

Abstract
For T cell activation, three signals have to be provided from the antigen presenting cell; Signal 1 (antigen recognition), signal 2 (co-stimulation) and signal 3 (cytokine priming). Blocking negative co-stimulation during antigen presentation to T cells is becoming a promising therapeutic strategy to enhance cancer immunotherapy. Here we will focus on interference with PD-1/PD-L1 negative co-stimulation during antigen presentation to T cells as a therapeutic approach. We will discuss the potential mechanisms and the therapeutic consequences by which interference/inhibition with this interaction results in anti-tumour immunity. Particularly, we will comment on whether blocking negative co-stimulation provides differentiation signals to T cells undergoing antigen presentation. A major dogma in immunology states that T cell differentiation signals are given by cytokines and chemokines (signal 3) rather than co-stimulation (signal 2). We will discuss whether this is the case when blocking PD-L1/PD-1 negative co-stimulation.
Keywords: Cancer, Co-stimulation, Dendritic cell, Immunotherapy, PD-L1, PD-1, CD80
Introduction
The objective of anti-tumour immunotherapy is to stimulate immune responses that can identify and eliminate tumour cells. This approach is powerful, at least from a theoretical point of view [1-3]. Some classic anti-neoplastic treatments such as chemotherapy lack specificity and significantly affect the functions of normal, non-cancerous tissues and organs. Other approaches such as radiotherapy and surgical removal target tumour cells locally, by either inducing their direct destruction, or by removing them. However, small numbers of cancer cells can metastasise from the primary tumour and colonise other places in the organism. Immunotherapy, on the other hand, depends on the specific recognition of tumour-associated antigens (TAA), and the expansion of TAA-specific cytotoxic cells. These cells would potentially attack primary tumour cells as well as metastases. However, cancer immunotherapy has to overcome major obstacles. TAAs are frequently overexpressed auto-antigens, or poorly immunogenic mutated autoantigens (quasi-antigens) [3-7]. In either case, the immune system has in place strong tolerogenic mechanisms that prevent cytotoxic cells from attacking TAA-expressing cells. These mechanisms are essential to keep systemic tolerance and prevent the development of autoimmune diseases. Nevertheless, there is a significant amount of experimental evidence that suggests that tumours are in fact actively attacked by the immune system. As a consequence, cancer cells are constantly subjected to a strong selective pressure from the immune system [8]. To counteract the immune attack, cancer cells actively inhibit and escape from the immune system. They achieve this by a variety of mechanisms, including low expression of major histocompatibility molecules (MHC molecules associate to antigen peptides for antigen presentation to T cells), secretion of potent immunosuppressive cytokines, and expression of T cell inhibitory molecules such as some members of the B7 family of molecules (PD-L1, PD-L2, B7-H3, VISTA) [9-13].
Thus, current immunotherapy approaches are aimed at stimulating the expansion of effective TAA-cytotoxic T cells, and counteracting the strong immunosuppressive mechanisms exerted by cancer cells. Recently, the use of biological agents based on blocking/neutralising antibodies has “revolutionised” biomedicine [14]. These agents have been successfully applied for the treatment of autoimmune disease and cancer. Some examples for clinical use are Rituximab (B cell-depleting antibody used for leukaemia, rheumatoid arthritis, lupus), Infliximab (TNF-alpha-neutralising antibody used in rheumatic diseases) or Ipilimumab (blocking/depleting anti-CTLA4 antibody, used in cancer) [15-19]. In the case of cancer immunotherapy, these antibodies usually interfere with T cell inhibitory interactions, such as CTLA4 on the surface of T cells with CD80/CD86 on the surface of professional antigen presenting cells (APCs), or in an analogous way, PD-1 with PD-L1/PD-L2 [9].
It is widely accepted that T cell responses are essential for effective anti-tumour immunotherapy. However, T cell activities are controlled at multiple levels. These regulatory controls are necessary to prevent T cells from becoming hyperactivated, causing significant collateral damage to non-target tissue. One of these key regulatory T cell inhibitory interactions takes place between PD-L1 on APCs, and PD-1 on T cells. In fact, the use of therapeutic blocking antibodies in several recent clinical trials has highlighted the anti-tumour efficacy of blocking PD-L1/PD-1. It has to be noted that this interaction takes place at two different time points in the T cell life cycle; first, during antigen presentation to naïve T cells for their activation and differentiation, and second, during antigen recognition on the target cancer cell. This interaction is regarded as a major “T cell brake”, which inhibits T cell activities particularly during their cytotoxic attack in the tumour itself. This interaction plays a different role during naïve T cell activation, where it might also influence the differentiation pathway of activated T cells, leading to either cytotoxic, antibody or regulatory responses. As PD-L1/PD-1 blocking antibodies are systemically administered, it is highly likely that this interaction is inhibited both during naïve T cell activation and during the engagement of cytotoxic T cells to their targets.
Most of the published work does not differentiate between these two scenarios, but the specific inhibition of this interaction during naïve T cell activation or in already committed cytotoxic/effector T cells may lead to different outcomes. We propose in this commentary that future work should be targeted in assessing the consequences of blocking this interaction locally rather than systemically, which could result in better treatments with lower toxicity.
Antigen Presentation to T cells and T cell Differentiation
T cell responses are critical for the induction of long-lasting immunity, particularly against infectious diseases. However, if activated T cells get out of control, or they become hyperactivated, they will cause significant collateral damage to non-infected tissue. This type of responses will enhance inflammation, and release of autoantigens from necrotic tissue, increasing the chances for the induction of autoimmune Diseases. To prevent this situation, T cell activation is controlled at several levels. One of such is antigen presentation, the process by which T cells recognise their cognate antigens presented to them by APCs, such as dendritic cells (DCs) [3]. T cells recognise antigens by binding of their T cell receptor (TCR) with complexes between the antigenic peptides and MHC molecules (p-MCH), present on the surface of APCs [9]. However, for the T cell to be effectively activated, at least two different types of interactions have to occur in the immunological synapse [20,21]. The first one (signal 1) is antigen recognition, mediated by binding of p-MCH complexes by specific TCRs. For effective TCR-dependent signal transduction in the T cell, another interaction (signal 2) has to occur. This interaction is termed co-stimulation, and the main example of activatory (or “positive”) co-stimulation is that mediated by CD80 binding to CD28 between the APC and the T cell [22]. The combination of TCR engagement and CD28 binding strongly activates Zap-70, lck and PI3K, which will lead to T cell activation, expansion and acquisition of effector activities [21,22]. However, in reality, a variety of ligand-receptor interactions take place in the immunological synapse (Figure 1). Many of these interactions are also inhibitory. The final integration between activatory and inhibitory interactions will determine the type and strength of the co-stimulatory signal given to the T cells [23,24]. This will determine the “degree” of T cell activation.
