

Abstract
Background and Purpose
Oltipraz, a cancer chemopreventive agent, has an anti-steatotic effect via liver X receptor-α (LXRα) inhibition. Here we have assessed the biological activity of a major metabolite of oltipraz (M2) against liver steatosis and steatohepatitis and the underlying mechanism(s).
Experimental Approach
Blood biochemistry and histopathology were assessed in high-fat diet (HFD)-fed mice treated with M2. An in vitroHepG2 cell model was used to study the mechanism of action. Immunoblotting, real-time PCR and luciferase reporter assays were performed to measure target protein or gene expression levels.
Key Results
M2 treatment inhibited HFD-induced steatohepatitis and diminished oxidative stress in liver. It increased expression of genes encoding proteins involved in mitochondrial fuel oxidation. Mitochondrial DNA content and oxygen consumption rate were enhanced. Moreover, M2 treatment repressed activity of LXRα and induction of its target genes, indicating anti-lipogenic effects. M2 activated AMP-activated protein kinase (AMPK). Inhibition of AMPK by over-expression of dominant negative AMPK (DN-AMPK) or by Compound C prevented M2 from inducing genes for fatty acid oxidation and repressed sterol regulatory element binding protein-1c (SREBP-1c) expression. M2 activated liver kinase B1 (LKB1) and increased the AMP/ATP ratio. LKB1 knockdown failed to reverse target protein modulations or AMPK activation by M2, supporting the proposal that both LKB1 and increased AMP/ATP ratio contribute to its anti-steatotic effect.
Conclusion and Implications
M2 inhibited liver steatosis and steatohepatitis by enhancing mitochondrial fuel oxidation and inhibiting lipogenesis. These effects reflected activation of AMPK elicited by increases in LKB1 activity and AMP/ATP ratio.
Keywords: steatohepatitis, high-fat diet, LKB1, AMPK, LXRα, AMP
Introduction
In the liver, balanced metabolism of lipids is essential for maintaining energy homeostasis because the proper amount of lipid is critical for normal liver function. The amount of hepatic fat is determined by the balance between consumption of free fatty acid (FFA) through mitochondrial oxidation and its de novo synthesis through induction of lipogenic genes (Day and James,1998; Berk,2008; Fabbrini et al.,2010). Hepatic steatosis may occur as a result of reduced energy expenditure and/or oversupplied energy uptake and represents an excess fat accumulation within the liver as an initial stage of non-alcoholic fatty liver disease (NAFLD), a spectrum of diseases caused by abnormal fat deposition leading to steatohepatitis progression (Day and James,1998; Berk,2008; Fabbrini et al.,2010).
NAFLD frequently occurs in patients with metabolic syndrome. As NAFLD causes diverse dysfunctions including insulin resistance, obesity, hyperlipidaemia and cardiovascular disorders, it has emerged as a leading cause of morbidity and mortality (Day and James,1998; Berk,2008; Fabbrini et al.,2010). Many experimental studies and clinical trials have been made to discover drugs or drug candidates for the treatment of NAFLD. Metformin, thiazolidinediones, ursodeoxycholic acid and vitamin E have some beneficial outcomes (Bugianesi et al.,2005; Mahady et al.,2011; Ratziu et al.,2011). However, most of them have weak efficacy and/or limitations. Hence, targeting both the increase in FFA oxidation and the reduction in abnormal fat synthesis may be a promising strategy to treat NAFLD progression (Day and James,1998; Berk,2008; Zhang et al.,2009; Fabbrini et al.,2010).
