

Abstract
Epidemic Clostridium difficile (027/BI/NAP1) rapidly emerged in the past decade as the leading cause of antibiotic-associated diarrhea worldwide. However, the key moments in the evolutionary history leading to its emergence and subsequent patterns of global spread remain unknown. Here we define the global population structure of C. difficile 027/BI/NAP1 based on whole-genome sequencing and phylogenetic analysis. We demonstrate that two distinct epidemic lineages, FQR1 and FQR2, not one as previously thought, emerged in North America within a relatively short period after acquiring the same fluoroquinolone resistance mutation and a highly-related conjugative transposon. The two epidemic lineages displayed distinct patterns of global spread, and the FQR2 lineage spread more widely leading to healthcare outbreaks in the UK, continental Europe and Australia. Our analysis identifies key genetic changes linked to the rapid trans-continental dissemination of epidemic C. difficile 027/BI/NAP1 and highlights the routes by which it spreads through the global healthcare system.
Clostridium difficile is the most common infectious cause of antibiotic-associated diarrhea and healthcare infection in the developed world1,2. Antibiotic treatment and hospitalization are major risk factors for C. difficile colonization leading to asymptomatic carriage, diarrhea, pseudomembraneous colitis or death3. C. difficile pathogenesis is associated with the production of the enterotoxins TcdA and TcdB that are encoded within a pathogenicity locus (PaLoc)4. C. difficile produces highly resistant and infectious spores, which promote environmental transmission within the healthcare setting5, and potentially facilitate spread over greater distances by those in the carrier state6.
The global emergence of C. difficile infection (CDI) in the past decade followed highly-publicized C. difficile outbreaks in the USA7 and Canada8 that were associated with increased rates of disease recurrence and mortality7-10. The outbreaks were caused by a previously uncommon, fluoroquinolone resistant variant of C. difficile genotyped as 027/BI/NAP17,8. Fluoroquinolone resistant (FQR) C. difficile 027/BI/NAP1 is still the most common variant causing CDI throughout North America11. During 2004-2006 there were severe C. difficile 027/BI/NAP1 outbreaks in the UK and in subsequent years FQR C. difficile 027/BI/NAP1 accounted for >40% of cases UK-wide12, and is now commonly found in continental Europe13,14 and more recently in Australia15. Although FQR C. difficile 027/BI/NAP1 is widespread in healthcare facilities worldwide the underlying reasons for its rapid emergence and the subsequent patterns of global spread remain unknown.
To address these questions, we sequenced the genomes of a global collection of C. difficile 027/BI/NAP1 (n=151) isolated primarily from hospital patients between 1985 and 2010 (Supplementary Table 1). Illumina reads were aligned to the genome of the C. difficile 027/BI/NAP1 strain R2029116. We identified a total of 3,686 single nucleotide polymorphisms (SNPs) within the 3.8 Mb non-repetitive, core genome (representing 95% of genome). Of these, 3,150 (85%) of the SNPs were clustered and private to 8 individual isolates, suggesting that these genomic regions were imported from outside the 027/BI/NAP1 lineage (Supplementary Fig. 1; Supplementary Note). These SNPs were removed as they could mask the true phylogenetic signal, leaving 536 SNPs for downstream phylogenetic analysis. Other than these events, homologous recombination has not played a major role in shaping the phylogeny of the global C. difficile 027/BI/NAP1 collection (Supplementary Fig. 2; Supplementary Note).
Figure 1a shows a maximum-likelihood phylogeny representing the C. difficile 027/BI/NAP1 global population structure. The phylogeny discriminates between >100 distinct genotypes within the global collection and shows limited geographical clustering, implying frequent, long-range transmission between humans, and in a limited number of cases two-way transmission between human and animal or food sources (Supplementary Note). The core genome of C. difficile 027/BI/NAP1 exhibits a relatively low-level of genetic diversity, consistent with its recent emergence, with very few SNPs defining the major branches in the phylogeny (Supplementary Fig. 3). Using three methods, we estimated the mutation rate of C. difficile 027/BI/NAP1 to be 1.47×10−7-5.33×10−7 (95% confidence intervals) substitutions per site per year, equivalent to 1-2 mutations per genome per year (Supplementary Fig. 4; Supplementary Table 2; Supplementary Note). Interestingly, this rate is ~10 times slower than Streptococcus pneumoniae17 and Staphylococcus aureus18 over similar timescales and may be due to the fact that metabolically dormant spores do not accumulate mutations19.
