

Abstract
Biomarkers which are indicative of acute physiological and emotional states are studied in a number of different areas in cognitive neuroscience. Currently, many cognitive studies are conducted based on programmed tasks followed by timed biofluid sampling, central laboratory processing, and followed by data analysis. In this work, we present a sensor platform capable of rapid biomarker detection specific for detecting neuropeptide orexin A, found in blood and saliva and known as an indicator of fatigue and cognitive performance. A peptide recognition element that selectively binds to orexin A was designed, characterized, and functionalized onto a zinc oxide field effect transistor to enable rapid detection. The detection limit using the sensor platform was sub-picomolar in water, and picomolar to nanomolar levels in saliva and serum. The transistor and recognition element sensor platform can be easily expanded, allowing for multiple biomarkers to be detected simultaneously, lending itself to complex biomarker analysis applicable to rapid feedback for neuroscience research and physiological monitoring.
Keywords: Orexin A, molecular recognition elements, binding peptide, field effect transistor, rapid detection, human performance monitoring
The distribution of orexin peptides and their receptors in the brain suggests that they may play a key role in multiple regulatory systems, including feeding, autonomic control, sleep, memory, and the reward system (for recent review, see ref (1)). Experiments revealed that orexin levels in the lateral hypothalamic area of rats gradually increased during the active period and exponentially declined in the rest phase.2 The levels of orexin A in the cerebrospinal fluid3 or plasma4 of narcoleptic patients is abnormally decreased (reduction 80–100%). Additionally, a correlation between orexin A levels and individuals with post traumatic stress disorder (PTSD) was made where the level was noticeably decreased.5 There is a considerable amount of evidence suggesting a regulatory role of orexin peptides in cognition performance impaired by sleep deprivation. It was found that intranasal application of orexin A reduces the effect of sleep deprivation on the cognitive performance in nonhuman primates.6 On the other hand, the administration of an orexin-1 receptor antagonist decreased the attentional performance in rats,7 suggesting that orexins contribute to attention. Therefore, it is hypothesized that measuring the level of orexin A in blood of human subjects can allow us to predict a vigilance state and cognitive performance.
Orexin A and B are both produced from a common precursor polypeptide by proteolytic cleavage. The sequences of orexin A are completely conserved among several mammalian species while orexin B is highly conserved (93%) with two residues substituted in rat and mouse comparing with the human sequence.8 Human orexin A neuropeptide has 33 amino acids and contains two disulfide bonds in the N-terminal region. The concentration of orexin A in tissue and blood is much higher than the concentration of orexin B, where orexin A can be found at pg/mL levels and orexin B is undetectable.9 Furthermore, orexin A is highly lipophilic and can rapidly cross the blood-brain barrier.10 In contrast, orexin B has low lipophilicity and is rapidly metabolized in blood which makes it difficult to detect. Orexin neuropeptides act via two G protein-coupled cell surface receptors named orexin receptor-1 (Ox1R) and orexin receptor two (Ox2R). The activation of Ox1R by orexin A is 10–100 times higher than that of orexin B, whereas the activity at the Ox2R is in the same range for both peptides.11
Current techniques for measuring orexin A levels in blood and saliva require fluid sampling and transportation to a laboratory for analysis such as separation through high performance liquid chromatography (HPLC),12 ELISA,13 or via radioimmunoassay (RIA) kit,14 which can detect down to levels of pg/mL. Each of these techniques requires trained technicians in a clinical laboratory setting, expensive supplies and analysis equipment, and, in the case of RIA, radioactive materials. Additionally, due to the sampling efforts and time needed for analysis, instant feedback of orexin A levels of the individual are not possible using these techniques. Rapid detection of biomarkers, such as orexin A, without time-consuming and labor-intensive laboratory analysis is a true need for multiple applications. Sensors using an electrical signal for transduction, such as the field effect transistor15 (FET) and chemiresistor,16 have recently been studied for detecting a variety of molecules.17,18 These sensors can be easily integrated into a modular chip design and programmed to sense a specific molecular target by functionalizing the semiconductor with a biorecognition element (BRE). The BRE is typically an antibody,19 DNA or RNA aptamer,20 or peptide21 specifically selected and designed to bind the target of interest. Peptides have advantages over antibodies in a number of ways, including improved stability, no need for harvesting in animals as peptides can be synthesized, and most importantly for electronic surface charge based platforms, peptides are much smaller than antibodies so they do not have Debye length issues41 as antibodies do. The binding event changes the local electron density on the surface of the semiconductor, resulting in a change in the current flowing through the device, signaling the presence of the target, hence the need for reducing Debye length issues. Additionally, antibodies can be prone to nonspecific binding due to Fc-receptor binding.42 In this work, we present a rapid electronic based sensor platform for detecting orexin A in biofluids at physiologically relevant concentrations.
Results and Discussion
We have developed an electronic based (FET) label-free biosensor with a novel integrated biological recognition element that provides rapid detection of orexin A in saliva and serum. The biological recognition element which binds orexin A was selected using a phage display technique, detailed in the Methods section, and verified through both binding kinetics and computational modeling. The BRE is integrated into an electronic based sensor platform where the target binding event is reported discretely and rapidly (Supporting Information Figure 1).
Phage Display and Binding Kinetics
Candidate orexin A binding peptides (OABP) were obtained through phage display using the Ph.D. 12 phage display from New England BioLabs. Two sequences of interest,DQSNKIISLQRL and FQWHQWNLN, referred to as OABP1 and OABP2, respectively, were identified. To accurately determine the binding kinetics of the peptide-orexin A complex, several concentrations of orexin A binding peptides were injected over a low-density surface of biotin-labeled orexin A immobilized on a streptavidin surface plasmon resonance (SPR) chip using a Biacore T-200. Kinetic fits of the OABP1 and OABP2 SPR sensorgrams (Supporting Information Figure 2) indicate that OAPB1 binds with higher affinity than OABP2 (Table 1). From these rate constants, Kd is calculated as 74 ± 2.1 nM for OABP1, which is comparable to the affinities determined from ELISA. As a negative control, the OABP1 sequence was randomized to QRQLNDKLSIIS (termed NCP) and tested for binding to orexin A, showing no binding (Supporting Information Figure 2). OABP1 was tested for its ability to bind native protein in solution and used in a peptide pull-down experiment in which it was bound to streptavidin magnetic beads and incubated overnight with 50 μM solutions of orexin A. The beads were washed extensively, and the bound protein was eluted. Samples were run on a SDS-PAGE gel and detected by Western Blot with an anti-orexin A monoclonal antibody (Supporting Information Figure 3). Elution samples were split with half of the sample analyzed by silver stain and half analyzed by Western Blot. From the silver stained gel, it can be seen orexin A was precipitated along with one other prominent protein from the sample. As a preliminary test of specificity in complex biological matrices, 200–1665 ng of orexin A was spiked into human brain cell lysate (1 mg/mL in 100 μL of HBS-EP), and streptavidin beads coated with 1 μg/mL of OABP1 were used to pull down orexin A using the same PPD procedure and Western Blot protocol as described above with the exception that BSA was removed from the final wash buffer (Supporting Information Figure 4).
