

Abstract
BACKGROUND AND PURPOSE
The free fatty acid FFA1 receptor and GPR120 are GPCRs whose endogenous ligands are medium- and long-chain FFAs, and they are important in regulating insulin and GLP-1 secretion respectively. Given that the ligands of FFA1 receptor and GPR120 have similar properties, selective pharmacological tools are required to study their functions further.
EXPERIMENTAL APPROACH
We used a docking simulation approach using homology models for each receptor. Biological activity was assessed by phosphorylation of ERK and elevation of intracellular calcium ([Ca2+]i) in cells transfected with FFA1 receptor or GPR120. Insulin secretion from murine pancreatic beta cells (MIN6) was also measured.
KEY RESULTS
Calculated hydrogen bonding energies between a series of synthetic carboxylic acid compounds and the homology models of the FFA1 receptor and GPR120, using docking simulations, correlated well with the effects of the compounds on ERK phosphorylation in transfected cells (R2= 0.65 for FFA1 receptor and 0.76 for GPR120). NCG75, the compound with the highest predicted selectivity for FFA1 receptors from this structure-activity relationship analysis, activated ERK and increased [Ca2+]i as potently as the known FFA1 receptor-selective agonist, Compound 1. Site-directed mutagenesis analysis based on the docking simulation showed that different amino acid residues were important for the recognition and activation by FFA1 receptor agonists. Moreover, NCG75 strongly induced ERK and [Ca2+]i responses, and promoted insulin secretion from MIN6 cells, which express endogenous FFA1 receptors.
CONCLUSION AND IMPLICATIONS
A docking simulation approach using FFA1 receptor and GPR120 homology models could be useful in predicting FFA1 receptor-selective agonists.
Keywords: G-protein coupled receptor, FFA1, GPR120, agonist, homology modelling
Introduction
The free fatty acids (FFAs) are not only essential nutritional components, but they also function as signalling molecules, activating specific receptors. One of these, the FFA1 receptor, was previously designated as an orphan receptor, GPR40 (receptor nomenclature follows Alexander et al., 2011). The FFA1 receptor is a GPCR activated by medium- to long-chain fatty acids and expressed abundantly in pancreatic beta cells, where it mediates the FFA-enhanced, glucose-stimulated, secretion of insulin (Briscoe et al., 2003; Itoh et al., 2003; Poitout, 2003; Steneberg et al., 2005; Feng et al., 2006; Stoddart et al., 2008). Based on this role in insulin secretion, the FFA1 receptor has received increasing attention as an attractive drug target in type 2 diabetes (Milligan et al., 2006; Hirasawa et al., 2008; Suzuki et al., 2008). GPR120 is another GPCR whose endogenous ligands are medium- to long-chain FFAs (Hirasawa et al., 2005). Although human FFA1 receptor share only 10% amino acid identity with human GPR120, the properties of their endogenous ligands (FFAs) are very similar (Itoh et al., 2003; Hirasawa et al., 2005) and many synthetic ligands of FFA1 receptor (Briscoe et al., 2003; Tikhonova et al., 2007; Bharate et al., 2008; Davi and Lebel, 2008; Hirasawa et al., 2008; Hara et al., 2009a; Sasaki et al., 2011) interact with GPR120 as well. Therefore, for further pharmacological analysis of these long-chain FFA receptors, the development of new ligands, selective for each receptor, is required.
Given that GPCRs are integral membrane proteins, the experimental determination of their tertiary structure is highly challenging. Detailed structural information about specific receptors can be inferred by homology from the recently defined higher resolution structures of GPCRs (Kobilka, 2007; Rosenbaum et al., 2009). In recent years, homology models of GPCRs that are based on the template structure of bovine rhodopsin have been successfully utilized for structure-based virtual screening for drug discovery (Radestock et al., 2008; Sun et al., 2010). We have recently succeeded in identifying agonists of GPR120 using docking simulation in a GPR120 homology model and demonstrated the usefulness of this approach to predict the agonist activity of compounds (Sun et al., 2010). Furthermore, Tikhonova et al. reported a homology model for the FFA1 receptor and used the model to identify residues that are important for agonist recognition and the activation of these receptors (Sum et al., 2007; Tikhonova et al., 2007). However, it remained unclear whether such an approach could be applied to the development of agonists that were selective for one of the two FFA receptors with similar pharmacological properties. Hence, in the present study, we investigated whether docking simulation using both the FFA1 receptor and GPR120 homology models might be useful in predicting the FFA1 receptor-selective agonist activity of a range of synthetic compounds.
Methods
Compounds
We examined NCG75(12-(phenyl-2-pyridinylamino) dodecanoic acid), together with 46 other synthetic carboxylic acid derivatives (denoted as NCG-II compounds and shown in Table 1). Some of the NCG-II compounds (30 compounds) have been reported previously (Sun et al., 2010). α-Linolenic acid (α-LA) and MEDICA 16 (3,3,14,14-tetramethylhexadecanedioic acid) were purchased from Sigma (St. Louis, MO, USA). GW9508 (4-[[(3-phenoxyphenyl)methyl]amino]benzenepropanoic acid) was synthesized according to the previously reported procedure (Garrido et al., 2006) and was purchased from Namiki Shoji Co., Ltd. (Tokyo, Japan). Compound 1 (3-{4-(3-phenoxybenzyloxy) phenyl}hex-4-ynoic acid) was synthesized according to the previously filed patent application (Akerman et al., 2005). All compounds were dissolved in dimethyl sulfoxide (DMSO) in stock solutions of 100 mM and stored at −20°C.
Table 1.