Figure 1. Immunological synapse between a dendritic cell (DC) and a CD4 T cell.
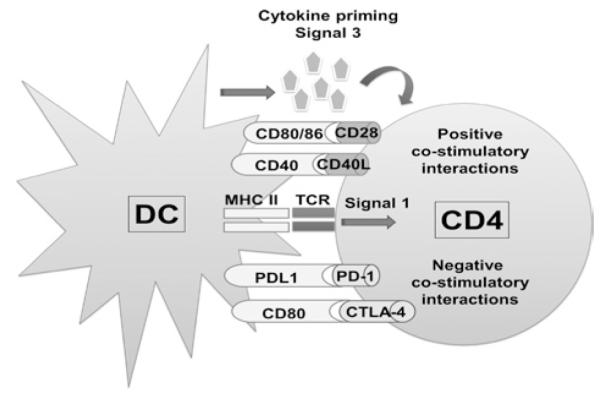
The scheme depicts the three signals between antigen presenting cells and T cells leading to T cell activation. Signal 1 is shown as binding between the peptide-MHC complex with the TCR, as shown in the center of the DC-T cell interaction. In the upper part, positive co-stimulatory interactions are shown, specifically CD80/CD28 and CD40/CD40L, while in the lower part of the DC-T cell interaction, negative co-stimulatory interactions are shown. In this case, PD-L1/PD-1 and CD90/CTLA-4. The integration within the T cell of these two types of interactions will determine the activation state of the T cell. On the upper part of the scheme, signal 3, or cytokine priming, is indicated. Depending on the combination of cytokines delivered by DC and T cells during their interaction, will result in different types of immune responses.
Pathogens exhibit a wide variety of life cycles and pathogenic pathways that require very different immune responses to eliminate them. A different strategy has to be employed to fight a viral infection than bacterial or parasitic infections. Depending on the particular pathogen encountered by DCs and other APCs, these APCs will secrete different cytokines while undertaking antigen presentation to T cells. The particular cytokine combinations (signal 3) will drive the differentiation profile of T cells while signals 1 and 2 are being delivered [9,25-27] (Figure 1). T cells then differentiate to specific types of effector cells such as cytotoxic CD8 and CD4 T cells, and CD4 T helper (Th) cells. These Th cells will preferentially produce particular cytokine profiles that control T and B cell responses in different ways. Th cells can be classified into Th1, Th2, Th17 and regulatory T cells (Tregs), producing mainly IFN-γ, IL4/IL10, IL17 and TGF-β/IL10, respectively. Th1 and Th17 will induce and regulate inflammatory responses, effective for anti-viral, anti-bacterial immunity and cancer [28,29]. Th2 will stimulate antibody responses, relevant for anti-viral and anti-parasitic responses [30], while Tregs will suppress immune responses and keep systemic tolerance [31].
PD-1/PD-L1 Negative Co-Stimulation and Its Role in T cell Activation and Differentiation
A variety of different ligand/receptor interactions take place in the immunological synapse while DCs and T cells are undergoing antigen presentation. Some of these interactions are clearly inducing inhibitory signals towards T cell activation. One of these inhibitory interactions is PD-L1/PD-1 binding [32]. PD-L1 is a member of the B7 family of co-stimulatory/inhibitory molecules that play a key part in immune regulation [33]. PD-L1/PD-1 binding strongly inhibits T cells and induces Treg differentiation. This interaction was found to be essential for maintaining peripheral tolerance [34-36]. In fact, the elimination in KO mouse models of any of these molecules demonstrates the critical role of PD-L1 in controlling immune responses [37] and these KO mice become more susceptible to the development of autoimmune disorders (Table 1).
Table 1.
Interference with PD-L1/PD-1 negative co-stimulation or Cbl-b favours autoimmune disorders.
| Immunopathology | Character | Species | |
|---|---|---|---|
| PD-1 −/− | Lupus-like autoimmune disease | Spontaneous | Mouse [38] |
| Cardiomyopathy | Spontaneous | Mouse [39] | |
| Type 1 diabetes | Predisposition | Mouse [40] | |
| PD-1 blockade | Type I diabetes | Spontaneous | Mouse [40] |
| Graft vs host disease | Worsened | Mouse [41] | |
| Experimental Autoimmune Encephalomyelitis | Worsened | Mouse [42] | |
| PD-1 polymorphisms | Multiple sclerosis | Susceptibility | Human [43] |
| Systemic Lupus Erythematosus | Susceptibility | Human [44] | |
| Lupus Nephritis | Susceptibility | Human [45] | |
| Rheumatoid Arthritis | Susceptibility | Human [46] | |
| Ankylosing Spondylitis | Susceptibility | Human [47] | |
| Cbl-b −/− | Type I diabetes | Susceptibility | Rat [48] |
| Multisystem autoimmune disease | Spontaneous | Mouse [49] | |
| Tumour rejection | Spontaneous | Mouse [50] | |
| Experimental Autoimmune Encephalomyelitis | Susceptibility | Mouse [22] |
PD-L1/PD-1 negative co-stimulation takes place at two different time points during T cell responses, with two different purposes. Firstly, during antigen presentation by professional APCs to naïve uncommitted T cells, and secondly during the cytotoxic T cell attack, as a way of containing collateral tissue damage.
