remove them from the well using a wide bore glass Pasteur pipette and pipette pump, and place them one by one onto the methylcellulose with their heads on the 45° angle of the groove and their tails laid toward the 90° angle of the groove. Gently press them into the methylcellulose with a blunt dissection probe to stabilize their position.
Adjust the Nanoject II microinjection rig such that the needle is tilted at a shallow angle that is approximately parallel to the 45° angle of the agarose mold. Make sure that the extension range of the needle will be sufficient to reach past the bottom of the mold well so that fewer adjustments have to be made once fish are in place.
Set the microinjection unit to release 4.6 nl (maximum) on the slow inject setting (23 nl/sec). At these settings, the delivered volume should just fill the anterior bulb of the intestine and not leak out of the esophagus or cloaca. Smaller volumes may be used depending on the purpose of the experiment.
Use one hand to make minor adjustments of the plate containing the immobilized larvae while simultaneously using the other to control the manipulator of the microinjection unit.
Gently maneuver the gavage needle into the mouth of anesthetized fish, through the esophagus, and slightly depress the esophageal sphincter to introduce the tip of the gavage needle just inside the anterior intestinal bulb (Figure 2, step 1-2, a&b).
Once inside the anterior bulb, gently depress the foot pedal or inject button to administer the material. Retract the needle quickly and smoothly trying not to release significant quantities of material into the esophagus (Figure 2, step 3-4).
Following gavage, use a wide-bore glass Pasteur pipette and pipette pump to gently move the zebrafish larvae to fresh media. Fill the pipette with water to about 1 inch above the neck bend, make a small water pocket in the methylcellulose by the larva's head to release it, lift the head gently into the mouth of the pipette, and draw the larva out of the methylcellulose. Gently expel fish and all water from the pipette into a dish of fresh media.
Rinse the larvae in fresh media by pipetting up and down gently or splashing with media several times to remove methylcellulose and revive the larvae from anesthesia. Transfer larvae into a Petri dish or 6-well plate until needed for subsequent imaging and analysis.
4. Dextran Assay to Test Gavage Safety and Intestinal Barrier Integrity
Microgavage can be used to deliver diverse materials into the intestine. As an example, we provide a protocol below for gavaging a 10 kD dextran conjugated to Texas Red to analyze barrier integrity of the intestinal epithelium. At least two potential routes of paracellular permeability exist, with differences in selectivity based on charge and size of the solutes, termed the "pore" or "leak" pathways29. Since this dextran is too large to be absorbed across the intestinal epithelium through the paracellular pore pathway (size limitation ~4Å, 10kD dextran= ~23Å)9,30-32, it should be retained within the lumen if intestinal barrier integrity is not compromised. Although 10 kD dextran could be transported via the paracellular leak pathway, this pathway has lower capacity and slower kinetics29,33, and, therefore, is not likely to occur within the time-frame of this assay. A 'positive control' treatment to disrupt barrier integrity is provided by inclusion of EDTA which disrupts epithelial tight junctions34,35. This dextran gavage assay can be used as a quality control test to demonstrate that the gavage procedure is being performed safely without unintended tissue damage. Additionally, this assay can be used to test intestinal barrier integrity as a function of genotype or treatment.
Prepare the following gavage solutions: 1% dextran/1x PBS/0.05% phenol red and 1% dextran/20 mM EDTA/1x PBS/0.05% phenol red (positive control). The 5% dextran stock in ddH2O is stored in 50 μl aliquots at -20 °C to reduce the number of freeze-thaw cycles. Also, prepare a mock gavage solution of 1x PBS/0.05% phenol red.
| + control (EDTA) | no EDTA | ||||
| stock | final | dilution factor | 10 μl | 10 μl | |
| EDTA, pH 8 | 200 mM | 20 mM | 10 | 1 | 0 |
| Dextran | 5% | 1% | 5 | 2 | 2 |
| PBS | 10x | 1x | 10 | 1 | 1 |
| Phenol Red | 0.50% | 0.05% | 10 | 1 | 1 |
| subtotal: | 5 | 4 | |||
| ddH2O: | 5 | 6 | |||
| Total: | 10 | 10 |
Use the protocol in section 3 to gavage groups of 10-20 zebrafish larvae for each experimental condition. Repeat this on duplicate or triplicate groups if possible.
After gavage, recover larvae from anesthesia and allow them to swim freely in fresh media.
At 18-20 min post-gavage, re-anesthetize the larvae as in step 3.3. Position the larvae in 3% methylcellulose on top of a 3% agarose block (this reduces background glare during imaging).
Image the larvae with a fluorescence stereoscope (e.g., Leica M205C) and a Texas Red filter set at a magnification that displays the larvae from the snout to immediately posterior to the end of the cloaca.
Acquire all images at the same exposure settings so that they can be quantified and compared later. Fluorescence will be intense in the intestinal lumen of all dextran-injected larvae, but barrier function is evaluated by the level of dextran that appears in extraintestinal tissues (i.e., the trunk and blood vessels) (Figure 5). Adjust the exposure to settings that enable visualization of trunk fluorescence even if this over-exposes lumen fluorescence.
