

Abstract
Background and Purpose
The discovery of DP2 as a second receptor for PGD2 has prompted the search for antagonists as potential novel therapies based on the associations between PGD2 and disease. Here we describe the biochemical and pharmacological properties of 4-(acetylamino)-3-[(4-chlorophenyl)thio]-2-methyl-1H-indole-1-acetic acid (AZD1981), a novel DP2 receptor antagonist.
Experimental Approach
Binding to DP2, functional receptor pharmacology and selectivity were studied in both human and animal systems.
Key Results
AZD1981 displaced radio-labelled PGD2 from human recombinant DP2 with high potency (pIC50 = 8.4). Binding was reversible, non-competitive and highly selective against a panel of more than 340 other enzymes and receptors, including DP1 (>1000-fold selective). AZD1981 inhibited DP2-mediated shape change and CD11b up-regulation in human eosinophils, shape change in basophils and chemotaxis of human eosinophils and Th2 cells with similar potency. AZD1981 exhibited good cross-species binding activity against mouse, rat, guinea pig, rabbit and dog DP2. Evaluation in mouse, rat or rabbit cell systems was not possible as they did not respond to DP2 agonists. Agonist responses were seen in guinea pig and dog, and AZD1981 blocked DP2-mediated eosinophil shape change. Such responses were more robust in the guinea pig, where AZD1981 also blocked DP2-dependent eosinophil emigration from bone marrow.
Conclusions and Implications
AZD1981 is a DP2 antagonist that blocks functional responses in eosinophils, Th2 cells and basophils. It exhibited similar potency irrespective of the cell type, DP2 agonist or species used. This selective orally active agent is currently under clinical evaluation as a potential therapeutic agent in respiratory diseases including asthma.
Keywords: AZD1981, DP2 antagonist, asthma
Introduction
PGD2 is a major product of the COX pathway and has long been implicated in diseases such as asthma and allergic rhinitis. High levels are seen in the bronchoalveolar lavage fluid of asthmatic patients, both constitutively and following acute antigen challenge (Murray et al., 1986; Wenzel et al., 1989; Liu et al., 1990; Crea et al., 1992; Nowak et al., 1993). Elevated levels of PGD2 have also been measured in allergen-challenged rhinitis patients (Horak et al., 1998). The major source of PGD2 in allergic disease is thought to be the mast cell that releases the prostanoid in response to allergen activation of high-affinity IgE receptors (Anhut et al., 1978; Lewis et al., 1982). More recently, it has been reported that patients with severe asthma have higher sputum PGD2 levels relative to other steroid-treated asthmatic patients (Balzar et al., 2011). Interestingly, accumulation of a particular subtype of PGD2-producing mast cells in the airway submucosa and epithelium is found in such patients (Balzar et al., 2011). Activation of these cells may therefore contribute to increases in local PGD2 levels in severe asthmatic patients.
Two distinct receptors are activated by PGD2: DP1 and DP2. DP1 was the first PGD2 receptor to be identified and has been proposed as a target for therapy of allergic disease and asthma (Matsuoka et al., 2000). Clinical trials of selective DP1 antagonists (laropiprant and S-5751) have so far failed to show any benefit in asthma or rhinitis (Philip et al., 2009; Arimura, 2010). Suboptimal properties may be responsible for the poor efficacy of S-5751 as a follow-up compound (S-555739) is still in clinical development. However, this cannot be said for laropiprant, which shows a clear benefit in niacin-mediated flushing, a response dependent on systemic PGD2 (Sanyal et al., 2010) and indicates that laropiprant achieved adequate systemic exposure to fully inhibit the receptor. These findings suggest that PGD2 activation of the DP1 receptor is not involved in the pathogenesis of asthma or rhinitis. Indeed, evidence has been presented indicating that DP1 rather than being a pro-inflammatory receptor may mediate a number of anti-inflammatory actions of PGD2 (Angeli et al., 2004; Spik et al., 2005). However, the properties of the second high-affinity receptor for PGD2, DP2 (chemoattractant receptor-homologous molecule expressed on Th2 cells, also called DP2 or GPR44) (Hirai et al., 2001) suggest that it may be responsible for pro-inflammatory activities of PGD2.
DP2 is a class A GPCR that, in humans, is expressed on the surface of eosinophils, basophils and a subset of Th2 lymphocytes (Nagata et al., 1999a; 1999b; Hirai et al., 2001). Activation of DP2 on these cells promotes shape change, increased CD11b expression (a cell surface protein that facilitates cell adhesion to the vascular cell wall and movement of cells from the circulation to the site of inflammation) and chemotaxis (Monneret et al., 2001; Gyles et al., 2006). DP2 promotes additional responses besides chemotaxis including cytokine production by Th2 lymphocytes (Xue et al., 2005; 2009a; Pettipher and Hansel, 2008), prevention of Th2 cell apoptosis (Xue et al., 2009b) and priming/degranulation of eosinophils (Gervais et al., 2001; Schuligoi et al., 2010).
PGD2 in the lungs of asthmatic patients acting through DP2 may, therefore, play a central role in the pathogenic inflammation that typifies asthma by promoting the accumulation and activation of inflammatory cells, including Th2 lymphocytes, eosinophils and basophils (Pettipher and Hansel, 2008; Schuligoi et al., 2010). Some support for this hypothesis comes from preclinical models of airway inflammation that show blockade of DP2 activation significantly reduces experimental allergic airway inflammation (Ulven et al., 2006; Uller et al., 2007; Lukacs et al., 2008; Stebbins et al., 2010). However, there are contradictory reports in DP2 knock-out mice with Chevalier et al. (2005), concluding that DP2 plays a restrictive role in IL5 production and eosinophil recruitment; whereas a second group using independently derived knock-out mice (Satoh et al., 2006), suggesting that DP2 plays an essential role in chronic allergic inflammation. Several DP2 antagonists have progressed into man (Norman, 2010; Ulven and Kostenis, 2010; Pettipher and Whittaker, 2012) and preliminary reports describe positive effects in allergen induced eosinophil numbers in the lung (Singh et al., 2012) and in asthma, improvements in forced expiratory volume in 1 s (FEV1), quality of life and nighttime symptoms (Barnes et al., 2012). Blockade of DP2 has therefore emerged as an interesting oral non-steroidal therapeutic approach to the treatment of asthma (Schuligoi et al., 2010).
Shortly after PGD2 was found to be a natural ligand for DP2, several groups, including ourselves, independently made the observation that indomethacin had partial agonist activity at this receptor (Hirai et al., 2002; Stubbs et al., 2002). Using indomethacin as a chemical starting point, we embarked on a programme to discover novel selective antagonists. Here we report the preclinical in vitro biochemical and pharmacological properties of AZD1981, a novel DP2 receptor antagonist currently under clinical evaluation as a potential therapeutic agent in respiratory diseases including asthma.
Methods
AZD1981
AZD1981, 4-(acetylamino)-3-[(4-chlorophenyl)thio]-2-methyl-1H-indole-1-acetic acid, was synthesized by the Department of Medicinal Chemistry of AstraZeneca R&D Charnwood, Loughborough, UK (Bonnert and Rasul, 2004; Luker et al., 2011). Determinations of the physical properties of the compound were made by the Department of Physical Chemistry of AstraZeneca R&D Charnwood, Loughborough, UK. The logD7.4 (distribution coefficient between 1-octanol and aqueous buffer, logDO/W, at pH 7.4) was measured using a method based on the traditional shake flask technique, but with the modification of measuring compounds in mixtures of up to five at a time using HPLC with quantitative MS to measure the relative octanol and aqueous concentrations. Plasma protein binding was determined using equilibrium dialysis of the compound between plasma and buffer at 37°C. The concentrations of compound in the plasma and buffer were then determined using HPLC with UV quantification and MS identification. Solubility was determined by generation of a saturated solution of the compound, followed by assaying the solution using HPLC with UV quantification and MS identification. A Sirius GLpKa instrument with dip probe absorption spectroscopy (DPAS) attachment was used to measure the acid dissociation constant (pKa).
DP2 binding studies
A scintillation proximity assay (SPA) following 3H]PGD2 binding to membranes of HEK cells expressing recombinant DP2 was used. The potency of AZD1981 as an antagonist was determined by quantifying its ability to displace specific radio-ligand binding (Royer et al., 2008). Briefly, membranes from HEK293 expressing recombinant human DP2 were pre-bound to Wheat Germ Agglutinin-coated PVT-SPA beads (Amersham, Little Chalfont, UK) for 18 h at 4°C. Assays were started by the addition of 25 μL of membrane-coated beads (10 mg mL−1 of beads) to an assay buffer (50 mm HEPES pH 7·4 containing 5 mm MgCl2) containing 2·5 nM [3H]PGD2 in the absence or the presence of increasing concentrations of the tested compounds (50 μL final volume). Non-specific binding was determined in the same conditions but in the presence of 10 μM DK-PGD2. Plates were incubated for 2 h at room temperature, and bead-associated radioactivity was measured using a Wallac Microbeta counter (Perkin Elmer, Beaconsfield, UK). The concentration of the compounds causing 50% inhibition of binding of [3H]PGD2 to the receptor was calculated (IC50). Ki values have not been derived from IC50, as there is no evidence of a simple competitive interaction with PGD2 (see below).