Figure 1.

Phylogeny of C. difficile 027/BI/NAP1 based on core genome SNPs. a, Global phylogeny (n=151 isolates). Colored nodes indicate the geographical source of the isolates. The position of the inferred root is indicated by a dashed line. b, Phylogeny of UK isolates (n=188) colored according to geographical source of isolates (gray - non-UK isolates). Dashed-line circled areas indicate the isolates with Thr82Ile substitution in gyrA associated with fluoroquinolone resistance. Black arrows show insertion of selected mobile elements.
Importantly, the global phylogeny demonstrates the presence of two genetically distinct lineages which have independently acquired an identical mutation (Thr82Ile) in DNA gyrase subunit A (gyrA), by either mutation or recombination, leading to high-level FQR20 (Fig. 1). Although both lineages share this mutation, which is the most frequent in FQR isolates20, both lineages are highly supported by maximum-likelihood and Bayesian methods (Supplementary Figs. 3, 5), unequivocally demonstrating that FQR has arisen in two epidemic C. difficile 027/BI/NAP1 lineages in two separate events. We named these lineages FQR1 and FQR2. Bayesian analysis estimates that FQR1 and FQR2 have emerged recently, with most recent common ancestors in ~1993 and ~1994, respectively (median estimates; 95% highest posterior density intervals are 1984-1999 for FQR1 and 1986-1999 for FQR2). Near the base of the C. difficile 027/BI/NAP1 phylogeny and outside of both FQR lineages are isolates from various global locations (Singapore, Japan, Korea, Canada, the USA, the UK, Germany and France) and sampling times (1985-2009). To our knowledge, none of the isolates in this part of the phylogeny are associated with major hospital outbreaks, suggesting that these isolates represent the pre-epidemic C. difficile 027/BI/NAP1 genetic background from which FQR epidemic lineages emerged.
Lineage FQR1 contains epidemic isolates associated with healthcare outbreaks in the USA (Pennsylvania (2001), Oregon (2003), New Jersey (2004), Arizona (2006 and 2007) and Maryland (2007))7 and isolates associated with sporadic infections in South Korea21 and Switzerland between 2007 and 2010. Bayesian phylogeographic analysis22 indicates that the FQR1 lineage originated from the USA (99% probability). The earliest isolate in the FQR1 lineage is from Pittsburgh, Pennsylvania in 2001, representing one of the earliest reports of an increase in CDI caused by FQR C. difficile 027/BI/NAP19,23. Thus, FQR1 appears to represent an epidemic lineage that originally emerged in the North East of the USA and subsequently transmitted to South Korea and Switzerland.
The FQR2 lineage contains the majority of epidemic isolates that are widely spread geographically (Figure 1a). The most striking feature in FQR2 is a star-like topology in the early part of the lineage generally consistent with rapid population expansion from a common progenitor (Supplementary Fig. 5). Two isolates in our collection were found on the node at the base of the star-like topology, one associated with an outbreak in Montreal in 200324 and the other from the Netherlands in 2006. Bayesian analysis suggests the FQR2 lineage also originated in North America (with 59% probability from the USA and 33% probability from Canada). Interestingly, all but one Canadian isolates found in this lineage were from Montreal where FQR C. difficile 027/BI/NAP1 outbreaks were initially reported in Canada8,10.
The Bayesian phylogeny25 contains multiple highly-supported sub-lineages of FQR2 associated with distinct geographic locations suggesting rapid trans-continental dissemination from North America to continental Europe, the UK and Australia. Our analysis demonstrates a single introductory event of FQR2 into Australia and at least four separate introductions into continental Europe, including two trans-Atlantic transmission events (one of which reached the Netherlands) and two from the UK, giving rise to present-day isolates in Austria and Poland26 (Fig. 2a; Supplementary Fig. 5). Similarly, the descendants of FQR2 were introduced into the UK on at least four occasions, including three trans-Atlantic transmission events from North America and one from continental Europe, leading to a series of highly-publicized outbreaks in UK hospitals during 2004-200627.
Figure 2.