Stagger the gavage times so that each group spends approximately the same amount of time in recovery after gavage and is imaged at the same approximate time post-gavage. For this experiment, each group takes approximately 1 hr for one person to complete. Fish are anesthetized at 18-20 min post-gavage, and imaging takes ~1-2 min per fish.
Use an image analysis program such as ImageJ to quantify the relative mean fluorescence intensity in a region of the trunk just above the intestine. Normalize these values by subtracting a background measurement taken in a region of the image outside of the fish.
Graph and analyze data from image analysis using GraphPad Prism or similar statistical software.
5. Fluorescent Microsphere Assay to Test Intestinal Motility
Gavage provides enhanced control over timing and amount of material delivery to the intestine which makes it an ideal alternative method for intestinal motility studies. Previously published techniques for measuring intestinal motility in zebrafish larvae include direct observation of intestinal contractions and waves by microscopy1,21,36, or feeding larvae with food blended with yellow-green fluorescent polystyrene microspheres26. In some studies, quantitation of intestinal motility patterns has been enhanced by immersing larvae in media containing food dye37 or a specialized video analysis technique called spatiotemporal mapping38,39. Here we provide a proof-of-concept study to show that it is possible to gavage fluorescent microspheres into the intestinal anterior bulb, and tracking of microsphere movement can be utilized to assess intestinal motility (Figure 6). The primary difference between this method and the approach of Field and colleagues26 is that the beads are introduced alone and cannot be incorporated into food due to the gavage needle diameter limits.
Maintain zebrafish larvae in 10 cm plates with 35 ml media until the experiment is performed at 7 dpf. Begin feeding larvae once each morning starting at 5 dpf with ~0.6 mg powdered food (larvae diet, as previously described40; administered using a sterile, 1 mm inoculating loop).
Gently shake the bottle of Fluoresbrite YG 2.0 μm polystyrene microspheres (~2.5% aqueous suspension) to mix the suspension. Aliquot a drop from the bottle onto parafilm so that a specific volume can be measured by pipetting.
In an eppendorf tube, centrifuge the beads at low speed in a benchtop microfuge, remove the supernatant, and replace with 1x PBS at the original volume. Repeat this process to wash the microspheres 2 times.
Prepare a gavage suspension containing 0.25% microspheres (1:10 dilution of stock suspension)/1x PBS/0.05% phenol red. There are ~2.6x103 microspheres in the 4.6 nl gavage volume administered per larvae.
Use the protocol in section 3 to gavage the desired number of larvae. Microspheres are somewhat buoyant; therefore, gavage should be performed as rapidly as possible to maximize consistency of the suspension.
In this experiment, 15 larvae were gavaged and then maintained in single wells of a 12-well plate following recovery from anesthesia in order to follow transit in individuals. However, larger groups can also be gavaged and transit assessed as a population rather than individually. Refer to Field et al. for standard practices26.
Assess intestinal motility by scoring the most rostral (or anterior) location of the fluorescent microspheres in live larvae at different times using a fluorescent stereomicroscope. Briefly anesthetize the larvae in 1x (0.017%) tricaine, and score the zones of microsphere location or capture images to score later.
Graph and compare the percent of total larvae with microspheres in a certain intestinal region over time.
Representative Results
When gavage is performed properly, the delivered material should be entirely contained within the anterior intestine with little to no residual material in the esophagus (Figure 3). A delivery volume should be chosen that can be accommodated within the anterior bulb and does not leak out through cloaca or esophagus. If the volume of material or pressure of delivery is too high, then the physical force of gavage may lead to damage of or leakage through the epithelium. It may also push out or alter other components of the luminal milieu (e.g., bile or mucus) that could change the local biochemical environment and potentially confound results. In general, quick retraction of the needle following administration of the material should allow the esophageal sphincter to close and prevent significant leakage into the esophagus.
This gavage method provides improved control over the timing and route of material exposure to larval zebrafish tissues compared to the immersion method (Figure 3). In larvae gavaged with the fluorescent endocytic tracer FM4-64FX, the amount of material being delivered to intestinal tissues is more consistent than the larvae that are immersed in the probe (Figure 3A-B). In larvae immersed for ~1 hr, FM4-64FX was detectable in external epithelial tissues throughout the body, but there was relatively little signal detected in the intestinal lumen (Figure 3A.ii) compared to the gavaged larvae (Figure 3B.ii-iv). Detectable levels of FM4-64FX were eventually observed in the intestinal epithelium of soaked larvae, but only after greater than 2 hr of immersion (Figure 3A.iv). For this reason, microgavage may be a better method to deliver probes for studying kinetics of intestinal transit and motility.
Microgavage also permits delivery of high concentrations of compounds that would otherwise cause extra-intestinal toxicity and decreased survival of zebrafish larvae in immersion experiments. When immersed in FM4-64FX at concentrations of ≥10 μM, larvae begin to die within 10-15 min of treatment, and all of the larvae are dead within 60 min of initiating exposure. In contrast, 100% of larvae survived gavage of FM4-64FX at concentrations up to 100-fold higher within the same time frame (Figure 3C). For those larvae surviving at 100 min post- gavage, subsequent survival through 24 hr after treatment was almost 100% (data not shown) establishing that the gavage technique itself is not harmful to the animals.