Structure of compounds, relative maximal ERK activity and hydrogen bonding energy between compounds and the homology model of the FFA1 receptor or GPR120
| Compound | Structure | Relative maximum ERK response | H-bonding energy (arbitrary units) | ||
|---|---|---|---|---|---|
| FFA1 | GPR120 | FFA1 | GPR120 | ||
| α-LA | 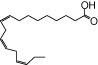 |
1.00 | 1.00* | −5.73 | −3.03* |
| AMGEN-1 | 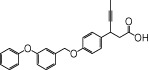 |
1.25 | 0.13 | −7.48 | −3.15 |
| GW9508 | 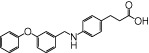 |
0.80 | 0.28 | −4.79 | −2.83 |
| MEDICA16 | 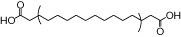 |
0.90 | 0.21* | −5.91 | −1.73* |
| NCG20 | 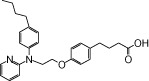 |
0.93 | 1.38* | −4.85 | −4.04* |
| NCG21 | 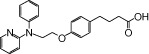 |
0.67 | 1.54* | −5.09 | −5.92* |
| NCG22 | 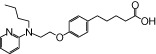 |
0.64 | 0.93* | −5.17 | −5.07* |
| NCG23 | 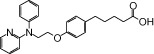 |
1.07 | 1.20* | −6.58 | −4.10* |
| NCG25 | 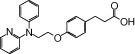 |
1.26 | 1.25* | −7.16 | −4.73* |
| NCG26 | 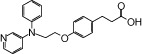 |
1.10 | 1.02* | −5.87 | −4.54* |
| NCG27 | 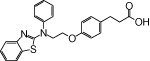 |
0.61 | 0.55* | −3.50 | −2.23* |
| NCG28 | 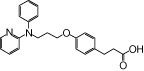 |
1.20 | 0.80* | −5.69 | −3.66* |
| NCG29 | 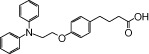 |
0.17 | 1.46* | −4.19 | −3.56* |
| NCG30 | 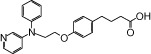 |
0.36 | 1.45* | −3.46 | −5.57* |
| NCG31 | 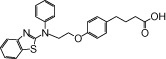 |
0.13 | 1.40* | −3.22 | −4.06* |
| NCG32 | 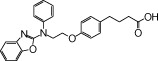 |
0.87 | 1.03* | −5.34 | −3.86* |
| NCG33 | 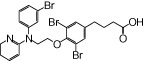 |
0.04 | 0.39* | −3.80 | −2.88* |
| NCG34 | 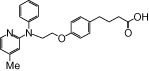 |
1.17 | 1.37* | −5.78 | −5.36* |
| NCG35 | 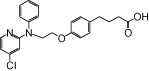 |
1.30 | 1.48* | −7.53 | −4.75* |
| NCG38 | 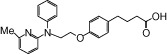 |
1.03 | 1.24* | −6.87 | −4.87* |
| NCG39 | 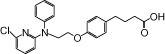 |
0.42 | 1.23* | −4.09 | −4.75* |
| NCG40 | 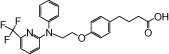 |
1.03 | 0.62* | −5.42 | −2.73* |
| NCG41 | 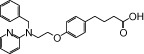 |
1.61 | 0.96* | −7.95 | −2.61* |
| NCG42 | 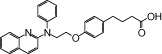 |
0.79 | 0.54* | −3.46 | −2.92* |
| NCG44 | 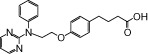 |
0.32 | 0.64* | −4.30 | −3.10* |
| NCG45 | 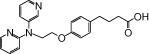 |
0.06 | 0.76* | −3.33 | −3.39* |
| NCG46 | 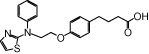 |
0.53 | 1.20* | −5.21 | −5.41* |
| NCG48 | 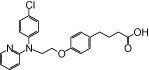 |
1.04 | 0.92* | −6.15 | −3.98* |
| NCG49 | 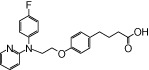 |
0.89 | 0.75* | −5.23 | −2.61* |
| NCG50 | 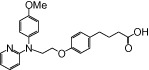 |
1.02 | 1.14* | −3.96 | −4.93* |
| NCG51 | 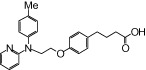 |
0.95 | 1.32* | −5.75 | −4.77* |
| NCG52 | 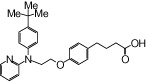 |
0.90 | 0.29* | −4.29 | −2.70* |
| NCG53 | 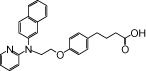 |
1.47 | 1.27* | −5.82 | −4.56* |
| NCG57 | 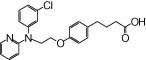 |
1.21 | 1.09* | −6.98 | −4.11* |
| NCG74 | 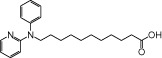 |
1.64 | 0.66 | −7.05 | −1.67 |
| NCG75 | 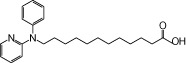 |
1.63 | 0.27 | −6.77 | −2.19 |
| NCG85 | 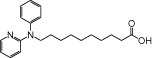 |
1.45 | 0.86 | −6.85 | −3.52 |
| NCG86 | 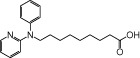 |
0.62 | 0.74 | −3.37 | −2.84 |
| NCG87 | 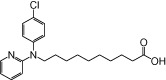 |
0.99 | 0.10 | −5.13 | −1.11 |
| NCG88 | 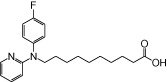 |
0.91 | 0.20 | −6.89 | −3.42 |
| NCG89 | 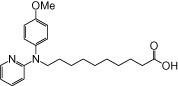 |
0.86 | 0.11 | −6.87 | −0.76 |
| NCG97 | 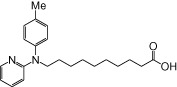 |
1.20 | 0.22 | −6.57 | −1.21 |
| NCG98 | 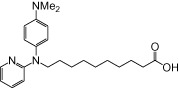 |
0.89 | 0.19 | −5.24 | −1.65 |
| NCG99 | 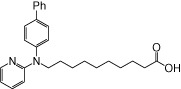 |
0.87 | 0.18 | −5.82 | −2.34 |
| NCG100 | 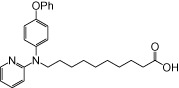 |
0.18 | 0.11 | −2.22 | −3.08 |
| NCG101 | 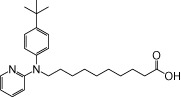 |
0.70 | 0.04 | −4.36 | −1.50 |
| NCG102 | 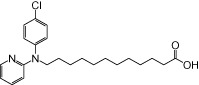 |
0.80 | 0.05 | −5.36 | −1.27 |
| NCG103 | 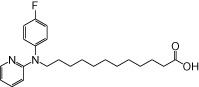 |
0.66 | 0.12 | −3.24 | −2.54 |
| NCG104 |  |
0.63 | 0.02 | −5.93 | −1.00 |
| NCG105 |  |
0.38 | 0.04 | −3.19 | −1.45 |
| NCG106 |  |
0.66 | 0.07 | −4.24 | −0.18 |
The relative maximal ERK response was calculated as the relative value of the agonist response elicited by each compound (100 µM), with respect to the response evoked by 100 µM α-LA in FFA1 receptor- or GPR120-expressing cells.
Values from Sun et al. (2010).
Cell lines
We used an inducible and stable human embryonic kidney (HEK293) cells expressing FFA1 receptors (T-REx FFA1) and cells expressing GPR120 (Flp-in GPR120), which were established previously (Hara et al., 2009b). T-REx FFA1 and Flp-in GPR120 cells were cultured as described previously (Hirasawa et al., 2005; Hara et al., 2009b). Briefly, T-REx FFA1 cells were cultured in DMEM supplemented with 10% FBS, 100 µg·mL−1 hygromycin (Invitrogen, Carlsbad, CA, USA) and 10 µg·mL−1 blasticidin S. Flp-in GPR120 cells were cultured in DMEM supplemented with 10% FBS and 100 µg·mL−1 zeocin. The murine pancreatic beta cell line (MIN6 cells) was maintained in DMEM (high glucose) containing 10% FBS, 60 µM β-mercaptoethanol, 100 U·mL−1 penicillin and 100 µg·mL−1 streptomycin. All cells were grown at 37°C in a humidified atmosphere of 5% CO2/95% air.
Site-directed mutagenesis
Site-directed mutagenesis was performed using the QuikChange II XL mutagenesis kit (Stratagene, La Jolla, CA, USA). Sequences on the promoter and the full insert in all constructs were verified by sequencing.
siRNA transfection
Cells were grown in antibiotic-free medium for 24 h and transfected with 60 nM siRNA against mouse FFA1 (target sequences of 5′-GGCCTGGAGTGTGGTACTCAA-3′; Qiagen, Valencia, CA, USA) or control siRNA (AllStars Negative Control siRNA; Qiagen) using Lipofectamine 2000 (Invitrogen), according to the manufacturer's protocol. Experiments were performed 24 h after transfection.