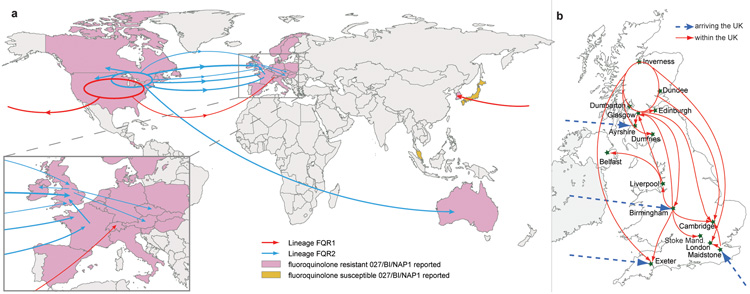
Transmission events inferred for epidemic C. difficile 027/BI/NAP1. a, Global spread of lineages FQR1 (red arrows) and FQR2 (blue arrows) inferred from phylogeographic analysis (Supplementary Fig. 5). Global map demonstrating the countries where fluoroquinolone-resistant (pink) and fluoroquinolone-sensitive (yellow) C. difficile 027/BI/NAP1 has been reported6. Width of the arrow is approximately proportional to the number of descendents from each sub-lineage. b, Inferred arrivals (blue dashed arrows) and transmissions (red arrows) of FQR2 into and within the UK based on phylogeographic analysis (Supplementary Fig. 5) and maximum likelihood phylogeny (Fig. 1b).
To investigate the introduction and subsequent spread of C. difficile 027/BI/NAP1 within the UK we analyzed the genomes of a collection of a further 145 UK isolates from healthcare patients (Supplementary Table 3) (in addition to the 43 UK isolates in our global collection). A maximum-likelihood phylogeny (Fig. 1b) demonstrates that long-range geographical transmission was frequent and extensive within the UK (Fig. 2b). This collection confirms that FQR2 C. difficile 027/BI/NAP1 likely reached the UK on at least four separate occasions, arriving independently in Exeter, Ayrshire, and Birmingham from North America and arriving in Maidstone from continental Europe (Fig. 1b and Fig. 2b). The introduction into the Maidstone area likely triggered a large-scale hospital outbreak that affected London and Cambridge27. Despite multiple introductions, the majority of present-day UK isolates (including those from Liverpool, Belfast, Birmingham, London, Cambridge, Exeter and multiple sites in Scotland) are descendants of one sub-lineage whose earliest representative in our collection is a 2002 Birmingham isolate. This dominant sub-lineage also underlies the Stoke Mandeville outbreak27 (Fig. 1b and Fig. 2b).
The emergence of epidemic C. difficile 027/BI/NAP1 has been proposed to be due to genetic changes in the tcdC gene of the PaLoc4,7,8,28. However, we discovered only 2 SNPs in the entire 19.6 kb PaLoc region in our 027/BI/NAP1 collection, both private to a single isolate. Consistent with a previous proposal based on two C. difficile 027/BI/NAP1 genomes16, there is no change within the entire PaLoc region between pre- and post-epidemic isolates of C. difficile 027/BI/NAP1 that could explain the emergence (Supplementary Note). As described above, we provide strong evidence that the acquisition of FQR in two distinct lineages is associated with the emergence of C. difficile 027/BI/NAP1. We next determined if there are other genetic changes linked to the acquisition of FQR that could underlie a presumed increase of fitness contributing to the emergence of FQR1 and FQR2. Only 2 and 7 SNPs define the branches leading to lineages FQR1 and FQR2, respectively. However, besides the gyrA mutation (Figure 1) there are no SNPs shared by both lineages (Supplementary Table 4). Further, there is little evidence that a significant change in phenotype could result from any of the SNPs that define the FQR1 and FQR2 lineages (Supplementary Table 4; Supplementary Note), except perhaps the gyrA mutation itself. Besides these SNPs and the gyrA mutation we also identified 10 non-synonymous SNPs conflicting with the phylogeny (homoplasic, 1.9% of the total number) that are present mainly within FQR1 and FQR2. These 10 SNPs affect 8 genes associated with antimicrobial resistance or cell surface modification (Table 1; Supplementary Fig. 3; Supplementary Note). However, these SNPs are only present within a small fraction of the FQR1 and FQR2 isolates so their presence cannot explain the emergence of C. difficile 027/BI/NAP1.
Table 1.