The same methodology was used for recombinant human, murine, rat, guinea pig, dog and rabbit DP2. Reversibility of binding to the human receptor was assessed by recovery of [3H]PGD2 binding after removal of AZD1981 by washing of the membrane-coated SPA beads. HEK-membrane-coated beads were incubated in the presence of AZD1981 for 2 h at room temperature to bind the compound to DP2. To remove the bound AZD1981, beads were centrifuged (1 min at 1300× g), and the pellet resuspended in 1 mL of assay buffer. This was repeated four times. Aliquots (30 μL) were transferred to 96-well plates, and [3H]PGD2 binding was evaluated as above. Parallel samples containing (i) 10 μM DK-PGD2 during the 2 h incubation and in the wash buffer; (ii) AZD1981 at 2 μM in the wash buffer; and (iii) vehicle were processed alongside to determine non-specific binding and the ‘no wash’ condition whilst controlling for loss of beads during the washing process. The time from first wash to end of first reading was approximately 13 min.
Receptor and enzyme selectivity studies
The drug/molecular target nomenclature used below conforms to the British Journal of Pharmacology's Guide to Receptors and Channels (Alexander et al., 2011).
DP1 receptor binding
The potency of AZD1981 as an antagonist at the human DP1 receptor was determined by quantifying its ability to displace specific binding of [3H]PGD2 from membranes of HEK cells expressing recombinant human DP1 receptors, as described above for DP2.
General selectivity
The general selectivity of AZD1981 was also assessed against enzymes or receptors at a single test concentration of 10 μM by Ricerca Biosciences (formally MDS Pharma, http://www.ricerca.com/discovery-pharmacology.asp) and CEREP (http://www.cerep.fr/Cerep/Users/index.asp) according to their standard protocols. IC50 determinations were made where greater than 50% inhibition was seen at the 10 μM concentration.
Aldose and aldehyde reductase
Inhibition of human recombinant aldose reductase and aldehyde reductase was determined by quantifying its effects on enzyme-catalysed conversion of dl-glyceraldehyde to glycerol and d-glucuronic acid to l-gulonic acid respectively. Human recombinant enzymes were obtained from Dr K Bohren, Department of Pediatrics, Baylor College of Medicine, Texas Children's Hospital, Houston, Texas, USA. Assays were performed in UV clear 96-well plates in a final volume of 200 μL. Each well contained AZD1981, recombinant human enzyme (10 μg·mL−1 aldose reductase diluted in 5 mM sodium phosphate buffer pH 7.5 containing 5 mM 2-mercaptoethanol or 2.5 μg·mL−1 aldehyde reductase diluted in 5 mM sodium phosphate buffer pH 7.5), substrate (0.2 mM dl-glyceraldehyde for aldose reductase or 2 mM d-glucuronic acid for aldehyde reductase) and NADPH (0.2 mM) in 0.1 M sodium phosphate buffer pH 7.0. The rate of reaction was measured by monitoring the decrease in absorbance at 340 nm.
Functional activity studies
CD11b up-regulation on eosinophils in a mixed leukocyte preparation
Human leukocytes were prepared from blood taken by venipuncture from healthy volunteers using Polymorphprep (Axis Shield, Oslo, Norway). Plasma was retained and centrifuged at 725× g for 10 min at room temperature to remove platelets and any contaminating red blood cells for use during the cell fixation step later in the procedure. Granulocytes were washed in HBSS containing 20 mM HEPES pH 7.4 (HBSS/HEPES), re-suspended at 3.5 ×106 cells·mL−1 in HBSS/HEPES and rested at room temperature for 30 min before use.
Assays contained AZD1981 or vehicle control [2 μL at 50 times the required final concentration in HBSS/HEPES containing 5% dimethyl sulphoxide (DMSO)], 78 μL of cell suspension, 10 μL of antibody mix or isotype control and 10 μL of agonist [13,14-dihydro-15-keto-PGD2 (DK-PGD2, Cayman Chemical Co., Ann Arbor, MI, USA) in HBSS/HEPES containing 0.1% DMSO]. The antibody mix was prepared by diluting FITC-labelled murine anti-human CD11b antibody (MHCD11b01 4, CALTAG Medsystems, Burlingame, CA, USA) and PE-labelled murine anti-human CD16 antibody (MHCD1604 4 CALTAG Medsystems) 1 in 5 in PBS containing 2 mM sodium azide and 0.5% w/v BSA. A solution of the respective isotype control immunoglobulins (MG101 and MG104 CALTAG Medsystems) was prepared by dilution in the same buffer. AZD1981 was pre-incubated with cells for 15 min before addition of the antibody mix and agonist. After incubation for 15 min at 37°C, cells were fixed by addition of 10 μL of ice-cold autologous plasma followed by 100 μL of ice-cold 0.05% formaldehyde in HBSS/HEPES and left in the dark for 15 min at room temperature. Fixed cells were transferred to tubes suitable for use with the flow cytometer, red blood cell lysis solution (150 mM NH4Cl, 10 mM KHCO3 1.27 mM EDTA pH 7.0, 800 μL) added, and the cells were incubated at room temperature for 10 min. Cells were finally pelleted by centrifugation (530× g for 5 min room temperature) and re-suspended in 0.3 mL of PBS containing 0.1% v/v CellFIX™ (Beckton Dickinson, Cowley, UK). CD11b expression was determined by flow cytometry. The eosinophil population within the granulocytes was gated on the basis of forward scatter/side scatter profile and low CD16 expression. CD11b expression was measured as the median peak fluorescence (MdX value) through FL-1.
Human eosinophil shape change assay
Human blood was taken by venipuncture from healthy volunteers into lithium heparin tubes and pre-treated at room temperature for 60 min with AZD1981 or vehicle by adding AZD1981 or vehicle directly to the tube from 100-fold concentrated stocks. Each well in a 96-well deep-well polypropylene plate contained 15R-methyl PGD2 (10 μL at 10 times the required final concentration) or vehicle (assay buffer containing 1.12% DMSO) and 90 μL of blood pre-treated with compound or vehicle. Plates were incubated for 15 min at 37°C, after which cells were fixed by addition of 100 μL of Optilyse B (Beckman Coulter, UK). After 10 min at room temperature, 1 mL of de-ionized water was added to each well, the samples allowed to stand at room temperature for 30 min and centrifuged for 5 min at 500× g at 15°C. Cells were finally re-suspended in 500 μL PBS containing 1% (v : v) Cyto-Chex (Alpha Labs, Eastleigh, UK). Shape change was analysed using a Coulter FC500 flow cytometer, and the eosinophil population within the granulocytes was gated on the basis of Forward Scatter/Side Scatter profile and high autofluorescence.
Human basophil shape change assay
Peripheral venous blood was drawn from healthy volunteers of either sex aged 20 to 40 years, after written informed consent as approved by the Institutional Review Board of the Medical University of Graz. Samples of citrated whole blood were labelled with FITC-conjugated HLA-DR and phycoerythrine (PE)-conjugated CD123 monoclonal antibodies (1:50 each) and pre-incubated with vehicle or AZD 1981 for 10 min at 37°C. Ninety-microlitre aliquots of whole blood were stimulated with 10 μL PGD2 for 4 min at 37°C. The samples were then transferred to ice and fixed with 250 μL of fixative solution followed by NH4Cl-induced lysis of red blood cells. Cells were then washed and re-suspended in 250 μL of fixative solution. Samples were immediately analysed on a FACSCalibur flow cytometer (Becton Dickinson, Mountain View, CA, USA). Basophils were gated as CD123-positive and HLA-DR-negative cells. Responses were quantified as percent of cells that which moved into a higher forward scatter gate initially defined to contain <20% of basophils in a non-stimulated sample.
Guinea pig and dog leukocyte shape change assays
Leukocyte shape change assays using guinea pig blood were performed as described in Royer et al. (2008). For shape change assays on dog cells, dog blood (9 mL) was taken from the jugular vein into Li-Heparin as an anticoagulant. AZD1981 (10 μL at 10 times the final required concentration) or vehicle, Dulbecco's PBS pH 7.4 containing 10 mM HEPES, 10 mM glucose, 0.1% BSA (assay buffer) and 1% DMSO, was mixed with agonist (10 μL at 10 times final concentration required) or vehicle (assay buffer) and 80 μL blood. After incubation with shaking at 37°C for 15 min, tubes were transferred to an ice bath and cells fixed by the addition of 200 μL fixative (10 times CellFIX™ diluted 1:10 in distilled water and then 1:4 in Isoton), and erythrocytes were lysed by the addition of 1 mL of ammonium chloride lysis solution (150 mM NH4Cl, 10 mM KHCO3 and 1.27 mM EDTA pH 7.0) and left at room temperature for at least 20 min. The tubes were centrifuged at 375× g for 5 min, the supernatant discarded and cells re-suspended in 200 μL of cell fixative (a 1 in 25 dilution of CellFIXTM in distilled water, then a 1 in 4 dilution in Isoton II). Within 1 h, shape change was determined using a Becton Dickinson FACScan. Eosinophils were identified as described for guinea pig blood (Royer et al., 2008).
Chemotaxis assays
Eosinophil chemotaxis studies were performed using purified human eosinophils as previously described (Royer et al., 2008).