Oltipraz [4-methyl-5-(2-pyrazinyl)-1,2-dithiole-3-thione] has been extensively studied as a cancer chemopreventive agent (Bolton et al.,1993; Velayutham et al.,2007). In addition, oltipraz may inhibit the development of insulin resistance and prevent hepatic steatosis in animals fed a high-fat diet (HFD) by activating the AMP-activated protein kinase (AMPK)-p70 S6-kinase 1 (S6K1) pathway (Bae et al.,2007; Hwahng et al.,2009). Oltipraz has a tendency to be accumulated in the liver and the large intestine due to its lipid solubility (Bae et al.,2003; Kim et al.,2010). At high concentrations, it stays longer in the body and undergoes biotransformation (Bae et al.,2003). When administered to mammalian species, oltipraz is metabolized to yield pyrrolopyrazine metabolites (Bae et al.,2003) and a major conversion of oltipraz into 7-methyl-6,8-bis(methylthio)pyrrolo[1,2-a]pyrazine (M2) was also observed in humans (Kim et al.,2010; RM = M2). M2 has an antioxidant effect (Ko et al.,2006; Kwon et al.,2009)and it activates nuclear factor (erythroid-derived)-like-2 (Nrf2) to increase its antioxidant capacity (Ko et al.,2006). Moreover, M2 may have a cytoprotective effect against severe oxidative stress (Kwon et al.,2009).
In view of the considerable biotransformation of oltipraz to M2 in vivo and the therapeutic potential of M2 against metabolic liver diseases (Bae et al.,2003; Kim et al.,2010), this study investigated if M2 inhibited steatohepatitis, and the underlying molecular basis of any such effects. In particular, we were interested in the effects of M2 on mitochondrial fuel oxidation and de novo fat synthesis in hepatocytes. Here, we report that M2 effectively inhibited non-alcoholic steatosis and steatohepatitis by dually activating AMPK. We also demonstrated its anti-inflammatory and antioxidant effects. Our findings provide key information on the therapeutic effect of M2, as an active metabolite of oltipraz, for hepatic steatosis and steatohepatitis.
Methods
Materials
Oltipraz and its metabolites were synthesized at the CJ Central Laboratories (Ichon City, Korea) and its chemical structure was verified by a variety of spectroscopic analyses (Bae et al.,2007; Hwahng et al.,2009; Kwon et al.,2009). M2 was additionally synthesized and purified by chromatography for in vivo study, and the chemical authenticity was confirmed by NMR analysis in Dr. YS Lee's laboratory, as previously described (Ko et al.,2006; Kwon et al.,2009; Kang et al.,2012). T0901317 (T090) and Compound C were obtained from Calbiochem (San Diego, CA, USA). Antibodies recognizing sterol regulatory element binding protein-1 (SREBP-1), PPARα and PPARγ coactivator 1α (PGC-1α) were purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA). Antibodies directed against phospho-AMPK, AMPK, phospho-liver kinase B1 (phospho-LKB1), LKB1, phospho-acetyl CoA carboxylase (phospho-ACC), ACC, and lamin A/C were supplied from Cell Signaling Technology (Beverly, MA, USA). Anti-nitrotyrosine antibody was obtained from Millipore (Billerica, MA, USA). Antibody directed against inducible nitric oxide synthase (iNOS) was purchased from BD Bioscience (San Jose, CA, USA). Polyclonal anti-liver X receptor α (LXRα) antibody was supplied from Thermo Scientific Pierce (Rockford, IL, USA). Mouse anti-carnitine palmitoyltransferase-1 (CPT-1) antibody and human polyclonal anti-CPT1A antibody were purchased from Alpha Diagnostics (San Antonio, TX, USA) and Proteintech (Chicago, IL, USA), respectively. Anti-β-actin antibody, GW3965, palmitate, bovine serum albumin (BSA), and carbonyl cyanide 3-chlorophenylhydrazone (CCCP) were obtained from Sigma-Aldrich Chemicals (St. Louis, MO, USA). Horseradish peroxidase-conjugated goat anti-rabbit, rabbit anti-goat, and goat anti-mouse IgGs were supplied from Zymed Laboratories (San Francisco, CA, USA).
