

Abstract
The E46K genetic missense mutation of the wild-type α-synuclein protein was recently identified in a family of Spanish origin with hereditary Parkinson’s disease. Detailed understanding of the structures of the monomeric E46K mutant-type α-synuclein protein as well as the impact of the E46K missense mutation on the conformations and free energy landscapes of the wild-type α-synuclein are required for gaining insights into the pathogenic mechanism of Parkinson’s disease. In this study, we use extensive parallel tempering molecular dynamics simulations along with thermodynamic calculations to assess the secondary and tertiary structural properties as well as the conformational preferences of the monomeric wild-type and E46K mutant-type α-synuclein proteins in an aqueous solution environment. We also present the residual secondary structure component conversion stabilities with dynamics using a theoretical strategy, which we most recently developed. To the best of our knowledge, this study presents the first detailed comparison of the structural and thermodynamic properties of the wild-type and E46K mutant-type α-synuclein proteins in an aqueous solution environment at the atomic level with dynamics. We find that the E46K mutation results not only in local but also in long-range changes in the structural properties of the wild-type α-synuclein protein. The mutation site shows a significant decrease in helical content as well as a large increase in β-sheet structure formation upon E46K mutation. In addition, the β-sheet content of the C-terminal region increases significantly in the E46K mutant-type αS in comparison to the wild-type αS. Our theoretical strategy developed to assess the thermodynamic preference of secondary structure transitions indicates that this shift in secondary structure is the result of a decrease in the thermodynamic preference of turn to helix conversions while the coil to β-sheet preference increases for these residues. Long-range intramolecular protein interactions of the C-terminal with the N-terminal and NAC regions increase upon E46K mutation, resulting in more compact structures for the E46K mutant-type rather than wild-type αS. However, the E46K mutant-type αS structures are less stable than the wild-type αS. Overall, our results show that the E46K mutant-type αS has a higher propensity to aggregate than the wild-type αS and that the N-terminal and C-terminal regions are reactive toward fibrillization and aggregation upon E46K mutation and we explain the associated reasons based on the structural properties herein. Small molecules or drugs that can block the specific residues forming abundant β-sheet structure, which we report here, might help to reduce the reactivity of these intrinsically disordered fibrillogenic proteins toward aggregation and their toxicity.
Keywords: α-synuclein, genetic missense mutation, free energy landsape, molecular dynamics
Parkinson’s disease (PD) is a debilitating neurodegenerative disorder that affects approximately 1% of the population above age 65 and up to 5% of the population by age 85.1 PD as well as some other neurodegenerative disorders are characterized by the formation of Lewy bodies, which are large proteinaceous inclusions composed mainly of the intrinsically disordered protein α-synuclein (αS).2−7 In addition, some inherited forms of PD are associated with genetic missense or replication mutations of the αS protein.8−11 Recently, a genetic missense mutation of the 140 amino acid residue αS protein resulting in the mutation of the 46th amino acid residue from glutamic acid to lysine (E46K) was isolated in a family of Spanish origin.8 Zarranz et al. linked this mutation to both familial PD and Lewy body dementia.8 The aggregation of αS is proposed to play a crucial role in the pathogenic mechanism of PD, especially the formation of the reportedly neurotoxic oligomeric species.12,13 Furthermore, the monomeric and oligomeric structures reportedly determine the aggregation mechanism of αS.14,15
The E46K genetic missense mutation alters the aggregation mechanism and neurotoxicity of the wild-type αS, but the structural properties and exact impact have been debated in the current literature. It was reported that the E46K mutant-type αS is neurotoxic whereas the wild-type αS is neuroprotective toward rat brains as well as for SH-SY5Y cells in the presence of environmental toxins.16 Circular dichroism (CD), electron microscopy, and thioflavin T (ThT) fluorescence studies as well as sedimentation assays report that the E46K mutant-type αS fibrillize more rapidly than the wild-type αS.17−20 However, Kamiyoshihara et al. reported that the E46K mutant-type αS formed fibrils at a slower rate than the wild-type αS even though the total number of fibrils formed is greater upon E46K mutation.21 While some experimental studies report that the E46K mutant-type αS fibril structure is morphologically similar to that of the wild-type αS,17−19 Choi et al. reported a more pronounced twisted appearance for the E46K mutant-type αS in comparison to the wild-type αS.20 Understanding the monomeric and oligomeric wild- and mutant-type αS structures as well as the effect of the E46K genetic missense mutation on these structures provides knowledge directly linked to the aggregation mechanism of αS.18,22,23 For example, specific secondary structure component formations, such as helix and β-sheet structures, in the structures of the monomeric intrinsically disordered fibrillogenic proteins are known to determine the fibrillization and aggregation mechanisms.18,24−26 Gaining insights into these structural properties at the monomeric level can then be used for developing therapeutic agents that target the aggregation mechanism of αS via blocking specific residues reactive toward aggregation. A detailed description of the dynamic structural properties of these species remains challenging with conventional experimental techniques due in part to complications provided by fast aggregation, rapid structural changes, and solvent effects. Despite, some experimental studies have reported the structural properties of the wild-type and E46K mutant-type αS proteins. Regarding oligomeric structures, Fredenburg et al. reported that the formation of protofibrillar structures decreases upon E46K mutation of αS but that the morphological characteristics of these structures are similar to those of its wild-type form.18 Nuclear magnetic resonance (NMR) and CD studies reported that the secondary structure components of the free wild-type αS protein are not greatly impacted upon E46K mutation.18,19,27,28 In contrast, single molecule force (SMF) spectroscopy measurements by Brucale et al. indicated the β-sheet content of the E46K mutant-type αS is significantly increased over the wild-type αS.25 NMR measurements also indicated that interactions between the N-terminal (Met1-Lys60) and C-terminal (Lys96-Ala140) regions are increased upon E46K mutation of the wild-type αS.27
Theoretical studies can provide insight into the atomic level dynamic structural changes of the wild-type αS upon E46K mutation at the monomeric level in solution that are not easily observable with conventional experimental tools. However, theoretical studies investigating the E46K mutant-type αS are mostly lacking or limited in their contents in the currently existing literature. For example, Sundar and co-workers reported the aggregation propensity of the E46K mutant-type αS is intermediate of the other two genetic missense mutations of αS via aggregation propensity calculations on structures obtained from classical molecular dynamics (MD) simulations in explicit water.29 Nevertheless, the impact of the E46K mutation on the structural and thermodynamic properties of the wild-type αS as well as sampling of conformations are not presented in their study. Perlmutter et al. also presented classical MD simulations of the wild-type and three genetic missense mutant αS proteins in the presence of either an sodium dodecyl sulfate (SDS) micelle or a phospholipid bilayer.30 Based on these simulations, they reported that the E46K mutation added a hydrogen bond between αS and the lipid or micelle that may be related to the increased bilayer affinity of E46K mutant-type αS.20,30 To the best of our knowledge, a detailed study presenting the impact of the E46K mutation on the structural and thermodynamic properties of the wild-type αS in the free state at atomic level with dynamics has not yet been presented in the literature. Therefore, we performed all-atom replica exchange molecular dynamics (REMD) simulations on the full-length monomeric wild-type and E46K mutant-type αS proteins in aqueous solution. Using these simulated structures, we calculated the secondary and tertiary structural components as well as the thermodynamic properties of the monomeric wild-type and E46K mutant-type αS in an aqueous solution environment. Furthermore, we used our recently developed new theoretical strategy for gaining knowledge about the residual secondary structure component conversions at the atomic level with dynamics. Based on these results, we find that the E46K mutation greatly impacts the structural and thermodynamic properties of the monomeric wild-type αS in aqueous solution.
Results and Discussion
The calculated secondary structure components per residue of the wild-type and E46K mutant-type αS are depicted in Figure 1. Helical and β-sheet structures are proposed to play a crucial role in the aggregation process of intrinsically disordered fibrillogenic proteins in neurodegenerative diseases, and therefore, these secondary structure components are of special interest. The most abundant α-helix formation in the N-terminal region of the wild-type αS occurs at Glu20-Gln24 with 20%–35% abundance for the wild-type αS (Figure 1). Even though the abundance of α-helix structure of Glu20-Gln24 is similar in the E46K mutant-type αS, the most abundant α-helix structure in the N-terminal region of the E46K mutant-type αS occurs at Ser9-Glu13, which is at least 15% more abundant than in the wild-type αS. Furthermore, Ala27-Ala29 and Thr54-Thr59 also present an increase in α-helical structure upon E46K mutation of the wild-type αS. However, the vice versa trend is observed for residues Tyr39-Ser42. For the nonamyloid β component region (NAC; Glu61-Val95), residues Glu61-Thr64 of the wild-type αS protein form abundant α-helical structure. The formation of helical structure in the N-terminal and NAC regions is associated with lipid or vesicle binding of the αS protein.31−34 Therefore, differences in the α-helix formation of the αS protein as a result of the E46K mutation of the wild-type αS may affect the binding of αS with lipids or vesicles. Previous experimental studies reported that the E46K mutant-type αS has a higher affinity for binding to negatively charged vesicles than the wild-type.20 Our results support these experiments and further show that this reported higher affinity may be associated with the higher α-helix content at Ser9-Glu13, Ala27-Ala29, and Thr54-Thr59 in the N-terminal region upon E46K mutation. Within the C-terminal region (Lys96-Ala140), the most prominent α-helix formation appears at Lys96-Gly101 for both wild-type and E46K mutant-type αS; however, the abundance at Gln99-Lys101 increases by up to 20% in E46K mutant-type αS rather than in the wild-type αS conformations. In addition, the α-helical structure at Pro120-Glu123 almost disappears upon E46K mutation in the structures of the wild-type αS protein.