DP2+ T-cell lines were expanded from the peripheral blood of healthy volunteers. The initial step involved isolation of DP2+ cells from nylon wool purified human peripheral blood T cells using anti-DP2-specific antibodies coupled to magnetic beads (Anti-DP2 Microbead kit 130-091-274, Miltenyi Biotec, Surrey, UK). Purified DP2+ T cells were expanded in culture using a non-specific stimulus [anti-CD3/anti-CD28-coated microbeads, Dynabeads, Invitrogen, Paisley, Scotland in RPMI 1640 medium containing 10% human AB serum Penicillin (100 U·mL−1), streptomycin (100 μg·mL−1), 2 mM L-glutamine and 20 U·mL−1 human recombinant IL-2]. Expanded cells had a type 2 phenotype as indicated by high IL-4 production and low IFNγ production after stimulation with anti-CD3/anti-CD28 or PHA/PMA. DP2 expression was monitored by flow cytometry using PE-labelled anti-DP2 antibodies (clone BM16, Beckman Coulter) and was stable for at least five rounds of expansion (data not shown). Chemotaxis assays, cells were performed at least 5 days after the removal of the anti-CD3/anti-CD28 beads. T cells in RPMI 1640 containing 20 mM HEPES pH 7.4 and 5% human AB serum were applied to the upper surface of 96-well Chemo Tx™ microplates (101-5), 5 μm pore size, 3.2 mm diameter well (Neuroprobe). The lower wells contained DK-PGD2. An equal concentration of AZD1981 was also present in the upper and lower solutions. After incubation for 1 h at 37°C/5% CO2, migrated cells were transferred to a fresh 96-well plate and quantified by cell-associated LDH using a commercially available kit (Cytotox 96, Promega, Southhampton, UK). A standard curve relating cell number to absorbance was constructed on a separate 96-well plate.
In situ perfusion of the guinea pig hind limb
Eosinophil mobilization in isolated perfused guinea pig hind limb was measured as described previously (Royer et al., 2008).
Data analysis
Agonist and antagonist concentration–effect curves were fitted to a 4-parameter logistic equation to estimate [A]50 and [IC]50 values, both of which were assumed to be log-normally distributed and quoted as p[A]50 and pIC50 values. In experiments investigating effects on CD11b up-regulation on human eosinophils, agonist concentration effect (E/[A]) data was fitted to the following model of non-competitive antagonism:
| (1) |
in which Em is the maximum possible effect; n determines the steepness of the occupancy–effect relationship; KB is the dissociation constant of the non-competitive antagonist; τ is the efficacy of the agonist. The fitting procedure provides an estimate of KB.
Equation 1 describes non-competitive antagonism (see Kenakin, 2009) in terms of the operational model of agonism (Black and Leff, 1983). It assumes that antagonist binding precludes binding of the agonist.
In the shape changes assays, E/[A] curve data were fitted to the following equation to estimate the affinity (pA2) of AZD1981:
| (2) |
in which [B] is the concentration of AZD1981 and r is the concentration ratio calculated from the [A]50 obtained in the presence and absence of AZD1981. In circumstances where there is a substantial receptor reserve such that there is measurable dextral displacement of the E/[A] curves, the pA2 value estimated from equation (2) is a reliable estimate of pKB for non-competitive antagonists (Kenakin, 2009).
Curve fitting procedures were performed using Excel, Graph Pad Prism or Origin graphics packages. All data are expressed as mean ± SEM.
Results
AZD1981
The structure of AZD1981 is shown in Figure 1. The compound is an indole acetic acid with high aqueous solubility, relatively high plasma protein binding and moderately low logD (Table 1).
Figure 1.
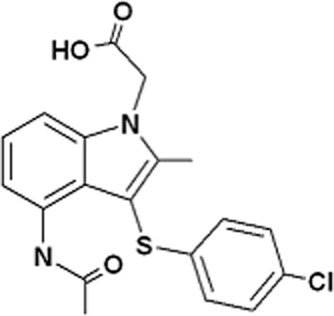
Structure ofAZD1981.
Table 1.
Physical properties of AZD1981
| Property | Value |
|---|---|
| Molecular weight (free acid), Da | 388.9 |
| Log D7.4 | −0.22 |
| Plasma protein binding (% bound), human/rat/mouse/dog/rabbit/guinea pig | 97.2/98.3/97.5/97.4/98.4/96.5 |
| Solubility (in 10 mM sodium phosphate pH7.4 at 20°C) | 1.87 mM |
| pKa | 2.64 |
AZD1981 blocks PGD2 binding to human DP2
The potency of AZD1981 at human DP2 was measured with a radioligand binding assay using membranes from HEK 293 cells expressing recombinant receptor. AZD1981 produced a concentration-dependent displacement of the [3H]PGD2-specific binding with a mean pIC50 of 8.4 ± 0.1 (n = 25, geometric mean IC50 of 4 nM, Figure 2A). The displacement curve had a Hill slope of unity with no evidence of more than one binding site. Binding to human DP2 was fully reversible as assessed by the recovery of [3H]PGD2 binding within 13 min (the shortest possible time period in which a measurement could be made) after removal of the compound (Figure 2B). The [3H]PGD2 concentration used in the binding assay was 2.5 nM. This was two- to threefold below its pKd, which we measured to be 8.3 ± 0.1 (n = 4) (Carrillo et al., 2005).
Figure 2.
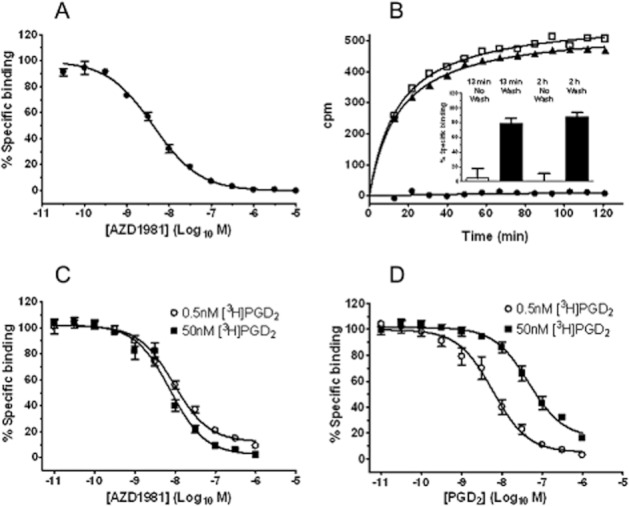
AZD1981 is a potent antagonist at DP2. (A) Displacement of specific binding of [3H]PGD2 (2.5 nM) to HEK cell membranes expressing recombinant human DP2 by AZD1981. Values are mean ± SEM (n = 25). (B) Reversibility of inhibition by AZD1981 of specific binding of [3H]PGD2 to HEK cells transfected with human DP2. The inset shows the percentage recovery of [3H]PGD2 binding 13 min and 2 h after removal of AZD1981 compared with control samples where membranes were washed in buffer containing AZD1981. Values are displayed as mean ± SEM for duplicate values from four separate experiments. The main panel shows results from one of the replicates contributing to the data in the inset and depicts [3H]PGD2 association to washed beads in comparison with control DP2 membrane-coated beads, which had not been treated AZD1981. The closed circles show [3H]PGD2 association to beads washed with AZD1981 and define non-specific binding. (C) Displacement of specific binding of [3H]PGD2 (0.5 and 50 nM) to HEK cell membranes expressing recombinant human DP2 by AZD1981. (D) Displacement of specific binding of [3H]PGD2 (0.5 and 50 nM) to HEK cell membranes expressing recombinant human DP2 by unlabelled PGD2. For (C) and (D), values are displayed as mean ± SEM for duplicate values from six separate experiments.
In a separate set of experiments, we investigated the potency of AZD1981 at different concentrations of [3H]PGD2. The experimental binding windows at 0.5 and 50 nM [3H]PGD2 were sevenfold and threefold respectively. At a radioligand concentration of 0.5 nM the pIC50 value for this displacement was 8.2 ± 0.1 (n = 12 from six separate experiments) (Figure 2C). A similar pIC50 value (8. 0 ± 0.1, n = 12 from 6 separate experiments) was obtained at a 100-fold higher radioligand concentration of 50 nM (Figure 2C), indicating a non-competitive interaction. This behaviour was in contrast to that seen with unlabelled PGD2 which was investigated in parallel. As expected, unlabelled PGD2 also produced a concentration-dependent displacement of [3H]PGD2 binding to human DP2. At the low radioligand concentration of 0.5 nM the pIC50 value for this displacement was 8.3 ± 0.2 (n = 12 from six separate experiments) (Figure 2D), but at the higher radioligand concentration of 50 nM, the same preparation of unlabelled PGD2 generated a pIC50 value of 7.3 ± 0.1 (n = 12 from six separate experiments). The difference in pIC50 values for unlabelled PGD2 were exactly in line with the prediction by the Cheng–Prusoff relationship (Cheng and Prusoff, 1973, using the KD valued for PGD2 quoted in Table 3), suggesting that unlabelled PGD2 displaced the radioligand competitively.
Table 3.
In vitro profile of AZD1981 across species
| Species | [3H]PGD2 binding | AZD1981 |
|---|---|---|
| pKd | pIC50 | |
| Human | 8.3 ± 0.1 (n = 4a | 8.4 ± 0.1 (n = 25) |
| Rat | 8.3 ± 0.0 (n = 2a | 8.5 ± 0.1 (n = 4) |
| Mouse | 8.1 ± 0.2 (n = 2a | 8.1 ± 0.2 (n = 4) |
| Dog | 8.2 ± 0.1 (n = 3a | 8.1 ± 0.2 (n = 4) |
| Guinea pig | 8.3 ± 0.4 (n = 2a | 7.8 ± 0.2 (n = 6) |
| Rabbit | 8.0 ± 0.1 (n = 3) | 8.7 ± 0.1 (n = 8) |
Data from Carrillo et al. (2005).