Table 1. Equilibrium Dissociation Constants and Kinetic Constants for Orexin Binding Peptidesa.
| peptide | sequence | Ka (1/Ms) | Kd (1/s) | KD |
|---|---|---|---|---|
| orexin binding peptide (OABP1) | DQSNKIISLQRL | 2.62 × 105 | 0.0194 | 74 ± 2.1 nM |
| orexin binding peptide (OABP2) | TPWFQWHQWNLN | 3.28 × 104 | 1.38 | 42 ± 1.2 μM |
| scrambled orexin peptide (NCP) | QRQLNDKLSIIS | NB | NB | NB |
On-rates (Ka), off-rates (Kd), and dissociation constants (KD) shown were determined from kinetic fits of several sensorgrams across the same concentration series used in equilibrium binding response analysis. NB = no binding.
Computational Modeling
Due to the higher binding affinity of OABP1, this peptide sequence was used for all computational modeling and sensor platform studies detailed in the remaining sections of this paper. The molecular modeling approach of characterizing the orexin A neuropeptide and binding peptide (OA-OABP1) interactions has two objectives. The first is to predict the three-dimensional arrangement of an OA-OABP1 complex given the known orexin-A three-dimensional structures.22 The second objective is to estimate the OA-OABP1 binding energy. One of the most difficult problems in peptide–peptide docking is the flexibility of peptides which have many more degrees of freedom compared with typical small molecules. To address this issue, a two-step process is typically used for predicting the structure of protein–protein complexes.23 Initially, the proteins are considered as rigid molecules and a global search of the rotational and translational space is performed to generate a set of possible complexes. The second stage, called refinement, optimizes and rescores the generated rigid-body conformations by allowing small backbone and side-chain movements for both proteins.24 We applied a similar approach to study the binding of peptides to the orexin A molecule. The rigid docking was performed by using the PatchDock package which is based on the molecular shape complementarity algorithm.25 The generated structures were processed with the FireDock package for flexible docking.26 FireDock performs refinement of each candidate complex by allowing side-chain flexibility and adjustment of the relative orientation of the molecules.
The experimental NMR structure of the orexin A molecule was retrieved from the Protein Data Bank (code 1WSO). Coordinates from the first NMR structure were used in computational docking. Figure 1a shows the unbound structure of orexin A. The residues drawn in red (Leu16, Leu19, Leu20, His26, Gly29, Ile30, Leu31, Thr32, and Leu33) correspond to residues which are involved in binding to orexin-1 receptor.27 It was shown by Darker et al.27 that replacement of these residues with alanine resulted in a significant drop in functional potency at the Ox1R receptor.
Figure 1.
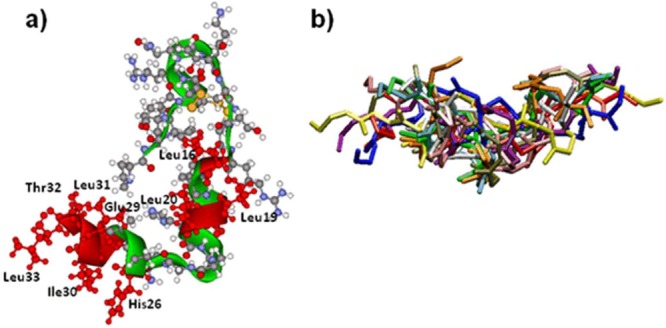
Unbound configurations of molecules used in docking: (a) Experimental structure of orexin A.22 The residues drawn in red are involved in binding to orexin-1 receptor. (b) Ten representative unbound conformations for orexin A binding peptide (only backbone atoms are shown). Structures were visualized with Accelrys’s Discovery Studio Visualizer (http://accelrys.com/products/discovery-studio/).
Short peptides, such as OABP1 and a negative control binding peptide (NCP) used in this study, are very flexible and often lack a well-defined conformation in their unbound state. Therefore, we generated an ensemble of unbound configurations for OABP1 and NCP using replica exchange molecular dynamics.28 A total of 1000 generated conformations for each peptide were used in docking calculations. The representative 10 unbound conformations for OABP1 are presented in Figure 1b (only backbone atoms are shown). The backbone atom root-mean-square deviations were in the range of 1–5 Å, which indicates a significant variability in peptide conformations.
The docking of binding peptides to orexin A was performed in two steps. Initially, the PatchDock package, that considers molecules as rigid, was used to perform a global search of the rotational and translational space based on shape complementary to generate a set of possible complexes. The generated structures were refined using the FireDock software for each candidate complex by allowing the side chains flexibility and adjustments of the relative orientation of the molecules. The refined complexes were scored and ranked according to the FireDock energy function. Figure 2 shows the highest ranked conformation of the OA-OABP1 complex. It can be easily seen that OABP1 peptide binds into the same region of the orexin A molecule as the Ox1R receptor through a combination of electrostatics, hydrogen bonding, and van der Waals interactions. In the proposed structure of the complex, residue Leu12 (which is drawn in yellow) is located at the side of orexin A molecule that allows it to link to the FET surface without the interruption of a complex. NCP also binds to the orexin A molecule although at a much lower level compared with the OAPB1 (Table 2).
Figure 2.
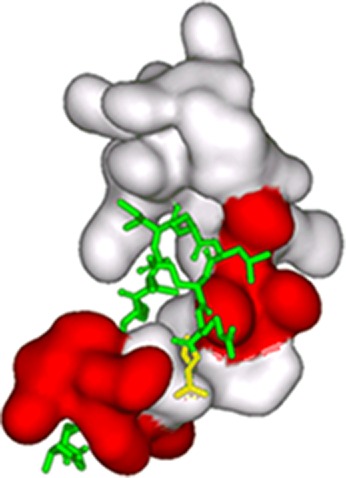
Proposed docking conformation of orexin A with OABP1. The binding peptide is presented as sticks on the surface of orexin A molecule. The part of orexin surface that corresponds to residues that are involved in binding to orexin-1 receptor is drawn in red. The residue Leu12 that forms a bond with G4 linker is drawn in yellow.