Figure 1.
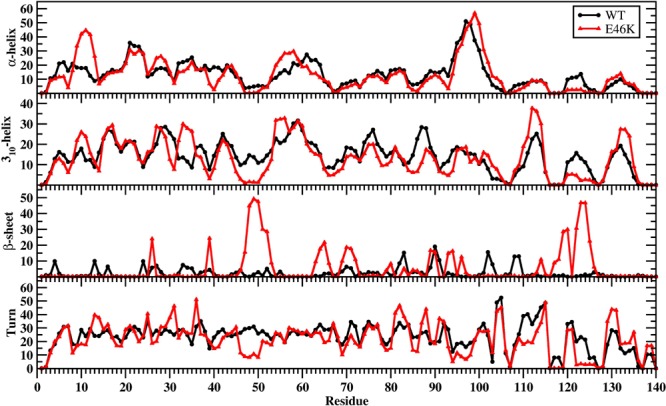
WT and E46K αS secondary structure components. Secondary structure abundances per residue for the wild-type (black) and E46K mutant-type (red) αS. The abundances for the π-helix and coil structures are not displayed.
Both the wild-type and E46K mutant-type αS display abundant 310-helix formation (>20% abundance) in the N-terminal region at Val15-Ala18, Glu20-Thr22, Ala27-Gly31, Gly41-Ser42, and Thr54-Lys60. Upon E46K mutation, we observe an increase in the 310-helix formation at Ser9-Ala11, Lys32-Glu35, and Thr54-Ala56 by up to 15%. On the other side, the 310-helix structure formation at residues Glu46-Gly51 almost disappears when Glu46 is replaced by Lys. In the NAC region, residues Val74-Val77 and Gly86-Ala89 of the wild-type αS form abundant 310-helix structure (>20%). Overall, the abundance of 310-helix structure in the NAC region decreases upon E46K mutation except at residues Thr64, Asn65, Thr72, Thr81-Glu83, Ala90, and Ala91. Residues Ile112-Glu114 present the most abundant 310-helix formation for both the wild-type and E46K mutant-type αS; however, the abundance is slightly increased for the E46K mutant-type in comparison to the wild-type αS. We also note an increase in 310-helix formation at Gly132-Gln134 upon E46K mutation while the vice versa trend is observed for Pro120-Ala124 in the C-terminal region.
For the wild-type αS, the most abundant β-sheet structures (up to 20%) are formed in parts of the NAC and C-terminal regions (Val70, Val71, Val82, Glu83, Ala89-Ala91, Lys102, Asn103, Pro108, and Gln109). We also observe β-sheet structure formation in the N-terminal region of the wild-type αS at Phe4, Glu13, Val16, Gln24, Val26, Ala27, Thr33-Glu35, Val37, and Val52. Interestingly, the NAC and N-terminal residues (except Phe4) are located in regions considered part of the 11-mer repeats. There are seven regions of 11-mer repeats, sequences that assume right handed coiled coil conformations, in the N-terminal and NAC region of α-synuclein.35 It is suggested these repeats lower the propensity of α-synuclein to form β-sheet due to their preference for α-helix formation.35 In comparison, the most prominent β-sheet structures (20%–50%) for the E46K mutant-type αS are located in the N-terminal and C-terminal regions at residues Val26, Tyr39, Gly47-Ala53, Asp119, Pro120, and Asn122-Tyr125. In addition, Val63-Val66, Ala69-Thr72, Lys80, Gly93, Phe94, Lys96, Glu114, Pro117, and Val118 in the NAC and C-terminal regions present β-sheet structure formation with 5%–20% abundances. Kessler et al. reported αS was more fibrillogenic upon deletion of the N-terminal and C-terminal, thus implying the NAC region is more prone to aggregation.35 In comparison with our results, the NAC region had minor β-sheet structure formation (5%–20% abundance), whereas the N-terminal and C-terminal exhibited a 20%–50%. Moreover, specific residues (Val37–Lys43, Val52–Thr59, Gln62–Val66, Gly68–Val77, and Ala90–Val95) were reported in NMR study conducted by Vilar et al. as the main regions to form β-sheet structures that compose the wild-type αS fibrils.36 The most abundant (up to 55%) turn structure formations occur in the C-terminal region of the wild-type but this is shifted to the N-terminal region in the E46K mutant-type αS. Large discrepancies in the tendencies to form turn structure in the N-terminal region are noticed at Glu13, Gly14, Ala30, Gly31, Gly36, and Lys46-His50; in the NAC region at Val82, Ala85, and Gly86; and in the C-terminal region at Glu110-Ile112, Pro117, Val118, Asn122-Ala124, and Glu130, and Glu131 with a difference up to 20%.
We also calculated the free energy change values associated with the transitions between two different secondary structure components for each residue of the wild-type and E46K mutant-type αS in an aqueous solution medium (Figure 2) using a new theoretical strategy that we most recently developed.37 These calculations overall indicate that turn or helix to coil transitions as well as turn to helix transitions are the most preferred transitions for the wild-type and E46K mutant-type α-synuclein proteins. Regarding the formation of helical and β-sheet structures, transitions between turn to helix structures are thermodynamically most preferred for residues Val3-Val95 (N-terminal and NAC regions) of the wild-type αS protein. Coil to β-sheet transitions at Phe4, Glu13, Ala90, and Ala91 are significantly less stable upon E46K mutation of the wild-type form. Helix to β-sheet transition per residues is slightly more stable in parts of the N-terminal and NAC regions in the structures of the E46K mutant-type αS in comparison to the same regions in the wild-type αS protein. Similar characteristics are also detected for the turn to β-sheet conversion per residues. Interestingly, β-sheet to helix conversion per residue with dynamics is more preferred in parts of the N-terminal and NAC regions of the E46K mutant-type rather than the same regions of the wild-type αS protein. In the C-terminal region, turn to helix transitions for the wild-type αS are more stable at Val95-Lys102, Gln109-Pro117, Pro120-Glu123, and Glu130-Tyr133 in the E46K mutant-type αS structures. In addition, the preferred coil to β-sheet transitions at Lys102, Asn103, Pro108, and Gln109 in the C-terminal region of the wild-type αS are less stable in wild-type αS. Overall, CD and NMR measurements indicated that the secondary structural properties of the wild-type and E46K mutant-type αS are similar to one another.18,19,27,28 In contrast, SMF spectroscopy measurements indicated an number of β-sheet conformations upon E46K mutation of the wild-type αS.25 Furthermore, E46K mutant-type αS is reported to convert to β-sheet conformations more readily than the wild-type αS.17,19 Our results agree with those experiments that reported an increased β-sheet conformation for the E46K mutant-type rather than wild-type αS.17,19,25 Moreover, we identify that this increase in β-sheet conformation occurs in the N-terminal and in the C-terminal regions of the E46K mutant-type αS. The most preferred transition to a β-sheet conformation for these residues occurs from a coil structure based on our thermodynamic calculations (Figure 2). The increased β-sheet content of the E46K mutant-type αS also agrees with the reported increased aggregation propensity of αS upon E46K mutation that was reported by some experiments given that the fibrillization and aggregation processes have been related to the formation of β-sheet.17−20 Due to the proposed importance of β-sheet formation in the aggregation process of αS, our results indicate that residues Val26, Tyr39, Gly47-Ala53, Asp119, Pro120, and Asn122-Tyr125 are most likely to be involved in the fibrillization and aggregation mechanisms of the E46K mutant-type αS. This is in contrast to the findings of Kumar et al., who reported that residues Gly68-Ala78 and Gly36-Gly41 have the highest aggregation propensity in the E46K mutant-type αS.29 While some β-sheet formation occurs in these regions in our simulated structures of the E46K mutant-type αS, we predict the highest aggregation propensity to be for residues Gly47-Ala53, Asp119, Pro120, and Asn122-Tyr125.
Figure 2.