Docking simulation
Test compound structures were built systematically using the software PyMol (DeLano Scientific, San Carlos, CA, USA). We prepared two homology models, for the FFA1 receptor and for GPR120. The GPR120 homology model was developed based on a photoactivated model derived from the crystal structure of bovine rhodopsin (Ishiguro et al., 2004; Sun et al., 2010). The FFA1 receptor homology model was based on the structure of bovine rhodopsin and has already been reported (Tikhonova et al., 2007). We employed Tikhonova's constructed model according to the procedure of Lu et al., (2010). The molecular docking of compounds against the FFA1 receptor and GPR120 models was performed by the molecular docking algorithm MolDock using Molegro Virtual Docker software (Molegro ApS, Aarhus, Denmark) respectively (Thomsen and Christensen, 2006). The models and the compounds to be docked were imported into the docking program, following the software instructions. Potential ligand binding sites of proteins were calculated using the Molegro cavity detection algorithm. The hydrogen bonding energy, which is considered to be one of the important parameters in characterizing the interaction between GPCRs and their ligands (Shim et al., 2003; Xhaard et al., 2006; Sun et al., 2010), was estimated in arbitrary units using the Molegro program.
Phosphorylation of ERK
Phosphorylation of ERK (ERK activation) induced by various compounds in T-REx FFA1 cells, Flp-in GPR120 cells and MIN6 cells was measured as described previously (Hirasawa et al., 2005). Briefly, cells were serum-starved for 20 h (T-REx FFA1 and Flp-in GPR120 cells) and 2 h (MIN6 cells) respectively. The cells were then treated with each compound that was being tested at concentrations of 1, 3, 10 or 100 µM. After 10 min of incubation, total cell extracts were prepared and subjected to Western blotting using anti-phospho- and anti-total-kinase antibodies (Cell Signaling Technology, Boston, MA, USA).
[Ca2+]i measurement
Cells were seeded at a density of 2 × 105 cells·per well on collagen-coated 96-well plates, incubated at 37°C for 21 h and then incubated in HBSS (pH 7.4) containing Calcium Assay Kit Component A (Molecular Devices, Sunnyvale, CA, USA) for 1 h at room temperature. Compounds used in the fluorometric imaging plate reader (FLIPR; Molecular Devices) assay were dissolved in HBSS (1% DMSO) and prepared in another set of 96-well plates. These plates were set on the FLIPR, and mobilization of [Ca2+]i evoked by agonists was monitored. Data analysis was performed using Igor Pro (WaveMetrics, Lake Oswego, OR, USA).
Insulin secretion in MIN6 cells
MIN6 cells were seeded into each well of 96-well plates at a density of 4 × 104 cells·per well. The cells were subsequently cultured for 24 h at 37°C in a humidified atmosphere of 5% CO2/95% air. The media were removed from the cells and the cells washed twice with glucose-free Krebs [119 mM NaCl; 4.74 mM KCl; 2.54 mM CaCl2; 1.19 mM MgCl2; 1.19 mM KH2PO4; 25 mM NaHCO3; 10 mM HEPES (pH 7.4) ]. A sample (100 µL) of the same Krebs solution but containing 2.5 mM glucose, was added to each well and the plates were returned to the incubator for 1 h. Subsequently, the plates were washed a further twice with glucose-free Krebs and 100 µL of Krebs containing 25 mM glucose, and test compounds at the desired concentration were added. Following 2 h of incubation, the Krebs solution in each well was removed and the insulin content of each sample was determined with an elisa kit (Shibayagi, Gunma, Japan).
Single-cell [Ca2+]i measurement
The [Ca2+]i was measured and recorded by conventional Ca2+ imaging methods, using an image processor (Argus 50; Hamamatsu Photonics, Hamamatsu, Japan). Briefly, MIN6 cells were loaded with fura-2 acetoxymethyl ester (fura-2 AM; Dojindo, Tokyo, Japan) by incubation in 2 µM fura-2 acetoxymethyl ester for 30 min at 37°C. Changes in [Ca2+]i were measured at 30°C in Tyrode's solution. Fluorescence of fura-2 was measured by applying UV light at 340 and 380 nm alternately and by passing the light emitted through a 505-nm dichroic mirror (DCLP; Omega Optical, Brattleboro, VT, USA). Fluorescence was detected by an SPD-CCD camera (MC681APD-R0B0; Texas Instruments, Dallas, TX, USA). Ca2+ images were acquired at an interval of 20 s and processed later to calculate the ratio of fluorescence intensity at 340 and 380 nm, using NIH Image program (http://rsb.info.nih.gov/nih-image/).
Data analysis
In the present study, we investigated the relationship between the calculated hydrogen bonding energy and the relative effect on ERK at 100 µM of each ligand. The hydrogen bonding energy is considered to be a measure of the goodness of fit between the ligand structure and the modelled virtual cavity. As it was not possible to obtain full concentration–response curves for all the NCG compounds, the relative ERK activity at a single ligand concentration (100 µM) was used for the correlation with the calculated hydrogen bonding energy. It is important to note that under such conditions, changes in the ERK activity may reflect with equal chance a difference in EC50 or a difference in Emax among the ligands.
One-way anova was used to evaluate treatment effects. If the anova value was significant, comparisons between the control and treatment groups were performed using anova, followed by Dunnett's t-test to localize the significant difference. P < 0.05 was considered statistically significant.
Results
Docking simulation of FFA1 receptor-selective agonists using homology models
To develop FFA1 receptor-selective agonists and investigate the structure-activity relationships (SARs) of agonists at the FFA1 receptor, a series of 47 carboxylic acid derivatives (denoted as NCG-II compounds) was synthesized, and a docking simulation was conducted using the homology models of the FFA1 receptor and GPR120 that were reported by Tikhonova et al. (2007) and our group (Sun et al., 2010) respectively. In addition to the NCG-II compounds, we used several reference compounds: (i) α-LA, an endogenous ligand for FFA1 receptors and GPR120 (Hirasawa et al., 2005); (ii) Compound 1, known as a selective agonist for FFA1 receptors (Akerman et al., 2005); (iii) GW9508, known as a dual agonist for FFA1 receptors and GPR120; and (iv) MEDICA 16, known as a selective agonist for FFA1 receptor (Hara et al., 2009b). All compounds were docked individually into either the FFA1 receptor or GPR120 model using the Molegro Virtual Docker, and the in silico hydrogen bonding energies of these compounds were calculated (Table 1). To examine the agonist activities of the compounds, their ability to activate ERK by phosphorylation was assessed in human embryonic kidney (HEK293) cells that expressed either FFA1 receptor or GPR120 and compared with the ERK activation induced by a standard concentration of the known activator, phorbol myristate acetate (PMA). All of the NCG-II compounds tested activated ERK, but their potency, expressed as activation relative to that by PMA, differed with their structure. However, as shown in Figure 1, a plot of this relative ERK activation against the calculated hydrogen bonding energy showed a high correlation (R2= 0.65 for the FFA1 receptor and R2= 0.76 for GPR120 respectively). Among a group of NCG-II compounds that showed potent ERK activation in cells expressing FFA1 receptor, NCG75 was found to have the highest selectivity for these receptors on the basis of the ratio of the in silico-calculated hydrogen bonding energy in the FFA1 receptor homology model to that in the GPR120 model (−6.77 vs. −2.19; ratio = 3.09) (Figure 1). Hence, on the basis of the docking simulation using the homology models, NCG75 was predicted to be a potent FFA1 receptor-selective agonist.