During antigen presentation to naïve T cells, this interaction acts as a brake in TCR signal transduction. PD-1 is transiently up-regulated during antigen presentation as a consequence of T cell activation [32] and PD-L1/PD-1 co-stimulation results in ligand-induced TCR down-modulation [9,38-41]. TCR down-modulation is a fundamental immunological process that regulates TCR signalling, although its precise physiological role is still under debate. There is evidence suggesting that TCR down-modulation is absolutely required for T cell activation [42,43]. However, most of the studies show that this process prevents T cell hyperactivation by terminating TCR signal transduction [44-49]. PD-1 associates to the TCR at the immunological synapse and controls its signal transduction as well as its presence on the T cell surface [39,41]. We demonstrated that TCR down-modulation in mouse CD8 T cells was largely reduced when PD-L1 was silenced in antigen-presenting DCs, or when PD-L1/PD-1 was blocked using antibodies during antigen presentation [39]. Therefore, we proposed the “extrinsic signal model” for antigen-induced TCR down-modulation, triggered by PD-L1/PD-1 co-stimulation (Figure 2).
Figure 2. Extrinsic model for antigen-induced T cell receptor down-modulation.
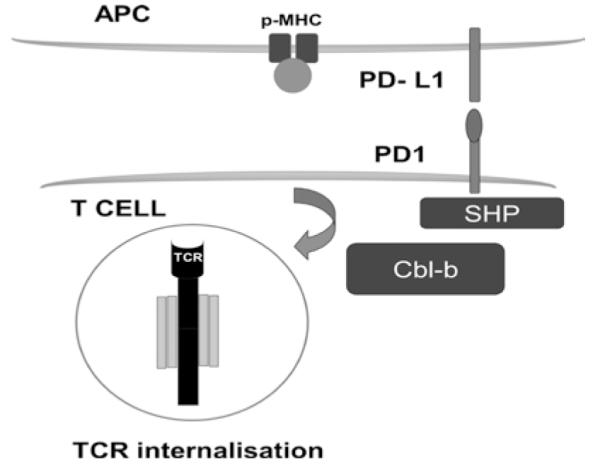
The scheme depicts the internalisation of the TCR during antigen presentation after its interaction with the p-MHC complex as depicted. After T cell activation, PD-1 is expressed on the surface of the T cell undergoing antigen presentation, where it binds to PD-L1 expressed by the APC as shown in the figure. Engaged PD-1 recruits phosphatases (SHP) that will terminate TCR signal transduction. Additionally, the E3 ubiquitin Cbl-b is up-regulated and triggers TCR internalisation.
At the molecular level, the lack of PD-L1/PD-1 co-stimulation during physiological antigen presentation blocked Cbl-b expression in CD8 T cells, an E3 ubiquitin ligase of the casitas-B lymphoma family (Cbl) critical for TCR down-modulation and T cell activation [22,50,51]. Rewrite as: Cbl E3 ubiquitin ligases are transcriptionally up-regulated in the presence of PD-L1/PD-1 co-stimulation, following TCR/CD28 engagement [23]. In fact, elimination of Cbl genes in KO mice results in hyperactivated T cells, in which TCR down-modulation is severely impaired [48,50] (Table 1). These T cells exhibited persistent TCR signal transduction and enhanced anti-viral immunity [52], in agreement with our observations in CD8 T cells undergoing antigen presentation by PD-L1-silenced DCs [39]. CD8 T cells activated by DCs in the absence of PD-L1/PD-1 co-stimulation were clearly hyperactivated, with high TCR surface levels, and with a significant increase in production of pro-inflammatory cytokines IFN-γ and IL17. Therefore, this data would apparently suggest that PD-L1/PD-1 signal transduction commits CD8 T cell differentiation towards cytotoxic responses, by a yet undefined mechanism.
PD-L1/PD-1 interactions also occur during the T cell cytotoxic attack exerted towards tumour cells, possibly as a way of containing unwanted excessive collateral damaged to normal tissue. In this situation, T cells are already differentiated and committed towards particular effector activities. PD-L1/PD-1 interaction may not control T cell differentiation in this instance, but rather TCR signal transduction leading to their anti-tumour activities. PD-L1/PD-1 interaction is therefore used by several tumours to avert the cytotoxic attack of effector T cells [53-58]. Thus, its blockade augments anti-cancer immune responses and improves immunotherapy [59,60]. However, in many instances PD-L1/PD-1 blockade or silencing using siRNA results in limited therapeutic activities, unless given in combination with other treatments such as co-administration with anti-CTLA4 antibodies [61,62], PD-L2-blocking antibodies [63], TLR ligands [62], chemotherapy [64], cytokine treatments [65] or modulators of intracellular signalling pathways in DCs [39]. It is yet unclear why PD-L1/PD-1 blockade on its own does not achieve optimal therapeutic effects in these experimental models.
Blocking PD-L1/PD-1 Negative Co-Stimulation may Commit T cells Towards Cytotoxic T cell Responses
According to the experimental evidence, blocking PD-L1/PD-1 interaction potentially acts at two levels. Firstly, it prolongs TCR signal transduction, and thus, increases the degree of T cell activation. Second, it may skew T cell differentiation towards pro-inflammatory responses. In our opinion it is critical to clarify this issue, as an adequate T cell differentiation will endow T cells with their cytotoxic anti-tumour potential.
Most of the experimental data suggests that PD-L1/PD-1 blockade hyperactivates cytotoxic T cells. Hyperactivated T cells exhibit a higher proliferation rate, increased cytokine production, and possibly differentiation of multifunctional T cells [32,37,39,60,66]. Particularly the capacity of PD-L1/PD-1 blockade to stimulate polyfunctional T cells has important therapeutic implications [67-69]. These observations suggest that rather than committing T cells towards a pro-inflammatory differentiation pathway, PD-L1/PD-1 blockade may potentially enhance other types of immune responses.
In advanced melanoma patients, systemic administration of PD-1 blocking antibodies leads to differentiation of Th1/Th17 cells and a decrease in Th2 cells. However, this has been tested in the context of superantigen stimulation or recall responses towards tetanus toxoid [70]. In this way, possibly already “committed” memory T cells could be activated. In agreement with this study, most of the experimental evidence suggests that PD-L1/PD-1 blockade results in hyperactivated T cells, which exhibit increased proliferation and production of pro-inflammatory cytokines, and enhanced effector T cell infiltration into tumours [9,32,36,38-40,63,64,71]. However, is it really true that T cells undergoing antigen presentation in the absence (or reduction) of PD-L1/PD-1 co-stimulation are truly committed to Th1/Th17-polarisation? This is difficult to reconcile with the idea that T cell polarisation is driven by cytokine priming, or signal three [9].