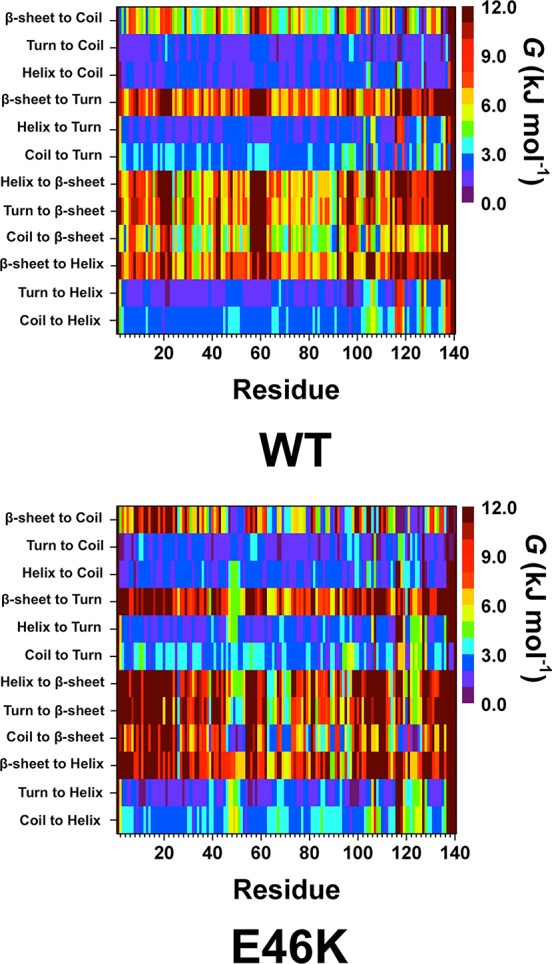
WT and E46K αS secondary structure component transition stability. The stability of secondary structure transitions between two specific secondary structure components per residue for the wild-type (WT) and E46K mutant-type (E46K) αS proteins. The color scale corresponds to the free energy value associated with specific secondary structure transition between two secondary structure components for a specific residue.
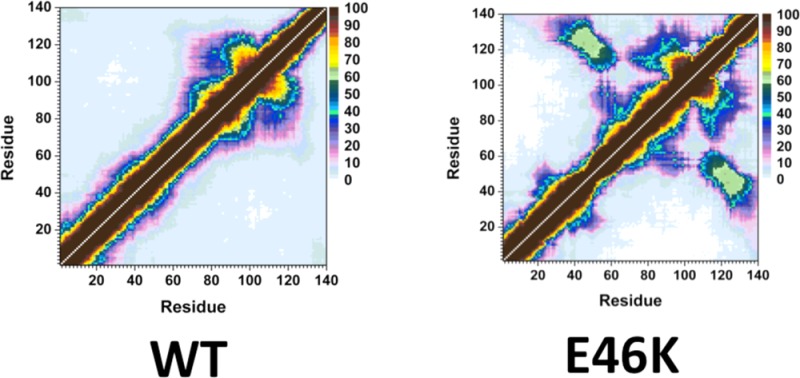
The probability of intramolecular protein interactions between residues in the structures of the wild-type and E46K mutant-type αS proteins are presented in Figure 3. The NAC and C-terminal regions of the wild-type αS protein interact strongly (>50%) via residues Gly86-Asn103 and Glu104-Asn122 (Figure 3, top). Strong intramolecular interactions between the NAC and C-terminal regions of the E46K mutant-type αS are also observed between residues Val82-Asp98 and Glu104-Ala124 (Figure 3, bottom). In addition, an increase in moderately abundant interactions (≤50%) between Val70-Val82 and Glu110-Val118 as well as between Val82-Glu105 and Glu126-Ala140 occurs upon E46K mutation. Both the wild-type and E46K mutant-type αS present strong intramolecular interactions with an abundance between residues Val70-Gly84 and Ala85-Leu100 within the NAC region. The E46K mutation also results in an increase in interactions between Val48-Gly67 and Val82-Lys102 by up to 40%. Furthermore, intramolecular interactions between the N-terminal and NAC regions (Gly7-Val66) with the C-terminal region (Gly106-Ala140) are up to 50% more abundant in the E46K mutant-type αS than the wild-type αS. In agreement with these findings, Rospigliosi et al. presented that interactions between the C-terminal region with the NAC and N-terminal regions are enhanced upon E46K mutation of the wild-type αS.27 Interactions within the N-terminal region are also increased in the E46K mutant-type αS in comparison to the wild-type αS between residues Val16-Ala30 and Gly36-Lys58. However, the weak intramolecular interactions (<10%) between the N-terminal region (Met1-Glu20) with the NAC and C-terminal regions (Thr64-Pro120) completely disappear upon E46K mutation of the wild-type αS protein. Interestingly, interactions between this region of the N-terminal (Met1-Glu20) and the NAC region have been used to identify the two different possible vesicle binding structures.38 Experimental measurements have proposed that the αS binds to vesicle and/or lipid structures in either a broken or extended helical conformation that can depend on the structure of the vesicle or lipid as well as on the protein conformation.38 Specifically, the extended helical structure is reported to more likely bind to a flat micellar surface whereas the broken helix structure is more likely to bond to a spherical micellar structure.38 The broken helical structure presents a distance of ∼34 Å between the N-terminal and NAC regions whereas this distance increases to ∼67 Å in the extended helix conformation.38 Our reported results for the interactions between the NAC and N-terminal regions indicate that, upon E46K mutation, the distance is increased and therefore an extended conformation is more likely in the E46K mutant-type rather than wild-type αS. As a result, we predict that the E46K mutant-type αS may show a higher binding to the flat micellar surfaces than the wild-type whereas the vice versa trend is expected for spherical micellar surfaces.
Figure 3.
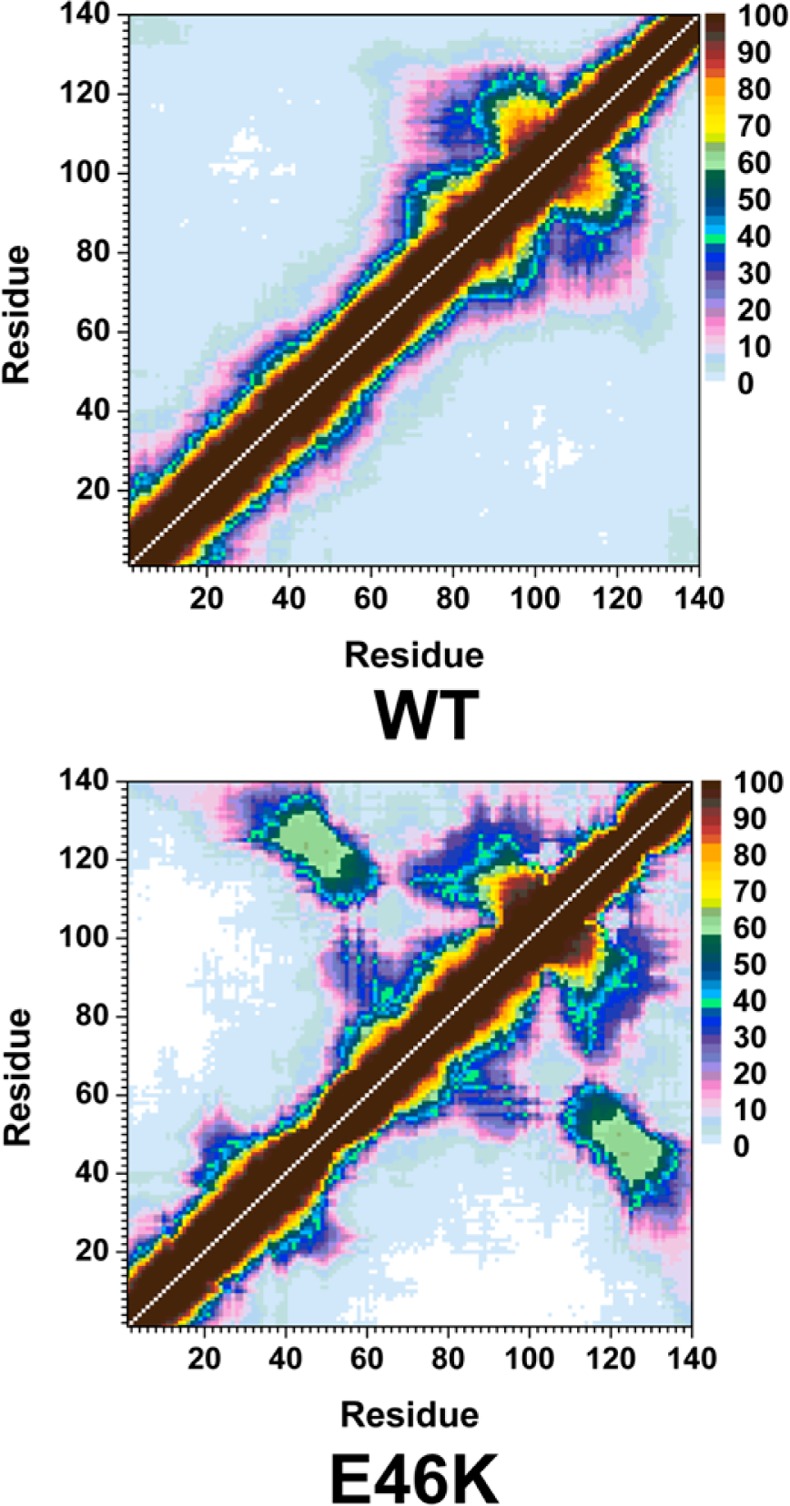
WT and E46K αS tertiary structures. Calculated intramolecular interactions of the wild-type (WT) and the E46K mutant-type (E46K) αS. The color scale corresponds to the probability (P) of the distance between the heavy atoms (C, N, O, S) of a residue being ≤20 Å from each other.