Figure 1.
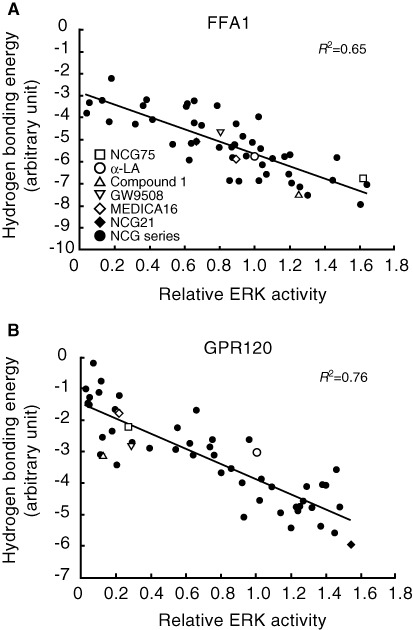
Correlation between the relative ERK activity and the calculated hydrogen bond energy of interaction based on modelling. The straight line represents (A) the line y=−2.75 ×−2.90 (FFA1 receptor) and (B) y=−2.49 ×−1.37 (GPR120). The coefficient of determination (R2= 0.65, FFA1 receptor and 0.76, GPR120) reflects a high correlation between the hydrogen bonding energy and the relative ERK activity.
To test the selectivity of NCG75, together with the activity of α-LA and Compound 1, we examined the responses of ERK and [Ca2+]i induced by these compounds in HEK293 cells expressing FFA1 receptor or GPR120. As shown in Figure 2a, α-LA, which is an endogenous ligand for FFA1 receptors as well as GPR120, and the FFA1 receptor-selective agonist, Compound 1 (Akerman et al., 2005) stimulated ERK in cells expressing FFA1 receptors. NCG75 also activated ERK in a dose-dependent manner, with a potency similar to α-LA and Compound 1 (Figure 2a). In cells that expressed GPR120, α-LA strongly stimulated ERK, but neither Compound 1 nor NCG75 were potent agonists (Figure 2b). In addition to ERK activation, α-LA, Compound 1 and NCG75 showed similar dose–response relationships for elevation of [Ca2+]i in cells expressing FFA1 receptor (EC50 values: 7.97 µM for α-LA, 2.50 µM for compound 1 and 5.11 µM for NCG75) (Figure 3). In contrast, in the Flp-in GPR120 cells, only α-LA was a potent agonist in terms of effects on [Ca2+]i (EC50 values: 3.63 µM for α-LA, 18.5 µM for Compound 1 and 46.2 µM for NCG75) (Figure 3). These results with respect to the ERK and [Ca2+]i responses appeared to support the prediction that NCG75 was a FFA1 receptor-selective agonist.
Figure 2.
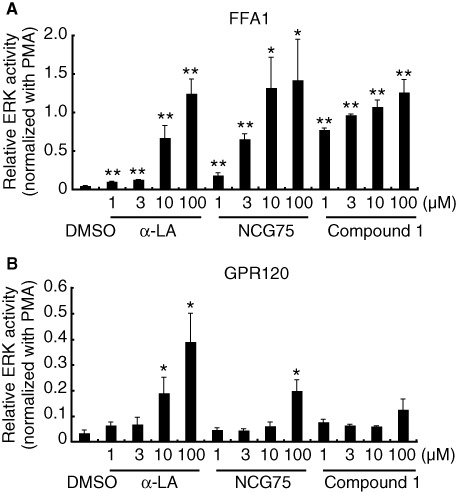
Effect of compounds on ERK activity in (A) T-REx FFA1 and (B) Flp-in GPR120 cells. Cells were serum-starved for 20 h and then treated with various compounds at 1–100 µM. Cell lysates were analyzed by immunoblotting using anti-phospho- and anti-total-kinase antibodies. The amount of phosphorylated ERK was normalized to the amount of total ERK. The data were then presented as -fold difference relative to the amount of ERK phosphorylation that was obtained in the presence of phorbol 12-myristate 13-acetate (PMA). Results represent means ± SEM of three independent experiments. *P < 0.05, **P < 0.01, significantly different from control (DMSO).
Figure 3.
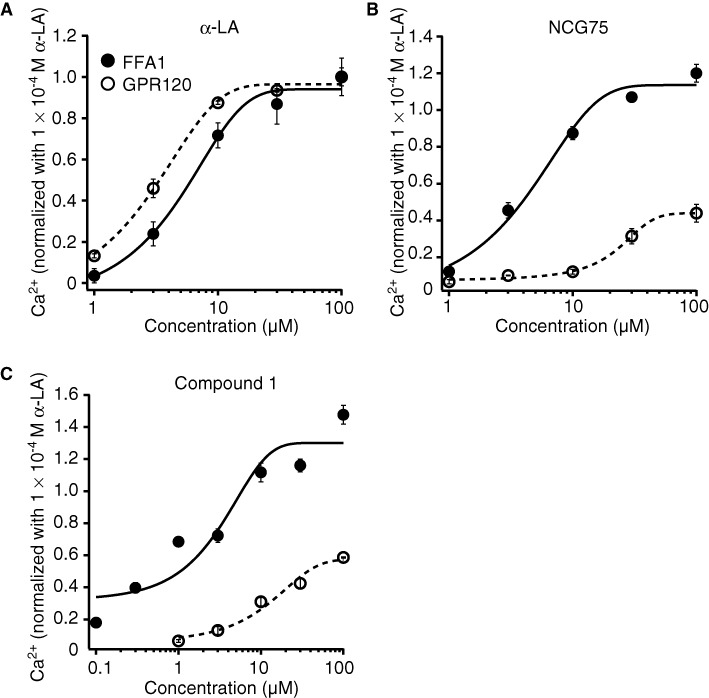
Effects of compounds on [Ca2+]i in T-REx FFA1 cells and Flp-in GPR120 cells. T-REx FFA1 cells and Flp-in GPR120 cells were stimulated with α-LA (A), NCG75 (B) or compound 1 (C). Cells were seeded at a density of 2 × 105 cells per·well on collagen-coated 96-well plates, incubated at 37°C for 21 h and then incubated in HBSS (pH 7.4) that contained Calcium Assay Kit Component A for 1 h at room temperature. Compounds that were used in the FLIPR assay were dissolved in HBSS (1% DMSO) and prepared in another set of 96-well plates. These plates were set on the FLIPR, and mobilization of [Ca2+]i evoked by agonists was monitored.