PD-L1/PD-1 may “simply” control the timing of TCR stimulation by removing TCRs (or contributing to) from the T cell surface, and by terminating the intracellular signal transduction pathways by recruiting phosphatases (SHP1 and SHP2). This would not necessarily imply that T cells undergoing antigen presentation in the absence of PD-L1/PD-1 co-stimulation may be committed to pro-inflammatory responses. This might be especially true in the tumour itself, where T cells have been already committed towards specific subsets, such as Tregs, Th2s or Th1s. In this situation, PD-L1/PD-1 acts mainly as a brake in TCR signal transduction. The ultimate polarisation of T cell differentiation may still be provided by the combination of cytokines secreted during antigen presentation, or by the lack of PD-L1/PD-1 during antigen presentation to naïve T cells. The consequences of whether it commits T cells to pro-inflammatory differentiation are in any case important for the design of therapeutic treatments.
If PD-L1/PD-1 blockade/interference provides a Th1/Th17 differentiation signal, blocking antibodies and other interference systems would surely improve anti-tumour and anti-viral immune responses. However, if PD-L1/PD-1 signalling is just a T cell “brake”, blocking antibodies in immunosuppressive settings (such as advanced cancers, or locally in the tumour itself) may not be as effective as other blocking antibodies such as anti-CTLA4. Blocking this interaction may hyperactivate Th2 or Treg cells in the tumour microenvironment, rather than expanding pro-inflammatory effector T cells.
Application of PD-L1/PD-1 Blockade in Clinical Trials
Recently, the first results of successful human clinical trials have been published, using either systemic administration of a PD-L1 [72] or a PD-1 blocking antibody [73,74]. The importance of these trials is their application in a wide number of advanced cancers, including melanoma, colon, renal, pancreatic, ovarian, gastric, lung and breast cancer. In addition, toxicity studies were carried out, taking into consideration the potential high risk in the development of inflammatory disorders/complications. The anti-PD-L1 clinical trial showed that although the percentage of patients exhibiting objective responses was relatively low (durable tumor regression 6 to 17%), the authors demonstrated that immunotherapy could be successfully applied even in cancers that were previously thought to be unresponsive to such therapies [72]. However, 9% of the patients exhibited serious toxic effects as a direct result of the treatment, highlighting the importance of minimising the adverse effects of this potent immunotherapeutic approach [72].
The same authors carried out clinical trials using systemic administration of a PD-1 blocking antibody, in a similar array of cancers [74]. In this anti-PD-1 clinical trial, objective responses were observed only in those tumours with PD-L1 expression, as expected. Interestingly, seemingly better responses than with anti-PD-L1 blocking antibodies were observed (1/4 to 1/5 patients showed objective responses) and objective responses were also observed in various sites of metastasis. Compared to the anti-PD-L1 clinical trial the percentage of patients exhibiting drug-related serious adverse events was higher (14% of patients) and they were all related to immune aetiology and directly related to the treatment [74].
Overall, these clinical trials highlight the applicability and efficacy of interference with negative co-stimulation, in particular PD-L1/PD-1 interaction, even if associated to serious adverse effects.
Considering the published data, it would seem that interference with PD-L1/PD-1 blockade stimulates anti-viral and anti-cancer immune responses and rescues T cells from exhaustion [71,75-79]. In fact, there is evidence that shows skewing towards a Th1/Th17 response in advanced cancer [70]. However, other reports show that the therapeutic improvements are modest [80] and in some cases restricted to antigen presentation in the context of minor histocompatibility complexes [63]. In most of the experimental systems and clinical trials, PD-L1/PD-1 blockade works best in combination with other immune-stimulatory approaches, or even chemotherapy [62,64,81,82]. Even though PD-L1/PD-1 blockade may in fact lead to inflammatory responses (Table 1), we propose that reinforcing T cell differentiation by providing strong signals three will certainly improve PD-L1/PD-1 blocking as an effective therapeutic strategy.
Where to Block, at the Beginning or at the End?
Possibly the major problem of systemic application of PD-L1/PD-1 blocking antibodies is the inhibition of this key regulatory interaction not only in the tumour environment, but also during antigen presentation. It has to be noted that myeloid DCs actively present autoantigens and innocuous xenoantigens to T cells for the induction and maintenance of immunological tolerance. In this context, abrogation of this interaction may enhance the risk of developing inflammatory disorders and even autoimmune disease [38]. Therefore, efforts have to be made to improve this promising strategy to maximise anti-tumour therapeutic activities while reducing toxicity. So, where would it be therapeutically more relevant to inhibit PD-L1/PD-1 co-stimulation? During antigen presentation or in the tumour microenvironment?
So far, to our knowledge, there is not a specific published study addressing this question. In a recent study from our group, we inhibited PD-L1/PD-1 co-stimulation in APCs using RNA interference, and applied this strategy in a mouse model of lymphoma. Interestingly, although T cell responses were accelerated as a result of T cell hyperactivation, this strategy did not improve the anti-tumour effects compared to vaccination with PD-L1 non-silenced DCs [39]. To achieve a significant therapeutic effect PD-L1 KO had to be combined with constitutive activators of intracellular signalling pathways in DCs [2,39,83]. These results showed that inhibition of PD-L1/PD-1 co-stimulation during antigen presentation in the absence of additional immunostimulatory strategies is not sufficient per se.
Consequently, it seems clear that PD-L1/PD-1 co-stimulation has to be targeted at the tumour microenvironment. Tumour cells up-regulate PD-L1 to dampen down the cytotoxic T cell attack. This upregulation is possibly a consequence of pro-inflammatory cytokine production by tumour infiltrating immune cells. IFN produced by inflammatory cells, for example, acts as potent PD-L1 up-regulator.
In summary, we propose that to achieve therapeutic significant effects of interference with PD-L1/PD-1 negative co-stimulation while reducing its toxicity would be to target inhibition, (1) during antigen presentation in combination with DC stimulators [9], or (2) specifically in the tumour microenvironment, avoiding the expansion of autoreactive T cells that may arise during antigen presentation.