Non-synonymous homoplasic SNPs identified in the core genomes of C. difficile 027/BI/NAP1 isolates. The positions refer to those in the R20291 genome. The alleles listed relate to the forward strand.
| Position | Region | SNP | Substitution | Antibiotic |
|---|---|---|---|---|
| 5420 | DNA gyrase subunit B (gyrB) |
1276G> A |
Asp426Asn | Fluoroquinolone |
| 6310 | DNA gyrase subunit A (gyrA) |
245C>T | Thr82Ile | Fluoroquinolone |
| 95412 | DNA-directed RNA polymerase beta chain (rpoB) |
1504C> A |
His502Asn | Rifampicin |
| 95422 | rpoB | 1514G> A |
Arg505Lys | Rifampicin |
| 103867 | translation elongation factor G (fusA) |
1363C> A/1363 C>T |
His455Asn/His 455Tyr |
Fusidic acid |
| 104117 | fusA | 1613C> T |
Pro538Leu | Fusidic acid |
| 1800920 | two-component response regulator |
31G>A | Glu11Lys | |
| 1802086 | two-component sensor histidine kinase |
446C>T | Thr149Ile | |
| 3170481 | S-layer precursor protein (slpA) |
467G>A /467G> T |
Pro156Leu/Pr o156Gln |
|
| 3170482 | slpA | 466G>A /466G> T |
Pro156Ser/Pro 156Thr |
|
| 3938789 | putative membrane protein |
996A>C | Tyr332* |
Horizontal gene transfer is a key mechanism driving C. difficile evolution29,30. We therefore assembled the C. difficile 027/BI/NAP1 genomes and identified widespread acquisition of a range of mobile elements associated with erythromycin, chloramphenicol, tetracycline and aminoglycoside resistance throughout the phylogeny (Supplementary Fig. 3; Supplementary Note). Importantly, we found a class of CTn5-like elements29 in almost all isolates in FQR1 and FQR2 lineages, where they are inserted in the same genome location in the majority of isolates (Supplementary Fig. 6; Supplementary Note). The FQR1 and FQR2 lineages both share a common CTn5-like element, which we named Tn6192, that contains genes encoding an ABC transport system, a two-component transcriptional regulator and a predicted DNA-binding protein (Supplementary Table 5). The version found in the FQR2 lineage differs from that in the FQR1 lineage by harboring a contiguous insertion of 15.7 kb, Tn610531, which contains 14 genes, four of which are predicted to be DNA-binding proteins or transcriptional regulators (Supplementary Table 5). This element has also acted as a hot-spot for further integrations throughout the FQR2 lineage (Supplementary Fig. 6). The presence of Tn6192 is the only other significant genetic trait, aside from the fluoroquinolone resistance SNP, that differentiates FQR1 and FQR2 from the pre-epidemic isolates (Supplementary Table 6). It is unknown whether the genes carried by this element have any phenotypic effect on the core genome. However, Tn6192 may have played a role in the success of these lineages.
We demonstrate that the separate acquisitions of FQR and a conjugative transposon in two distinct lineages of C. difficile 027/BI/NAP1 are the key genetic changes linked to its rapid emergence during the early 2000s7,8. Further, our data suggest that the acquisition of resistance to commonly used antibiotics is a major feature of the continued evolution and persistence of C. difficile 027/BI/NAP1 in the healthcare settings. It is notable that fluoroquinolone antibiotics were one of the most commonly prescribed antibiotic classes in North America during the late 1990s and early 2000s32, so it is during this time that the selective pressure for the acquisition and maintenance of FQR within healthcare settings would have been at its highest, explaining the near simultaneous emergence of more than one clone of FQR C. difficile 027/BI/NAP1. We also demonstrate the ease and rapidity with which FQR C. difficile 027/BI/NAP1 has transmitted internationally highlighting the interconnectedness of the global healthcare system facilitated by rapid human travel. Whole-genome sequencing and phylogenetic analysis has been used successfully for clinical transmission studies of methicillin resistant S. aureus within a hospital33 and although the low mutation rate may pose some limitations we believe this approach will have great potential for C. difficile. Our analysis provides a genomic framework for understanding the population structure, geographical source, epidemiology and evolution of this highly transmissible healthcare pathogen.
Online Methods
C. difficile strain collection
The C. difficile isolates chosen for this study were characterized as PCR-ribotype 027 or 176, REA type BI or PFGE type NAP1 and include 151 isolates from a global collection and 188 isolates from the UK, with an overlap of 43 isolates between the collections. The global collection includes 25 isolates from two previous studies16,30. New genome sequencing data was generated for 6 of these isolates. Supplementary Table 1 and Supplementary Table 3 summarize the isolate details. Genomic DNA was extracted as previously described30.