Determination of amino acid residues in FFA1 required for agonist recognition and activation
Docking simulation predicted the presence of hydrogen bonds between the oxygen of the carboxylate group on the ligands (Compound 1, GW9508, NCG75 and α-LA) and the guanidine groups of Arg183 and Arg258 (Figure 4). We noticed that the calculated distances of the hydrogen bonds between the ligand and Arg183, and between the ligand and Arg258 were similar for Compound 1 and GW9508, whereas for α-LA and NCG75, the two calculated distances were significantly different. Thus, as shown in Figure 4, the distances of the hydrogen bond between compound 1 and either Arg183 or Arg258 were 2.61 and 2.74 (or 2.81) Å, respectively (Figure 4c), and the equivalent distances for GW9508 were 2.84 and 2.91 (or 2.65) Å respectively (Figure 4d). In contrast, the distances of the hydrogen bond between α-LA and either Arg183 or Arg258 were 4.46 and 2.63 (or 3.47) Å, respectively (Figure 4a), and the equivalent distances for NCG75 were 4.60 and 2.58 (or 2.98) Å respectively (Figure 4b). The different distances of the hydrogen bond might indicate the different extent to which the residues contribute to agonist recognition and activation (Shim et al., 2003; Xhaard et al., 2006; Sun et al., 2010). Site-directed mutagenesis confirmed that α-LA and NCG75 activated ERK in cells expressing the mutant FFA1 receptor Arg183Ala (R183A) in a dose-dependent manner, similar to that in cells with the wild-type (WT) receptor. However, these compounds did not activate ERK in cells expressing the Arg258Ala (R258A) mutant receptor (Figure 5a). In contrast, Compound 1 did not activate ERK in cells bearing either of the mutant FFA1 receptors. As observed with ERK, α-LA and NCG75 strongly promoted [Ca2+]i responses in a dose-dependent manner in R183A cells, but not in R258A cells (Figure 5b,c), but Compound 1 did not elevate [Ca2+]i in either R183A or R258A cells (Figure 5d). In order to investigate the selectivity of these four compounds (Compound 1, GW9508, NCG75 and α-LA) between FFA1 receptors and GPR120 from the structural point of view, we carried out further docking simulations to GPR120 for these compounds. As shown in Figure 4e–h, the simulation predicted the presence of hydrogen bonds between the oxygen of the carboxylate on α-LA and GW9508, but not that on NCG75 and Compound 1, and the guanidine of Arg99, which had been shown to be involved in the interaction of ligands (Sun et al., 2010).
Figure 4.
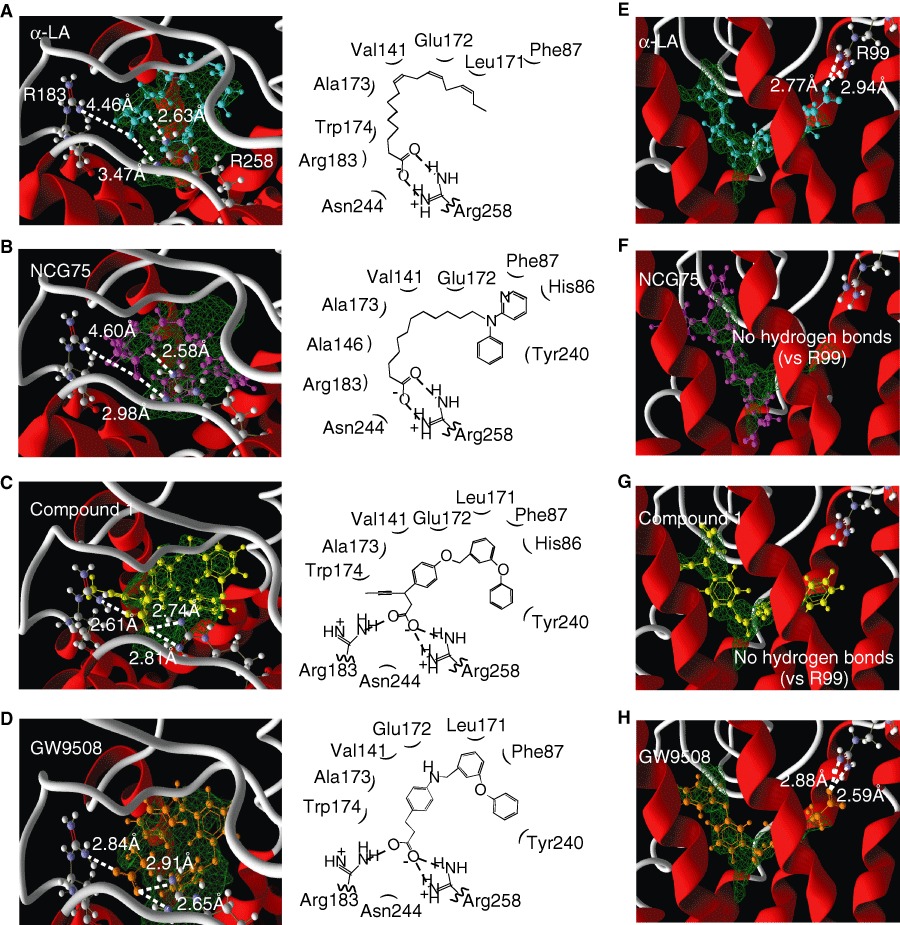
The FFA1 receptor and GPR120 homology models docked with each compound tested (A–D, for FFA1 receptor; E–H, for GPR120). α-LA (A,E), NCG75 (B,F), compound 1 (C,G) and GW9508 (D,H) were docked into the binding pocket of the FFA1 receptor or the GPR120. Cyan, α-LA; magenta, NCG75; yellow, compound 1; orange, GW9508; green: the binding pocket predicted by the Molegro cavity detection algorithm. The dashed lines indicate the distances between the oxygen of the carboxylate groups of the compounds and the guanidine nitrogen of arginine residues.
Figure 5.