Acknowledgements
ID is funded by an ERASMUS (EU) scholarship. KB is funded by the Fund for Scientific Research-Flandes. DE is funded by an arthritis research UK career development fellowship (18433).
Footnotes
The authors declare no conflicts of interest.
References
- 1.Mellman I, Coukos G, Dranoff G. Cancer immunotherapy comes of age. Nature. 2011;480:480–489. doi: 10.1038/nature10673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Arce F, Kochan G, Breckpot K, Stephenson H, Escors D. Selective activation of intracellular signalling pathways in dendritic cells for cancer immunotherapy. Anticancer Agents Med Chem. 2012;12:29–39. doi: 10.2174/187152012798764679. [DOI] [PubMed] [Google Scholar]
- 3.Breckpot K, Escors D. Dendritic cells for active anti-cancer immunotherapy: targeting activation pathways through genetic modification. Endocr Metab Immune Disord Drug Targets. 2009;9:328–343. doi: 10.2174/187153009789839156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Perro M, Tsang J, Xue SA, Escors D, Cesco-Gaspere M, et al. Generation of multi-functional antigen-specific human T-cells by lentiviral TCR gene transfer. Gene Ther. 2010;17:721–732. doi: 10.1038/gt.2010.4. [DOI] [PubMed] [Google Scholar]
- 5.Boon T, van der Bruggen P. Human tumor antigens recognized by T lymphocytes. J Exp Med. 1996;183:725–729. doi: 10.1084/jem.183.3.725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chen YT, Scanlan MJ, Sahin U, Türeci O, Gure AO, et al. A testicular antigen aberrantly expressed in human cancers detected by autologous antibody screening. Proc Natl Acad Sci USA. 1997;94:1914–1918. doi: 10.1073/pnas.94.5.1914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Van den Eynde BJ, van der Bruggen P. T cell defined tumor antigens. Curr Opin Immunol. 1997;9:684–693. doi: 10.1016/s0952-7915(97)80050-7. [DOI] [PubMed] [Google Scholar]
- 8.DuPage M, Mazumdar C, Schmidt LM, Cheung AF, Jacks T. Expression of tumour-specific antigens underlies cancer immunoediting. Nature. 2012;482:405–409. doi: 10.1038/nature10803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Liechtenstein T, Dufait I, Lanna A, Breckpot K, Escors D. Modulating Co-Stimulation During Antigen Presentation to Enhance Cancer Immunotherapy. Immunol Endocr Metab Agents Med Chem. 2012;12:224–235. doi: 10.2174/187152212802001875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wang L, Rubinstein R, Lines JL, Wasiuk A, Ahonen C, et al. VISTA, a novel mouse Ig superfamily ligand that negatively regulates T cell responses. J Exp Med. 2011;208:577–592. doi: 10.1084/jem.20100619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chapoval AI, Ni J, Lau JS, Wilcox RA, Flies DB, et al. B7-H3: a costimulatory molecule for T cell activation and IFN-gamma production. Nat Immunol. 2001;2:269–274. doi: 10.1038/85339. [DOI] [PubMed] [Google Scholar]
- 12.Tekle C, Nygren MK, Chen YW, Dybsjord I, Nesland JM, et al. B7-H3 contributes to the metastatic capacity of melanoma cells by modulation of known metastasis-associated genes. Int J Cancer. 2012;130:2282–2290. doi: 10.1002/ijc.26238. [DOI] [PubMed] [Google Scholar]
- 13.Sica GL, Choi IH, Zhu G, Tamada K, Wang SD, et al. B7-H4, a molecule of the B7 family, negatively regulates T cell immunity. Immunity. 2003;18:849–861. doi: 10.1016/s1074-7613(03)00152-3. [DOI] [PubMed] [Google Scholar]
- 14.Dienstmann R, Markman B, Tabernero J. Application of monoclonal antibodies as cancer therapy in solid tumors. Curr Clin Pharmacol. 2012;7:137–145. doi: 10.2174/157488412800228929. [DOI] [PubMed] [Google Scholar]
- 15.Ehrenstein MR, Evans JG, Singh A, Moore S, Warnes G, et al. Compromised function of regulatory T cells in rheumatoid arthritis and reversal by anti-TNFalpha therapy. J Exp Med. 2004;200:277–285. doi: 10.1084/jem.20040165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Nadkarni S, Mauri C, Ehrenstein MR. Anti-TNF-alpha therapy induces a distinct regulatory T cell population in patients with rheumatoid arthritis via TGF-beta. J Exp Med. 2007;204:33–39. doi: 10.1084/jem.20061531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Trinh VA, Hwu WJ. Ipilimumab in the treatment of melanoma. Expert Opin Biol Ther. 2012;12:773–782. doi: 10.1517/14712598.2012.675325. [DOI] [PubMed] [Google Scholar]
- 18.Tomasini P, Khobta N, Greillier L, Barlesi F. Ipilimumab: its potential in non-small cell lung cancer. Ther Adv Med Oncol. 2012;4:43–50. doi: 10.1177/1758834011431718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.de la Torre I, Leandro MJ, Edwards JC, Cambridge G. Baseline serum immunoglobulin levels in patients with rheumatoid arthritis: relationships with clinical parameters and with B-cell dynamics following rituximab. Clin Exp Rheumatol. 2012;30:554–560. [PubMed] [Google Scholar]
- 20.Horgan KJ, Van Seventer GA, Shimizu Y, Shaw S. Hyporesponsiveness of “naive” (CD45RA+) human T cells to multiple receptor-mediated stimuli but augmentation of responses by co-stimuli. Eur J Immunol. 1990;20:1111–1118. doi: 10.1002/eji.1830200525. [DOI] [PubMed] [Google Scholar]
- 21.Janeway CA, Jr, Bottomly K. Signals and signs for lymphocyte responses. Cell. 1994;76:275–285. doi: 10.1016/0092-8674(94)90335-2. [DOI] [PubMed] [Google Scholar]