Selectivity of AZD1981
Activity against the other high-affinity receptor for PGD2 was assessed in an identical binding assay where membranes from HEK cells expressing recombinant human DP1 were used in place of DP2. AZD1981 had no significant affinity towards recombinant human DP1 receptors with only a mean 27% (range 14–50%; n = 4) displacement of [3H]PGD2-specific binding observed at the highest concentration tested (10 μM) (Figure 3A).
Figure 3.
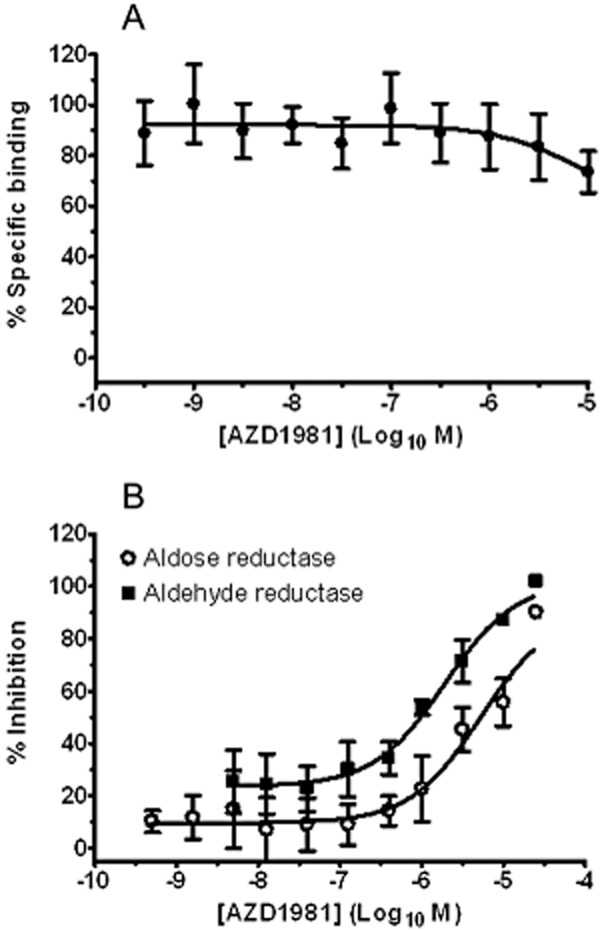
AZD1981 is a selective DP2 antagonist: (A) Effect of AZD1981 on specific binding of [3H]PGD2 to HEK cells transfected with human DP1 receptors. Values are displayed as mean ± SEM (n = 4). (B) Effect of AZD1981 on human recombinant aldose reductase and aldehyde reductase enzyme activities. Values are displayed as mean ± SEM (n = 4).
General selectivity was assessed against a panel of 338 in vitro radioligand binding and enzyme assays, covering a diverse range of receptors, ion channels, transporters and enzymes, initially at a single concentration of 10 μM. This included agents known to induce eosinophil and basophil chemotaxis, CD11b up-regulation and shape change and T-cell chemotaxis. Concentration–effect curves were generated for hits defined as >50% inhibition. Significant activity was detected at two targets, rat aldose reductase and rat steroid 5α-reductase (Table 2), while no activity was seen against COX-1, COX-2 or the thromboxane A2 (TP) receptor. Compared with the binding potency for DP2, AZD1981 showed 10-fold selectivity over rat aldose reductase and 1700-fold selectivity over rat steroid 5α-reductase. Further characterization of the activity of AZD1981 as inhibitor of human recombinant aldose reductase and aldehyde reductase enzyme activities revealed pIC50 values of 5.2 ± 0.1 (n = 4) and 5.8 ± 0.1 (n = 3) respectively (Figure 3B). The corresponding selectivity of AZD1981 for DP2 was, therefore, 1600-fold for human aldose reductase and 400-fold for human aldehyde reductase.
Table 2.
Hits from general selectivity testing
| Target | % inhibition at 10 μM | pIC50 (μM) |
|---|---|---|
| Aldose reductase (rat) | 98% | 7.4 |
| Steroid 5α-reductase (rat) | 88% | 5.2 |
AZD1981 blocks DP2-mediated CD11b up-regulation in human eosinophils
Increasing concentrations of DK-PGD2 induced an increase in expression of CD11b in eosinophils isolated from human peripheral blood with a p[A]50 of 7.8 ± 0.1 (n = 3, Figure 4A). AZD1981 caused rightward shifts of the control DK-PGD2 concentration–effect curve and a depression of the maximum responses at higher concentrations (Figure 4A). Analysis of these data using a model of non-competitive antagonism yielded an affinity (pKB) value for AZD1981 of 8.55 ± 0.03, (n = 3). This value was consistent with the potency determined for displacement of 3H-PGD2 binding to human recombinant DP2. Analysis of one of the replicates contributing to this data is shown in Figure 4B.
Figure 4.
AZD1981 blocks DP2-mediated up-regulation of CD11b expression in human eosinophils in vitro in the absence of plasma. (A) Effect of increasing concentrations of AZD1981 on DK-PGD2-stimulated CD11b expression on partially purified human eosinophils in vitro. Values are mean ± SEM (n = 3). (B) Data from one of the replicates in (A) fitted to the equation for non-competitive antagonism (equation 1). The estimated pKB was 8.5.
AZD1981 blocks DP2-mediated shape change in human eosinophils and basophils in blood
The activity of AZD1981 was also investigated in whole blood. In these experiments, DP2-mediated shape change was chosen as the readout (Heinemann et al., 2003) as this response is more suited for use as a clinical biomarker than the CD11b assay relying on isolated leukocytes. In eosinophils, a single concentration of 1 μM, AZD1981 caused a large (20-fold) rightward parallel shift in the 15R-methyl PGD2 E/[A] curve with no evidence of a decrease in the maximal response (Figure 5A). Estimation of a pA2 from this data (equation 2) gave a value of 7.3 ± 0.02 (n = 4). In basophils, 1 μM AZD1981 caused a slightly larger (70-fold) rightward parallel shift in the PGD2E/[A] curve with no evidence of a decrease in the maximal response (Figure 5B). Estimation of a pA2 from this data (equation 2) gave a value of 7.5 ± 0.47 (n = 5). As can be seen from the larger SEM value, the response in basophils was not as robust as that seen in eosinophils.
Figure 5.
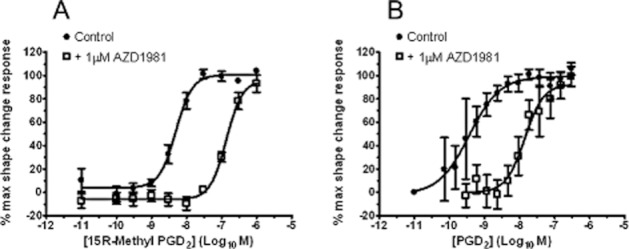
AZD1981 blocks DP2-mediated shape change in (A) human eosinophils and (B) human basophils in blood in vitro. Effect of 1 μM AZD1981 on the E/[A] curve for 15R-methyl-PGD2 is shown in panel A and on the E/[A] curve for PGD2 in panel B. Values are mean ± SEM (n = 4 for eosinophils, n = 5 for basophils).
AZD1981 blocks DP2-mediated chemotaxis of human Th2 cells and eosinophils
The ability of AZD1981 to block chemotaxis was investigated in Th2 cells and eosinophils. PGD2 induced a concentration-dependent chemotaxis of eosinophils isolated from human peripheral blood, but as typical of such systems, the E/[A] curve was bell shaped (data not shown). The effect of AZD1981 was therefore investigated using a single sub-maximal concentration of agonist (1 μM). AZD1981 produced a concentration-dependent inhibition of eosinophil migration with a pIC50 value of 7.6 ± 0.1 (n = 4) (Figure 6A). A similar bell-shaped E/[A] curve was obtained with chemotaxis of Th2 cells (data not shown) so as with human eosinophils, a single submaximal concentration of DK-PGD2 (330 nM) was used to investigate the effects of AZD1981. Using this format, the pIC50 of AZD1981 for inhibition of chemotaxis of DP2+ T-cell lines was 7.5 ± 0.1 (n = 5) (Figure 6B). This value is in close agreement with the value obtained with human eosinophil chemotaxis.
Figure 6.
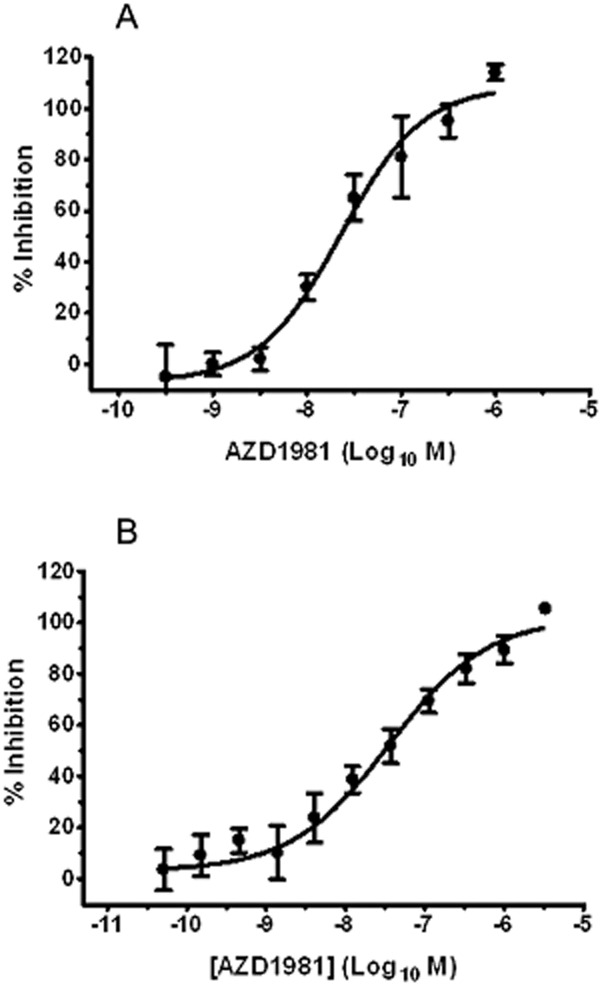
AZD1981 blocks DP2-mediated chemotaxis. (A) Human eosinophils. Increasing concentrations of AZD1981 were investigated against a single concentration of PGD2 (1 μM), generating a pIC50. Values are mean ± SEM (n = 4). (B) Human Th2 cells. Increasing concentrations of AZD1981 were investigated against a single concentration of DK-PGD2 (330 nM), generating a pIC50. Values are mean ± SEM (n = 5).