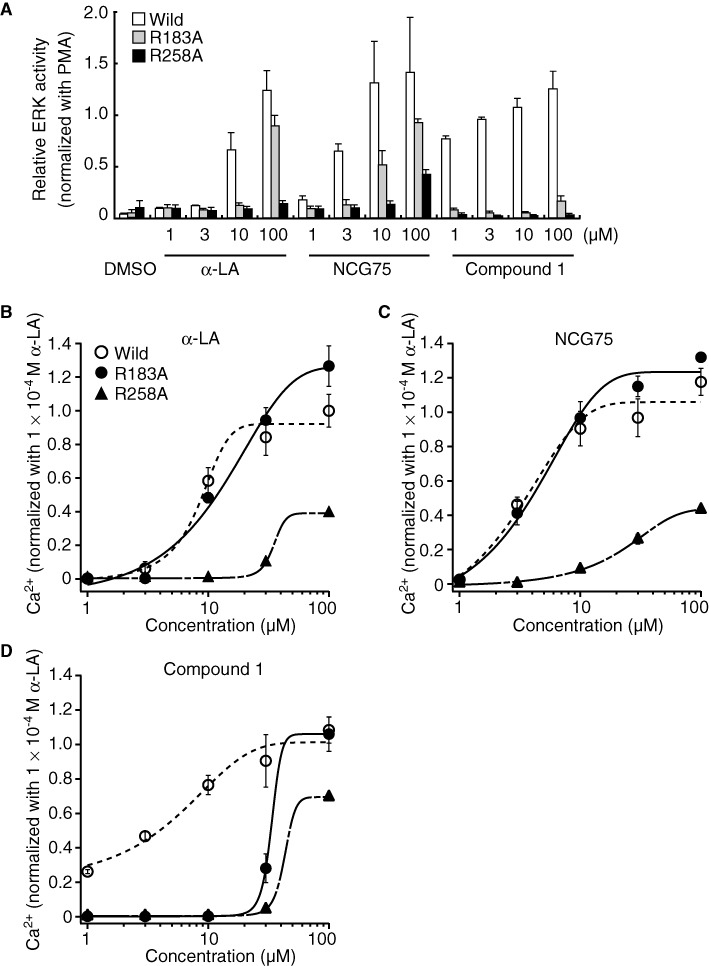
Effect of compounds in cells bearing mutant FFA1 receptors. (A) Relative ERK activity was measured in each mutant cell line, as described for Figure 2. Mobilization of [Ca2+]i evoked by agonists was monitored in each mutant cell line (B–D), as described for Figure 3. Cells were stimulated with α-LA (B), NCG75 (C) or compound 1 (D). Cells were seeded at a density of 2 × 105 cells·well−1 on collagen-coated 96-well plates, incubated at 37°C for 21 h and then incubated in HBSS (pH 7.4) that contained Calcium Assay Kit Component A (Molecular Devices, Sunnyvale, CA, USA) for 1 h at room temperature. Compounds that were used in the fluorometric imaging plate reader (FLIPR; Molecular Devices) assay were dissolved in HBSS (1% DMSO) and prepared in another set of 96-well plates. These plates were set on the FLIPR, and mobilization of [Ca2+]i evoked by agonists was monitored.
Pharmacological effects of NCG75 on MIN6 cells
We examined the pharmacological effects of NCG75 on the murine pancreatic beta cell line MIN6, which expresses FFA1 receptors endogenously. NCG75 stimulated ERK responses in MIN6 cells in a dose-dependent manner and with greater potency and efficacy than α-LA (Figure 6a). In addition, similar to α-LA, NCG75 stimulated [Ca2+]i responses in MIN6 cells (Figure 6b). Furthermore, both NCG75 and α-LA stimulated insulin secretion in MIN6 cells and such stimulation was inhibited significantly by an siRNA against mouse FFA1 receptors (Figure 6c).
Figure 6.
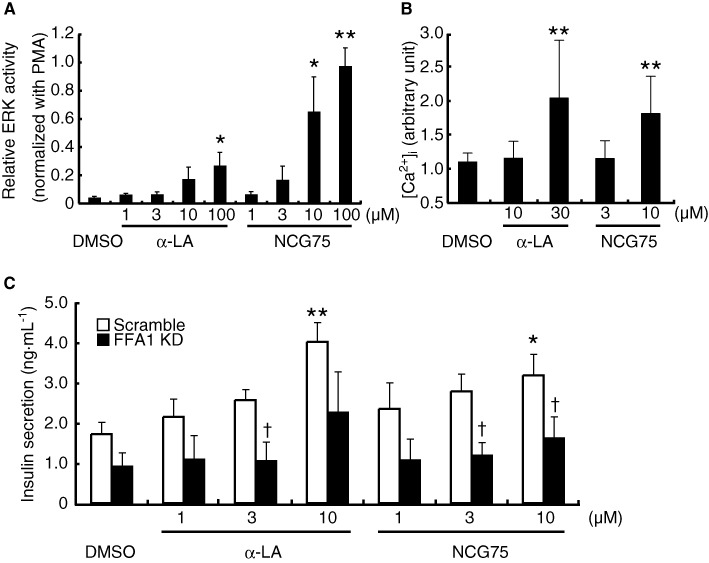
Effect of NCG75 in MIN6 cells. (A) Relative ERK activity was measured in MIN6 cells, as described for Figure 2. Results are means ± SEM of five independent experiments. *P < 0.05, **P < 0.01, significantly different from control (DMSO). (B) The maximum [Ca2+]i response induced by the indicated compounds between 0 and 10 min is shown. Results are means ± SEM of three independent experiments. The data were normalized against the maximum response observed in the presence of DMSO. *P < 0.05, **P < 0.01, significantly different from control (DMSO). (C) Effect of siRNA for the FFA1 receptor (FFA1 KD) on insulin secretion in MIN6 cells was measured after treatment with compounds at 1–10 µM for 120 min at 37°C in Krebs buffer that contained 25 mM glucose. Results are means ± SEM of five independent experiments. *P < 0.05, significantly different from control (DMSO); †P < 0.05, significantly different from control (scramble) siRNA.
Discussion
Previously, we showed that a docking simulation using a homology model for GPR120 might be useful in predicting the agonist activity of compounds and identified a GPR120 agonist (NCG21) (Sun et al., 2010). Combining the GPR120 homology model with the previously reported homology model for FFA1 receptors, the present study showed that a docking simulation successfully predicted FFA1 receptor-selective agonist activity, and, as a consequence, we developed the FFA1 receptor-selective agonist NCG75. First, a series of carboxylic acid derivatives (denoted as NCG-II compounds) was synthesized, and, together with several known FFA1 receptor-selective agonists (MEDICA 16, Compound 1) and non-selective agonists (α-LA and GW9508), were docked individually into either the FFA1 receptor or GPR120 homology model. On the other hand, as it was not possible to obtain full concentration–response curves for all of NCG-II compounds, we used the relative effect on ERK at a single ligand concentration for the correlation. The simulations showed that the calculated hydrogen bonding energies for the compounds docked into the FFA1 receptor or GPR120 homology models correlated well with their relative effect on ERK at 100 µM of ligand for the relevant FFA receptor. Hence, these results suggested that this method could be useful to investigate the SARs of FFA1 receptor and GPR120 agonists. Furthermore, we predicted the selectivity of the compounds for the receptors by using the ratio of the in silico-calculated hydrogen bonding energy in the FFA1 receptor homology model to that in the GPR120 model, which identified NCG75 as a FFA1 receptor-selective agonist.
The results of the present study showed that computational docking simulations could accurately predict amino acid residues that are important for the recognition and activation of FFA1 receptor by an agonist. Using FFA1 receptor homology modelling and site-directed mutagenesis, Sum et al. (2007) had shown previously that linoleic acid and GW9508 are anchored to FFA1 receptors via interactions between their carboxylate groups and Arg183, Asn244 and Arg258. In good agreement with these findings, our docking simulation confirmed that hydrogen bonds between the oxygen of the carboxylate group on the ligands and the guanidine groups of Arg183 and Arg258 were important for agonist recognition and the activation of FFA1 receptor in the case of Compound 1 and GW9508. However, our analyses also showed that Arg258 alone, and not both Arg183 and Arg258, was critical for the activation of FFA1 receptors by NCG75 or α-LA. The results might also support the hypothesis that the calculated distance of a hydrogen bond in a docking simulation reflects the extent of contribution of the residue to agonist recognition and activation.