Sequencing, mapping, and SNP detection
Paired-end multiplex libraries were created as described18, followed by sequencing on Illumina GAIIx and HiSeq2000 platforms. The read length was 54 bp for samples Liv1-Liv21, 108 bp for samples Gla001-Gla022, and 76 bp for the rest. All isolates were sequenced to an average coverage of 110-fold across the isolates. Sequencing reads were aligned with BWA34 against the genome sequence of the ribotype 027 reference strain R2029116. SNPs were identified with SAMtools35. A coverage cut-off of >5-fold and <three times the average coverage was set for each individual isolate during SNP detection. Repetitive regions in the reference genome sequence were characterized using REPuter36 and the repeat finder functions in the MUMmer package37. The boundaries of repetitive regions were extended to include the mobile elements in R20291. SNPs falling within these repetitive regions were excluded. To confirm the alleles for each variant position, SNPs were checked at each position in all sequencing reads in all isolates. An allele was only considered to be valid if supported by all reads (if 5<coverage<=40) or >92.5% of the total reads (if coverage >40) covering the position; otherwise it was treated as missing data. Our method allows no false positive SNPs and a false negative rate of ~8% (Supplementary Fig. 7; Supplementary Note).
Phylogenetic analysis
An appropriate evolutionary model (simple GTR) was determined using jModelTest 0.1.138. Phylogenetic relationships were inferred using three methods: 1), split-decomposition and neighbor-net methods in SplitsTree439; 2), the program PHYML40, and 3), the program BEAST25. In the first two cases, a simple GTR model was used. Neighbor-joining tree (Supplementary Fig. 2b) were also constructed with PHYML, and the results were compared. Two non-027/BI/NAP1 C. difficile isolates 63029 and CF530 were used to root the global phylogeny.
In the BEAST analysis, three clock models (strict, relaxed lognormal and relaxed exponential) and two population models (constant and skyline) were tested initially. The relaxed exponential clock model in combination with skyline population model was determined as more suitable based on Bayes Factor calculations41 and was used for later BEAST runs. The program was specified to estimate tMRCA (time to the most recent common ancestor) of taxon groupings. All other parameters were set to default. These analyses were carried out with a chain length of 400,000,000 states, and re-sampling every 20,000 states. The phylogeograpic history was also inferred with BEAST using a Bayesian method as described22. The ESS values were >200 for all parameters.
Accessory genome
For each genome, the unaligned sequencing reads were assembled using Velvet42. To assess whether the resulting contigs were unique, each contig with a length >1kb was searched using BLASTN against the current pan-genome, which was made by concatenating the draft genome sequence of M7404 and already determined unique contigs. Any unique contigs were added to the pan-genome. If the match was of >80% identity and covered >40% of the contig length, this contig was not considered to be unique, and was not added to the current pan-genome. The resulting unique contigs were individually searched against the NCBI bacterial genome database to check for contamination. The filtered unique contigs were added to the genome sequence of M7404 to create a pan-genome. Finally, the sequencing reads from each strain were aligned against the constructed pan-genome to assess for the presence and absence of genomic regions in each isolate. The raw Illumina data were also assembled de novo. Draft genome sequences were produced for isolates by ordering contigs against a suitable reference sequence. Pairwise genome comparisons were made in Artemis Comparison Tool43. Key mobile elements and their nomenclature are described in Supplementary Note.
Identification of homoplasic characters and homologous recombination
Homoplasic SNPs were identified by examining the SNP allele pattern across all isolates in relation to the phylogenetic tree. A SNP was considered homoplasic if the allele pattern did not agree with the tree topology. Genomic regions affected by homologous recombination were identified by a) clusters of SNPs within 2,000 bp windows and b) an iterative method to eliminate recombination sites as described17 The identified homologous recombination blocks were excluded from phylogenetic and population genetic analysis.
Mutation rate
The apparent mutation rate was estimated using three methods: 1), A full maximum-likelihood model assuming a rapid expansion which results in perfect star genealogies, implemented in an R script44 (Supplementary Note; Supplementary Fig. 8); 2), the program BEAST25; and 3), the program Path-O-Gen v1.3 (See URL). A maximum-likelihood tree was used in this analysis. BEAST analyses were carried out as stated above. The dataset used for 2) and 3) is the final SNP alignment of the global collection. A final mutation rate was determined by combining the single estimate from method 3) and 95% confidence intervals from methods 1) and 2)45.