- 22.Chiang YJ, Kole HK, Brown K, Naramura M, Fukuhara S, et al. Cbl-b regulates the CD28 dependence of T-cell activation. Nature. 2000;403:216–220. doi: 10.1038/35003235. [DOI] [PubMed] [Google Scholar]
- 23.Nurieva R, Thomas S, Nguyen T, Martin-Orozco N, Wang Y, et al. T-cell tolerance or function is determined by combinatorial costimulatory signals. EMBO J. 2006;25:2623–2633. doi: 10.1038/sj.emboj.7601146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Krummel MF, Allison JP. Pillars article: CD28 and CTLA-4 have opposing effects on the response of T cells to stimulation. The journal of experimental medicine. 1995. 182: 459-465. J Immunol. 2011;187:3459–3465. [PubMed] [Google Scholar]
- 25.Curtsinger JM, Schmidt CS, Mondino A, Lins DC, Kedl RM, et al. Inflammatory cytokines provide a third signal for activation of naive CD4+ and CD8+ T cells. J Immunol. 1999;162:3256–3262. [PubMed] [Google Scholar]
- 26.Curtsinger JM, Johnson CM, Mescher MF. CD8 T cell clonal expansion and development of effector function require prolonged exposure to antigen, costimulation, and signal 3 cytokine. J Immunol. 2003;171:5165–5171. doi: 10.4049/jimmunol.171.10.5165. [DOI] [PubMed] [Google Scholar]
- 27.Curtsinger JM, Lins DC, Mescher MF. Signal 3 determines tolerance versus full activation of naive CD8 T cells: dissociating proliferation and development of effector function. J Exp Med. 2003;197:1141–1151. doi: 10.1084/jem.20021910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bettelli E, Carrier Y, Gao W, Korn T, Strom TB, et al. Reciprocal developmental pathways for the generation of pathogenic effector TH17 and regulatory T cells. Nature. 2006;441:235–238. doi: 10.1038/nature04753. [DOI] [PubMed] [Google Scholar]
- 29.Macatonia SE, Hosken NA, Litton M, Vieira P, Hsieh CS, et al. Dendritic cells produce IL-12 and direct the development of Th1 cells from naive CD4+ T cells. J Immunol. 1995;154:5071–5079. [PubMed] [Google Scholar]
- 30.Kalinski P, Hilkens CM, Snijders A, Snijdewint FG, Kapsenberg ML. IL-12-deficient dendritic cells, generated in the presence of prostaglandin E2, promote type 2 cytokine production in maturing human naive T helper cells. J Immunol. 1997;159:28–35. [PubMed] [Google Scholar]
- 31.Sakaguchi S, Yamaguchi T, Nomura T, Ono M. Regulatory T cells and immune tolerance. Cell. 2008;133:775–787. doi: 10.1016/j.cell.2008.05.009. [DOI] [PubMed] [Google Scholar]
- 32.Freeman GJ, Long AJ, Iwai Y, Bourque K, Chernova T, et al. Engagement of the PD-1 immunoinhibitory receptor by a novel B7 family member leads to negative regulation of lymphocyte activation. J Exp Med. 2000;192:1027–1034. doi: 10.1084/jem.192.7.1027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Greenwald RJ, Freeman GJ, Sharpe AH. The B7 family revisited. Annu Rev Immunol. 2005;23:515–548. doi: 10.1146/annurev.immunol.23.021704.115611. [DOI] [PubMed] [Google Scholar]
- 34.Francisco LM, Sage PT, Sharpe AH. The PD-1 pathway in tolerance and autoimmunity. Immunol Rev. 2010;236:219–242. doi: 10.1111/j.1600-065X.2010.00923.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wang L, Pino-Lagos K, de Vries VC, Guleria I, Sayegh MH, et al. Programmed death 1 ligand signaling regulates the generation of adaptive Foxp3+CD4+ regulatory T cells. Proc Natl Acad Sci U S A. 2008;105:9331–9336. doi: 10.1073/pnas.0710441105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Fife BT, Pauken KE, Eagar TN, Obu T, Wu J, et al. Interactions between PD-1 and PD-L1 promote tolerance by blocking the TCR-induced stop signal. Nat Immunol. 2009;10:1185–1192. doi: 10.1038/ni.1790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Latchman YE, Liang SC, Wu Y, Chernova T, Sobel RA, et al. PD-L1-deficient mice show that PD-L1 on T cells, antigen-presenting cells, and host tissues negatively regulates T cells. Proc Natl Acad Sci U S A. 2004;101:10691–10696. doi: 10.1073/pnas.0307252101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Karwacz K, Arce F, Bricogne C, Kochan G, Escors D. PD-L1 co-stimulation, ligand-induced TCR down-modulation and anti-tumor immunotherapy. Oncoimmunology. 2012;1:86–88. doi: 10.4161/onci.1.1.17824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Karwacz K, Bricogne C, MacDonald D, Arce F, Bennett CL, et al. PD-L1 co-stimulation contributes to ligand-induced T cell receptor down-modulation on CD8+ T cells. EMBO Mol Med. 2011;3:581–592. doi: 10.1002/emmm.201100165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Escors D, Bricogne C, Arce F, Kochan G, Karwacz K. On the Mechanism of T cell receptor down-modulation and its physiological significance. J Biosci Med. 2011;1 [PMC free article] [PubMed] [Google Scholar]
- 41.Yokosuka T, Takamatsu M, Kobayashi-Imanishi W, Hashimoto-Tane A, Azuma M, et al. Programmed cell death 1 forms negative costimulatory microclusters that directly inhibit T cell receptor signaling by recruiting phosphatase SHP2. J Exp Med. 2012;209:1201–1217. doi: 10.1084/jem.20112741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hansen AK, Regner M, Bonefeld CM, Boding L, Kongsbak M, et al. TCR down-regulation boosts T-cell-mediated cytotoxicity and protection against poxvirus infections. Eur J Immunol. 2011;41:1948–1957. doi: 10.1002/eji.201141413. [DOI] [PubMed] [Google Scholar]
- 43.Valitutti S, Müller S, Cella M, Padovan E, Lanzavecchia A. Serial triggering of many T-cell receptors by a few peptide-MHC complexes. Nature. 1995;375:148–151. doi: 10.1038/375148a0. [DOI] [PubMed] [Google Scholar]