The radius of gyration (Rg) of a protein yields information about the relative compactness of the overall protein structure, which has been linked to the stability as well as the amount of intramolecular interactions within a protein structure. Figure 4 depicts the probability distribution of the Rg values for the structures of the wild-type and E46K mutant-type αS. Based on this data, the probability of finding structures with Rg values between 25.5 and 32.5 Å is higher for the E46K mutant-type rather than wild-type αS. The vice versa trend is observed for structures with Rg values varying between 35.5 and 55.0 Å. Overall, these trends agree with the E46K mutant-type αS protein having a more compact structure than the wild-type αS, as stated above. In agreement with these findings as well as those from the tertiary structures of these two proteins, the increased retention time of the E46K mutant-type αS in comparison to the wild-type αS in SEC suggests that the conformations of the E46K mutant-type αS are more compact than those of the wild-type αS.18 Overall, these findings suggest that the E46K mutant-type αS structures possess a greater number of intramolecular interactions and are more stable than the wild-type αS structures in aqueous solution. In order to quantitatively assess the relative stability of the wild-type and E46K mutant-type αS structures, conformational Gibbs free energy calculations were performed on the wild-type and E46K mutant-type αS structures. The difference between these Gibbs free energy values yields an estimate of the degree of difference in the stability of the two protein structures. The average thermodynamic values, presented in Table 1, indicate that the conformations of the wild-type αS are thermodynamically more stable over the structures of the E46K mutant-type αS protein by 114.5 kJ mol–1. Additionally, the entropic (TS) and enthalpic (H) contributions to the conformational Gibbs free energies indicate that the wild-type αS is entropically and enthalpically more preferred than the E46K mutant-type αS protein by 52.5 and 61.8 kJ mol–1, respectively. The decreased thermodynamic preference of the E46K mutant-type αS structures in comparison to the wild-type αS conformations indicates that the aggregation rate of the wild-type αS increases upon E46K mutation. This result is in agreement with those experimental studies that also reported an increased aggregation rate of the E46K mutant-type αS in comparison to the wild-type αS.17−20
Figure 4.
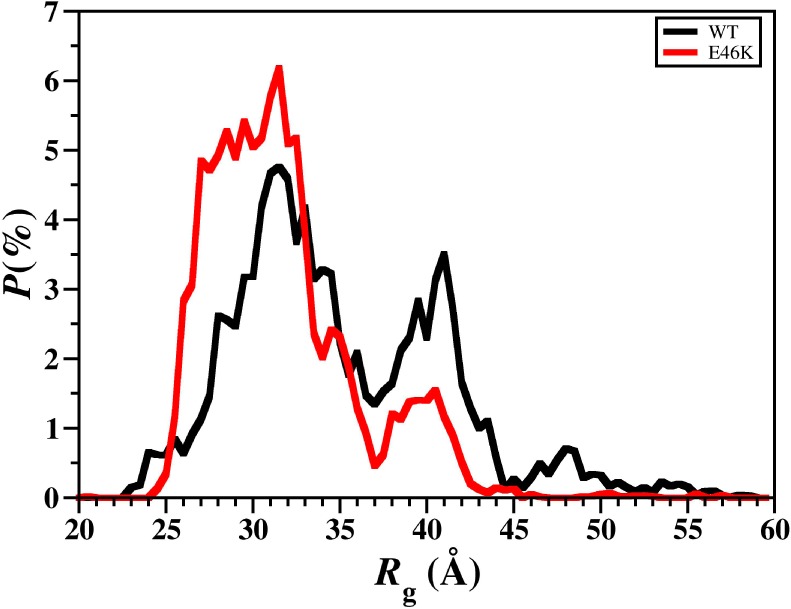
WT and E46K αS radius of gyration values. Distribution of the radius of gyration (Rg) values of the wild-type (black) and E46K mutant-type (red) αS for the structures obtained after convergence.
Table 1. Calculated Total Energy (Etotal), Solvation Free Energy (Gsol), Average Enthalpy (H), Entropy (TS), and Gibbs Free Energy (G) Values for the Wild-Type and E46K Mutant-Type α-Synuclein Proteins.
| peptide | ⟨Etotal⟩ (kJ mol-1) | ⟨Gsol⟩ (kJ mol-1) | ⟨H⟩ (kJ mol-1) | –T⟨S⟩ (kJ mol-1) | ⟨G⟩ (kJ mol-1) |
|---|---|---|---|---|---|
| WT | 7146.5 | –16704.1 | –9557.4 | –7043.8 | –16601.4 |
| (±174.3) | (±169.7) | (±18.4) | (±8.4) | (±13.0) | |
| E46K | 5260.6 | –14756.2 | –9495.6 | –6991.3 | –16486.9 |
| (±450.8) | (±499.2) | (±60.9) | (±13.7) | (±54.2) |
We also assessed the conformational preferences of the wild-type and E46K mutant-type αS proteins using potential of mean force (PMF) surfaces along the coordinates of Rg and end-to-end distance (RE-E), which is the distance between the N-terminus backbone N atom and the C-terminal backbone carboxyl C atom (Figure 5), following our previous studies on intrinsically disordered fibrillogenic proteins.37,39,40 The PMF surfaces determine the most favorable protein conformations based on specific structural parameters, such as Rg and RE-E via capturing the solvent effects. Different intrinsically disordered proteins structures may have similar thermodynamic properties, which results in the presence of multiple favorable basins along the PMF surface. Analyzing the structures in these different basins provides insights into the conformational properties based on specific structural parameters that influence the favorability of the wild-type and E46K mutant-type αS conformations in aqueous solution. The PMF surface of the wild-type αS shows two favorable basins located at Rg values varying between 28.0 and 35.5 Å (basin IA) and between 37.4 and 42.0 Å (basin IB) and at RE-E values varying between 51 and 98 Å (basin IA) and between 108 and 148 Å (basin IB). The difference in PMF values of the region between these two basins (>1 kBT) indicates that transitions between these two favorable PMF basins might require overriding large energy barriers (Figure 5, top). Two favorable PMF basins are also observed for the E46K mutant-type αS on the PMF surface (Figure 5, bottom). The first basin (basin IA) is located at Rg values between 25.8 and 33.2 Å and RE-E values between 50 and 108 Å. The second favorable PMF basin (basin IB) is located at Rg values between 33.3 and 36.4 Å and RE-E values between 68 and 79 Å. Interestingly, the Rg and RE-E values of basin IA are similar for the wild-type and E46K mutant-type αS proteins; however, the location of basin IB is shifted upon E46K mutation of the wild-type αS. Furthermore, our results suggest that conformational transitions these two favorable PMF basins do not require overriding large energy barriers upon E46K mutation. We also observe significant differences in the secondary structural properties of the structures located in the favorable basins for both the wild-type and E46K mutant-type αS (Figure 6). For example, the C-terminal region of the wild-type αS structures located in basin IA presents the most abundant α-helix formation at residues Phe94-Gly101 (up to 56%), which is decreased by 30% in the basin IB structures (Figure 6, top). In contrast, the most abundant α-helix structure is observed in the N-terminal and NAC regions of the basin IB structures of the wild-type αS at residues Glu20-Val26, Lys32-Gly36 and Lys28-Thr64 (up to 46%). Although α-helix structure is also present at these residues in the basin IA structures, the abundance is 10% less than the abundance in the basin IB wild-type αS structures. Furthermore, the α-helix content in the N-terminal and NAC regions of the basin IA wild-type αS structures at Met5, Lys6, Glu28, Ala29, Leu38, Val40-Lys43, Glu61, Lys80-Glu83, and Ala89-Thr92 decreases in the basin IB structures. Furthermore, we detect that the α-helix abundance at Glu20-Gln24 and Lys32-Glu35 decreases with decreasing preference along the PMF surface for the basin IB structures (basin IB > basin II > basin III) of the wild-type αS. The formation of 310-helix structure also varies between structures of the two favorable PMF basins. The 310-helix content at residues Val15-Ala17, Gly25-Gly31, Gly36-Leu38, Val40-Ser42, Gly86-Ile88, Val95-Gln99, and Gly101 increases by up to 40% in the basin IB structures rather than the basin IA structures of the wild-type αS. Despite, residues Lys43-Lys45, Gln79-Ala85, and Asp121-Tyr125 present the vice versa trend for 310-helix abundance between the basin IA and basin IB structures of the wild-type αS. However, we do not note any significant trends in the 310-helix content along the PMF surface for either favorable PMF basin. We also note large variations in the β-sheet structure formed in the basin IA and basin IB structures of the wild-type αS. For example, the basin IA structures present abundant β-sheet formation (up to 22%) at residues Val16, Gln24, Glu35, Val70, Val71, Lys102, and Pro108. However, the basin IB structures present more abundant β-sheet structure (up to 60%) at residues Phe4, Glu13, Val82, Glu83, Ala90, Ala91, and Gln109. Furthermore, the β-sheet structure at Phe4, Glu13, Val82, Glu83, Ala90, and Ala91 of the basin IB structures decreases along the PMF surface (basin IB > basin II > basin III).