Table 2. Binding Free Energy ΔG and Dissociation constants KD for the Five Top Conformations of OA-OABP1 and OA-NCP.
| binding peptide |
negative control |
|||
|---|---|---|---|---|
| complex no. | ΔG(kcal/mol) | KD | ΔG(kcal/mol) | KD |
| 1 | –11.46 | 3.9 nM | –6.87 | 9.1 μM |
| 2 | –9.19 | 180 nM | –6.45 | 18.5 μM |
| 3 | –6.12 | 32.3 μM | –4.51 | 500 μM |
| 4 | –2.73 | 10 mM | –2.77 | 9 mM |
| 5 | –2.08 | 30 mM | +0.14 | |
Initially, the docking configurations for orexin A with OABP1 and NCP were scored with the FireDock energy function.26 That function takes into account atomic contact energy (ACE), van der Waals interactions, partial electrostatics, hydrogen and disulfide bonds, p-stacking and aliphatic interactions, and additional terms.24 Although the FireDock scoring function allows a ranking of binding complexes based on their binding affinity, it does not allow the calculation of the binding free energy. Therefore, we refined the top predicted complex of orexin A with OABP1 using molecular dynamics (MD) simulations. We used the Amber1029 package to perform MD simulations, and the absolute binding free energy was calculated using the molecular mechanics/Poisson–Boltzmann surface area (MM/PBSA) methodology:30 ΔGbinding = ⟨Gcomplex⟩ – ⟨GorexinA⟩ – ⟨Gpeptide⟩. To prepare a system for MM/PBSA calculations, the OA-OABP1 complex was solvated in a rectangular box of TIP3P water molecules. The system was initially minimized for 1000 steps followed by 35 ps of equilibration at 300 K with the restrain on all solute molecules. Next, we performed energy minimization for the entire system. Then, the system was equilibrated for 50 ps at 300 K, and finally, we run a production run for 1 ns. The snapshots of the system trajectory were saved every 10 ps of MD simulations. These snapshots were used to extract the structure of the OA-OABP1 complex as well as unbound molecules. The entropic contribution was calculated using a normal-mode analysis.31 Table 2 shows the binding free energy for the 5 top conformations of OA-OABP1 and OA-NCP. As the data indicates, complexes of OA-OABP1 have lower binding free energies that correspond to significantly higher binding affinity comparing to complexes of OA-NCP. The calculated dissociation constants KD for the OA-OABP1 complexes are in the low nanomolar range, compared with low to high micromolar range for the OA-NCP complexes.
Sensor Development and Testing
The electronic sensor platform is based on a zinc oxide field effect transistor (ZnO-FET), which has several device and biosensor advantages. First, ZnO is deposited via pulsed laser deposition at room temperature conditions which lends to less expensive fabrication demands and ability to use a variety of substrates, including flexible and lightweight plastics.32 Second, ZnO deposited under the correct conditions naturally forms a vertically aligned nanostructure giving the semiconductor enhanced surface area which is ideal for FET sensors. High performance ZnO FETs have been developed for high speed electronic applications33 and also used as a sensor platform for small molecule liquid state sensing using a DNA aptamer as a BRE.34 The selectivity of the system reported here is based on a bifunctional peptide designed to bind to both the ZnO35 semiconductor as an anchor to the FET and to the biomarker target, orexin A. This anchor-sensor motif was successfully used in peptide BRE FETs based on carbon nanotubes21 and graphene36 for trinitrotoluene sensing. The ZnO FET structures are based on an interdigitated electrode design with a 2 μm gap and back gate electrode (Figure 3a). The device stack up is a doped Si wafer, SiO2 insulator, ZnO semiconductor, and gold electrodes (from bottom up). The ZnO is etched (dark region in Figure 3a) for confinement to the active electrode area and to keep each FET electrically isolated from its neighbor. There is no additional material covering the ZnO semiconductor in these devices to ensure direct coupling of the BRE to the ZnO. The diced FET chips (15 mm × 5 mm) were incubated in the peptide solution for 6 min, washed, and dried. A common approach for attaching BREs to surfaces is through covalent linker chemistry.37 This approach is effective, although it can be difficult to control the density of linker and BRE densities on the surface, which directly affects sensor reproducibility. The anchoring peptide domain is based on the sequence developed by Tomczak et al.35 which binds to ZnO. This sequence is added to the orexin A binding peptide (OABP1) sequence with a four glycine residue linker to provide flexibility and separation between the two domains, resulting in a sequence of DQSNKIISLQRL-GGGG-LHVMHKVAPPRGGGC (OABP1–4G linker–ZnO binder). The placement of the leucine end of the OABP1 sequence to the 4G linker was specifically chosen based on the computational modeling data showing the importance of the amino acids on the aspartic acid end of OABP1 to the binding of orexin A.
Figure 3.

Attachment and dry state performance of the ZnO PeptiFET. (a) Optical micrograph of interdigitated electrode ZnO-FET structure. (b) Atomic force micrographs of functionalized PeptiFET overall device structure (i), SiO2 region morphology (ii), and ZnO region morphology. (c) PeptiFET dry state performance with modulation of VG and VSD.
The bifunctional peptide approach is very effective in this design, with the ZnO anchor sequence preferentially binding to the semiconductor over any other surface on the FET array. This is shown in Figure 3b where the surface morphology of the SiO2 region is completely flat (Figure 3b-i), whereas the ZnO region shows a large surface morphology change (Figure 3b-ii) due to the attachment of the bifunctional peptide to the semiconductor. The functionalized ZnO PeptiFET maintains a high performance after processing with ON/OFF ratios of 108 and good current response with modulation of both the gate (VG) and source/drain (VSD) voltages (Figure 3c).
Rapid sensor response was obtained by exposing the PeptiFET to various concentrations of orexin A target solutions while monitoring the source/drain current (ISD) at a constant VG and VSD. For liquid state sensing, the PeptiFET must be rehydrated in order for effective binding of orexin A to take place. This is done by exposing the device to 20 μL of sterile water (Gibco ultrapure DNase RNase free water, 18.2 MΩ) and monitoring the ISD until it is stabilized. At this time, the sensor can be exposed to solutions containing the target or negative control molecules. The sensor was exposed to a solution containing 10 nM orexin A for 1 min, and the source drain current was monitored. A rapid response in the current was observed that reaches a plateau within 60 s. Orexin A has an overall positive charge (pI = 7.8), so the addition of a more positive charge distribution on the surface of the n-type semiconductor causes an increase in current upon binding. The sensor was exposed to varying concentrations of orexin A (10 nM to 1 μM) and monitored over several minutes. Exposure of the sensor to increasing concentrations of orexin A also resulted in a concomitant increase in current (Figure 4a). The magnitude of current increase at higher concentrations begins to decrease due to saturation of the peptide binding sites. For these rapid static measurements, (single drop, no flow through) the response for each concentration of orexin A is determined by difference in current change from the stabilized rehydration step to the stabilized target exposure step. For a given VG and VSD, a response curve can be generated as overall increase in current (A) as a function of concentration of orexin A. The detection limit of the orexin A PeptiFET in water can be estimated at sub-picomolar levels (Supporting Information Figure 6), which is well within the physiologically relevant range.3−5 The rapid nature of the detection of orexin A in sterile water is shown in Figure 4b where a current increase is immediately seen after application of a 10 fM droplet of orexin A. There was no normalization of the data for any of the devices, and great care was taken to control incubation time and device processing to maximize consistency.
Figure 4.

Rapid sensor response of the PeptiFET to orexin A. (a) Increase in ISD within multiple additions of orexin A at concentrations of 10 nM, 100 nM, and 1 μM for VG = 5 V and VSD = 0.5 V. (b) Rapid response to 10 fM orexin A in water, for VG = 5 V and VSD = 0.2 V. (c) Rapid detection of 10 fM orexin A in saliva. (d) Rapid detection of 1 nM orexin A in fetal bovine serum.