We developed an assay to monitor damage of the intestinal barrier during microgavage by using a TxRed-labeled dextran (10,000 MW). The gavage procedure itself does not generally lead to disruption or damage of the intestinal barrier in zebrafish larvae (Figure 4). When 10 kD dextran is gavaged alone, it is largely maintained within the intestinal lumen (Figure 4A, top panel), and some is absorbed into enterocytes in the mid-intestine (data not shown). As a positive control, dextran is co-gavaged with EDTA which disrupts epithelial tight junctions and allows the dextran to reach the basolateral side of the epithelium where it is dispersed to other tissues (Figure 4A, bottom panel). High magnification confocal imaging of EDTA treated larvae reveals that dextran enters circulation (i.e. in the lumen of the dorsal aorta (DA), posterior cardinal vein (PCV), and some intersomitic vessels (ISV)) and infiltrates spaces adjacent to the vasculature (Figure 5). Some variability is seen with this assay (e.g., some extra-intestinal fluorescence in dextran only-gavaged larvae, and low body fluorescence in dextran-EDTA gavaged) (Figure 4B). However, when averaged over 10-20 larvae, statistically significant differences are consistently observed between the dextran and dextran-EDTA gavaged groups (Figure 4B). Using a microforge to produce consistently blunted and fire-polished gavage needles decreases the risk of damage and reduces the number of larvae in which dextran alone reaches extra-intestinal sites (Figure 4B, right panel).
To evaluate the potential for using this method to study intestinal motility, we gavaged a suspension of fluorescent microspheres into the anterior bulb and followed their movement through the intestine over time (Figure 6A). We followed the scoring system of Field and colleagues26 to facilitate comparison with published intestinal transit rates. Our data show that microsphere transit through the intestine occurred at a similar rate and frequency distribution when compared to similar time points from the Field et al.26 study (Figure 6B). Within 24 hr, ~50% of the larvae had completely evacuated all of the microspheres, and in the remaining larvae, residual beads were in more caudal transit zones (Figure 6B). These data suggest that gavage alone with the volumes presented here does not cause gut stasis or significantly alter the overall rate of intestinal motility.
 Figure 1. Gavage needle production, clipping, and calibration. (A) Comparison of needles manufactured for embryo injection versus gavage. Needles for gavage are shorter and have an increased slope angle. They are easier to measure for clipping and they are slightly less flexible which is necessary to depress the esophageal sphincter for access to the intestine. Red lines indicate ~1 mm from tip where the needle is clipped. Scale bar = 1 mm. (B) Examples of clipped gavage needles. The diameter of the gavage needles ranges between 27-30 μm, and should be as blunt and smooth as possible. Microforging produces consistently blunted needles. (C) Calibration of gavage volume by measuring diameter of droplets formed in mineral oil. In this protocol, our gavage volume is ~4.6 nl. Scale bar = 100 μm.
Figure 1. Gavage needle production, clipping, and calibration. (A) Comparison of needles manufactured for embryo injection versus gavage. Needles for gavage are shorter and have an increased slope angle. They are easier to measure for clipping and they are slightly less flexible which is necessary to depress the esophageal sphincter for access to the intestine. Red lines indicate ~1 mm from tip where the needle is clipped. Scale bar = 1 mm. (B) Examples of clipped gavage needles. The diameter of the gavage needles ranges between 27-30 μm, and should be as blunt and smooth as possible. Microforging produces consistently blunted needles. (C) Calibration of gavage volume by measuring diameter of droplets formed in mineral oil. In this protocol, our gavage volume is ~4.6 nl. Scale bar = 100 μm.
 Figure 2. Larval zebrafish and gavage diagram. (A) Diagram of the larval zebrafish at 6 dpf. NC=notochord, SB=swim bladder, ES=esophageal sphincter, eso=esophagus, OV=otic vesicle, ISV= intersomitic vessels. (B) Diagram of gavage procedure. 1. Gavage needle is maneuvered into the mouth of the larvae. 2. Needle is advanced up to the esophageal sphincter, and then the needle is used to depress the sphincter in a ventral direction (a) and further advanced to just inside the anterior intestine (b). 3. Material is administered into the intestinal lumen. The volume should be just enough to fill the anterior bulb of the intestine without significant leakage out through the cloaca or esophagus. 4. The needle is quickly and gently retracted from the animal. Material should be mostly contained in the intestine, although negligible amounts are sometimes left in the esophagus.