Animal treatment
All animal care and experimental studies were conducted in accordance with the institutional guidelines for the care and use of laboratory animals. All studies involving animals are reported in accordance with the ARRIVE guidelines for reporting experiments involving animals (Kilkenny et al.,2010; McGrath et al.,2010). A total of 36 animals were used in the experiments described here. Male C57BL/6 mice were obtained from Charles River Orient (Seoul, Korea) and were acclimatized for 1 week in a clean room at the Animal Center for Pharmaceutical Research, Seoul National University. At 5 weeks of age, C57BL/6 mice were started on either a normal diet (ND; n = 8) or a HFD (fat 60% w/w, Dyets Inc., Bethlehem, PA, USA) for 8 weeks. The mice fed on an HFD were evenly distributed based on the body weight into three treatment groups (HFD alone, HFD + M2 10 mg·kg−1·day−1 and HFD + M2 30 mg·kg−1·day−1, n = 7–12 each). M2 (10 or 30 mg·kg−1) dissolved in 40% polyethylene glycol 400 (Duksan Pure Chemicals, Ansan, Korea) was orally administered to mice, four times per week, during the last 4 weeks of the diet feeding. Control animals received vehicle only.
Haematoxylin & eosin or Oil Red O staining
The left lateral lobe of the liver was sliced, and tissue slices were fixed in 10% buffered-neutral formalin for 6 h. The liver slices were stained with haematoxylin and eosin (H&E). Oil Red O staining was used to visualize neutral triglyceride (TG) and lipids in frozen sections of the left lateral lobe of the liver. For Oil Red O staining, 4 μm sections were cut from frozen optimal cutting temperature samples, affixed to microscope slides and allowed to air-dry overnight at room temperature. The liver sections were then stained in fresh Oil Red O for 10 min and rinsed with water (Hwahng et al.,2009).
TG measurement
Samples of mouse liver (0.3 g) or HepG2 cells were homogenized in 0.1 M Tris–acetate buffer (pH 7.4) containing 0.1 M potassium chloride and 1 mM EDTA. Six volumes of chloroform/methanol (2:1) were then added. After vigorous stirring, the mixtures were kept on ice for 1 h and then centrifuged at 800× g for 3 min. The resulting lower phase was aspirated. The TG content was determined using Sigma Diagnostic Triglyceride Reagents (Hwahng et al.,2009).
Blood chemistry
Plasma alanine aminotransferase (ALT) activity, TG, FFA and total cholesterol contents were analysed using Spectrum, an automatic blood chemistry analyzer (Abbott Laboratories, Abbott Park, IL, USA). Fasting blood glucose was assessed using Accu-Chek Active (Roche Diagnostics, Heidelberg, Germany).
RNA isolation and quantitative real-time PCR assays
Total RNA was extracted using Trizol (Invitrogen, Carlsbad, CA, USA) without using DNase I and was reverse-transcribed. The resulting cDNA was amplified by quantitative real-time PCR (qRT-PCR). PCR was performed with the Light Cycler 1.5 (Roche, Mannheim, Germany) using a Light Cycler DNA master SYBR green-I kit. The cDNA was amplified by PCR. The primer sequences and melting temperatures (Tm) used for qRT-PCR assays were provided in Supporting Information Table S1. The conditions for PCR were 95°C for 3 min, followed by 45–55 cycles of 10 s at 95°C, 10 s at each Tm, and 15 s at 72°C.
Determination of reduced GSH
Reduced GSH content in the liver tissue was quantified using a commercially available GSH determination kit (Oxis International, Portland, OR, USA) as previously described (Han et al.,2011).
Cell culture
HepG2 cell line was purchased from ATCC (American Type Culture Collection, Manassas, VA, USA). The cells were plated at 1 × 105 per well in six-well plates, and cultures with 70–80% confluence were used. All cells were maintained in DMEM containing 10% FBS. Total cell lysates and nuclear extracts were prepared as previously described (Bae et al.,2007; Hwahng et al.,2009; Kwon et al.,2009).