In addition to the two arginine residues described earlier (Arg183 and Arg258), our docking simulation indicated that other amino acids could interact importantly with NCG75 (Figure 4b). For example, the pyridine and phenyl ring of NCG75 could form π-π interactions with His86, Phe87 and Tyr240. In addition, the hydrophobic amino acid residues (Val141, Ala146 and Ala173) that are located around the methylene group of NCG75 might contribute to stabilizing the binding of NCG75 to FFA1 receptors through hydrophobic interaction. Furthermore, the SAR with ERK showed that the length of the methylene chain of NCG75 was critical for FFA1 receptors selectivity because the chain length of 11 carbon atoms allowed the carboxylate group of NCG75 to dock into a position that was closer to Arg258 in the binding site of the FFA1 receptor than in GPR120. In fact, NCG74 and NCG85, which are NCG-II compounds whose structures are similar to that of NCG75 but have different lengths of methylene chain (lengths of 10 and 9 carbon atoms for NCG74 and NCG85, respectively), showed non-selective activation of ERK in both FFA1 receptor- and GPR120-expressing cells (Table 1). In addition, experiments with analogues of NCG75 with various lengths of methylene chain (9–13 carbon atoms) confirmed that NCG75 had the highest selectivity for FFA1 receptor (data not shown).
We noticed a potential discrepancy between the ERK (or [Ca2+]i) activity of NCG75 and its effect on insulin release, as shown in Figure 6. Thus, although NCG75 seemed to be equivalent or even more efficient than α-LA in stimulating ERK and [Ca2+]i in both transfected cells and MIN6 cells, the ability of NCG75 to enhance glucose-induced insulin release from MIN6 cells was lower than that of α-LA. This is not a characteristic limited to NCG ligands, as similar discrepancies have been reported for other synthetic FFA1 receptor agonists, such as TAK-875 (Tsujihata et al., 2011) and GW9508 (Briscoe et al., 2006). TAK-875 enhanced the intracellular production of inositol monophosphate in CHO-hFFA1 cells at lower concentrations than those of the endogenous ligand (oleic acid), but the maximum effect of TAK-875 on insulin secretion in INS-1832/13 cells appeared to be similar to that of oleic acid (Tsujihata et al., 2011). Also, GW9508 elicited [Ca2+]i responses in HEK-293 cells expressing the hFFA1 receptor at lower concentrations compared with the endogenous ligands (α-LA and linoleic acid), but the maximum effect of GW9508 on insulin secretion in MIN6 cells appeared to be similar to that of linoleic acid (Briscoe et al., 2006). One possible explanation for this discrepancy may be that some residual activity of the compounds at PPARγ might interfere with their effects on insulin secretion (Nakamichi et al., 2003). However, this probably did not apply here, as MIN6 cells do not express PPARγ, and PPARγ activators have little effect on insulin secretion from MIN6 cells (Nakamichi et al., 2003). Further studies would be required to understand these discrepant effects of FFA1 receptor agonists.
In conclusion, we report here the SARs of a series of NCG-II compounds with agonist activities at FFA1 receptors and GPR120 that correlated with hydrogen bonding energies, calculated using docking simulations. Among the NCG-II compounds, NCG75 was predicted to be the most potent FFA1 receptor-selective agonist, based on SARs. Thus, the present study showed that a docking simulation using FFA1 receptor and GPR120 homology models might be useful in predicting the receptor-selective agonist activity of a series of compounds. In addition, NCG75, which was confirmed to be a potent FFA1 receptor-selective agonist, might become an important pharmacological tool to investigate the biological functions of these receptors.
Acknowledgments
This work was supported, in part, by research grants from the Japan Science and Technology Agency; the Funding Program for World-Leading Innovative R&D on Science and Technology (FIRST Program), initiated by the Council for Science and Technology Policy; the Targeted Proteins Research Program of MEXT, Japan; Grant-in-Aid for Scientific Research [19109001, 21390021]; the Pharmacological Research Foundation, Tokyo; and the Hou-ansha Foundation.
Glossary
- α-LA
α-linolenic acid
- [Ca2+]i
intracellular calcium concentration
- Compound
1, 3-{4-(3-phenoxybenzyloxy) phenyl}hex-4-ynoic acid
- DMEM
Dulbecco's modified Eagle's medium
- FBS
fetal bovine serum
- FFA
free fatty acid
- MEDICA
16, 3,3,14,14-tetramethylhexadecanedioic acid
- MIN6
murine pancreatic beta cell line
- NCG
75, 12-(phenyl-2-pyridinylamino) dodecanoic acid
- SAR
structure–activity relationship
Conflicts of interest
None.
References
- Akerman M, Houze J, Lin DCH, Liu J, Luo J, Medina JC, et al. 2005. Arylmethoxyphenyl-alkylcarboxylic acids and related derivatives for use in treating metabolic disorders. Patent Application No. WO 2005/086661.
- Alexander SPH, Mathie A, Peters JA. Guide to Receptors and Channels (GRAC), 5th Edition. Br J Pharmacol. 2011;164(Suppl. 1):S1–S324. doi: 10.1111/j.1476-5381.2011.01649_1.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bharate SB, Rodge A, Joshi RK, Kaur J, Srinivasan S, Kumar SS, et al. Discovery of diacylphloroglucinols as a new class of GPR40 (FFAR1) agonists. Bioorg Med Chem Lett. 2008;18:6357–6361. doi: 10.1016/j.bmcl.2008.10.085. [DOI] [PubMed] [Google Scholar]
- Briscoe CP, Tadayyon M, Andrews JL, Benson WG, Chambers JK, Eilert MM, et al. The orphan G protein-coupled receptor GPR40 is activated by medium and long chain fatty acids. J Biol Chem. 2003;278:11303–11311. doi: 10.1074/jbc.M211495200. [DOI] [PubMed] [Google Scholar]
- Briscoe CP, Peat AJ, McKeown SC, Corbett DF, Goetz AS, Littleton TR, et al. Pharmacological regulation of insulin secretion in MIN6 cells through the fatty acid receptor GPR40: identification of agonist and antagonist small molecules. Br J Pharmacol. 2006;148:619–628. doi: 10.1038/sj.bjp.0706770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davi M, Lebel H. One-pot approach for the synthesis of trans-cyclopropyl compounds from aldehydes. Application to the synthesis of GPR40 receptor agonists. Chem Commun (Camb) 2008;40:4974–4976. doi: 10.1039/b810708d. [DOI] [PubMed] [Google Scholar]