- 44.Baniyash M. TCR zeta-chain downregulation: curtailing an excessive inflammatory immune response. Nat Rev Immunol. 2004;4:675–687. doi: 10.1038/nri1434. [DOI] [PubMed] [Google Scholar]
- 45.Boding L, Bonefeld CM, Nielsen BL, Lauritsen JP, von Essen MR, et al. TCR down-regulation controls T cell homeostasis. J Immunol. 2009;183:4994–5005. doi: 10.4049/jimmunol.0901539. [DOI] [PubMed] [Google Scholar]
- 46.Bonefeld CM, Haks M, Nielsen B, von Essen M, Boding L, et al. TCR down-regulation controls virus-specific CD8+ T cell responses. J Immunol. 2008;181:7786–7799. doi: 10.4049/jimmunol.181.11.7786. [DOI] [PubMed] [Google Scholar]
- 47.Liu H, Rhodes M, Wiest DL, Vignali DA. On the dynamics of TCR:CD3 complex cell surface expression and downmodulation. Immunity. 2000;13:665–675. doi: 10.1016/s1074-7613(00)00066-2. [DOI] [PubMed] [Google Scholar]
- 48.Naramura M, Jang IK, Kole H, Huang F, Haines D, et al. c-Cbl and Cbl-b regulate T cell responsiveness by promoting ligand-induced TCR down-modulation. Nat Immunol. 2002;3:1192–1199. doi: 10.1038/ni855. [DOI] [PubMed] [Google Scholar]
- 49.Wang H, Holst J, Woo SR, Guy C, Bettini M, et al. Tonic ubiquitylation controls T-cell receptor: CD3 complex expression during T-cell development. EMBO J. 2010;29:1285–1298. doi: 10.1038/emboj.2010.10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Bachmaier K, Krawczyk C, Kozieradzki I, Kong YY, Sasaki T, et al. Negative regulation of lymphocyte activation and autoimmunity by the molecular adaptor Cbl-b. Nature. 2000;403:211–216. doi: 10.1038/35003228. [DOI] [PubMed] [Google Scholar]
- 51.Jeon MS, Atfield A, Venuprasad K, Krawczyk C, Sarao R, et al. Essential role of the E3 ubiquitin ligase Cbl-b in T cell anergy induction. Immunity. 2004;21:167–177. doi: 10.1016/j.immuni.2004.07.013. [DOI] [PubMed] [Google Scholar]
- 52.Shamim M, Nanjappa SG, Singh A, Plisch EH, LeBlanc SE, et al. Cbl-b regulates antigen-induced TCR down-regulation and IFN-gamma production by effector CD8 T cells without affecting functional avidity. J Immunol. 2007;179:7233–7243. doi: 10.4049/jimmunol.179.11.7233. [DOI] [PubMed] [Google Scholar]
- 53.Blank C, Gajewski TF, Mackensen A. Interaction of PD-L1 on tumor cells with PD-1 on tumor-specific T cells as a mechanism of immune evasion: implications for tumor immunotherapy. Cancer Immunol Immunother. 2005;54:307–314. doi: 10.1007/s00262-004-0593-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Wilcox RA, Feldman AL, Wada DA, Yang ZZ, Comfere NI, et al. B7-H1 (PD-L1, CD274) suppresses host immunity in T-cell lymphoproliferative disorders. Blood. 2009;114:2149–2158. doi: 10.1182/blood-2009-04-216671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Zhang L, Gajewski TF, Kline J. PD-1/PD-L1 interactions inhibit antitumor immune responses in a murine acute myeloid leukemia model. Blood. 2009;114:1545–1552. doi: 10.1182/blood-2009-03-206672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Samimi S, Benoit B, Evans K, Wherry EJ, Showe L, et al. Increased programmed death-1 expression on CD4+ T cells in cutaneous T-cell lymphoma: implications for immune suppression. Arch Dermatol. 2010;146:1382–1388. doi: 10.1001/archdermatol.2010.200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Zhou Q, Munger ME, Highfill SL, Tolar J, Weigel BJ, et al. Program death-1 signaling and regulatory T cells collaborate to resist the function of adoptively transferred cytotoxic T lymphocytes in advanced acute myeloid leukemia. Blood. 2010;116:2484–2493. doi: 10.1182/blood-2010-03-275446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Andorsky DJ, Yamada RE, Said J, Pinkus GS, Betting DJ, et al. Programmed death ligand 1 is expressed by non-hodgkin lymphomas and inhibits the activity of tumor-associated T cells. Clin Cancer Res. 2011;17:4232–4244. doi: 10.1158/1078-0432.CCR-10-2660. [DOI] [PubMed] [Google Scholar]
- 59.Blank C, Kuball J, Voelkl S, Wiendl H, Becker B, et al. Blockade of PD-L1 (B7-H1) augments human tumor-specific T cell responses in vitro. Int J Cancer. 2006;119:317–327. doi: 10.1002/ijc.21775. [DOI] [PubMed] [Google Scholar]
- 60.Pilon-Thomas S, Mackay A, Vohra N, Mulé JJ. Blockade of programmed death ligand 1 enhances the therapeutic efficacy of combination immunotherapy against melanoma. J Immunol. 2010;184:3442–3449. doi: 10.4049/jimmunol.0904114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Curran MA, Montalvo W, Yagita H, Allison JP. PD-1 and CTLA-4 combination blockade expands infiltrating T cells and reduces regulatory T and myeloid cells within B16 melanoma tumors. Proc Natl Acad Sci U S A. 2010;107:4275–4280. doi: 10.1073/pnas.0915174107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Mangsbo SM, Sandin LC, Anger K, Korman AJ, Loskog A, et al. Enhanced tumor eradication by combining CTLA-4 or PD-1 blockade with CpG therapy. J Immunother. 2010;33:225–235. doi: 10.1097/CJI.0b013e3181c01fcb. [DOI] [PubMed] [Google Scholar]
- 63.Hobo W, Maas F, Adisty N, de Witte T, Schaap N, et al. siRNA silencing of PD-L1 and PD-L2 on dendritic cells augments expansion and function of minor histocompatibility antigen-specific CD8+ T cells. Blood. 2010;116:4501–4511. doi: 10.1182/blood-2010-04-278739. [DOI] [PubMed] [Google Scholar]