Figure 5.
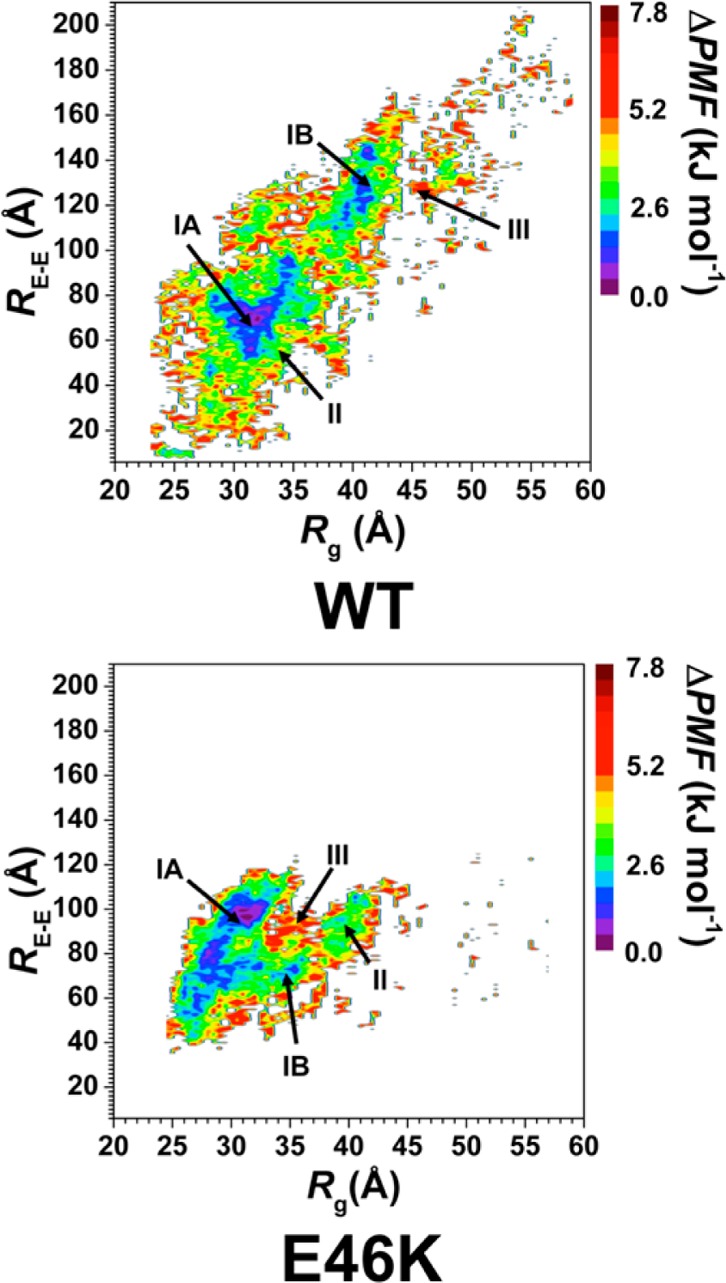
WT and E46K αS PMF surfaces. Change in the potential of mean force (ΔPMF) of the wild-type (WT) and E46K mutant-type (E46K) αS along the coordinates of radius of gyrations (Rg) and end-to-end distance (RE-E) in units of kJ mol–1. The reference values for the PMF surfaces are 6.73 and 6.48 kJ mol–1 for the wild-type and E46K mutant-type αS, respectively.
Figure 6.

WT and E46K αS secondary structure components of the PMF basin structures. Calculated secondary structure abundances per residue for the structures of the wild-type (WT) and E46K mutant-type (E46K) αS located in the basin IA (purple), basin IB (blue), basin II (green), and basin III (red) along the PMF surface. The abundances for the π-helix and coil structures are not displayed.
Regarding the secondary structure components of the E46K mutant-type αS in the two most favorable PMF basins (Figure 6B), we note that the abundant (up to 50%) α-helix formation at Gly7-Lys12, Gly25-Glu28, Gly31-Val37, and Ala53-Glu57 in the basin IA structures decreases by up to 20% in the basin IB structures. On the other hand, the α-helix structure at Glu20-Gln24, Thr72-Thr75, and Lys96-Gly101 is up to 40% more abundant in basin IB compared to the basin IA structures. Moreover, residues Leu8-Ala11 and Ala53-Glu57 of the basin IA structures and Glu20-Gln24 and Lys96-Gln99 of the basin IB structures present decreasing α-helix content along the PMF surface (basin IA > basin II > basin III or basin IB > basin II or basin III, respectively). The formation of 310-helix structure also displays significant variances between the basin IA and basin IB structures. For example, the 310-helix content at residues Ser9-Ala11, Val15-Ala19, Gly25, Val26, Gly36-Lys45, Thr54-Ala56, Tyr133, and Gln134 is at least 5% more abundant in the basin IA rather than basin IB structures of the E46K mutant-type αS. However, the basin IB structures present up to 50% more abundant 310-helix structure at residues Ala27-Ala29, Lys32-Glu35, Lys58-Glu61, Ala69-Glu83, Val95-Lys97, Glu110-Glu114, and Pro128-Gly132 than in the basin IA structures. We also note that residues Ala27-Ala29 and Lys32-Glu35 display decreasing 310-helix content along the PMF surface while the same trend is only observed for residues Thr54-Ala56 in the basin IA structures. The formation of β-sheet structure also presents large variations between the basin IA and basin IB structures of the E46K mutant-type αS. The basin IB structures present abundant (up to 85%) β-sheet structure at residues Val26, Tyr39, Lys80, Glu83, Ala89, Ala90, Gly93, Phe94, and Val118 that is decreased by 60% in the basin IA structures. On the other side, β-sheet formation occurs at Gly47-Val52, Val63-Val66, Ala69-Thr72, and Asp119-Glu126 with up to 62% more abundance in the basin IA rather than basin IB structures of the E46K mutant-type αS. Moreover, these residues also display decreasing β-sheet content along the PMF surface for each of their respective basins.
Notable differences in the tertiary structural properties of the basin IA and basin IB structures of the wild-type and E46K mutant-type αS are also evident (Figure 7). The basin IA structures of the wild-type αS (Figure 7, top left) show abundant intramolecular interactions between Gly86-Glu104 in the NAC and C-terminal regions with Glu105-Ala124 in the C-terminal region and Val70-Glu83 in the NAC region (between 50% and 95%). The N-terminal and C-terminal regions also interact with the NAC region with up to 45% abundance via residues Val55-Gly86 and Glu104-Tyr136. The N-terminal (Leu8-Val16) and C-terminal (Gly132-Ala140) interactions are also observed with low abundance (5%) in the basin IA structures of the wild-type αS. In parallel with the basin IA structures, the NAC and C-terminal regions of the basin IB structures of the wild-type αS interact abundantly via residues Thr72-Ile88 with Ile88-Val118 and residues Ala90-Asn103 with Glu104-Pro120. However, the low abundance interactions between residues Met1-Ala18 in the N-terminal region with the NAC and C-terminal regions in the basin IA structures completely disappear in the basin IB structures. Instead, residues Met1-Lys10 interact with residues Gly14-Gly25 with up to 40% abundance in the basin IB structures of the wild-type αS. The tertiary structure properties of the basin IA (Figure 7, bottom left) and basin IB (Figure 7, bottom right) structures of the E46K mutant-type αS also display significant differences. The highly abundant intramolecular interactions between residues Val66-Gly106 and Ala107-Ala140 in the basin IB structures decrease by up to 70% in the basin IA structures of the E46K mutant-type αS. We also note the same trend for interactions between residues Lys10-Val37 and Leu38-Lys60 as well as between Met1-Lys10 and Val36-Val49 in the N-terminal region of the E46K mutant-type basin IB structures. On the other side, we observe that the interactions of residues Met1-Asn65 with Gly106-Ala140 and residues Val48-Gly68 with Val82-Lys102 are up to 65% more abundant in the structures of basin IA rather than basin IB.