Rapid Detection in Biological Fluids
The response of the PeptiFET to orexin A in water is an important study for the proof of concept of the device scheme, but the ultimate application of this sensor is in the rapid detection in biological fluids such as saliva and serum, where sub-nM detection is needed.2−5,12,13 Orexin A has a relatively low molecular weight (3561 g/mol) compared to many of the other molecules in the blood and saliva, such as blood cells, proteins, and enzymes. Therefore, a simple preprocessing step of size exclusion filtration using a 0.2 μm filter can be applied to both saliva and serum to lower the background noise of the biological fluid systems and increase the sensitivity on the PeptiFET. This is likely due to the reduction of large and high viscosity contaminants in the biofluids, such as mucus and large proteins. Before the filtration process, the sensitivity of the PeptiFET in a saliva matrix was seen at 1.4 μM (Supporting Information Figure 7). After filtration, the PeptiFET showed significant response to 10 fM (1 × 10–14) of orexin A when spiked into a 20 μL drop of human saliva, as shown in Figure 4c. Likewise, the PeptiFET can detect down to 1 nM of orexin A when spiked in filtered fetal bovine serum (Figure 4d). A rapid initial increase in the current is observed in the presence of orexin A followed by a stabilization period. The rapid increase is likely due to the initial disruption of charged species near the semiconductor surface after dropwise addition of the target solution.
Negative control experiments are essential to FET sensor evaluation since many semiconductor materials can show current response to a wide variety of molecules and environments. Orexin A tested on an unfunctionalized ZnO FET showed a negligible current change (Supporting Information Figure 8). Thus, the effects of nonspecific binding of the target to the FET are minimal. The BREs add specificity to the sensor, but must also be evaluated against negative control target molecules. Since orexin A is a 33 amino acid peptide, the most stringent negative control that can be tested is a peptide with the same 33 amino acids, but with a different sequence from that of native orexin A. Orexin A sequence was randomized with the following sequence: TGRLKLQCGALPICHQTPHALSLLEAYGRDCCN (MW = 3561, pI = 7.8), as compared to orexin A: QPLPDCCRQKTCSCRLYELLHGAGNHAAGILTL (MW = 3561, pI = 7.8). The randomized sequence is labeled as the negative control target (NCT). This molecule was exposed to the PeptiFET sensor at a concentration of 100 nM, where a slight increase in current (2 × 10–7 A) was detected (Figure 5a). The same negative control peptide (NCP) used in binding kinetics and modeling (Table 1) was also used in the evaluation of the sensor performance. The following sequence was used: QRQLNDKLSIIS-GGGG-Z1 (NCP-Z1), where Z1 is the ZnO binding sequence, compared to the positive control: DQSNKIISLQRL-GGGG-Z1 (OABP1-Z1). In this case, 100 nM orexin A was exposed to a ZnO FET which was functionalized with NCP, and again only a slight increase in current (2 × 10–8A) was observed (Figure 5b). However, when the negative control responses are overlaid with positive control responses (Figure 5c), they become negligible.
Figure 5.
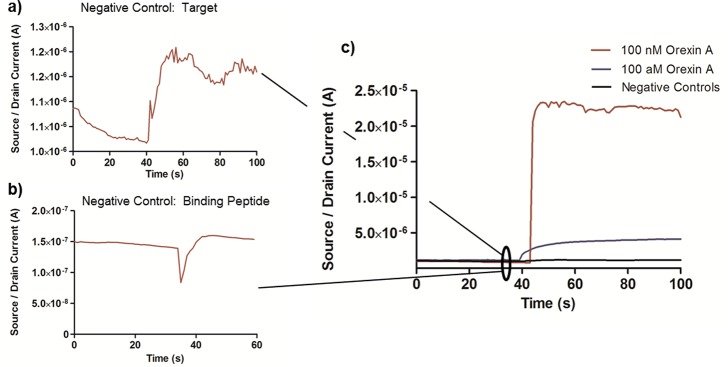
Negative control sensor testing. (a) PeptiFET device functionalized with positive control binding peptide and exposed to negative control scrambled target peptide. (b) PeptiFET device functionalized with negative control scrambled binding peptide and exposed to 100 nM aqueous orexin A. (c) Overlay of positive and negative control rapid sensor response.
Direct comparison of 100 nM exposures shows an increase in signal of 115× over the negative control target and 1115× over the negative control binding peptide. Additionally, the response of the positive control PeptiFET to 10 fM orexin A is also overlaid in Figure 5c, also showing the minimization of negative control response in water. Similarly, minimal response was seen in negative control testing in saliva (Supporting Information Figure 9). The data obtained in the PeptiFET with the OABP1-Z1 (Figure 5b and c) showed an ∼1000× increase in signal over the PeptiFET functionalized with the NCP, corresponding with the ∼1000× increase in KD shown in Table 2.
Discussion
Biomarker analysis in biofluids is a powerful tool for understanding the relationship between different types of events and the physiological and emotional state of a subject. Current methods to study biomarker levels involve timed intervals of biofluid sampling during a tasking, offline analysis, and data correlation after the fact. Studies are limited due to the inability to include rapid biofeedback in order to modify the tasks in situ or to provide any augmentation to counter the detrimental events. One example of a proposed augmentation is the intranasal administration of orexin A6 when decreases in attentional performance are observed indicated by lower orexin A levels in the subject. Thus, we have developed a sensor platform approach to provide rapid biomarker analysis in biofluids.
Rapid biomarker sensing in biological fluids is complicated by the diversity and number of different molecules present in the biological fluids. An effective method to sensing in high background environments is by utilizing biological recognition elements (BRE), which are designed to selectively bind a target of interest. We report the evaluation of an experimentally selected and fully characterized (Table 1) binding peptide for the neuropeptide orexin A, which is a biomarker linked to fatigue and cognitive level.1−5 Molecular modeling approaches were taken to further understand and characterize the interactions between the selected binding peptide and the neuropeptide orexin A. This is an important step in the design of BRE based sensing devices to optimize the system for maximum sensitivity and selectivity. For instance, since the binding peptide is tethered to a semiconductor surface, we must know which portion of the sequence is most involved in binding to make sure it is not hindered from binding. A two step process23 was taken for this study involving rigid docking of the two peptide sequences using PatchDock25 to generate a set of possible complex structures, followed by flexible docking of these structures via FireDock.26 These experiments, along with a previous study of orexin A receptors, show that the following amino acids are essential to the binding complex: Leu16, Leu19, Leu20, His26, Gly29, Ile30, Leu31, Thr32, and Leu33. The top binding complexes were ranked according to the FireDock scoring function and then refined and further analyzed using molecular dynamics simulations. Through these final simulations, the binding energy of the top scoring complexes was determined at levels of low nanomolar range, corresponding well to the sensor data. Additionally, the negative control binding peptide was analyzed the same manner, and showed a significant decrease in binding affinity of 1000× to high μM range, again correlating well to experimental values, where ∼1000× greater signal was seen in positive control sensors over negative control.