- 64.Sierro SR, Donda A, Perret R, Guillaume P, Yagita H, et al. Combination of lentivector immunization and low-dose chemotherapy or PD-1/PD-L1 blocking primes self-reactive T cells and induces anti-tumor immunity. Eur J Immunol. 2011;41:2217–2228. doi: 10.1002/eji.201041235. [DOI] [PubMed] [Google Scholar]
- 65.Yu P, Steel JC, Zhang M, Morris JC, Waitz R, et al. Simultaneous inhibition of two regulatory T-cell subsets enhanced Interleukin-15 efficacy in a prostate tumor model. Proc Natl Acad Sci U S A. 2012;109:6187–6192. doi: 10.1073/pnas.1203479109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Wang W, Lau R, Yu D, Zhu W, Korman A, et al. PD1 blockade reverses the suppression of melanoma antigen-specific CTL by CD4+ CD25(Hi) regulatory T cells. Int Immunol. 2009;21:1065–1077. doi: 10.1093/intimm/dxp072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Bhadra R, Gigley JP, Khan IA. PD-1-mediated attrition of polyfunctional memory CD8+ T cells in chronic toxoplasma infection. J Infect Dis. 2012;206:125–134. doi: 10.1093/infdis/jis304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Brooks DG, Ha SJ, Elsaesser H, Sharpe AH, Freeman GJ, et al. IL-10 and PD-L1 operate through distinct pathways to suppress T-cell activity during persistent viral infection. Proc Natl Acad Sci U S A. 2008;105:20428–20433. doi: 10.1073/pnas.0811139106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Velu V, Titanji K, Zhu B, Husain S, Pladevega A, et al. Enhancing SIV-specific immunity in vivo by PD-1 blockade. Nature. 2009;458:206–210. doi: 10.1038/nature07662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Dulos J, Carven GJ, van Boxtel SJ, Evers S, Driessen-Engels LJ, et al. PD-1 blockade augments Th1 and Th17 and suppresses Th2 responses in peripheral blood from patients with prostate and advanced melanoma cancer. J Immunother. 2012;35:169–178. doi: 10.1097/CJI.0b013e318247a4e7. [DOI] [PubMed] [Google Scholar]
- 71.Barber DL, Wherry EJ, Masopust D, Zhu B, Allison JP, et al. Restoring function in exhausted CD8 T cells during chronic viral infection. Nature. 2006;439:682–687. doi: 10.1038/nature04444. [DOI] [PubMed] [Google Scholar]
- 72.Brahmer JR, Tykodi SS, Chow LQ, Hwu WJ, Topalian SL, et al. Safety and activity of anti-PD-L1 antibody in patients with advanced cancer. N Engl J Med. 2012;366:2455–2465. doi: 10.1056/NEJMoa1200694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Brahmer JR, Drake CG, Wollner I, Powderly JD, Picus J, et al. Phase I study of single-agent anti-programmed death-1 (MDX-1106) in refractory solid tumors: safety, clinical activity, pharmacodynamics, and immunologic correlates. J Clin Oncol. 2010;28:3167–3175. doi: 10.1200/JCO.2009.26.7609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Topalian SL, Hodi FS, Brahmer JR, Gettinger SN, Smith DC, et al. Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N Engl J Med. 2012;366:2443–2454. doi: 10.1056/NEJMoa1200690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Dai B, Xiao L, Bryson PD, Fang J, Wang P. PD-1/PD-L1 Blockade Can Enhance HIV-1 Gag-specific T Cell Immunity Elicited by Dendritic Cell-Directed Lentiviral Vaccines. Mol Ther. 2012;20:1800–1809. doi: 10.1038/mt.2012.98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Sakthivel P, Gereke M, Bruder D. Therapeutic intervention in cancer and chronic viral infections: antibody mediated manipulation of PD-1/PD-L1 interaction. Rev Recent Clin Trials. 2012;7:10–23. doi: 10.2174/157488712799363262. [DOI] [PubMed] [Google Scholar]
- 77.Wherry EJ. T cell exhaustion. Nat Immunol. 2011;131:492–499. doi: 10.1038/ni.2035. [DOI] [PubMed] [Google Scholar]
- 78.Aubert RD, Kamphorst AO, Sarkar S, Vezys V, Ha SJ, et al. Antigen-specific CD4 T-cell help rescues exhausted CD8 T cells during chronic viral infection. Proc Natl Acad Sci U S A. 2011;108:21182–21187. doi: 10.1073/pnas.1118450109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Brown KE, Freeman GJ, Wherry EJ, Sharpe AH. Role of PD-1 in regulating acute infections. Curr Opin Immunol. 2010;22:397–401. doi: 10.1016/j.coi.2010.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Breton G, Yassine-Diab B, Cohn L, Boulassel MR, Routy JP, et al. siRNA knockdown of PD-L1 and PD-L2 in monocyte-derived dendritic cells only modestly improves proliferative responses to Gag by CD8(+) T cells from HIV-1-infected individuals. J Clin Immunol. 2009;29:637–645. doi: 10.1007/s10875-009-9313-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Vezys V, Penaloza-MacMaster P, Barber DL, Ha SJ, Konieczny B, et al. 4-1BB signaling synergizes with programmed death ligand 1 blockade to augment CD8 T cell responses during chronic viral infection. J Immunol. 2011;187:1634–1642. doi: 10.4049/jimmunol.1100077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Nakamoto N, Cho H, Shaked A, Olthoff K, Valiga ME, et al. Synergistic reversal of intrahepatic HCV-specific CD8 T cell exhaustion by combined PD-1/ CTLA-4 blockade. PLoS Pathog. 2009;5:e1000313. doi: 10.1371/journal.ppat.1000313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Escors D, Lopes L, Lin R, Hiscott J, Akira S, et al. Targeting dendritic cell signaling to regulate the response to immunization. Blood. 2008;111:3050–3061. doi: 10.1182/blood-2007-11-122408. [DOI] [PubMed] [Google Scholar]