Immunoblot analysis
SDS-PAGE and immunoblot analyses were performed according to the previously published procedures (Hwahng et al.,2009). In brief, cells were centrifuged at 3000× g for 3 min and were allowed to expand osmotically to the point of lysis after the addition of lysis buffer. Lysates were centrifuged at 10 000× g for 10 min to obtain supernatants and were stored at −70°C until use. Protein concentrations were measured by the Bradford assay (Bio-Rad, Hercules, CA, USA) using Ultrospec 6300 pro UV/visible spectrophotometer (GE Healthcare, Biochrom Ltd, Cambridge, UK). Proteins were resolved through gel electrophoresis and were transferred onto nitrocellulose membrane. The protein-bound nitrocellulose membrane was incubated with primary antibodies overnight at 4°C and then reacted with HRP-conjugated secondary antibodies. For immunoblotting, antibodies directed against SREB-1, PPARα, and PGC-1α were diluted at a ratio of 1:1000, whereas other antibodies recognizing phospho-AMPK, AMPK, phospho-LKB1, LKB1, phospho-ACC, ACC, lamin A/C, β-actin, nitrotyrosine, LXRα, CPT-1 and iNOS were performed at a ratio of 1:5000–10 000. Protein bands of interest were developed using an enhanced chemiluminescence system (Amersham, Chalfont St. Giles, Buckinghamshire, UK). Equal protein loading was verified by immunoblotting for β-actin.
Measurements of mtDNA and mtCOX II
Total DNA was extracted from tissues according to the manufacturer's instructions (Nucleogen, Siheung, Korea). Mitochondrial DNA (mtDNA) was assessed by SYBR green qRT-PCR assays. Fluorescence intensities during PCR were recorded and analysed in Light Cycler 1.5. Levels of cytochrome c oxidase subunit II (mtCOX II) transcribed from mtDNA were quantified by qRT-PCR and were normalized against that of nuclear-encoded receptor-interacting protein 140 (RIP140).
Preparation of mitochondria from mouse liver
Functional mitochondria were isolated from mouse liver according to the standard procedures (Frezza et al.,2007). After mincing of the liver into small pieces, the liver was homogenized, and the homogenate was then fractionated by centrifugation at 600× g for 10 min at 4°C. The supernatant was centrifuged at 7000× g for 10 min at 4°C to obtain the pellet containing mitochondria. Oxygen consumption rate was measured using the mitochondria. All the procedures were performed at 4°C to minimize damage from phospholipases and proteases.
Assays of oxygen consumption rate
Oxygen consumption rate was measured using the functional mitochondria prepared from mouse liver or in HepG2 cells. Mitochondrial respiratory rates were assessed using a Clark-type electrode in a continuously stirred sealed and thermostatically controlled chamber (Oxytherm System, Hansatech Instruments Ltd, Norfolk, UK) maintained at 37.8°C. For cell-based assays, HepG2 cells were suspended at a concentration of 2 × 106 cells·mL–1 of culture medium. Total cells were loaded into the Oxytherm chamber, and oxygen consumption within the chamber was measured using normal media as a control.
Reporter gene assays
HepG2 cells were transiently transfected with TK-CYP7A1-LXREx3-LUC (1 μg) or pGL2-FAS (1 μg) for 12 h in the presence of FuGENE® reagent (Roche, Nutley, NJ, USA). The activity of luciferase was measured by adding luciferase assay reagent (Promega, Madison, WI, USA).