AZD1981 blocks binding to mouse, rat, rabbit and dog DP2
Saturation binding experiments showed that the dissociation constant (pKd) for [3H]PGD2 binding to mouse, rat, guinea pig, rabbit and dog recombinant DP2 was similar to the pKd for binding to human DP2 (Table 3). AZD1981 displaced [3H]PGD2 binding from all the species tested with similar pIC50 values (Table 3).
DP2-mediated functional response in non-human cells
Neither PGD2 nor DK-PGD2 induced an increase in CD11b or shape change in mouse, rat or rabbit eosinophils (data not shown). Functional responses in Th2 cells could only be evaluated in mice as these cells could not be isolated from any other preclinical species. However, no PGD2 or DK-PGD2 induced Ca2+ or chemotactic responses could be demonstrated in murine Th2 cells, even though they expressed DP2 mRNA (data not shown).
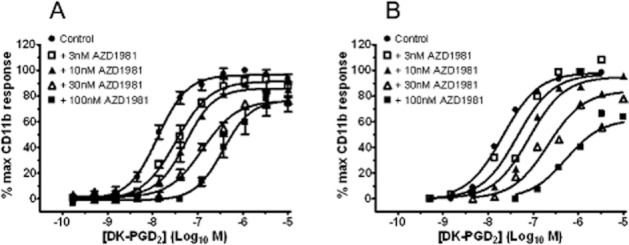
In contrast, a positive shape change response was seen in eosinophils in guinea pig whole blood and in eosinophils in dog blood using either DK-PGD2 or 15R-methyl PGD2 stimulation (Figure 7A,B). The rank order in potency and the p[A]50 values for both DK-PGD2 and 15R-methyl PGD2 were consistent with that seen with human cells (Hirai et al., 2001; Monneret et al., 2003); but, as can be seen from the large SEM values obtained in dog whole blood, the response in guinea pig blood was more robust.
Figure 7.
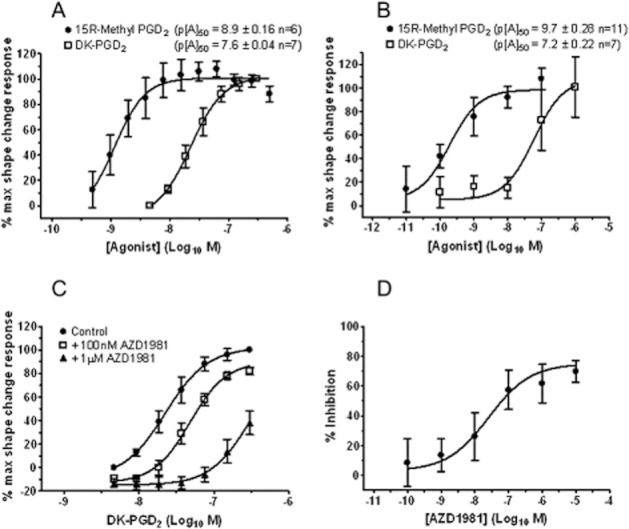
AZD1981 blocks DK-PGD2-mediated shape change in guinea pig and dog blood granulocytes. (A,B) Induction of a shape change response with the selective DP2 agonists DK-PGD2 and 15R-methyl PGD2 in whole blood taken from (A) guinea pigs and (B) dogs. (C) Effect of 100 nM and 1 μM AZD1981 on the E/[A] curve for DK-PGD2-stimulated shape change of eosinophils in guinea pig blood in vitro. (D) Effect of increasing concentrations of AZD1981 on 15R-methyl PGD2 (1 nM)-stimulated shape change of eosinophils in dog blood in vitro. Values are mean ± SEM (n = 5–11).
AZD1981 blocks DP2-mediated shape change in guinea pig and dog granulocytes and DP2-mediated ex vivo induced release of eosinophils in guinea pig hind limb
In guinea pig blood, AZD1981 induced a rightward shift of the DK-PGD2 E/[A] curve (Figure 7C). The pA2 value generated from this data using equation 2 was 6.9 ± 0.12 (n = 5).
As a result of the increased variability in the DP2 mediated response in dog blood a Schild-type analysis of AZD1981 was not practical. A pIC50 for AZD1981 was therefore generated using a single submaximal concentration of (1 nM) of 15R-methyl PGD2 (Figure 7D) yielding a value of 7.5 ± 0.4 (n = 6).
Using the previously described guinea pig hind limb model (Royer et al., 2008), 10 nM AZD1981 significantly inhibited DK-PGD2-induced eosinophil mobilization by approximately 50%, and the response was completely inhibited with 100 nM AZD1981 (Figure 8). This level of inhibition is consistent with the measured affinity of AZD1981 for displacement of [3H]PGD2 binding to guinea pig recombinant DP2 (see above).
Figure 8.
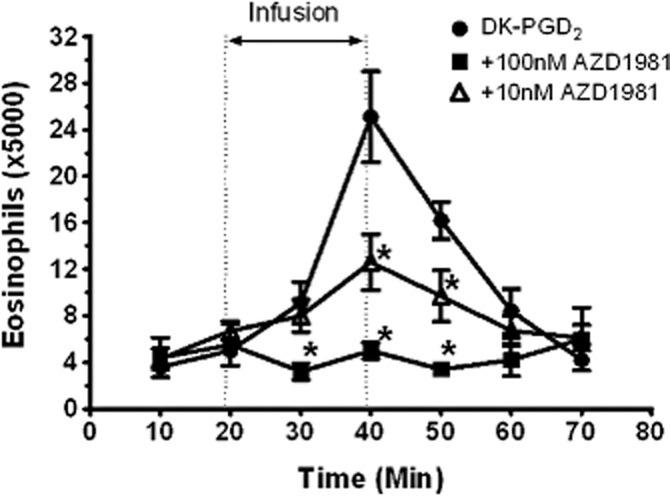
Effect of AZD1981 on the release of eosinophils from the isolated perfused guinea pig hind limb in response to 30 nM DK-PGD2. AZD1981 or vehicle was present in the perfusate throughout the experiment while DK-PGD2 was added during the 20 to 40 min period. Values are mean ± SEM (n = 5–7). *P < 0.05 versus vehicle by two-way anova for repeated measurements.
Discussion
Our pharmacological studies have demonstrated that AZD1981 is a potent, selective and reversible DP2 antagonist. However, the observation that the pIC50 for AZD1981 displacement of [3H]PGD2 was the same at 0.5 nM as at 50 nM radioligand demonstrates that it is not behaving as a simple competitive antagonist. The insurmountable antagonism that AZD1981 exhibited in the eosinophil CD11b assay (Figure 4) supports this finding. Limited data exist on the mode of action of other DP2 antagonists, but there are two publications (Mathiesen et al., 2006; Gervais et al., 2011) that highlight potential insurmountable antagonist profiles. Both have attributed their compound profiles to slow dissociation kinetics and hence equilibrium not being achieved in the time frame of their experiments. Data from our reversibility studies with AZD1981 highlight that the compound rapidly dissociates and hence issues of hemi-equilibrium are unlikely to explain the data. In addition, we were able to confirm with the SPA binding format that steady-state conditions had been achieved, indicating that the non-competitive profile of AZD1981 in this system cannot be the result of hemi-equilibrium. The simplest interpretation of our data is that AZD1981 binds to a site distinct from PGD2 that precludes agonist binding and activation. Further studies are required to confirm this hypothesis, but such a mode of action has potential advantages in pathophysiological situations where agonist concentrations are high, as the effects of the antagonist are less likely to be overcome.
Having characterized the mode of action of AZD1981 in binding studies against DP2, we next evaluated binding at DP1, the other high-affinity receptor for PGD2. DP1 has also been proposed as a target for therapy of allergic disease and asthma (Matsuoka et al., 2000), so understanding selectivity was important for interpretation of functional responses with AZD1981. AZD1981 had no significant activity at DP1 and hence has a different profile from the dual DP2/DP1 antagonist AMG853 (Banfield et al., 2010).
The starting point for identification of reversible DP2 antagonists was an observation we made that the NSAID indomethacin had partial agonist activity at DP2. This finding was subsequently published by two other independent groups (Hirai et al., 2002; Stubbs et al., 2002), which reinforced the view that this pharmacophore was a strong chemical starting point. Since indomethacin is a potent inhibitor of COX, the optimization programme leading to the discovery of AZD1981 involved removal of this activity at the same time as converting agonist properties into antagonist activity and maintaining favourable drug-like features. In agreement with our previously published DP2 antagonists derived from indomethacin (Birkinshaw et al., 2006), AZD1981 showed no inhibition of COX activity.