This binding peptide was integrated into a field effect transistor (FET) structure where the binding of the target was transduced into an electrical signal. The advantages to this electrical based approach is (relative) ease of fabrication, ability of FETs to be highly arrayed, low power requirements (coin cell battery), and simple reporting that is amenable to wireless communication. This sensor architecture resulted in selective detection down to 100 aM concentrations in water (Figure 4b), 10 fM in filtered human saliva (Figure 4c), and 1 nM in filtered fetal bovine serum (Figure 4d). We have demonstrated the sensitivity and selectivity of the BRE by testing the sensor performance in complex fluids and using a variety of peptide controls.
We believe the sensor platform presented in this study can be used for analysis of a complex signature of biomarkers in multiple biofluids in rapid by arraying the FET elements and functionalizing each with a BRE specific to each biomarker, and applicable to a wide variety of neuroscience research applications. While we have shown effective detection of orexin A at physiologically relevant levels in both clean solutions and biofluids, much development remains before this can be a feasible method for rapid to real-time detection of biomarkers. Some of these tasks identified are currently being worked on in this research group, including preprocessing of biofluid samples to increase the signal-to-noise ratio of detection by removing interfering molecules, passivation of functionalized semiconductor surfaces, and controlled fluid flow for both functionalization and testing via microfluidic fluid transport.
Methods
Peptides were custom synthesized and purchased from GenScript (New Jersey) and purified via HPLC to >95% purity with the following sequences: (i) Orexin A binding peptide + ZnO peptide (OABP1-Z1): DQSNKIISLQRL-GGGG-LHVMHKVAPPRGGGC. (ii) Orexin A negative control binding peptide + ZnO peptide (NCP-Z1): QRQLNDKLSIIS-GGGG-LHVMHKVAPPRGGGC. (iii) Target negative control (TNC): QPLPDCCRQKTCSCRLYELLHGAGNHAAGILTL.
Sterile water (UltraPure distilled DNase RNase free) was purchased from Invitrogen, orexin A (human, rat, mouse) was purchased from Sigma Aldrich and used without further purification, normal human saliva was purchased from Innovative Research (Novi, MI), and fetal bovine serum was purchased from Invitrogen. Saliva and serum were used either in the native state or filtered in a 0.2 μm PTFE syringe filter. ZnO FETs were fabricated on a doped Si substrate with a insulating layer of 30 nm SiO2. ZnO was deposited on the insulator via pulsed laser deposition (PLD) at room temperature to a thickness of 100 nm, and patterned using photolithography and etching for electrical separation between FETs. Source/drain electrodes of titanium/platinum/gold (20/30/50 nm) were deposited by evaporation and liftoff techniques.
Peptide Selection: Phage Display
Panning procedures were conducted following the New England Biolabs Ph.D.-12 Kit protocol for microscale phage selection and amplification. Orexin A target protein (150 μL) purchased from Sigma (10 nM orexin in 100 mM NaHCO3, pH 8.6) was adsorbed on to the surface of the well in a 96-well microtiter plate. After an overnight incubation at 40 °C, 200 μL of blocking buffer (100 mM NaHCO3, 5 mg/mL BSA, pH 8.6) was added and incubated for 1 h at 40 °C. The blocking buffer was removed, and the wells were washed six times with wash buffer (50 mM Tris-HCl (pH 7.5), 150 mM NaCl; TSB) at room temperature, and then the phage library (approximately 6×1010 phage in TBS with 0.1% Tween 20) was added to the well and mixed by gentle agitation. After incubation, the unbound phages were removed and the wells were washed 10 times with wash buffer. The bound phage were then eluted using 150 μL of 200 mM glycine pH 2.2 (Aldrich) containing 0.5% BSA (Sigma), rocked gently for 10 min at room temperature, and neutralized using 15 μL of 1 M Tris (Sigma) pH 9.1. The eluate was added to 20 mL E. coli ER2738 culture and incubated at 37 °C with vigorous shaking for 4.5 h. The culture was transferred to a centrifuge tube and spun for 10 min at 10 000g at 40 °C. The supernatant was collected, and 1/6 volume of PEG/NaCl (20% PEG8000, 2.5 M NaCl) was added. The phage was allowed to precipitate at 40 °C overnight. PEG precipitation was spun for 15 min at 10 000 g at 40 °C. The pellet was resuspended in 1 mL of TBS and reprecipitated with PEG/NaCl. Finally, the pellet was suspended in 200 μL TBS and stored at 40 °C. The procedure was repeated four times for a total of five rounds of panning with the exception that the Tween concentration was increased 0.05% per round. Phage titers were determined as described in the Ph.D.-12 manual.
After five rounds of panning, 50 phage plaques were randomly selected from fresh titer plates using a sterile inoculation loops, and were amplified individually for 4.5–5 h at 37 °C in 1.5 mL of E. coli ER2738 culture grown in LB broth with 20 mg/L tetracycline (Sigma) until OD600 = 0.4–0.6. After incubation, the amplified plaque solutions were centrifuged twice (6000 rpm, 10 min) and the supernatants were transferred to a sterile 1.5 mL microcentrifuge tubes. Phagemid DNA was then isolated from the purified phage stocks using the Qiagen QIAprep Spin M13 kit (Qiagen). DNA concentration and purity measurements were made using a Thermo Fisher Scientific NanoDrop spectrophotometer. DNA samples were then sent to The Ohio State University (http://pmgf.biosci.ohio-state.edu/index.html) for sequencing. The random 12 amino acid sequence (12-mer peptide) was determined by translating the DNA sequence.
Binding Peptide Characterization
Each orexin A peptide was synthesized by AnaSpec (Fremont, CA) with a mini PEG2-Lys-Biotin on the carboxy group. The synthesis scale was 2 μmol with >98% purity. Lyophilized peptide was dissolved with molecular grade water at pH 7.4 to a concentration of 1 mg/mL. The peptides were further diluted to a ∼0.1 mg/mL concentration in HBS-EP (10 mM HEPES pH 7.4, 150 mM NaCl, 50 mM EDTA, 0.005% (v/v) Surfactant P20 at pH 7.2), before being further diluted to give an SPR assay concentration of ∼50 μg/mL. Orexin binding peptides, OABP 1 and 2, were synthesized by AnaSpec at the 2 μmol scale with 98% purity. Orexin scrambled peptide QRQLNDKLSIIS (NCP) was used as a negative control.
Binding Kinetics
Surface plasmon resonance (SPR) experiments were done on a Biacore T200 SPR system (GE Healthcare, Piscataway, NJ). All buffers and reagents were purchased from G.E. Healthcare unless otherwise noted. A standard coupling protocol was employed to immobilize a streptavidin capture surface as described by the manufacturer. The immobilization was performed at 25 °C using sodium acetate (10 mM in 150 mM NaCl, pH 4.5) as the running buffer. Biotinylated orexin A was flowed over flow cell 2 at10 μL/min until the desired level of capture was achieved (100 RU). Flow cells 1 and 4 were used as a reference spots. Predicted Rmax values based on protein capture levels for all assays were kept within the range of 50–150 RU.