Preparation of palmitate solution and treatment
Palmitate solution was prepared as previously described with minor modification (Karaskov et al.,2006). Briefly, 10 mM palmitate stock was prepared in sterile PBS and filtered. Fatty acid-free BSA solution (5%) was made in MEM/EBSS medium and was filtered. A palmitate solution was obtained by diluting 10 mM palmitate stock with 5% BSA solution plus MEM/EBSS medium without FBS to a final concentration of 1 mM. After overnight serum starvation, HepG2 cells were incubated with 0.5 mM palmitate for 24 h. Cells were exposed to 30 μM M2 1 h prior to the addition of palmitate.
siRNA transfection
To knockdown LKB1, cells were transfected with either a siRNA directed against human LKB1 (Santa Cruz, CA, USA) or a non-targeting control siRNA (100 pmol·mL−1) using AMAXA nucleofection system according to the manufacturer's instructions (Lonza, Köln, Germany). After transfection for 24 h, the cells were exposed to T090 (T0901317) or T090 plus M2 (30 μM) for 12 h. The resultant samples were analysed by immunoblot analyses. LKB1 knockdown was confirmed by immunoblotting.
Measurement of AMP/ATP ratio
Information on the protocol of measurement of AMP/ATP ratio is provided in the Supporting Information (Hahn-Windgassen et al.,2005).
Data analyses
All data are reported as mean ± SE. For cell-based assays, data represent at least three separate experiments. One-way anova was used to assess significant differences among treatment groups. For each significant treatment effect, the Newman–Keuls test was utilized to compare multiple group means.
Results
Inhibition of liver fat accumulation and body weight gain
First, we assessed the effect of M2 on liver steatosis. HFD feeding for 8 weeks caused severe fat deposition, as indicated by increases in the intensity of Oil Red O staining (Figure 1A) and hepatic TG content (Figure 1B). Treatment of mice with M2 (10 or 30 mg·kg−1·day−1, for 4 days per week) for the last 4 weeks during the total 8 weeks of HFD feeding, notably attenuated the fat accumulation, compared with vehicle treatment. Mice fed HFD for 8 weeks exhibited a marked increase in body weight gain compared with those fed a normal diet (ND), whereas those treated with M2 (10 or 30 mg·kg−1·day−1) showed significantly reduced body weight gain (Figure 1C). No difference was observed in the amount of food intake among the treatment groups (data not shown). In addition, M2 treatment prevented increases in plasma TG and total cholesterol levels in mice fed a HFD (Figure 1D). Similarly, the increase in fasting plasma glucose content, which reflects basal insulin sensitivity (Matthews et al.,1985), was suppressed. These results indicated that M2 treatment inhibited hepatic fat accumulation with reduction in body weight gain, and improved blood metabolic parameters.