Figure 2. Larval zebrafish and gavage diagram. (A) Diagram of the larval zebrafish at 6 dpf. NC=notochord, SB=swim bladder, ES=esophageal sphincter, eso=esophagus, OV=otic vesicle, ISV= intersomitic vessels. (B) Diagram of gavage procedure. 1. Gavage needle is maneuvered into the mouth of the larvae. 2. Needle is advanced up to the esophageal sphincter, and then the needle is used to depress the sphincter in a ventral direction (a) and further advanced to just inside the anterior intestine (b). 3. Material is administered into the intestinal lumen. The volume should be just enough to fill the anterior bulb of the intestine without significant leakage out through the cloaca or esophagus. 4. The needle is quickly and gently retracted from the animal. Material should be mostly contained in the intestine, although negligible amounts are sometimes left in the esophagus.
 Figure 3. Gavage and immersion of FM4-64FX provide distinct routes of exposure and differ in associated toxicity. Representative images of wild-type TL zebrafish larvae at 6 dpf immersed or gavaged with the fluorescent endocytosis probe, FM4-64FX41. Unless noted otherwise, pictures were taken at ~1 hr following the start of immersion or post-gavage. (A) In immersed larvae, the probe was absorbed at sites throughout the body and resulted in abundant extra-intestinal fluorescence that obscured signal in the intestine (A.ii-iii). Larvae immersed in 50 μm FM4-64FX died within 10 min so the image shown is from 7 min post-gavage (A.iii). No observable signal was seen at 2 hr of immersion, but by 5 hr post-immersion intestinal staining was increased (A.iv). (B) In the gavaged larvae, fluorescence was restricted to the intestinal lumen and fluorescent signals were more consistent (B.ii-iv). (C) Comparison of larval survival in different concentrations of FM4-64FX. Survival of gavaged larvae was 100% in all concentrations while larvae immersed in concentrations up to 100-fold lower died within 30 min (N=39-41 larvae/condition over 3 individual experiments). Bars represent ± SEM.
Figure 3. Gavage and immersion of FM4-64FX provide distinct routes of exposure and differ in associated toxicity. Representative images of wild-type TL zebrafish larvae at 6 dpf immersed or gavaged with the fluorescent endocytosis probe, FM4-64FX41. Unless noted otherwise, pictures were taken at ~1 hr following the start of immersion or post-gavage. (A) In immersed larvae, the probe was absorbed at sites throughout the body and resulted in abundant extra-intestinal fluorescence that obscured signal in the intestine (A.ii-iii). Larvae immersed in 50 μm FM4-64FX died within 10 min so the image shown is from 7 min post-gavage (A.iii). No observable signal was seen at 2 hr of immersion, but by 5 hr post-immersion intestinal staining was increased (A.iv). (B) In the gavaged larvae, fluorescence was restricted to the intestinal lumen and fluorescent signals were more consistent (B.ii-iv). (C) Comparison of larval survival in different concentrations of FM4-64FX. Survival of gavaged larvae was 100% in all concentrations while larvae immersed in concentrations up to 100-fold lower died within 30 min (N=39-41 larvae/condition over 3 individual experiments). Bars represent ± SEM.
 Figure 4. Dextran assay for epithelial barrier integrity. (A) Zebrafish wild-type TL larvae at 6 dpf were gavaged with a 1% TxR-dextran (10,000 MW) solution with or without 20 mM EDTA. Without EDTA, the dextran is retained within the lumen of the intestine. In larvae co-gavaged with EDTA, dextran is seen in circulation (white arrow, dorsal aorta) and intersomitic vessels (white arrowheads) and spaces. Scale bar = 200 μm. All pictures were acquired with the same magnification and exposure settings. (B) ImageJ was used to quantitate the relative mean fluorescence in an ROI in the zebrafish larvae trunk above segment 2 and 3 of the intestine (trunk ROI; small white dashed rectangle in A), and normalized to an ROI outside of the fish (bkgd ROI) as an internal control to account for background glare. One representative experiment is presented using a regular needle (N = 20/group) or a microforged needle (N = 15/group). Each point denotes normalized mean fluorescence in the trunk for one larva. Bars represent mean ± SEM. A non-parametric Mann-Whitney t-test was performed (***p<0.001).
Figure 4. Dextran assay for epithelial barrier integrity. (A) Zebrafish wild-type TL larvae at 6 dpf were gavaged with a 1% TxR-dextran (10,000 MW) solution with or without 20 mM EDTA. Without EDTA, the dextran is retained within the lumen of the intestine. In larvae co-gavaged with EDTA, dextran is seen in circulation (white arrow, dorsal aorta) and intersomitic vessels (white arrowheads) and spaces. Scale bar = 200 μm. All pictures were acquired with the same magnification and exposure settings. (B) ImageJ was used to quantitate the relative mean fluorescence in an ROI in the zebrafish larvae trunk above segment 2 and 3 of the intestine (trunk ROI; small white dashed rectangle in A), and normalized to an ROI outside of the fish (bkgd ROI) as an internal control to account for background glare. One representative experiment is presented using a regular needle (N = 20/group) or a microforged needle (N = 15/group). Each point denotes normalized mean fluorescence in the trunk for one larva. Bars represent mean ± SEM. A non-parametric Mann-Whitney t-test was performed (***p<0.001).