- Feng DD, Luo Z, Roh SG, Hernandez M, Tawadros N, Keating DJ, et al. Reduction in voltage-gated K+ currents in primary cultured rat pancreatic beta-cells by linoleic acids. Endocrinology. 2006;147:674–682. doi: 10.1210/en.2005-0225. [DOI] [PubMed] [Google Scholar]
- Garrido DM, Corbett DF, Dwornik KA, Goetz AS, Littleton TR, McKeown SC, et al. Synthesis and activity of small molecule GPR40 agonists. Bioorg Med Chem Lett. 2006;16:1840–1845. doi: 10.1016/j.bmcl.2006.01.007. [DOI] [PubMed] [Google Scholar]
- Hara T, Hirasawa A, Sun Q, Koshimizu TA, Itsubo C, Sadakane K, et al. Flow cytometry-based binding assay for GPR40 (FFAR1; free fatty acid receptor 1) Mol Pharmacol. 2009a;75:85–91. doi: 10.1124/mol.108.052225. [DOI] [PubMed] [Google Scholar]
- Hara T, Hirasawa A, Sun Q, Sadakane K, Itsubo C, Iga T, et al. Novel selective ligands for free fatty acid receptors GPR120 and GPR40. Naunyn Schmiedebergs Arch Pharmacol. 2009b;380:247–255. doi: 10.1007/s00210-009-0425-9. [DOI] [PubMed] [Google Scholar]
- Hirasawa A, Tsumaya K, Awaji T, Katsuma S, Adachi T, Yamada M, et al. Free fatty acids regulate gut incretin glucagon-like peptide-1 secretion through GPR120. Nat Med. 2005;11:90–94. doi: 10.1038/nm1168. [DOI] [PubMed] [Google Scholar]
- Hirasawa A, Hara T, Katsuma S, Adachi T, Tsujimoto G. Free fatty acid receptors and drug discovery. Biol Pharm Bull. 2008;31:1847–1851. doi: 10.1248/bpb.31.1847. [DOI] [PubMed] [Google Scholar]
- Ishiguro M, Oyama Y, Hirano T. Structural models of the photointermediates in the rhodopsin photocascade, lumirhodopsin, metarhodopsin I, and metarhodopsin II. Chembiochem. 2004;5:298–310. doi: 10.1002/cbic.200300668. [DOI] [PubMed] [Google Scholar]
- Itoh Y, Kawamata Y, Harada M, Kobayashi M, Fujii R, Fukusumi S, et al. Free fatty acids regulate insulin secretion from pancreatic beta cells through GPR40. Nature. 2003;422:173–176. doi: 10.1038/nature01478. [DOI] [PubMed] [Google Scholar]
- Kobilka BK. G protein coupled receptor structure and activation. Biochim Biophys Acta. 2007;1768:794–807. doi: 10.1016/j.bbamem.2006.10.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu SY, Jiang YJ, Lv J, Wu TX, Yu QS, Zhu WL. Molecular docking and molecular dynamics simulation studies of GPR40 receptor-agonist interactions. J Mol Graph Model. 2010;28:766–774. doi: 10.1016/j.jmgm.2010.02.001. [DOI] [PubMed] [Google Scholar]
- Milligan G, Stoddart LA, Brown AJ. G protein-coupled receptors for free fatty acids. Cell Signal. 2006;18:1360–1365. doi: 10.1016/j.cellsig.2006.03.011. [DOI] [PubMed] [Google Scholar]
- Nakamichi Y, Kikuta T, Ito E, Ohara-Imaizumi M, Nishiwaki C, Ishida H, et al. PPARγ overexpression suppresses glucose-induced proinsulin biosynthesis and insulin release synergistically with pioglitazone in MIN6 cells. Biochem Biophys Res Commun. 2003;306:832–836. doi: 10.1016/s0006-291x(03)01045-3. [DOI] [PubMed] [Google Scholar]
- Poitout V. The ins and outs of fatty acids on the pancreatic beta cell. Trends Endocrinol Metab. 2003;14:201–203. doi: 10.1016/s1043-2760(03)00086-9. [DOI] [PubMed] [Google Scholar]
- Radestock S, Weil T, Renner S. Homology model-based virtual screening for GPCR ligands using docking and target-biased scoring. J Chem Inf Model. 2008;48:1104–1117. doi: 10.1021/ci8000265. [DOI] [PubMed] [Google Scholar]
- Rosenbaum DM, Rasmussen SG, Kobilka BK. The structure and function of G-protein-coupled receptors. Nature. 2009;459:356–363. doi: 10.1038/nature08144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sasaki S, Kitamura S, Negoro N, Suzuki M, Tsujihata Y, Suzuki N, et al. Design, synthesis, and biological activity of potent and orally available G protein-coupled receptor 40 agonists. J Med Chem. 2011;54:1365–1378. doi: 10.1021/jm101405t. [DOI] [PubMed] [Google Scholar]
- Shim JY, Welsh WJ, Howlett AC. Homology model of the CB1 cannabinoid receptor: sites critical for nonclassical cannabinoid agonist interaction. Biopolymers. 2003;71:169–189. doi: 10.1002/bip.10424. [DOI] [PubMed] [Google Scholar]
- Steneberg P, Rubins N, Bartoov-Shifman R, Walker MD, Edlund H. The FFA receptor GPR40 links hyperinsulinemia, hepatic steatosis, and impaired glucose homeostasis in mouse. Cell Metab. 2005;1:245–258. doi: 10.1016/j.cmet.2005.03.007. [DOI] [PubMed] [Google Scholar]
- Stoddart LA, Smith NJ, Milligan G. International Union of Pharmacology. LXXI. Free fatty acid receptors FFA1, -2, and -3: pharmacology and pathophysiological functions. Pharmacol Rev. 2008;60:405–417. doi: 10.1124/pr.108.00802. [DOI] [PubMed] [Google Scholar]
- Sum CS, Tikhonova IG, Neumann S, Engel S, Raaka BM, Costanzi S, et al. Identification of residues important for agonist recognition and activation in GPR40. J Biol Chem. 2007;282:29248–292455. doi: 10.1074/jbc.M705077200. [DOI] [PubMed] [Google Scholar]
- Sun Q, Hirasawa A, Hara T, Kimura I, Adachi T, Awaji T, et al. Structure-activity relationships of GPR120 agonists based on a docking simulation. Mol Pharmacol. 2010;78:804–810. doi: 10.1124/mol.110.066324. [DOI] [PubMed] [Google Scholar]
- Suzuki T, Igari S, Hirasawa A, Hata M, Ishiguro M, Fujieda H, et al. Identification of G protein-coupled receptor 120-selective agonists derived from PPARgamma agonists. J Med Chem. 2008;51:7640–7644. doi: 10.1021/jm800970b. [DOI] [PubMed] [Google Scholar]
- Thomsen R, Christensen MH. MolDock: a new technique for high-accuracy molecular docking. J Med Chem. 2006;49:3315–3321. doi: 10.1021/jm051197e. [DOI] [PubMed] [Google Scholar]
- Tikhonova IG, Sum CS, Neumann S, Thomas CJ, Raaka BM, Costanzi S, et al. Bidirectional, iterative approach to the structural delineation of the functional ‘chemoprint’ in GPR40 for agonist recognition. J Med Chem. 2007;50:2981–2989. doi: 10.1021/jm0614782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsujihata Y, Ito R, Suzuki M, Harada A, Negoro N, Yasuma T, et al. TAK-875, an orally available G protein-coupled receptor 40/free fatty acid receptor 1 agonist, enhances glucose-dependent insulin secretion and improves both postprandial and fasting hyperglycemia in type 2 diabetic rats. J Pharmacol Exp Ther. 2011;339:228–237. doi: 10.1124/jpet.111.183772. [DOI] [PubMed] [Google Scholar]
- Xhaard H, Rantanen VV, Nyrönen T, Johnson MS. Molecular evolution of adrenoceptors and dopamine receptors: implications for the binding of catecholamines. J Med Chem. 2006;49:1706–1719. doi: 10.1021/jm0511031. [DOI] [PubMed] [Google Scholar]