Data Analysis
Dissociation (kd) and association (ka) rate constants were obtained by nonlinear regression analysis of the primary sensorgram data according to a 1:1 binding model using the BiaEvaluation version 3.2 software provided by the manufacturer. The dissociation constant KD was calculated using the formula KD = kd/ka.
Binding Pull-Down Assays
Biotinylated orexin A peptide was immobilized onto prewashed streptavidin coated magnetic beads (DynaBeads M-280 streptavidin, Invitrogen, Carlsbad, CA) by incubating 1 μg of OABP1 with 25 μL of bead slurry at room temperature for 60 min. Beads without OABP1 were used as a control to evaluate nonspecific binding to the beads. A second control was conducted using human brain cell lysate (1 mg/mL, ProSci Incorporated, Poway, CA) to assess the potential for the orexin A peptide to bind with proteins present in the lysate. Cell lysate components that bound nonspecifically to the beads were removed by incubating cell lysate (1 mg/mL in 100 μL HBS-EP) with 50 μL of washed streptavidin beads and rotated at 40 °C overnight. The next day, the magnetic beads were collected and the precleared cell lysate was removed and used in the pull-down experiments. OABP1 coated beads were washed five times with 500 μL of 1× HBS-EP supplemented with 0.1% BSA and incubated overnight at 40 °C with rotation in the presence the desired amount of orexin A target protein (Sigma, St. Louis, MO) in HBS-EP buffer with 0.5 mg of precleared cell lysate. After binding, beads were washed five times with 500 μL of HBS-EP and heated at 70 °C in 20 μL of Tricine sample buffer (BioRad, Hercules, CA) for 10 min. One half of each elution sample was analyzed by 10–20% Tris-Tricine SDS-PAGE(BioRad) with 5–10 μL of Precision Plus protein dual Xtra strandard and Kalidoscope polypeptides markers (BioRad) for 45 min at 80 V and imaged after staining with the Silver SNAP staining kit (Pierce Thermo Scientific, Rockford, IL). The other half of the sample was analyzed by SDS-PAGE, and the target was detected by using Western blot (SuperSignal West Dura, Pierce Thermo Scientific) with anti-goat orexin A antibody (C19, Santa Cruz Biotechnology, Inc.). The Western blot was imaged on a GE Healthcare Typhoon Trio instrument.
Affinity Determination by ELISA
Immunosorbant assays (ELISA) were conducted by incubating 1000 ng of orexin A protein in 0.1 M sodium bicarbonate, pH 9.8, in a Maxisorb NUNC 96-well plate overnight at 4 °C in a humidifier. The solution was removed and replaced with 100 μL of blocking buffer (2% BSA in PBS, pH 7.4), which was incubated for 1 h at 37 °C in the humidifier. The solution was removed, and the plate was washed three times with PBS-Tween and tapped dry. The biotinylated OABP1 peptides were added to the plate at varying concentrations in PBS-Tween (0.05%). The peptides were incubated with the plate for 1 h at 37 °C. The solution was removed, and the plate was washed three times with PBS-Tween. Horseradish peroxidase conjugated streptavidin was diluted 1:1000 in 0.1% BSA in PBS-Tween, and 50 μL was added to each well and incubated for 1 h at 37 °C. The strepavidin solution was removed, and the plate was washed three times with buffer PBS-tween. A volume of 50 μL of TMB (3,3′,5,5′-tetramethylbenzidine) was added to each well, and the solution was incubated for 15 min at 24 °C. A 50 μL aliquot of 0.5 M HCl was added to stop the reaction, and the plate was scanned immediately using a SpectroMax plate reader. These assays were conducted in triplicate, and the data were then normalized by subtracting all fluorescent values from the no protein control, plotted, and fit using GraphPad Prism.
Sensor Platform: Equipment
Sensor device testing was done using a Keithley semiconductor parameter analyzer (SCS 4200) and MMR 4-probe station for addressing the FET source and drain electrodes. Atomic force microscopy images were taken with a Veeco BioScope AFM in tapping mode.
Functionalization
A 3″ silicon wafer with a dense array of ZnO FETs was diced into ∼15 mm × 5 mm FET arrays. These smaller arrays contain six FETs with identical geometries (Figure 3a) which can each be tested individually. The bifunctional peptides were solubilized in UltraPure water at a concentration of 20 μg/mL and aliquotted into 1.5 mL sterile (autoclaved) plastic vials at a volume of 1 mL. The diced FET arrays were then incubated in the vial, gently shaken for 6 min, and immediately washed with UltraPure water and dried with nitrogen. Both the peptide concentration and incubation times were optimized for this sensor system. The peptide functionalized ZnO FET (PeptiFET) was then evaluated for sensor performance (preferably within 4 h of functionalization).
Device Testing
The PeptiFET devices were tested in a rapid mode by applying a constant source/drain voltage (VSD) and gate voltage (VG) and monitoring the source/drain current (ISD) at a rate of one data point per second. In order for effective target binding to take place, the binding peptide must be rehydrated. This was done by applying an initial 20 μL drop of UltraPure water and monitoring the current (ISD) until stabilized (∼2–3 min). When ISD reached a stable value, the target solutions were applied and sensor performance analyzed.
Modeling
In our computational study, we used the 3D structure of orexin A determined using a two-dimensional nuclear magnetic resonance (NMR) spectroscopy and posted into the Protein Data Bank (PDB code 1WSO).22 The structures of OABP1 were generated using the Rosetta ab initio fragment assembly package.38 The 1000 top-scoring structural models were refined by energy minimization, rescored with an implicit solvent model, and clustered by a pairwise hierarchical method to identify a set of unique structures using a protein structure prediction pipeline (PSPP).39 To account for the peptide flexibility, an ensemble docking technique was applied. The models with the lowest energy in ten clusters as predicted by PSPP were selected as input structures for replica exchange molecular dynamics (REMD) simulations.40
The REMD simulations were performed with the Amber10 suite of programs29 as implemented in Automatic Protein Ensemble Generator (https://applications.bioanalysis.org/apeg). Eight replicas distributed over a temperature range from 270 to 600 K were used. The implicit water simulations for each replica were initially equilibrated for 200 ps at the corresponding temperature following by 5 ns of production run. The time step was set to 2 fs, and SHAKE was applied to constrain the bonds connecting hydrogen atoms. The temperature exchanges were attempted every 1 ps, and 5000 snapshots from the production run were used for cluster analysis. Clustering was performed with the Amber10 PTRAJ modules based on the pairwise backbone-atom only root-mean-square deviations (RMSD). Saved snapshot conformations were clustered into 100 clusters, and representatives from each cluster were taken for docking. A total of 1000 cluster representatives obtained for 10 conformations generated by Rosetta were used in docking calculations.