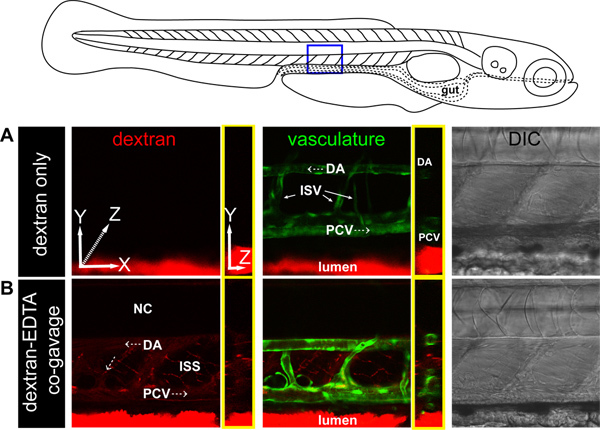 Figure 5. Dextran is released into extraintestinal sites after injury. Confocal imaging of 6 dpf Tg(kdrl:EGFP)s843 zebrafish larvae (green vasculature)42 gavaged with 1% dextran with or without 20 mM EDTA. (A) In single confocal slices of a region above the distal intestine, gavaged TxR-dextran remains restricted to the intestinal lumen without leaking into the vasculature or extra-intestinal spaces. (B) When co-gavaged with EDTA, TxR-dextran is observed in extraintestinal spaces and within the lumen of the dorsal aorta (DA), posterior cardinal vein (PCV), intersomitic vessels (ISV), and in intersomitic spaces (ISS) adjacent to ISVs. Blurred signal is due to blood flow (direction of blood flow indicated by white dotted arrows). All images are Z-axis projections of lateral views except the images bounded in yellow which show transverse projections along the Z-axis.
Figure 5. Dextran is released into extraintestinal sites after injury. Confocal imaging of 6 dpf Tg(kdrl:EGFP)s843 zebrafish larvae (green vasculature)42 gavaged with 1% dextran with or without 20 mM EDTA. (A) In single confocal slices of a region above the distal intestine, gavaged TxR-dextran remains restricted to the intestinal lumen without leaking into the vasculature or extra-intestinal spaces. (B) When co-gavaged with EDTA, TxR-dextran is observed in extraintestinal spaces and within the lumen of the dorsal aorta (DA), posterior cardinal vein (PCV), intersomitic vessels (ISV), and in intersomitic spaces (ISS) adjacent to ISVs. Blurred signal is due to blood flow (direction of blood flow indicated by white dotted arrows). All images are Z-axis projections of lateral views except the images bounded in yellow which show transverse projections along the Z-axis.
 Figure 6. Analysis of intestinal motility by fluorescent microsphere gavage. Fed wild-type TL zebrafish larvae were gavaged at 7 dpf with a suspension of 0.25% fluorescent microspheres/1x PBS/0.05% phenol red. Microsphere transit through the intestine was followed by live stereomicroscopy at the indicated time points post-gavage. (A) Example of intestinal transit within one larva over time. Intestinal zones are indicated in the top overlay image and follow the convention of Field et al26. Images below show green fluorescent signal and the location of the intestine is outlined (white dashed line). Scale bar = 200 μm. (B) The most rostral location of microspheres was used to determine the transit zone scores. Bars represent the percent of total larvae containing microspheres in that zone (N = 3-15), and numbers at the top of each graph indicate the time elapsed after gavage. Click here to view larger figure.
Figure 6. Analysis of intestinal motility by fluorescent microsphere gavage. Fed wild-type TL zebrafish larvae were gavaged at 7 dpf with a suspension of 0.25% fluorescent microspheres/1x PBS/0.05% phenol red. Microsphere transit through the intestine was followed by live stereomicroscopy at the indicated time points post-gavage. (A) Example of intestinal transit within one larva over time. Intestinal zones are indicated in the top overlay image and follow the convention of Field et al26. Images below show green fluorescent signal and the location of the intestine is outlined (white dashed line). Scale bar = 200 μm. (B) The most rostral location of microspheres was used to determine the transit zone scores. Bars represent the percent of total larvae containing microspheres in that zone (N = 3-15), and numbers at the top of each graph indicate the time elapsed after gavage. Click here to view larger figure.
Discussion
In this work, we describe a novel protocol for direct delivery of materials to the larval zebrafish intestine by microgavage. There are several critical steps throughout the procedure that should be kept in mind. First, the zebrafish larvae should be in good health before the gavage experiment to prevent death unrelated to treatment. Another important factor is the quality of the gavage needle. One of the most difficult parts of the protocol is clipping the needle at the appropriate location without creating sharp, jagged edges. A micropipette microforge can be used to achieve more consistent breakage and fire-polish the needle tips to avoid sharp edges. Although they are not essential, the microforged needles do decrease the frequency of damaging the larvae during the procedure. Also, microgavage can be accomplished by mounting in 3% methylcellulose without an agarose mold. However, the mold helps to properly position the fish which increases the speed of gavaging several fish at once and decreases errors that lead to physical damage.