Figure 1.
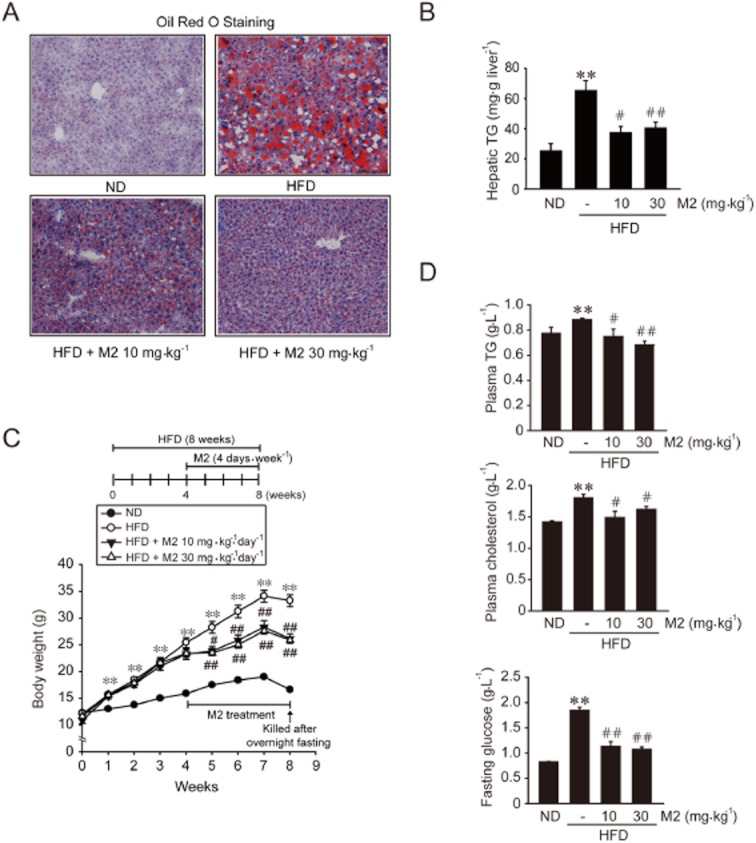
M2 inhibition of hepatic fat accumulation and body weight gain. (A) Oil Red O staining. Male C57BL/6 mice were fed on either a normal diet (ND) or high-fat diet (HFD) for 8 weeks. M2 (10 or 30 mg·kg−1·day−1) was orally administered to mice four times per week during the last 4 weeks of the diet feeding. Control animals received vehicle only. The representative pictures show Oil Red O staining of the liver sections (magnification 100×). (B) Hepatic TG contents (n = 5–9 animals per group). (C) Body weight gains. At the end of HFD feeding, mice were fasted overnight to measure fasting plasma glucose levels. Fasting caused decreases in body weight at the eighth week. (D) Plasma TG, total cholesterol and fasting glucose contents. For B, C and D, **P < 0.01, significantly different from ND alone; #P < 0.05, ##P < 0.01, significantly different from HFD alone. For C and D, n = 7–12 animals per group.
Inhibition of hepatic injury and inflammation
Excess fat accumulation provokes oxidation of FFAs and production of reactive oxygen species (ROS), which leads to oxidative injury (Day and James,1998; Fabbrini et al.,2010). Next, we examined whether M2 treatment inhibited steatohepatitis, inflammation and oxidative stress in the liver of mice fed HFD. HFD feeding for 8 weeks caused liver injury, as shown by lipid droplet formation, fatty degeneration of hepatocytes and inflammatory cell infiltration (Figure 2A). M2 treatment completely ameliorated HFD-induced liver injury, as also supported by an improvement in plasma ALT activity (Figure 2B). Healthy control mice fed a ND showed no changes. In addition, M2 treatment abolished increases in CD68 (a macrophage marker), TNFα and iNOS mRNA levels in the liver (Figure 2C), suggesting that it may inhibit infiltration of macrophages. Similarly, M2 prevented iNOS induction and reduced nitrotyrosinylation of proteins in the liver (Figure 2D). The antioxidant effect of M2 was confirmed by an increase in hepatic GSH content in mice fed a HFD (Figure 2E). These results demonstrate that M2 has the ability to inhibit hepatocyte injury, inflammation and oxidative stress elicited by excessive fat accumulation.
Figure 2.