The OA-OABP1 docking was performed in two steps. The rigid docking was performed using the PatchDock package25 with the default set of parameters. The generated structures were processed to the FireDock package26 for flexible docking. The refinement procedure was performed in three steps. Initially, we performed a coarse refinement using the restricted interface side-chain optimization (RISCO) mode with an atomic radii scaling of 0.8 and a 50 cycles of Monte Carlo rigid-body optimization (RBO). Next, the 25 best configurations were processed for a fine refinement using the full side-chain optimization (FISCO) mode, an atomic radii scaling of 0.8, and 50 cycles of RBO. Finally, the top 10 models were selected for a full-side optimization with the radii scaling of 0.85 but without RBO. The refined complexes were scored and ranked according to FireDock energy function.
Acknowledgments
We are grateful to B. Bayraktaroglu and K. Leedy of the Sensors Directorate for fabrication of zinc oxide FETs, S. N. Kim for valuable discussions and expertise in sensor testing, J. L. Chavez for continued helpful discussions, L. Narayanan for HPLC analysis, J. Slocik for QCM experiments, and J. Schlager for support in BRE identification.
Glossary
Abbreviations
- FET
field effect transistor
- BRE
biorecognition element
- OA
orexin A
- OABP(1 and 2)
orexin A binding peptide
- NCP
negative control peptide
- ZnO FET
zinc oxide field effect transistor
- PeptiFET
peptide functionalized field effect transistor
Supporting Information Available
Additional data and figures referenced in the text. This material is available free of charge via the Internet at http://pubs.acs.org.
Author Contributions
J.H. designed, tested, and analyzed the PeptiFETs. W.L. identified the orexin A binding peptide through phage display and characterized binding kinetics. Y.C. designed, tested, and analyzed all modeling data. M.T. identified the ZnO binding peptide and designed the fusion peptides. J.H., W.L., and Y.C. wrote the manuscript. R.N., M.S., and N.K.L. provided ideation, direction, and data/results discussion.
This work was supported by the Air Force Research Laboratory, Bio-X Strategic Technology Thrust, 711th Chief Scientist Seedling Fund, Air Force Office of Scientific Research, and RX Bio program in the Materials and Manufacturing Directorate.
The authors declare no competing financial interest.
Supplementary Material
References
- Tsujino T.; Sakurai T. (2009) Orexin/hypocretin: a neuropeptide at the interface of sleep, energy homeostasis, and reward system. Pharmacol. Rev. 61, 162–176. [DOI] [PubMed] [Google Scholar]
- Yoshida Y.; Fujiki N.; Nakajima T.; Ripley B.; Matsumura H.; Yoneda H.; Mignot E.; Nishino S. (2001) Fluctuation of extracellular hypocretin-1 (orexin A) levels in the rat in relation to the light-dark cycle and sleep-wake activities. Eur. J. Neurosci. 14, 1075–1081. [DOI] [PubMed] [Google Scholar]
- Peyron. C.; Faraco J.; Rogers W.; Ripley B.; Overeem S.; Charnay Y.; Nevsimalova S.; Aldrich M.; Reynolds D.; Albin R.; Li R.; Hungs M.; Pedrazzoli M.; Padigaru M.; Kucherlapati M.; Fan J.; Maki R.; Lammers G. J.; Bouras C.; Kucherlapati R.; Nishino S.; Mignot E. (2000) A mutation in a case of early onset narcolepsy and a generalized absence of hypocretin peptides in human narcoleptic brains. Nat. Med. 6, 991–997. [DOI] [PubMed] [Google Scholar]
- Higuchi S.; Usui A.; Murasaki M.; Matsushita S.; Nishioka N.; Yoshino A.; Matsui T.; Muraoka H.; Ishizuka Y.; Kanba S.; Sakurai T. (2002) Plasma orexin-A is lower in patients with narcolepsy. Neurosci. Lett. 318, 61–64. [DOI] [PubMed] [Google Scholar]
- Strawn J.; Pyne-Geithman G.; Ekhator N.; Horn P.; Uhde T.; Shutter L.; Baker D.; Geracioti T. (2010) Low cerebrospinal fluid and plasma orexin-A (hypocretin-1) concentrations in combat-related posttraumatic stress disorder. Psychoneuroendocrinology 35, 1001–1007. [DOI] [PubMed] [Google Scholar]
- Deadwyler S. A.; Porrino L.; Siegel J. M.; Hampson R. E. (2007) Systemic and nasal delivery of orexin-A (hypocretin-1) reduces the effects of sleep deprivation on cognitive performance in nonhuman primates. J. Neurosci. 27, 14239–14247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boschen K. E.; Fadel J. R.; Burk J. A. (2009) Systemic and intrabasalis administration of the orexin-1 receptor antagonist, SB-334867, disrupts attentional performance in rats. Psychopharmacology 206, 205–213. [DOI] [PubMed] [Google Scholar]
- Sakurai T.; Amamiya A.; Ishii M.; Matsuzaki I.; Chemelli R. M.; Tanaka H.; Williams S. C.; Richardson J. A.; Kozlowski G. P.; Wilson S.; Arch J. R.; Buckingham R. E.; Haynes A. C.; Carr S. A.; Annan R. S.; McNulty D. E.; Liu W. S.; Terrett J. A.; Elshourbagy N. A.; Bergsma D. J.; Yanagisawa M. (1998) Orexins and orexin receptors: a family of hypothalamic neuropeptides and G protein-coupled receptors that regulate feeding behavior. Cell 92, 573–585. [DOI] [PubMed] [Google Scholar]
- Baranowska B.; Wolinska E.; Martynska W.; Chmielowska M.; Baranowska-Bik A. (2005) Plasma orexin A, orexin B, leptin, neuropeptide Y (NPY) and insulin in obese women. Neuroendocrinol. Lett. 26, 293–296. [PubMed] [Google Scholar]
- Kastin A. J.; Akerstrom V. (1999) Orexin A but not orexin B rapidly enters brain from blood by simple diffusion. J. Pharmacol. Exp. Ther. 289, 219–223. [PubMed] [Google Scholar]
- Ammoun S.; Holmqvist T.; Shariatmadari R.; Oonk H. B.; Detheux M.; Parmentier M.; Akerman K. E.; Kukkonen J. P. (2003) Distinct recognition of OX1 and OX2 receptors by orexin peptides. J Pharmacol. Exp. Ther. 305, 507–514. [DOI] [PubMed] [Google Scholar]
- Arihara Z.; Takahashi K.; Murakami O.; Totsune K.; Sone M.; Satoh F. (2001) Immunoreactive orexin-A in human plasma. Peptides 22, 139–142. [DOI] [PubMed] [Google Scholar]
- Heinonen M. V.; Purhonen A. K.; Miettinen P.; Pääkkönen M.; Pirinen E.; Alhava E.; Åkerman K.; Herzig K. H. (2005) Apelin, orexin-A and leptin plasma levels in morbid obesity and effect of gastric banding. Regul. Pept. 130, 7–13. [DOI] [PubMed] [Google Scholar]