This technique provides several advantages over the other currently accepted methods for achieving intestinal loading in the zebrafish model. The most important advantage of this technique is that the amount of material introduced and the timing of delivery can be more tightly regulated. These factors are important for analyzing kinetics of absorption or transit through the intestine. In order for material to gain access to the intestinal lumen and be absorbed into the intestinal epithelium, the zebrafish larvae must swallow or feed on the substrates along with water. However, the rate and volume of swallowing is variable across individuals and cannot be controlled26,43. Therefore, if we aim to study processes that occur rapidly following intestinal loading, we may miss early kinetic differences if we are unable to rigorously control the timing of loading. This is also a problem when the process being studied is saturable. If we only look at static time points long after uptake has occurred, we cannot assess more subtle differences in the interaction of substrates with intestinal tissues.
To explore utility of using gavage to assess intestinal transit, we gavaged fluorescent microspheres and compared the results to the published method of Field et al. where fluorescent microspheres were mixed with the larval diet and supplied to the zebrafish media26. Since only ~50% of larvae consume the food/beads, pre-experiment sorting of animals with ingested microspheres is essential to reduce the variability and ensure that transit is truly being measured. The benefit of using gavage is that all fish receive a controlled amount of the motility tracer at a defined time and it abrogates the need for pre-sorting. Another difference between both the feeding and gavage methods compared with direct observation approaches21,36,38 is that is that a relatively large number of animals can be screened over many hours. However, this does come at the expense of not being able to discern divergent motility patterns.
The microgavage approach for delivering material to the intestine also allows control over the route of entry into the zebrafish system. During immersion, the material can be absorbed through external epithelia and other entry points (e.g., lateral line neuromasts) in addition to being swallowed. In some cases, compounds entering this way can be toxic (Figure 3C). Absorption into these tissues may also deplete the level of soluble compound available for uptake if and when it reaches the intestinal compartment. Alternatively, it is also possible that if external absorption occurs more quickly than ingestion it could lead to early changes in the animal that affect subsequent physiological processes.
In this protocol, we provide two examples of how this method can be used to evaluate intestinal physiology. First, we first show how to deliver large molecular weight dextran into the lumen and monitor passage into extraintestinal spaces. This method can be used to verify that the gavage procedure is being performed properly without physical damage to the epithelium, and as a novel assay for intestinal epithelial barrier integrity. Second, we demonstrate how to assess intestinal motility by gavaging fluorescent microspheres into the intestinal lumen and monitoring their transit. We anticipate that these assays can be applied to study intestinal barrier function and intestinal motility during different experimental conditions (e.g., genetic manipulation, drug treatment, or exposure to environmental factors).
Any material can be applied the zebrafish intestine by microgavage as long as a liquid solution or suspension can be prepared, and any particles are small enough to pass through the diameter of the microgavage needle tip. In addition to the uses already mentioned, we envision ample possibilities to utilize this technique. Introduction of pharmacological inhibitors by microgavage would limit extraintestinal activity and developmental effects compared to less exact approaches. Microbial communities with defined composition or purified microbial factors could be introduced to the intestine, removing the requirement for natural colonization to study microbe impact on intestinal cell function. In animals where transgene expression is under control of inducible promoters, regulatory agents could be applied such that only cells in the intestinal lumen would respond. Similarly, tools for disrupting gene expression or protein production, such as morpholino constructs or mRNA, could be introduced to intestinal cells at late developmental time points. Most of these options have not yet been tested, but we look forward to seeing how the zebrafish research community uses and benefits from the microgavage system.
Disclosures
No conflicts of interest declared.
Acknowledgments
We thank members of the Rawls laboratory for helpful suggestions on content and Dr. Alan Fanning for valuable discussions on tight junction size permeability and disruption methods. We also thank Dr. Michael Chua and Dr. Neal Kramarcy of the Michael Hooker Microscopy Facility for confocal microscope support. This work was supported by National Institutes of Health grants T32 DK007737-15 (J.L.C. trainee), F32 DK094592 (to J.L.C.), and R01 DK081426 (to J.F.R.).