Figure 7.

WT and E46K αS tertiary structures for the favorable PMF basin structures. Calculated intramolecular interactions of the wild-type αS structures located in basin IA (WT IA) and basin IB (WT IB) of the PMF surface and of the E46K mutant-type αS structures located in basin IA (E46K IA) and basin IB (E46K IB) of the PMF surface. The color scale corresponds to the probability (P) of the distance between the heavy atoms (C, N, O, S) of a residue being ≤20 Å from each other.
Herein we present the structures of the E46K mutant-type αS and the impact of the E46K mutation on the structural and thermodynamic properties of the wild-type αS protein in an aqueous solution environment. The helical content of the wild-type and E46K mutant-type αS shows an increase for a few residues in the N-terminal and C-terminal regions but a slight decrease in the NAC region upon E46K mutation. The formation of β-sheet structures is significantly increased for the E46K mutant-type αS in comparison to the wild-type αS, especially in parts of the N-terminal and in the C-terminal regions. This result indicates that the N-terminal and C-terminal regions are more likely to participate in the aggregation mechanism of the E46K mutant-type αS rather than the wild-type αS. The use of our newly developed theoretical strategy for identifying the thermodynamic preference of secondary structure component transitions reveal that helix to β-sheet as well as β-heet to helix conversions are more stable in parts of the N-terminal and NAC regions of the E46K mutant-type αS in comparison to the wild-type αS protein. Moreover, coil to β-sheet conversion is more stable in parts of the C-terminal region of the wild-type αS rather than the E46K mutant-type αS protein. Analyses of the tertiary structures present that the E46K mutation increases the likelihood of long-range intramolecular interactions of the C-terminal region with the N-terminal and NAC regions of αS. Due to the decrease in N-terminal and NAC region interactions as well as the increase in helical content of the N-terminal region upon E46K mutation, our results also predict that the E46K mutant-type αS is more likely to bind to vesicles and/or lipid membranes in an extended helical conformation than the wild-type αS protein. Furthermore, the tertiary structures and Rg calculations yield that the conformations of the E46K mutant-type αS are overall more compact than those of the wild-type form. The abundant intramolecular interactions and compactness of the E46K mutant-type αS, evaluation of the conformational thermodynamic preferences of the E46K mutant-type and wild-type αS proteins present that the E46K mutant-type αS is structurally and thermodynamically more stable than those of the wild-type form. Therefore, the E46K mutant-type αS structures are more likely to aggregate than the wild-type αS conformations, which is in agreement with some experiments reporting similar characteristics, but here we present the information at the atomic level with dynamics in an aqueous solution environment. Furthermore, the PMF surfaces of both proteins present two favorable conformational basins with different secondary and tertiary structural properties. However, transitions between these two basins are predicted to occur more readily for the E46K rather than the wild-type αS due to the decreased energy barrier upon E46K mutation of the wild-type αS. Overall, our results show that the E46K mutation significantly impacts the structural properties of the wild-type αS protein not only at the mutation site but also throughout the protein structure. Furthermore, the resulting differences in the structural and thermodynamic properties of the wild-type αS mutation can be associated with the observed differences in the conformational properties and aggregation kinetics of the two αS proteins. To the best of our knowledge, this study present the first detailed insights, at the atomic level with dynamics in an aqueous solution environment, about the structures of the E46K mutant-type αS and the impact of the E46K mutation on the structures of the wild-type αS in terms of secondary structure transition stabilities per residue and tertiary structure properties along with conformational free energy landscapes. The information presented herein fills the currently existing gap in our fundamental knowledge about the E46K mutant-type αS protein and how this mutation impacts the wild-type form. Given that the β-sheet formation has been linked directly to the aggregation process and toxicity of αS, we propose that the residues we report herein with abundant β-sheet formation should be blocked via small molecules or antibodies to reduce the toxicity.
Methods
REMD simulations enable increased conformational sampling of protein structures by using replicas at different temperatures to overcome energy barriers between different minimum energy conformations of the same protein.41,42 This special sampling is especially necessary in the case of intrinsically disordered proteins, such as αS, which can possess numerous minimum energy conformations due to their disordered nature. Therefore, we performed REMD simulations on the wild-type and E46K mutant-type full-length αS proteins using the AMBER ff99SB force field parameters with the AMBER 11 software package.43,44 The Onufriev-Bashford-Case Generalized Born implicit solvent model was utilized to avoid possible errors due to confined aqueous volume and heat capacity variations observed in explicit solvent REMD simulations.39,40,45 The replica temperatures were exponentially distributed between 283 and 490 K to yield an approximate exchange probability of 0.70 for both simulations, resulting in a total number of 56 replicas.46 Starting from fully extended structures, the wild-type and E46K mutant-type αS structures were equilibrated for 500 ns for each replica. REMD simulations were then performed for a total of 40 ns with exchanges between replicas attempted every 5 ps. The time step was set to 2 fs, and trajectories were saved every 500 steps. The temperature was controlled using Langevin dynamics with a collision frequency of 2 ps–1.47,48 Long-range interactions were treated with the particle mesh Ewald (PME) method with a cutoff value of 450 Å, and bonds to hydrogen atoms were constrained using the SHAKE algorithm. The convergence of both simulations was confirmed to occur after 20 ns of simulation per replica by the cumulative secondary structure abundance (see the Supporting Information), as shown in our previous studies.37,39,40,49 All structural and thermodynamic properties of the wild-type and E46K mutant-type αS were determined from the structures obtained from the 20 ns of simulation after convergence was reached for the replica closest to physiological temperature (310 K). The secondary structure components of both αS proteins were determined using the DSSP program, which includes hydrogen bond and dihedral angle criteria.50 In addition, we analyzed the thermodynamic preference of secondary structure transitions between two different secondary structure components with recently developed theoretical strategy.37 This new strategy utilizes conditional probability to justify the potential of mean force. Specifically, the probability of a secondary structure transition is defined as P(ti→j|Stj), where P(ti→j) is the probability of a transition from one specific (i) to another specific secondary structure component (j) per residue and P(Stj) is the probability of a transition between two secondary structure components leading to the specific formation of j for a specific residue. The secondary structure components are defined based on hydrogen bond and backbone dihedral angle criteria. The hydrogen bond criteria which we implements uses the EHB = ErEtEp relationship in which the hydrogen bond energy (EHB) depends on the spatial arrangements; the distance-dependent energy term (Er) is calculated via Er = (A/r8) + (B/r6) with A = 3Er8 kcal Å8 mol–1 and B = 4Er6 kcal Å8 mol–1, where r and E represent the hydrogen bond distance and its energy, respectively. The angular spatial energy terms are calculated using eq 1:
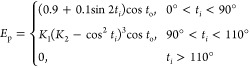 |
1 |
The spatial angles ti and to are computed based on the distance deviations between the hydrogen atom and the electron lone pair electron bisector that is located within the lone pair orbital plane and the distance from this plane, respectively. The values K1 and K2 are calculated via K1 = (0.9/cos6110°) and K2 = cos2110°, respectively. The method that we adopt for identifying the residual secondary structures has been used extensively. However, to the best of our knowledge, the current literature does not include a new theoretical strategy now a software package for determining the stability of secondary structure component transitions. We calculate these thermodynamic stabilities using our ProtMet software package eq 2:
| 2 |
where kB is the Boltzmann constant, T is the temperature, and Z(λ) is the ratio of the probability of transitions between two distinct secondary structure components per residue to the probability of overall transitions between two, three, or four components leading to the formation of a specific residual secondary structure.
Intramolecular interactions between residues were considered to occur if at least one heavy atom (C, O, N, S) of a specific residue was at least ≤20 Å from any heavy atom of another residue. A cutoff distance of 20 Å was utilized in order to account for any long-range intramolecular interactions observed experimental from residual dipolar coupling measurements. The thermodynamic properties of the wild-type and E46K mutant-type αS were assessed using two different methods: the MMPBSA method and PMF calculations.39,40,51 The MM/PBSA method calculates the conformational Gibbs free energy (G) of each structure based on the potential energy (Etotal), solvation free energy (Gsol), and estimated entropy (S) of each structure at a specific temperature (T) (eq 3).52
| 3 |
Gsol is calculated from the sum of the electrostatic and nonpolar contributions. In order to calculate the electrostatic contribution, dielectric constant values of 1 and 80 are used for the protein and solvent environment, respectively. The entropic contribution to G was estimated using normal-mode analysis (NMA).53 The PMF surfaces are determined along the coordinates of radius of gyration (Rg) and the end-to-end distance (RE-E), as shown in our previous studies on intrinsically disordered proteins.37,39,40
Acknowledgments
The calculations and simulations were performed on Kraken at the National Institute for Computational Sciences, Texas Advanced Computing Center (TACC) and the University of Texas at San Antonio Computational Biology Initiative.