- Abdo W.; Bloem B.; Kremer H.; Lammers G.; Verbeek M.; Overeem S. (2008) CSF hypocretin-1 levels are normal in multiple-system atrophy. Parkinsonism Relat. Disord. 14, 342–344. [DOI] [PubMed] [Google Scholar]
- Patolsky F.; Lieber C. (2005) Nanowire nanosensors. Mater. Today 8, 20. [Google Scholar]
- Aswal D., and Gupta S. (2007) Science and Technology of Chemiresistor Gas Sensors, Nova Science Publishers, New York. [Google Scholar]
- Cui Y.; Wei Q.; Park H.; Lieber C. (2001) Highly sensitive and selective detection of biological and chemical species. Science 293, 1289–1292. [DOI] [PubMed] [Google Scholar]
- Covington J.; Gardner J.; Briand D.; de Rooij N. (2001) A polymer gate FET sensor array for detecting organic vapours. Sens. Actuators, B 77, 155–162. [Google Scholar]
- North J. (1985) Immunosensors: Antibody-based biosensors. Trends Biotechnol. 3, 180–186. [Google Scholar]
- Maehashi K.; Katsura T.; Kerman K.; Takamura Y.; Matsumoto K.; Tamiya E. (2007) Label-free protein biosensor based on aptamer-modified carbon nanotube field effect transistors. Anal. Chem. 79, 782–787. [DOI] [PubMed] [Google Scholar]
- Kuang Z.; Kim S.; Crookes-Goodson W.; Farmer B.; Naik R. (2010) Biomimetic Chemosensor: Designing Peptide Recognition Elements for Surface Functionalization of Carbon Nanotube Field Effect Transistors. ACS Nano 4, 452–458. [DOI] [PubMed] [Google Scholar]
- Lee J. O.; So H. M.; Jeon E. K.; Chang H.; Won K.; Kim Y. H. (2008) Aptamers as molecular recognition elements for electrical nanobiosensors. Anal. Bioanal. Chem. 390, 1023–1032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buchwalow I.; Samoilova V.; Boecker W.; Tiemann M. (2011) Non-specific binding of antibodies in immunohistochemistry: fallacies and facts. Sci. Rep. 1, 1–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takai T.; Takaya T.; Nakano M.; Akutsu H.; Nakagawa. A.; Aimoto S.; Nagai K.; Ikegami T. (2006) Orexin-A is composed of a highly conserved C-terminal and a specific, hydrophilic N-terminal region, revealing the structural basis of specific recognition by the orexin-1 receptor. J. Pept. Sci. 12, 443–454. [DOI] [PubMed] [Google Scholar]
- Bonvin A. M. (2006) Flexible protein–protein docking. Curr. Opin. Struct. Biol. 16, 194–200. [DOI] [PubMed] [Google Scholar]
- Andrusier N.; Mashiach E.; Nussinov R.; Wolfson H. J. (2008) Principles of flexible protein–protein docking. Proteins 73, 271–289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duhovny D., Nussinov R., and Wolfson H. J. (2002) Efficient unbound docking of rigid molecules. Proceedings of the Fourth International Workshop on Algorithms in Bioinformatics, September 17–21, 2002, 2452, pp 185–200, Springer-Verlag GmbH, Rome, Italy. [Google Scholar]
- Andrusier N.; Nussinov R.; Wolfson H. J. (2007) FireDock: Fast Interaction Refinement in Molecular Docking. Proteins 69, 139–159. [DOI] [PubMed] [Google Scholar]
- Darker J. G.; Porter R. A.; Eggleston D. S.; Smart D.; Brough S. J.; Sabido-David C.; Jerman J. C. (2001) Structure-activity analysis of truncated orexin-A analogues at the orexin-1 receptor. Bioorg. Med. Chem. Lett. 11, 737–740. [DOI] [PubMed] [Google Scholar]
- Sugita Y.; Okamoto Y. (1999) Replica-exchange molecular dynamics method for protein folding. Chem. Phys. Lett. 314, 141–151. [Google Scholar]
- Case D. A., Darden T. A., Cheatham T. E. III, Simmerling C. L., Wang J., Duke R. E., Luo R., Crowley M., Walker R. C., Zhang W., Merz K. M., Wang B., Hayik S., Roitberg A., Seabra G., Kolossvary I., Wong K. F., Paesani F., Vanicek J., Wu X., Brozell S. R., Steinbrecher T., Gohlke H., Yang L., Tan C., Mongan J., Hornak V., Cui G., Mathews D. H., Seetin M. G., Sagui C., Babin V., Kollman P. A. (2008) AMBER 10, University of California, San Francisco. [Google Scholar]
- Kollman P. A.; Massova I.; Reyes C.; Kuhn B.; Huo S.; Chong L.; Lee M.; Lee T.; Duan Y.; Wang W.; Donini O.; Cieplak P.; Srinivasan J.; Case D. A.; Cheatham T. E. III. (2000) Calculating structures and free energies of complex molecules: Combining molecular mechanics and continuum models. Acc. Chem. Res. 33, 889–897. [DOI] [PubMed] [Google Scholar]
- Kottalam J.; Case D. A. (1990) Langevin modes of macromolecules: application to crambin and DNA hexamers. Biopolymers 29, 1409–1421. [DOI] [PubMed] [Google Scholar]
- Nomura K.; Ohta H.; Takagi A.; Kamiya T.; Hirano M.; Hosono H. (2004) Room-Temperature fabrication of transparent flexible thin-film transistors using amorphous oxide semiconductors. Nature 432, 488–492. [DOI] [PubMed] [Google Scholar]
- Bayraktaroglu B.; Leedy K.; Neidhard R. (2009) High-frequency ZnO thin-film transistors on Si substrates. IEEE Electron Device Lett. 30, 946–948. [Google Scholar]
- Hagen J.; Kim S.; Bayraktaroglu B.; Leedy K.; Chavez J.; Kelley-Loughnane N.; Naik R.; Stone M. (2010) Sensors 11, 6645–6655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomczak M.; Gupta M.; Drummy L.; Rozenzhak S.; Naik R. (2009) Morphological control and assembly of zinc oxide using a biotemplate. Acta Biomater. 5, 876–882. [DOI] [PubMed] [Google Scholar]
- Cui Y.; Kim S. N.; Jones S. E.; Wissler L. L.; Naik R. R.; McAlpine M. C. (2010) Chemical Functionalization of Graphene Enabled by Phage Displayed Peptides. Nano Lett. 10, 4559–4565. [DOI] [PubMed] [Google Scholar]
- Hermanson G. (2008) Bioconjugate Techniques, Elsevier, New York: . [Google Scholar]
- Simons K. T.; Kooperberg C.; Huang E.; Baker D. (1997) Assembly of protein tertiary structures from fragments with similar local sequences using simulated annealing and Bayesian scoring functions. J. Mol. Biol. 268, 209–225. [DOI] [PubMed] [Google Scholar]
- Lee M. S.; Bondugula R.; Desai V.; Zavaljevski N.; Yeh I. C.; Wallqvist A.; Reifman J. (2009) PSPP: a protein structure prediction pipeline for computing clusters. PLoS One 4, 6254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugita Y.; Okamoto Y. (1999) Replica-exchange molecular dynamics method for protein folding. Chem. Phys. Lett. 314, 141–151. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.