Supplementary Material
Acknowledgements
This project was funded by the Wellcome Trust (grant numbers 098051 and 086418), the Medical Research Council New Investigator Research Grant (TDL; grant number 93614) and the Scottish Infection Research Network. We acknowledge the funding from the NIHR Biomedical Research Centre in Liverpool. Both FM and PR were supported by the BRC. MP is a NIHR Senior Investigator. We are grateful to members of the European Study Group of Clostridium difficile (ESGCD), a working group of ESCMI, including F. Barbut, T. Eckmanns, M.L. Lambert, F. Fitzpatrick, C. Wiuff, H. Pituch, P. Reichert, A.F. Widmer, F. Allerberger, D.W. Notermans, M. Delmée, R. Frei, O. Lyytikäinen, A. Ingebretsen and I.R. Poxton. We thank the Wellcome Trust Sanger Institute sequencing and informatics teams.
Footnotes
Accession Codes
Illumina sequence reads for the C. difficile 027/BI/NAP1 global and UK collections were deposited in the European Nucleotide Archive under the accession codes listed in Supplementary Table 1 and Supplementary Table 3.
URL
Path-O-Gen, http://tree.bio.ed.ac.uk/software/pathogen/.
Author Contributions
M.H. analyzed data. T.D.L., M.H., G.Dougan, B.W.W. and J.P. were involved in the study design. F.M., P.R., L.E., D.J.P., M.J.M., D.F., K.B.B., S.D., J.B., D.B., J.E.C., G.Douce, D.G., H.J.K., T.H.K., H.K., M.S., T.L., S.M., E.B., S.J.P., N.M.B., T.R., G.S., M.W., M.P., E.K., P.H. and B.W.W. were involved in isolate collection and DNA extraction. T.R.C. contributed to Bayesian analysis. M.H., J.P., T.D.L., G.Dougan, T.R.C. and S.R.H. contributed to data interpretation. M.H., J.P., T.D.L. and G.Dougan wrote the paper.
REFERENCES
- 1.Bartlett JG. Clostridium difficile: progress and challenges. Ann N Y Acad Sci. 2010;1213:62–69. doi: 10.1111/j.1749-6632.2010.05863.x. [DOI] [PubMed] [Google Scholar]
- 2.Kelly CP, LaMont JT. Clostridium difficile--more difficult than ever. N Engl J Med. 2008;359:1932–1940. doi: 10.1056/NEJMra0707500. [DOI] [PubMed] [Google Scholar]
- 3.Kuijper EJ, Coignard B, Tull P. Emergence of Clostridium difficile-associated disease in North America and Europe. Clin Microbiol Infect. 2006;12(Suppl 6):2–18. doi: 10.1111/j.1469-0691.2006.01580.x. [DOI] [PubMed] [Google Scholar]
- 4.Warny M, et al. Toxin production by an emerging strain of Clostridium difficile associated with outbreaks of severe disease in North America and Europe. Lancet. 2005;366:1079–1084. doi: 10.1016/S0140-6736(05)67420-X. [DOI] [PubMed] [Google Scholar]
- 5.Riggs MM, et al. Asymptomatic carriers are a potential source for transmission of epidemic and nonepidemic Clostridium difficile strains among long-term care facility residents. Clin Infect Dis. 2007;45:992–998. doi: 10.1086/521854. [DOI] [PubMed] [Google Scholar]
- 6.Clements AC, Magalhaes RJ, Tatem AJ, Paterson DL, Riley TV. Clostridium difficile PCR ribotype 027: assessing the risks of further worldwide spread. Lancet Infect Dis. 2010;10:395–404. doi: 10.1016/S1473-3099(10)70080-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.McDonald LC, et al. An epidemic, toxin gene-variant strain of Clostridium difficile. N Engl J Med. 2005;353:2433–2441. doi: 10.1056/NEJMoa051590. [DOI] [PubMed] [Google Scholar]
- 8.Loo VG, et al. A predominantly clonal multi-institutional outbreak of Clostridium difficile-associated diarrhea with high morbidity and mortality. N Engl J Med. 2005;353:2442–2449. doi: 10.1056/NEJMoa051639. [DOI] [PubMed] [Google Scholar]
- 9.Muto CA, et al. A large outbreak of Clostridium difficile-associated disease with an unexpected proportion of deaths and colectomies at a teaching hospital following increased fluoroquinolone use. Infect Control Hosp Epidemiol. 2005;26:273–280. doi: 10.1086/502539. [DOI] [PubMed] [Google Scholar]