During the chemical programme leading to AZD1981, we identified that related structures had the potential to be inhibitors of aldose and/or aldehyde reductases. A general feature of many tight-binding aldose or aldehye reductase inhibitors is a polar group, usually a carboxylate, attached to a hydrophobic core consisting of one or more ring structures (Petrash, 2004), such as found in AZD1981 and indomethacin. Indeed, indomethacin itself has been reported to be a weak inhibitor of aldose reductase (Chaudhry et al., 1983). These two enzymes play important roles in osmoregulation and detoxification of endogenous and exogenous metabolites including alcohols and aldehydes (Petrash, 2004; Jin and Penning, 2007; Barski et al., 2008). AZD1981 showed high selectivity (400-fold) against these human enzymes although the selectivity margin against rat aldose reductase was only 10-fold. This reinforces the importance of evaluating activity against this family of enzymes using human counterparts (Chaudhry et al., 1983).
Broader selectivity testing revealed only one other significant hit (>50% activity). This was steroid 5α-reducase, but with a 1700-fold selectivity margin relative to DP2 binding, potency this was not seen as a concern. Interestingly, steroid reductases are NADPH-dependent enzymes forming part of the aldo-keto reductase superfamily (Barski et al., 2008), which may account for the weak activity. In summary, selectivity profiling of AZD1981 demonstrated it is a highly selective DP2 antagonist.
To characterize the profile of AZD1981 at the cellular level, we focused on physiologically relevant cell types that have the potential to be incorporated into clinical studies. None of our functional experiments revealed any evidence of agonist activity with this compound. Although we did not directly assess the selectivity of AZD1981 in the various functional assays our general selectivity testing revealed no significant affinity at other targets (e.g. CCR3, CCR4) known to induce eosinophil CD11b up-regulation, shape change or chemotaxis, basophil shape change and T-cell chemotaxis. Accordingly, it seems reasonable to assume that AZD1981 behaved as a selective DP2 antagonist in these assays.
In human eosinophils, we demonstrated suppression of CD11b expression and inhibition of migration towards DP2 agonists in both eosinophils and Th2 cells. CD11b expression has been shown to be important in the adhesion of DP2 cells to the vasculature, an important first step for these cells in leaving the circulation and migrating into inflamed tissue (Gyles et al., 2006). The profile of AZD1981 in the CD11b assay highlighted a depression of the maximal response. This feature coupled with the data from the binding assay led us to estimate the functional potency using a model of non-competitive antagonism (Figure 4). Importantly, the potency estimate obtained was identical to that generated in the binding assay. Equivalent analyses could not be undertaken in the eosinophil and Th2 cell chemotaxis assays as the agonist E/[A] curves were typically bell shaped. Nevertheless, pIC50 estimates in these assays were consistent with estimates obtained with both the binding and isolated eosinophil CD11b assays (taking into account the plasma protein binding of AZD1981; see Table 1).
Shape change in eosinophils and basophils was used to investigate the profile of AZD1981 in blood as it had the potential to be applied to clinical studies. Interestingly, the profile of AZD1981 did not show depression of the maximum response. This is not inconsistent with the mode of action described above as it can be explained by a higher receptor reserve related to either the assay system or the agonists employed (15R-methyl PGD2 and PGD2). These data demonstrate that AZD1981 is potent in whole blood systems and the pA2 values obtained are consistent with the values calculated from the binding potency adjusted for plasma protein binding.
In summary, in these human functional studies, which used several agonists across different cellular systems, we have demonstrated that the potency of AZD1981 is independent of the agonist, cell type or cell function. The profile described for AZD1981 suggests it will inhibit other published DP2-mediated responses in human cells including Th2 cell cytokine production (Xue et al., 2005), PGD2-mediated Th2 cell apoptosis (Xue et al., 2009b), basophil chemotaxis (Hirai et al., 2001) and eosinophil activation (Gervais et al., 2001; Schuligoi et al., 2010).
The insurmountable antagonism that AZD1981 exhibited in the eosinophil CD11b assay (Figure 4) supports the finding in binding studies that AZD18981 does not behave as a simple competitive antagonist. Although the surmountable antagonism observed in the human shape change assays (Figure 5) may appear at odds with this hypothesis, such behaviour can be explained by the use of a higher efficacy agonist (15R-methyl PGD2) in eosinophils and the presence of a higher receptor reserve in basophils. Other explanations for the pharmacological profile of AZD1981 across assays, such as the presence of different affinity states of DP2 seem less likely as our 3H]PGD2 saturation curves were monophasic and the eosinophil CD11b data revealed no evidence of complex AZD1981 binding, across a wide concentration range. Furthermore, to our knowledge, there is no literature evidence suggesting the existence of multiple receptor states of DP2 in functional assays. Thus, both binding and functional studies highlight AZD1981 is not a simple competitive antagonist, but further work will be required to determine the exact MoA of AZD1981.
Having assessed the profile of AZD1981 in human systems we went on to evaluate species cross-over. The affinity of AZD1981 for recombinant DP2 for mouse, rat, guinea pig, rabbit and dog was similar to that observed for human. However, despite clearly being able to show similar responses in the human counterparts, we were unable to show DP2-dependent responses in vitro in native cells (eosinophils and/or Th2 cells) from rats, mice or rabbits (data not shown). The reason for this is unknown but it is unlikely to be due to non-recognition of the receptor by DP2 agonists as we can clearly demonstrate binding of PGD2 and the selective agonists DK-PGD2 and 15R-methyl PGD2 to murine or rat recombinant receptor. Interestingly, there is only one paper that demonstrates an in vitro activity on rodent cells, isolated from IL-5 transgenic mice (Spik et al., 2005). Coupled with the observation that in mice DP2 is also expressed in Th1 cells (Abe et al., 1999) the relevance of rodent species in evaluating the activity of DP2 antagonists is unclear. As a result, we were therefore unable to characterize AZD1981 in murine, rat or rabbit cell systems.
In contrast, in vitro functional responses were seen with DP2 agonists in guinea pig and dog cells. The rank order of potency for the two selective DP2 agonists used (DK-PGD2 and 15R-methyl PGD2) supports that these responses are mediated through DP2. The potency of AZD1981 in these assay systems were similar to those seen in human systems. Based on the greater variability in the dog shape change assay and additional dog functional studies in vivo (Marshall et al., 2005; 2006) the guinea pig was chosen as the species for further evaluation of the role of AZD1981.
Eosinophil mobilization from guinea pig bone marrow can be elicited with DP2 agonists (Heinemann et al., 2003), and this response is sensitive to a selective DP2 antagonist (Royer et al., 2008). Here we show that AZD1981 also blocks DP2-dependent eosinophil emigration from bone marrow. The potential importance of this activity relates to the observation that in response to inflammatory signals in the lung, eosinopoiesis occurs in the bone marrow, and mature eosinophils migrate from this compartment via the blood to the bronchial mucosa (Foster, 1999). Although the guinea pig appears to have the most robust DP2-mediated responses, lack of tools available to characterize Th2 cells in this species precludes robust evaluation of DP2 antagonists in in vivo models. The potential therapeutic role of DP2 antagonists has, therefore, relied heavily on human studies.
In summary, AZD1981 is a selective reversible DP2 antagonist which in human systems consistently blocks DP2 functional responses independently of agonist, cell type or output measured. AZD1981 is a representative of a novel class of non-steroidal oral agents with an apparent distinct mode of action, which provides an ideal opportunity to study the pathophysiological role of DP2 in human disease.
Acknowledgments
We would like to thank Gwen McNicol and Carol Weyman Jones (AstraZeneca R&D Charnwood, Loughbrough, UK), Birgit Brodacz (Medical University of Graz, Graz, Austria) for help with generation of some of the data and Ray Hutchinson for comments on the manuscript. The research was funded by AstraZeneca. Editorial assistance was provided by Ian Wright of Wright Medical Communications Ltd, Hartford, UK, and was funded by AstraZeneca.
Glossary
- DP2
chemoattractant receptor-homologous molecule expressed on Th2 cells
- FEV1
forced expiratory volume in 1 s
- IC50
concentration of compound causing 50% inhibition of binding of [3H]PGD2 to the receptor
- logD7.4
distribution coefficient between 1-octanol and aqueous buffer at pH 7.4
- PE
phycoerythrine; pIC50, negative logarithm of the IC50
- pKa
acid dissociation constant
Conflict of interest
During the experimental work included in this manuscript, authors were employed by AstraZeneca R&D Charnwood, Loughborough UK (JAS, FMB, EA, CM, ID, IGD, RVB and CAS), or the Medical University of Graz, Graz, Austria (AH).