References
- Kuhlman J, Eisen JS. Genetic screen for mutations affecting development and function of the enteric nervous system. Dev. Dyn. 2007;236:118–127. doi: 10.1002/dvdy.21033. [DOI] [PubMed] [Google Scholar]
- Pack M, et al. Mutations affecting development of zebrafish digestive organs. Development. 1996;123:321–328. doi: 10.1242/dev.123.1.321. [DOI] [PubMed] [Google Scholar]
- Wallace KN, Pack M. Unique and conserved aspects of gut development in zebrafish. Dev. Biol. 2003;255:12–29. doi: 10.1016/s0012-1606(02)00034-9. [DOI] [PubMed] [Google Scholar]
- Wallace KN, Akhter S, Smith EM, Lorent K, Pack M. Intestinal growth and differentiation in zebrafish. Mech. Dev. 2005;122:157–173. doi: 10.1016/j.mod.2004.10.009. [DOI] [PubMed] [Google Scholar]
- Ng AN, et al. Formation of the digestive system in zebrafish: III. Intestinal epithelium morphogenesis. Dev. Biol. 2005;286:114–135. doi: 10.1016/j.ydbio.2005.07.013. [DOI] [PubMed] [Google Scholar]
- Hama K, et al. In vivo imaging of zebrafish digestive organ function using multiple quenched fluorescent reporters. Am. J. Physiol. Gastrointest. Liver Physiol. 2009;296:445–453. doi: 10.1152/ajpgi.90513.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carten JD, Bradford MK, Farber SA. Visualizing digestive organ morphology and function using differential fatty acid metabolism in live zebrafish. Dev. Biol. 2011;360:276–285. doi: 10.1016/j.ydbio.2011.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rich A. A new high-content model system for studies of gastrointestinal transit: the zebrafish. Neurogastroenterol Motil. 2009;21:225–228. doi: 10.1111/j.1365-2982.2008.01251.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bagnat M, Cheung ID, Mostov KE, Stainier DY. Genetic control of single lumen formation in the zebrafish gut. Nat. Cell Biol. 2007;9:954–960. doi: 10.1038/ncb1621. [DOI] [PubMed] [Google Scholar]
- Bagnat M, et al. Cse1l is a negative regulator of CFTR-dependent fluid secretion. Curr. Biol. 2010;20:1840–1845. doi: 10.1016/j.cub.2010.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shepherd I, Eisen J. Development of the zebrafish enteric nervous system. Methods Cell Biol. 2011;101:143–160. doi: 10.1016/B978-0-12-387036-0.00006-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brugman S, et al. Oxazolone-induced enterocolitis in zebrafish depends on the composition of the intestinal microbiota. Gastroenterology. 2009;137:1757–1767. doi: 10.1053/j.gastro.2009.07.069. [DOI] [PubMed] [Google Scholar]
- Oehlers SH, et al. The inflammatory bowel disease (IBD) susceptibility genes NOD1 and NOD2 have conserved anti-bacterial roles in zebrafish. Dis. Model Mech. 2011;4:832–841. doi: 10.1242/dmm.006122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oehlers SH, et al. A chemical enterocolitis model in zebrafish larvae that is dependent on microbiota and responsive to pharmacological agents. Dev Dyn. 2011;240:288–298. doi: 10.1002/dvdy.22519. [DOI] [PubMed] [Google Scholar]
- Faro A, Boj SF, Clevers H. Fishing for intestinal cancer models: unraveling gastrointestinal homeostasis and tumorigenesis in zebrafish. Zebrafish. 2009;6:361–376. doi: 10.1089/zeb.2009.0617. [DOI] [PubMed] [Google Scholar]
- Fleming A, Jankowski J, Goldsmith P. In vivo analysis of gut function and disease changes in a zebrafish larvae model of inflammatory bowel disease: a feasibility study. Inflamm. Bowel Dis. 2010;16:1162–1172. doi: 10.1002/ibd.21200. [DOI] [PubMed] [Google Scholar]
- Kanther M, et al. Microbial colonization induces dynamic temporal and spatial patterns of NF-kappaB activation in the zebrafish digestive tract. Gastroenterology. 2011. [DOI] [PMC free article] [PubMed]
- Rawls JF, Samuel BS, Gordon JI. Gnotobiotic zebrafish reveal evolutionarily conserved responses to the gut microbiota. Proc. Natl. Acad. Sci. U.S.A. 2004;101:4596–4601. doi: 10.1073/pnas.0400706101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rawls JF, Mahowald MA, Goodman AL, Trent CM, Gordon JI. In vivo imaging and genetic analysis link bacterial motility and symbiosis in the zebrafish gut. Proc. Natl. Acad. Sci. U.S.A. 2007;104:7622–7627. doi: 10.1073/pnas.0702386104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanther M, Rawls JF. Host-microbe interactions in the developing zebrafish. Curr. Opin. Immunol. 2010;22:10–19. doi: 10.1016/j.coi.2010.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bates JM, et al. Distinct signals from the microbiota promote different aspects of zebrafish gut differentiation. Dev. Biol. 2006;297:374–386. doi: 10.1016/j.ydbio.2006.05.006. [DOI] [PubMed] [Google Scholar]
- Cheesman SE, Guillemin K. We know you are in there: conversing with the indigenous gut microbiota. Res Microbiol. 2007;158:2–9. doi: 10.1016/j.resmic.2006.10.005. [DOI] [PubMed] [Google Scholar]