Glossary
Abbreviations
- Parkinson’s disease
PD
- α-synuclein
αS
- circular dichroism
CD
- thioflavin T
ThT
- nuclear magnetic resonance
NMR
- single molecule force
SMF
- molecular dynamics
MD
- sodium dodecyl sulfate
SDS
- replica exchange molecular dynamics
REMD
- nonamyloid-β component
NAC
- radius of gyration
Rg
- size exclusion chromatography
SEC
- end-to-end distance
RE-E
- potential of mean force
PMF
- Molecular Mechanics/Poisson–Boltzmann surface area
MM/PBSA
- normal-mode analysis
NMA
Supporting Information Available
Convergence figures for the wild-type and E46K mutant-type αS. Convergence figures for the secondary structure transition stabilities and the PMF surfaces of both peptides. Description of the tertiary structural properties of both peptides for the least favorable basins on the PMF surfaces. This material is available free of charge via the Internet at http://pubs.acs.org.
Author Contributions
O.C., O.W.-S., A.D., A.A., and I.S. wrote the paper. O.C. designed the simulations. A.D. and O.W.-S. performed the simulations. O.C. and O.W.-S. designed and coded the developed secondary structure transition software.
This research was supported by an allocation and computing resources provided by the National Science Foundation (Grant No. TG-CHE110044). The authors are thankful for the financial support from the University of Texas at San Antonio.
The authors declare no competing financial interest.
Author Contributions
∥ O. C., O.W.-S., and A.D. contributed equally.
Supplementary Material
References
- Eriksen J. L.; Dawson T. M.; Dickson D. W.; Petrucelli L. (2003) Caught in the act: alpha-synuclein is the culprit in Parkinson’s disease. Neuron 40, 453–456. [DOI] [PubMed] [Google Scholar]
- Spillantini M. G.; Schmidt M. L.; Lee V. M. Y.; Trojanowski J. Q.; Jakes R.; Goedert M. (1997) α-Synuclein in Lewy bodies. Nature 388, 839–840. [DOI] [PubMed] [Google Scholar]
- Goedert M. (2001) Alpha-synuclein and neurodegenerative diseases. Nat. Rev. Neurosci. 2, 492–501. [DOI] [PubMed] [Google Scholar]
- Polymeropoulos M. H.; Higgins J. J.; Golbe L. I.; Johnson W. G.; Ide S. E.; DiIorio G.; Sanges G.; Stenroos E. S.; Pho L. T.; Schaffer A. A.; Lazzarini A. M.; Nussbaum R. L.; Duvoisin R. C. (1996) Mapping of a gene for Parkinson’s disease to chromosome 4q21-q23. Science 274, 1197–1199. [DOI] [PubMed] [Google Scholar]
- Clayton D. F.; George J. M. (1998) The synucleins: a family of proteins involved in synaptic function, plasticity, neurodegeneration and disease. Trends Neurosci. 21, 249–254. [DOI] [PubMed] [Google Scholar]
- Kruger R.; Muller T.; Riess O. (2000) Involvement of alpha-synuclein in Parkinson’s disease and other neurodegenerative disorders. J. Neural Transm. 107, 31–40. [DOI] [PubMed] [Google Scholar]
- Maroteaux L.; Campanelli J. T.; Scheller R. H. (1988) Synuclein - a Neuron-Specific Protein Localized to the Nucleus and Presynaptic Nerve-Terminal. J. Neurosci. 8, 2804–2815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zarranz J. J.; Alegre J.; Gomez-Esteban J. C.; Lezcano E.; Ros R.; Ampuero I.; Vidal L.; Hoenicka J.; Rodriguez O.; Atares B.; Llorens V.; Tortosa E. G.; del Ser T.; Munoz D. G.; de Yebenes J. G. (2004) The new mutation, E46K, of alpha-synuclein causes Parkinson and Lewy body dementia. Ann. Neurol. 55, 164–173. [DOI] [PubMed] [Google Scholar]
- Singleton A. B.; Farrer M.; Johnson J.; Singleton A.; Hague S.; Kachergus J.; Hulihan M.; Peuralinna T.; Dutra A.; Nussbaum R.; Lincoln S.; Crawley A.; Hanson M.; Maraganore D.; Adler C.; Cookson M. R.; Muenter M.; Baptista M.; Miller D.; Blancato J.; Hardy J.; Gwinn-Hardy K. (2003) alpha-synuclein locus triplication causes Parkinson’s disease. Science 302, 841–841. [DOI] [PubMed] [Google Scholar]
- Kruger R.; Kuhn W.; Muller T.; Woitalla D.; Graeber M.; Kosel S.; Przuntek H.; Epplen J. T.; Schols L.; Riess O. (1998) Ala30Pro mutation in the gene encoding alpha-synuclein in Parkinson’s disease. Nat. Genet. 18, 106–108. [DOI] [PubMed] [Google Scholar]
- Polymeropoulos M. H.; Lavedan C.; Leroy E.; Ide S. E.; Dehejia A.; Dutra A.; Pike B.; Root H.; Rubenstein J.; Boyer R.; Stenroos E. S.; Chandrasekharappa S.; Athanassiadou A.; Papapetropoulos T.; Johnson W. G.; Lazzarini A. M.; Duvoisin R. C.; DiIorio G.; Golbe L. I.; Nussbaum R. L. (1997) Mutation in the alpha-synuclein gene identified in families with Parkinson’s disease. Science 276, 2045–2047. [DOI] [PubMed] [Google Scholar]
- Winner B.; Jappelli R.; Maji S. K.; Desplats P. A.; Boyer L.; Aigner S.; Hetzer C.; Loher T.; Vilar M. a.; Campioni S.; Tzitzilonis C.; Soragni A.; Jessberger S.; Mira H.; Consiglio A.; Pham E.; Masliah E.; Gage F. H.; Riek R. (2011) In vivo demonstration that α-synuclein oligomers are toxic. Proc. Natl. Acad. Sci. U.S.A. 108, 4194–4199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Outeiro T. F.; Putcha P.; Tetzlaff J. E.; Spoelgen R.; Koker M.; Carvalho F.; Hyman B. T.; McLean P. J. (2008) Formation of Toxic Oligomeric α-Synuclein Species in Living Cells. PloS One 3, e1867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J.; Uversky V. N.; Fink A. L. (2002) Conformational behavior of human alpha-synuclein is modulated by familial Parkinson’s disease point mutations A30P and A53T. Neurotoxicology 23, 553–567. [DOI] [PubMed] [Google Scholar]
- Li J.; Uversky V. N.; Fink A. L. (2001) Effect of familial Parkinson’s disease point mutations A30P and A53T on the structural properties, aggregation, and fibrillation of human alpha-synuclein. Biochemistry 40, 11604–11613. [DOI] [PubMed] [Google Scholar]
- Choong C. J.; Say Y. H. (2011) Neuroprotection of alpha-synuclein under acute and chronic rotenone and maneb treatment is abolished by its familial Parkinson’s disease mutations A30P, A53T and E46K. Neurotoxicology 32, 857–863. [DOI] [PubMed] [Google Scholar]
- Ono K.; Ikeda T.; Takasaki J.; Yamada M. (2011) Familial Parkinson disease mutations influence alpha-synuclein assembly. Neurobiol. Dis. 43, 715–724. [DOI] [PubMed] [Google Scholar]
- Fredenburg R. A.; Rospigliosi C.; Meray R. K.; Kessler J. C.; Lashuel H. A.; Eliezer D.; Lansbury P. T. (2007) The impact of the E46K mutation on the properties of alpha-synuclein in its monomeric and oligomeric states. Biochemistry 46, 7107–7118. [DOI] [PubMed] [Google Scholar]
- Greenbaum E. A.; Graves C. L.; Mishizen-Eberz A. J.; Lupoli M. A.; Lynch D. R.; Englander S. W.; Axelsen P. H.; Giasson B. I. (2005) The E46K mutation in alpha-synuclein increases amyloid fibril formation. J. Biol. Chem. 280, 7800–7807. [DOI] [PubMed] [Google Scholar]
- Choi W.; Zibaee S.; Jakes R.; Serpell L. C.; Davletov B.; Crowther R. A.; Goedert M. (2004) Mutation E46K increases phospholipid binding and assembly into filaments of human alpha-synuclein. FEBS Lett. 576, 363–368. [DOI] [PubMed] [Google Scholar]
- Kamiyoshihara T.; Kojima M.; Ueda K.; Tashiro M.; Shimotakahara S. (2007) Observation of multiple intermediates in alpha-synuclein fibril formation by singular value decomposition analysis. Biochem. Biophys. Res. Commun. 355, 398–403. [DOI] [PubMed] [Google Scholar]