References
- Abe H, Takeshita T, Nagata K, Arita T, Endo Y, Fujita T, et al. Molecular cloning, chromosome mapping and characterization of the mouse CRTH2 gene, a putative member of the leukocyte chemoattractant receptor family. Gene. 1999;227:71–77. doi: 10.1016/s0378-1119(98)00599-x. [DOI] [PubMed] [Google Scholar]
- Alexander SPH, Mathie A, Peters JA. Guide to Receptors and Channels (GRAC), 5th Edition. Br J Phamacol. 2011;164(Suppl. 1):S1–S324. doi: 10.1111/j.1476-5381.2011.01649_1.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Angeli V, Staumont D, Charbonnier AS, Hammad H, Gosset P, Pichavant M, et al. Activation of the D prostanoid receptor 1 regulates immune and skin allergic responses. J Immunol. 2004;172:3822–3829. doi: 10.4049/jimmunol.172.6.3822. [DOI] [PubMed] [Google Scholar]
- Anhut H, Peskar BA, Bernauer W. Release of 15-keto-13,14-dihydro-thromboxane B2 and prostaglandin D 2 during anaphylaxis as measured by radioimmunoassay. Naunyn Schmiedebergs Arch Pharmacol. 1978;305:247–252. doi: 10.1007/BF00498818. [DOI] [PubMed] [Google Scholar]
- Arimura A. Discovery of a First-in-Class Drug, a Prostaglandin D2 Antagonist, for the Treatment of Allergic Diseases. 2010. pp. 281–287. Molecular Imaging for Integrated Medical Therapy and Drug Development Part IV.
- Balzar S, Fajt ML, Comhair SA, Erzurum SC, Bleecker E, Busse WW, et al. Mast cell phenotype, location and activation in severe asthma, data from the Severe Asthma Research Program. Am J Respir Crit Care Med. 2011;183:299–309. doi: 10.1164/rccm.201002-0295OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banfield C, Parnes J, Emery M, Ni L, Zhang N, Hodsman P. Single-dose, first-in-human study of AMG 853: pharmacokinetics, pharmacodynamics, and safety in healthy adults. Am J Respir Crit Care Med. 2010;181:A5397. [Google Scholar]
- Barnes N, Pavord I, Chuchalin A, Bell J, Hunter M, Lewis T, et al. A randomised, double-blind, placebo-controlled study of the CRTH2 antagonist OC000459 in moderate persistent asthma. Clin Exp Allergy. 2012;42:38–48. doi: 10.1111/j.1365-2222.2011.03813.x. [DOI] [PubMed] [Google Scholar]
- Barski OA, Tipparaju SM, Bhatnagar A. The aldo-keto reductase superfamily and its role in drug metabolism and detoxification. Drug Metab Rev. 2008;40:553–624. doi: 10.1080/03602530802431439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Birkinshaw TN, Teague SJ, Beech C, Bonnert RV, Hill S, Patel A, et al. Discovery of potent DP2 (DP2) receptor antagonists. Bioorg Med Chem Lett. 2006;16:4287–4290. doi: 10.1016/j.bmcl.2006.05.062. [DOI] [PubMed] [Google Scholar]
- Black JW, Leff P. Operational models of pharmacological agonism. Proc Royal Soc Biol. 1983;84:141–162. doi: 10.1098/rspb.1983.0093. [DOI] [PubMed] [Google Scholar]
- Bonnert R, Rasul R. Preparation of 3-(hetero)arylthio-indoleacetic acids as DP2 receptor ligands for the treatment of respiratory disorders. PCT Int Appl. 2004 WO2004106302. [Google Scholar]
- Carrillo J, Weyman-Jones C, Beri R, Jupp R, Arrowsmith E, McNicol G, et al. Pharmacological characterisation of dog recombinant DP2. Br J Pharmacol. 2005;3:102P. [Google Scholar]
- Chaudhry PS, Cabrera J, Juliani HR, Varma SD. Inhibition of human lens aldose reductase by flavonoids, sulindac and indomethacin. Biochem Pharmacol. 1983;32:1995–1998. doi: 10.1016/0006-2952(83)90417-3. [DOI] [PubMed] [Google Scholar]
- Cheng Y-C, Prusoff WH. Relationship between the inhibition constant (K1) and the concentration of inhibitor which causes 50 per cent inhibition (I50) of an enzymatic reaction. Biochem Pharmacol. 1973;22:3099–3108. doi: 10.1016/0006-2952(73)90196-2. [DOI] [PubMed] [Google Scholar]
- Chevalier E, Stock J, Fisher T, Dupont M, Fric M, Fargeau H, et al. Cutting edge: chemoattractant receptor-homologous molecule expressed on Th2 cells plays a restricting role on IL-5 production and eosinophil recruitment. J Immunol. 2005;175:2056–2060. doi: 10.4049/jimmunol.175.4.2056. [DOI] [PubMed] [Google Scholar]
- Crea AE, Nakhosteen JA, Lee TH. Mediator concentrations in bronchoalveolar lavage fluid of patients with mild asymptomatic bronchial asthma. Eur Respir J. 1992;5:190–195. [PubMed] [Google Scholar]
- Foster PS. Allergic networks regulating eosinophilia. Am J Respir Cell Mol Biol. 1999;21:451–454. doi: 10.1165/ajrcmb.21.4.f167. [DOI] [PubMed] [Google Scholar]
- Gervais FG, Cruz RP, Chateauneuf A, Gale S, Sawyer N, Nantel F, et al. Selective modulation of chemokinesis, degranulation, and apoptosis in eosinophils through the PGD2 receptors CRTH2 and DP. J Allergy Clin Immunol. 2001;108:982–988. doi: 10.1067/mai.2001.119919. [DOI] [PubMed] [Google Scholar]
- Gervais FG, Sawyer N, Stocco R, Hamel M, Krawczyk C, Sillaots S, et al. Pharmacological characterization of MK-7246, a potent and selective CRTH2 (chemoattractant receptor-homologous molecule expressed on T-helper type 2 cells) antagonist. Mol Pharmacol. 2011;79:69–76. doi: 10.1124/mol.110.068585. [DOI] [PubMed] [Google Scholar]
- Gyles SL, Xue L, Townsend ER, Wettey F, Pettipher R. A dominant role for chemoattractant receptor-homologous molecule expressed on T helper type 2 (Th2) cells (CRTH2) in mediating chemotaxis of CRTH2+ CD4+ Th2 lymphocytes in response to mast cell supernatants. Immunology. 2006;119:362–368. doi: 10.1111/j.1365-2567.2006.02440.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heinemann A, Schuligoi R, Sabroe I, Hartnell A, Peskar BA. Delta 12-prostaglandin J2, a plasma metabolite of prostaglandin D2, causes eosinophil mobilization from the bone marrow and primes eosinophils for chemotaxis. J Immunol. 2003;170:4752–4758. doi: 10.4049/jimmunol.170.9.4752. [DOI] [PubMed] [Google Scholar]
- Hirai H, Tanaka K, Yoshie O, Ogawa K, Kenmotsu K, Takamori Y, et al. Prostaglandin D2 selectively induces chemotaxis in T helper type 2 cells, eosinophils and basophils via seven-transmembrane receptor DP2. J Exp Med. 2001;193:255–261. doi: 10.1084/jem.193.2.255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirai H, Tanaka K, Takano S, Ichimasa M, Nakamura M, Nagata K. Agonistic effect of indomethacin on a prostaglandin D2 receptor, CRTH2. J Immunol. 2002;168:981–985. doi: 10.4049/jimmunol.168.3.981. [DOI] [PubMed] [Google Scholar]
- Horak F, Toth J, Hirschwehr R, Marks B, Stubner UP, Jager S, et al. Effect of continuous allergen challenge on clinical symptoms and mediator release in dust-mite-allergic patients. Allergy. 1998;53:68–72. doi: 10.1111/j.1398-9995.1998.tb03775.x. [DOI] [PubMed] [Google Scholar]
- Jin Y, Penning TM. Aldo-keto reductases and bioactivation/detoxication. Annu Rev Pharmacol Toxicol. 2007;47:263–292. doi: 10.1146/annurev.pharmtox.47.120505.105337. [DOI] [PubMed] [Google Scholar]
- Kenakin TP. Orthosteric drug antagonism. In: Kenakin TP, editor. A Pharmacology Primer: Theory, Applications and Methods. 3rd edn. Burlington, MA: Elsevier Academic Press; 2009. pp. 101–127. [Google Scholar]
- Lewis RA, Soter NA, Diamond PT, Austen KF, Oates JA, Roberts LJ., 2nd Prostaglandin D2 generation after activation of rat and human mast cells with anti-IgE. J Immunol. 1982;129:1627–1631. [PubMed] [Google Scholar]
- Liu MC, Bleecker ER, Lichtenstein LM, Kagey-Sobotka A, Niv Y, McLemore TL, et al. Evidence for elevated levels of histamine, prostaglandin D2, and other bronchoconstricting prostaglandins in the airways of subjects with mild asthma. Am Rev Respir Dis. 1990;142:126–132. doi: 10.1164/ajrccm/142.1.126. [DOI] [PubMed] [Google Scholar]
- Lukacs NW, Berlin AA, Franz-Bacon K, Sásik R, Sprague LJ, Ly TW, et al. CRTH2 antagonism significantly ameliorates airway hyperreactivity and downregulates inflammation-induced genes in a mouse model of airway inflammation. Am J Physiol Lung Cell Mol Physiol. 2008;295:L767–L779. doi: 10.1152/ajplung.90351.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luker T, Bonnert R, Brough S, Cook AR, Dickinson MR, Dougall I, et al. Substituted indole-1-acetic acids as potent and selective DP2 antagonists-discovery of AZD1981. Bioorg Med Chem Lett. 2011;21:6288–6292. doi: 10.1016/j.bmcl.2011.08.124. [DOI] [PubMed] [Google Scholar]