- Bates JM, Akerlund J, Mittge E, Guillemin K. Intestinal alkaline phosphatase detoxifies lipopolysaccharide and prevents inflammation in zebrafish in response to the gut microbiota. Cell Host Microbe. 2007;2:371–382. doi: 10.1016/j.chom.2007.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Camp JG, Jazwa AL, Trent CM, Rawls JF. Intronic cis-regulatory modules mediate tissue-specific and microbial control of angptl4/fiaf transcription. PLoS Genetics. 2012. In Press. [DOI] [PMC free article] [PubMed]
- Cheesman SE, Neal JT, Mittge E, Seredick BM, Guillemin K. Epithelial cell proliferation in the developing zebrafish intestine is regulated by the Wnt pathway and microbial signaling via Myd88. Proc. Natl. Acad. Sci. U.S.A. 2011;108:4570–4577. doi: 10.1073/pnas.1000072107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Field HA, Kelley KA, Martell L, Goldstein AM, Serluca FC. Analysis of gastrointestinal physiology using a novel intestinal transit assay in zebrafish. Neurogastroenterol. Motil. 2009;21:304–312. doi: 10.1111/j.1365-2982.2008.01234.x. [DOI] [PubMed] [Google Scholar]
- Westerfield M. The Zebrafish Book. 4 edn. University of Oregon Press; 2000. [Google Scholar]
- Pham LN, Kanther M, Semova I, Rawls JF. Methods for generating and colonizing gnotobiotic zebrafish. Nat. Protoc. 2008;3:1862–1875. doi: 10.1038/nprot.2008.186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen L, Weber CR, Raleigh DR, Yu D, Turner JR. Tight junction pore and leak pathways: a dynamic duo. Annu. Rev. Physiol. 2011;73:283–309. doi: 10.1146/annurev-physiol-012110-142150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Itallie CM, et al. The density of small tight junction pores varies among cell types and is increased by expression of claudin-2. J. Cell. Sci. 2008;121:298–305. doi: 10.1242/jcs.021485. [DOI] [PubMed] [Google Scholar]
- Van Itallie CM, Anderson JM. Measuring size-dependent permeability of the tight junction using PEG profiling. Methods Mol. Biol. 2011;762:1–11. doi: 10.1007/978-1-61779-185-7_1. [DOI] [PubMed] [Google Scholar]
- Watson CJ, Rowland M, Warhurst G. Functional modeling of tight junctions in intestinal cell monolayers using polyethylene glycol oligomers. Am. J. Physiol. Cell Physiol. 2001;281:388–397. doi: 10.1152/ajpcell.2001.281.2.C388. [DOI] [PubMed] [Google Scholar]
- Rodgers LS, Fanning AS. Regulation of epithelial permeability by the actin cytoskeleton. Cytoskeleton (Hoboken) 2011;68:653–660. doi: 10.1002/cm.20547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez-Mariscal L, Chavez de Ramirez B, Cereijido M. Tight junction formation in cultured epithelial cells (MDCK) J. Membr. Biol. 1985;86:113–125. doi: 10.1007/BF01870778. [DOI] [PubMed] [Google Scholar]
- Palant CE, Duffey ME, Mookerjee BK, Ho S, Bentzel CJ. Ca2+ regulation of tight-junction permeability and structure in Necturus gallbladder. Am. J. Physiol. 1983;245:C203–C212. doi: 10.1152/ajpcell.1983.245.3.C203. [DOI] [PubMed] [Google Scholar]
- Holmberg A, Schwerte T, Pelster B, Holmgren S. Ontogeny of the gut motility control system in zebrafish Danio rerio embryos and larvae. J. Exp. Biol. 2004;207:4085–4094. doi: 10.1242/jeb.01260. [DOI] [PubMed] [Google Scholar]
- Rich A, et al. Kit-like immunoreactivity in the zebrafish gastrointestinal tract reveals putative. 2007;236:903–911. doi: 10.1002/dvdy.21086. [DOI] [PubMed] [Google Scholar]
- Holmberg A, Olsson C, Hennig GW. TTX-sensitive and TTX-insensitive control of spontaneous gut motility in the developing zebrafish (Danio rerio) larvae. J. Exp. Biol. 2007;210:1084–1091. doi: 10.1242/jeb.000935. [DOI] [PubMed] [Google Scholar]
- Hennig GW, Costa M, Chen BN, Brookes SJ. Quantitative analysis of peristalsis in the guinea-pig small intestine using spatio-temporal maps. J. Physiol. 1999;517(Pt 2):575–590. doi: 10.1111/j.1469-7793.1999.0575t.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flynn EJ, 3rd, Trent CM, Rawls JF. Ontogeny and nutritional control of adipogenesis in zebrafish (Danio rerio. J. Lipid Res. 2009;50:1641–1652. doi: 10.1194/jlr.M800590-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clifton JD, et al. Identification of novel inhibitors of dietary lipid absorption using zebrafish. PLoS One. 2010;5:e12386. doi: 10.1371/journal.pone.0012386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin SW, Beis D, Mitchell T, Chen JN, Stainier DY. Cellular and molecular analyses of vascular tube and lumen formation in zebrafish. Development. 2005;132:5199–5209. doi: 10.1242/dev.02087. [DOI] [PubMed] [Google Scholar]
- Berghmans S, Hunt J, Roach A, Goldsmith P. Zebrafish offer the potential for a primary screen to identify a wide variety of potential anticonvulsants. Epilepsy Res. 2007;75:18–28. doi: 10.1016/j.eplepsyres.2007.03.015. [DOI] [PubMed] [Google Scholar]