- Bertoncini C. W.; Jung Y. S.; Fernandez C. O.; Hoyer W.; Griesinger C.; Jovin T. M.; Zweckstetter M. (2005) Release of long-range tertiary interactions potentiates aggregation of natively unstructured alpha-synuclein. Proc. Natl. Acad. Sci. U.S.A. 102, 1430–1435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertoncini C. W.; Fernandez C. O.; Griesinger C.; Jovin T. M.; Zweckstetter M. (2005) Familial mutants of α-synuclein with increased neurotoxicity have a destabilized conformation. J. Biol. Chem. 280, 30649–30652. [DOI] [PubMed] [Google Scholar]
- Zhu M.; Fink A. L. (2003) Lipid binding inhibits α-synuclein fibril formation. J. Biol. Chem. 278, 16873–16877. [DOI] [PubMed] [Google Scholar]
- Brucale M.; Sandal M.; Di Maio S.; Rampioni A.; Tessari I.; Tosatto L.; Bisaglia M.; Bubacco L.; Samori B. (2009) Pathogenic Mutations Shift the Equilibria of α-Synuclein Single Molecules towards Structured Conformers. ChemBioChem 10, 176–183. [DOI] [PubMed] [Google Scholar]
- Bussell R. Jr; Eliezer D. (2001) Residual structure and dynamics in Parkinson’s disease-associated mutants of α-synuclein. J. Biol. Chem. 276, 45996–46003. [DOI] [PubMed] [Google Scholar]
- Rospigliosi C. C.; McClendon S.; Schmid A. W.; Ramlall T. F.; Barre P.; Lashuel H. A.; Eliezer D. (2009) E46K Parkinson’s-Linked Mutation Enhances C-Terminal-to-N-Terminal Contacts in alpha-Synuclein. J. Mol. Biol. 388, 1022–1032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferreon A. C. M.; Moran C. R.; Ferreon J. C.; Deniz A. A. (2010) Alteration of the α-Synuclein Folding Landscape by a Mutation Related to Parkinson’s Disease. Angew. Chem., Int. Ed. 49, 3469–3472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar S.; Sarkar A.; Sundar D. (2009) Controlling aggregation propensity in A53T mutant of alpha-synuclein causing Parkinson’s disease. Biochem. Biophys. Res. Commun. 387, 305–309. [DOI] [PubMed] [Google Scholar]
- Perlmutter J. D.; Braun A. R.; Sachs J. N. (2009) Curvature Dynamics of alpha-Synuclein Familial Parkinson Disease Mutants. Molecular Simulations of the Micelle- And Bilayer-Bound Forms. J. Biol. Chem. 284, 7177–7189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eliezer D.; Kutluay E.; Bussell R.; Browne G. (2001) Conformational properties of alpha-synuclein in its free and lipid-associated states. J. Mol. Biol. 307, 1061–1073. [DOI] [PubMed] [Google Scholar]
- Davidson W. S.; Jonas A.; Clayton D. F.; George J. M. (1998) Stabilization of alpha-synuclein secondary structure upon binding to synthetic membranes. J. Biol. Chem. 273, 9443–9449. [DOI] [PubMed] [Google Scholar]
- Jo E. J.; McLaurin J.; Yip C. M. St; George-Hyslop P.; Fraser P. E. (2000) alpha-synuclein membrane interactions and lipid specificity. J. Biol. Chem. 275, 34328–34334. [DOI] [PubMed] [Google Scholar]
- Georgieva E. R.; Ramlall T. F.; Borbat P. P.; Freed J. H.; Eliezer D. (2010) The Lipid-binding Domain of Wild Type and Mutant alpha-Synuclein. Compactness and Interconversion between the Broken and Extended Helix Forms. J. Biol. Chem. 285, 28261–28274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kessler J. C.; Rochet J. C.; Lansbury P. T. (2003) The N-terminal repeat domain of alpha-synuclein inhibits beta-sheet and amyloid fibril formation. Biochemistry 42, 672–678. [DOI] [PubMed] [Google Scholar]
- Vilar M.; Chou H. T.; Luhrs T.; Maji S. K.; Riek-Loher D.; Verel R.; Manning G.; Stahlberg H.; Riek R. (2008) The fold of alpha-synuclein fibrils. Proc. Natl. Acad. Sci. U.S.A. 105, 8637–8642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coskuner O., Wise-Scira O., Perry G., and Kitahara T. (2012) The Structure of the E22Δ Mutant-Type Amyloid-β Alloforms and the Impact of E22Δ Mutation on the Structures of the Wild-Type Amyloid-β Alloforms. ACS Chem. Neurosci., published online October 25, 1012. DOI: 10.1021/cn300149j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferreon A. C. M.; Gambin Y.; Lemke E. A.; Deniz A. A. (2009) Interplay of alpha-synuclein binding and conformational switching probed by single-molecule fluorescence. Proc. Natl. Acad. Sci. U.S.A. 106, 5645–5650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wise-Scira O.; Xu L.; Kitahara T.; Perry G.; Coskuner O. (2011) Amyloid-beta peptide structure in aqueous solution varies with fragment size. J. Chem. Phys. 135, 205101. [DOI] [PubMed] [Google Scholar]
- Wise-Scira O.; Xu L.; Perry G.; Coskuner O. (2012) Structures and free energy landscapes of aqueous zinc(II)-bound amyloid-beta(1–40) and zinc(II)-bound amyloid-beta(1–42) with dynamics. J. Biol. Inorg. Chem. 17, 927–938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugita Y.; Okamoto Y. (1999) Replica-exchange molecular dynamics method for protein folding. Chem. Phys. Lett. 314, 141–151. [Google Scholar]
- Zhang W.; Wu C.; Duan Y. (2005) Convergence of replica exchange molecular dynamics. J. Chem. Phys. 123, 154105. [DOI] [PubMed] [Google Scholar]
- Simmerling C.; Hornak V.; Abel R.; Okur A.; Strockbine B.; Roitberg A. (2006) Comparison of multiple amber force fields and development of improved protein backbone parameters. Proteins 65, 712–725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Case D., Darden T., Cheatham T., Simmerling C., Wang J., Duke R., Luo R., Walker R., Zhang W., and Merz K. (2010) AMBER 11, University of California, San Francisco.
- Case D. A.; Onufriev A.; Bashford D. (2004) Exploring protein native states and large-scale conformational changes with a modified generalized born model. Proteins 55, 383–394. [DOI] [PubMed] [Google Scholar]
- Patriksson A.; van der Spoel D. (2008) A temperature predictor for parallel tempering simulations. Phys. Chem. Chem. Phys. 10, 2073–2077. [DOI] [PubMed] [Google Scholar]
- Allen M. P., and Tildesley D. J. (1999) Computer simulation of liquids, Clarendon Press: Oxford. [Google Scholar]
- Frenkel D., and Smit B. (2002) Understanding molecular simulation: from algorithms to applications, Vol. 1, Academic Press: San Diego, CA. [Google Scholar]
- Fawver J. N.; Duong K. T.; Wise-Scira O.; Petrofes Chapa R.; Schall H. E.; Coskuner O.; Zhu X.; Colom L. V.; Murray I. V. J. (2012) Probing and Trapping a Sensitive Conformation: Amyloid-β Fibrils, Oligomers, and Dimers. J. Alzheimer’s Dis. 32, 197–215. [DOI] [PubMed] [Google Scholar]
- Kabsch W.; Sander C. (1983) Dictionary of Protein Secondary Structure - Pattern-Recognition of Hydrogen-Bonded and Geometrical Features. Biopolymers 22, 2577–2637. [DOI] [PubMed] [Google Scholar]
- Lee M. R.; Duan Y.; Kollman P. A. (2000) Use of MM-PB/SA in estimating the free energies of proteins: Application to native, intermediates, and unfolded villin headpiece. Proteins: Struct., Funct., Genet. 39, 309–316. [PubMed] [Google Scholar]
- Kollman P. A.; Massova I.; Reyes C.; Kuhn B.; Huo S. H.; Chong L.; Lee M.; Lee T.; Duan Y.; Wang W.; Donini O.; Cieplak P.; Srinivasan J.; Case D. A.; Cheatham T. E. (2000) Calculating structures and free energies of complex molecules: Combining molecular mechanics and continuum models. Acc. Chem. Res. 33, 889–897. [DOI] [PubMed] [Google Scholar]
- Case D. A. (1994) Normal-Mode Analysis of Protein Dynamics. Curr. Opin. Struct. Biol. 4, 285–290. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.