- 10.Pepin J, Valiquette L, Cossette B. Mortality attributable to nosocomial Clostridium difficile-associated disease during an epidemic caused by a hypervirulent strain in Quebec. CMAJ. 2005;173:1037–1042. doi: 10.1503/cmaj.050978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.O’Connor JR, Johnson S, Gerding DN. Clostridium difficile infection caused by the epidemic BI/NAP1/027 strain. Gastroenterology. 2009;136:1913–1924. doi: 10.1053/j.gastro.2009.02.073. [DOI] [PubMed] [Google Scholar]
- 12.Brazier JS, et al. Distribution and antimicrobial susceptibility patterns of Clostridium difficile PCR ribotypes in English hospitals, 2007-08. Euro Surveill. 2008:13. doi: 10.2807/ese.13.41.19000-en. [DOI] [PubMed] [Google Scholar]
- 13.Bauer MP, et al. Clostridium difficile infection in Europe: a hospital-based survey. Lancet. 2011;377:63–73. doi: 10.1016/S0140-6736(10)61266-4. [DOI] [PubMed] [Google Scholar]
- 14.Kuijper EJ, et al. Update of Clostridium difficile infection due to PCR ribotype 027 in Europe, 2008. Euro Surveill. 2008:13. [PubMed] [Google Scholar]
- 15.Richards M, et al. Severe infection with Clostridium difficile PCR ribotype 027 acquired in Melbourne, Australia. Med J Aust. 2011;194:369–371. doi: 10.5694/j.1326-5377.2011.tb03012.x. [DOI] [PubMed] [Google Scholar]
- 16.Stabler RA, et al. Comparative genome and phenotypic analysis of Clostridium difficile 027 strains provides insight into the evolution of a hypervirulent bacterium. Genome Biol. 2009;10:R102. doi: 10.1186/gb-2009-10-9-r102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Croucher NJ, et al. Rapid pneumococcal evolution in response to clinical interventions. Science. 2011;331:430–434. doi: 10.1126/science.1198545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Harris SR, et al. Evolution of MRSA during hospital transmission and intercontinental spread. Science. 2010;327:469–474. doi: 10.1126/science.1182395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Pearson T, et al. Phylogenetic discovery bias in Bacillus anthracis using single-nucleotide polymorphisms from whole-genome sequencing. Proc Natl Acad Sci U S A. 2004;101:13536–13541. doi: 10.1073/pnas.0403844101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Spigaglia P, Carattoli A, Barbanti F, Mastrantonio P. Detection of gyrA and gyrB mutations in Clostridium difficile isolates by real-time PCR. Mol Cell Probes. 2010;24:61–67. doi: 10.1016/j.mcp.2009.10.002. [DOI] [PubMed] [Google Scholar]
- 21.Kim H, et al. Emergence of Clostridium difficile Ribotype 027 in Korea. Korean J Lab Med. 2011;31:191–196. doi: 10.3343/kjlm.2011.31.3.191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lemey P, Rambaut A, Drummond AJ, Suchard MA. Bayesian phylogeography finds its roots. PLoS Comput Biol. 2009;5:e1000520. doi: 10.1371/journal.pcbi.1000520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Dallal RM, et al. Fulminant Clostridium difficile: an underappreciated and increasing cause of death and complications. Ann Surg. 2002;235:363–372. doi: 10.1097/00000658-200203000-00008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.MacCannell DR, et al. Molecular analysis of Clostridium difficile PCR ribotype 027 isolates from Eastern and Western Canada. J Clin Microbiol. 2006;44:2147–2152. doi: 10.1128/JCM.02563-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Drummond AJ, Rambaut A. BEAST: Bayesian evolutionary analysis by sampling trees. BMC Evol Biol. 2007;7:214. doi: 10.1186/1471-2148-7-214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Nyc O, Pituch H, Matejkova J, Obuch-Woszczatynski P, Kuijper EJ. Clostridium difficile PCR ribotype 176 in the Czech Republic and Poland. Lancet. 2011;377:1407. doi: 10.1016/S0140-6736(11)60575-8. [DOI] [PubMed] [Google Scholar]