- Marshall C, Sargent C, Young S, Barker J. Is there a CRTH2 functional response in the dog? Br J Pharmacol. 2005;3:103P. (Winter Meeting) Available at: http://www.pa2online.org/abstract/abstract.jsp?abid=28258&author=Sargent&cat=-1&period=23 (accessed 6/12/12) [Google Scholar]
- Marshall C, Sargent C, Young S, Barker J. Functional effects of DP2 in the dog. Am J Respir Crit Care Med. 2006;3:A584. (abstract issue) [Google Scholar]
- Mathiesen JM, Christopoulos A, Ulven T, Royer JF, Campillo M, Heinemann A, et al. On the mechanism of interaction of potent surmountable and insurmountable antagonists with the prostaglandin D2 receptor CRTH2. Mol Pharmacol. 2006;69:1441–1453. doi: 10.1124/mol.105.017681. [DOI] [PubMed] [Google Scholar]
- Matsuoka T, Hirata M, Tanaka H, Takahashi Y, Murata T, Kabashima K, et al. Prostaglandin D2 as a mediator of allergic asthma. Science. 2000;287:2013–2017. doi: 10.1126/science.287.5460.2013. [DOI] [PubMed] [Google Scholar]
- Monneret G, Gravel S, Diamond M, Rokach J, Powell WS. Prostaglandin D2 is a potent chemoattractant for human eosinophils that acts via a novel DP receptor. Blood. 2001;98:1942–1948. doi: 10.1182/blood.v98.6.1942. [DOI] [PubMed] [Google Scholar]
- Monneret G, Cossette C, Gravel S, Rokach J, Powell WS. 15R-Methyl-prostaglandin D2 is a potent and selective DP2/DP2 in human eosinophils. J Pharmacol Exp Ther. 2003;304:349–355. doi: 10.1124/jpet.102.042937. [DOI] [PubMed] [Google Scholar]
- Murray JJ, Tonnel AB, Brash AR, Roberts LJ, 2nd, Gosset P, Workman R, et al. Release of prostaglandin D2 into human airways during acute antigen challenge. N Engl J Med. 1986;315:800–804. doi: 10.1056/NEJM198609253151304. [DOI] [PubMed] [Google Scholar]
- Nagata K, Hirai H, Tanaka K, Ogawa K, Aso T, Sugamura K, et al. DP2, an orphan receptor of T-helper-2-cells, is expressed on basophils and eosinophils and responds to mast cell-derived factor(s) FEBS Lett. 1999a;459:195–199. doi: 10.1016/s0014-5793(99)01251-x. [DOI] [PubMed] [Google Scholar]
- Nagata K, Tanaka K, Ogawa K, Kemmotsu K, Imai T, Yoshie O. Selective expression of a novel surface molecule by human Th2 cells in vivo. J Immunol. 1999b;162:1278–1286. [PubMed] [Google Scholar]
- Norman P. DP2 receptor antagonists in development. Exp Opin Invest Drugs. 2010;19:947–961. doi: 10.1517/13543784.2010.500019. [DOI] [PubMed] [Google Scholar]
- Nowak D, Grimminger F, Jorres R, Oldigs M, Rabe KF, Seeger W, et al. Increased LTB4 metabolites and PGD2 in BAL fluid after methacholine challenge in asthmatic subjects. Eur Respir J. 1993;6:405–412. [PubMed] [Google Scholar]
- Petrash JM. All in the family: aldose reductase and closely related aldo-keto reductases. Cell Mol Life Sci. 2004;61:737–749. doi: 10.1007/s00018-003-3402-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pettipher R, Hansel TT. Antagonists of the prostaglandin D2 receptor CRTH2. Drug News Perspect. 2008;21:317–322. doi: 10.1358/dnp.2008.21.6.1246831. [DOI] [PubMed] [Google Scholar]
- Pettipher R, Whittaker M. Update on the development of antagonists of chemoattractant receptor-homologous molecule expressed on Th2 Cells (CRTH2). From lead optimization to clinical proof-of-concept in asthma and allergic rhinitis. J Med Chem. 2012;55:2915–2931. doi: 10.1021/jm2013997. [DOI] [PubMed] [Google Scholar]
- Philip G, van Adelsberg J, Loeys T, Liu N, Wong P, Lai E, et al. Clinical studies of the DP1 antagonist laropiprant in asthma and allergic rhinitis. J Allergy Clin Immunol. 2009;124:942–948. doi: 10.1016/j.jaci.2009.07.006. [DOI] [PubMed] [Google Scholar]
- Royer JF, Schratl P, Carillo JJ, Jupp R, Barker J, Weyman-Jones C, et al. A novel antagonist of prostaglandin D2 blocks the locomotion of eosinophils and basophils. Eur J Clin Invest. 2008;38:663–671. doi: 10.1111/j.1365-2362.2008.01989.x. [DOI] [PubMed] [Google Scholar]
- Sanyal S, Kuvin JT, Karas RH. Niacin and laropiprant. Drugs Today (Barc) 2010;46:371–378. doi: 10.1358/dot.2010.46.6.1464844. [DOI] [PubMed] [Google Scholar]
- Satoh T, Moroi R, Aritake K, Urade Y, Kanai Y, Sumi K, et al. Prostaglandin D2 plays an essential role in chronic allergic inflammation of the skin via CRTH2 receptor. J Immunol. 2006;177:2621–2629. doi: 10.4049/jimmunol.177.4.2621. [DOI] [PubMed] [Google Scholar]
- Schuligoi R, Sturm E, Luschnig P, Konya V, Philipose S, Sedej M, et al. CRTH2 and d-type prostanoid receptor antagonists as novel therapeutic agents for inflammatory diseases. Pharmacology. 2010;85:372–382. doi: 10.1159/000313836. [DOI] [PubMed] [Google Scholar]
- Singh D, Cadden P, Hunter M, Collins LP, Perkins M, Pettipher R, et al. Inhibition of the asthmatic allergen challenge response by the CRTH2 antagonist OC000459. Eur Respir J. 2012 doi: 10.1183/09031936.00092111. (Epub April 2012) [DOI] [PubMed] [Google Scholar]
- Spik I, Brénuchon C, Angéli V, Staumont D, Fleury S, Capron M, et al. Activation of the prostaglandin D2 receptor DP2/CRTH2 increases allergic inflammation in mouse. J Immunol. 2005;174:3703–3708. doi: 10.4049/jimmunol.174.6.3703. [DOI] [PubMed] [Google Scholar]
- Stebbins KJ, Broadhead AR, Correa LD, Scott JM, Truong YP, Stearns BA, et al. Therapeutic efficacy of AM156, a novel prostanoid DP2 receptor antagonist, in murine models of allergic rhinitis and house dust mite-induced pulmonary inflammation. Eur J Pharmacol. 2010;638:142–149. doi: 10.1016/j.ejphar.2010.04.031. [DOI] [PubMed] [Google Scholar]
- Stubbs VE, Schratl P, Hartnell A, Williams TJ, Peskar BA, Heinemann A, et al. Indomethacin causes prostaglandin D(2)-like and eotaxin-like selective responses in eosinophils and basophils. J Biol Chem. 2002;277:26012–26020. doi: 10.1074/jbc.M201803200. [DOI] [PubMed] [Google Scholar]
- Uller L, Mathiesen JM, Alenmyr L, Korsgren M, Ulven T, Högberg T, et al. Antagonism of the prostaglandin D2 receptor CRTH2 attenuates asthma pathology in mouse eosinophilic airway inflammation. Respir Res. 2007;8:16–25. doi: 10.1186/1465-9921-8-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ulven T, Kostenis E. Novel CRTH2 antagonists: a review of patents from 2006 to 2009. Exp Opin Ther Patents. 2010;20:1505–1530. doi: 10.1517/13543776.2010.525506. [DOI] [PubMed] [Google Scholar]
- Ulven T, Receveur J-M, Grimstrup M, Rist O, Frimurer TM, Gerlach L-O, et al. Novel selective orally active CRTH2 antagonists for allergic inflammation developed from in silico derived hits. J Med Chem. 2006;49:6638–6641. doi: 10.1021/jm060657g. [DOI] [PubMed] [Google Scholar]
- Wenzel SE, Westcott JY, Smith HR, Larsen GL. Spectrum of prostanoid release after bronchoalveolar allergen challenge in atopic asthmatics and in control groups. An alteration in the ratio of bronchoconstrictive to bronchoprotective mediators. Am Rev Respir Dis. 1989;139:450–457. doi: 10.1164/ajrccm/139.2.450. [DOI] [PubMed] [Google Scholar]
- Xue L, Gyles SL, Wettey FR, Gazi L, Townsend E, Hunter MG, et al. Prostaglandin D2 causes preferential induction of proinflammatory Th2 cytokine production through an action on chemoattractant receptor-like molecule expressed on Th2 cells. J Immunol. 2005;175:6531–6536. doi: 10.4049/jimmunol.175.10.6531. [DOI] [PubMed] [Google Scholar]
- Xue L, Barrow A, Pettipher R. Interaction between prostaglandin D and chemoattractant receptor-homologous molecule expressed on Th2 cells mediates cytokine production by Th2 lymphocytes in response to activated mast cells. Clin Exp Immunol. 2009a;156:126–133. doi: 10.1111/j.1365-2249.2008.03871.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xue L, Barrow A, Pettipher R. Novel function of CRTH2 in preventing apoptosis of human Th2 cells through activation of the phosphatidylinositol 3-kinase pathway. J Immunol. 2009b;182:7580–7586. doi: 10.4049/jimmunol.0804090. [DOI] [PubMed] [Google Scholar]