- 27.Brazier JS. Clostridium difficile: The anaerobe that made the grade. Anaerobe. 2012 doi: 10.1016/j.anaerobe.2011.12.021. [DOI] [PubMed] [Google Scholar]
- 28.Carter GP, Awad MM, Kelly ML, Rood JI, Lyras D. TcdB or not TcdB: a tale of two Clostridium difficile toxins. Future Microbiol. 2011;6:121–123. doi: 10.2217/fmb.10.169. [DOI] [PubMed] [Google Scholar]
- 29.Sebaihia M, et al. The multidrug-resistant human pathogen Clostridium difficile has a highly mobile, mosaic genome. Nat Genet. 2006;38:779–786. doi: 10.1038/ng1830. [DOI] [PubMed] [Google Scholar]
- 30.He M, et al. Evolutionary dynamics of Clostridium difficile over short and long time scales. Proc Natl Acad Sci U S A. 2010;107:7527–7532. doi: 10.1073/pnas.0914322107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Brouwer MSM, Warburton PJ, Roberts AP, Mullany P, Allan E. Genetic Organisation, Mobility and Predicted Functions of Genes on Integrated, Mobile Genetic Elements in Sequenced Strains of Clostridium difficile. PLoS ONE. 2011;6:e23014. doi: 10.1371/journal.pone.0023014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Linder JA, Huang ES, Steinman MA, Gonzales R, Stafford RS. Fluoroquinolone prescribing in the United States: 1995 to 2002. Am J Med. 2005;118:259–268. doi: 10.1016/j.amjmed.2004.09.015. [DOI] [PubMed] [Google Scholar]
- 33.Koser CU, et al. Rapid whole-genome sequencing for investigation of a neonatal MRSA outbreak. N Engl J Med. 2012;366:2267–2275. doi: 10.1056/NEJMoa1109910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25:1754–1760. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Li H, et al. The Sequence Alignment/Map format and SAMtools. Bioinformatics. 2009;25:2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kurtz S, et al. REPuter: the manifold applications of repeat analysis on a genomic scale. Nucleic Acids Res. 2001;29:4633–4642. doi: 10.1093/nar/29.22.4633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kurtz S, et al. Versatile and open software for comparing large genomes. Genome Biol. 2004;5:R12. doi: 10.1186/gb-2004-5-2-r12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Posada D. jModelTest: phylogenetic model averaging. Molecular Biology and Evolution. 2008;25:1253. doi: 10.1093/molbev/msn083. [DOI] [PubMed] [Google Scholar]
- 39.Huson DH, Bryant D. Application of phylogenetic networks in evolutionary studies. Mol Biol Evol. 2006;23:254–267. doi: 10.1093/molbev/msj030. [DOI] [PubMed] [Google Scholar]
- 40.Guindon S, Gascuel O. A simple, fast, and accurate algorithm to estimate large phylogenies by maximum likelihood. Syst Biol. 2003;52:696–704. doi: 10.1080/10635150390235520. [DOI] [PubMed] [Google Scholar]
- 41.Suchard MA, Weiss RE, Sinsheimer JS. Bayesian selection of continuous-time Markov chain evolutionary models. Mol Biol Evol. 2001;18:1001–1013. doi: 10.1093/oxfordjournals.molbev.a003872. [DOI] [PubMed] [Google Scholar]
- 42.Zerbino DR, Birney E. Velvet: algorithms for de novo short read assembly using de Bruijn graphs. Genome Res. 2008;18:821–829. doi: 10.1101/gr.074492.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Carver TJ, et al. ACT: the Artemis Comparison Tool. Bioinformatics. 2005;21:3422–3423. doi: 10.1093/bioinformatics/bti553. [DOI] [PubMed] [Google Scholar]
- 44.Morelli G, et al. Yersinia pestis genome sequencing identifies patterns of global phylogenetic diversity. Nat Genet. 2010 doi: 10.1038/ng.705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Drummond AJ, Rambaut A, Shapiro B, Pybus OG. Bayesian coalescent inference of past population dynamics from molecular sequences. Mol Biol Evol. 2005;22:1185–1192. doi: 10.1093/molbev/msi103. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.