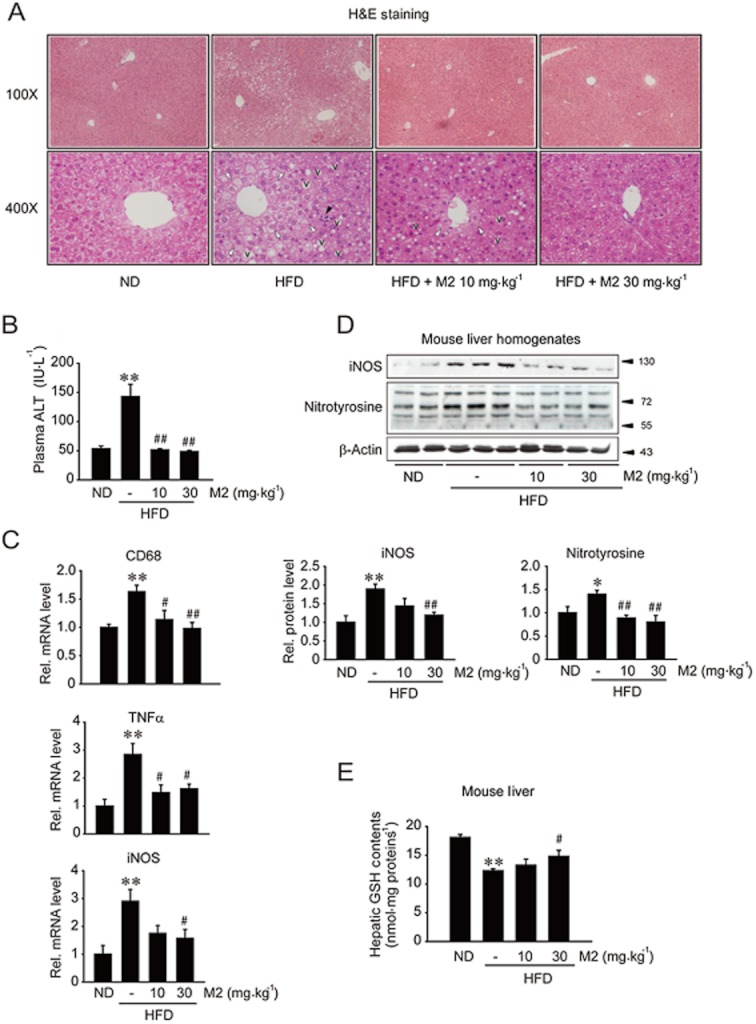
Inhibition of liver injury, inflammation and oxidative stress by M2. (A) H&E staining of the liver sections with lower (100×) and higher (400×) magnifications. Animals were treated as described in the legend of Figure 1A (M2, 10 or 30 mg·kg−1·day−1). In A, with higher magnification, Vs represent lipid droplet formation; white arrowheads indicate fatty degeneration of hepatocyte, whereas the black arrowhead denotes infiltration of inflammatory cells. (B) Plasma ALT activity (n = 7–12 animals per group). (C) qRT-PCR assays. The levels of CD68, TNFα and iNOS mRNAs were measured in liver tissue. The levels of GAPDH mRNA were used as a reference for data normalization. (D) Immunoblotting for iNOS and nitrotyrosinylated proteins in the liver homogenates. (E) Hepatic GSH contents. Reduced GSH levels were measured on the liver homogenates. For B–E, *P < 0.05, **P < 0.01, significantly different from ND alone; #P < 0.05, ##P < 0.01, significantly different from HFD alone. For C–E, n = 3–11 animals per group.
Enhancement of mitochondrial function and fuel oxidation
Next, we examined the effect of M2 on mitochondrial function by monitoring CPT-1, PPARα and PGC-1α transcript levels, whose protein products promote mitochondrial fuel oxidation and biogenesis (Lee et al.,2001; Patsouris et al.,2006; Crunkhorn et al.,2007; Hancock et al.,2008). In HepG2 cells, M2 treatment significantly increased the levels of CPT-1, PPARα and PGC-1α mRNA (Figure 3A). To confirm the effect of M2 on mitochondrial function, we assessed protein levels of the genes. In the time-course study, M2 treatment induced CPT-1, PPARα and PGC-1α, which was maintained up to 48 h (Figure 3B). M2 treatment (12 h) increased the protein levels with the maximal increase being noted at 3–30 μM (Figure 3C). These results showed that M2 has an ability to enhance mitochondrial fuel oxidation capacity. Treatment with M2 also prevented a decrease in hepatic CPT-1 level caused by HFD feeding (Figure 3D). In contrast, PPARα protein and mRNA levels were increased by HFD feeding, which was antagonized by M2 treatment (Supporting Information Figure S1A,B). HFD feeding caused differential changes in PGC-1α protein and mRNA levels, which were also reversed by M2 treatment. These compensatory changes induced by HFD feeding were comparable to previous observations (Lee et al.,2001; Patsouris et al.,2006; Crunkhorn et al.,2007; Hancock et al.,2008).
Figure 3.
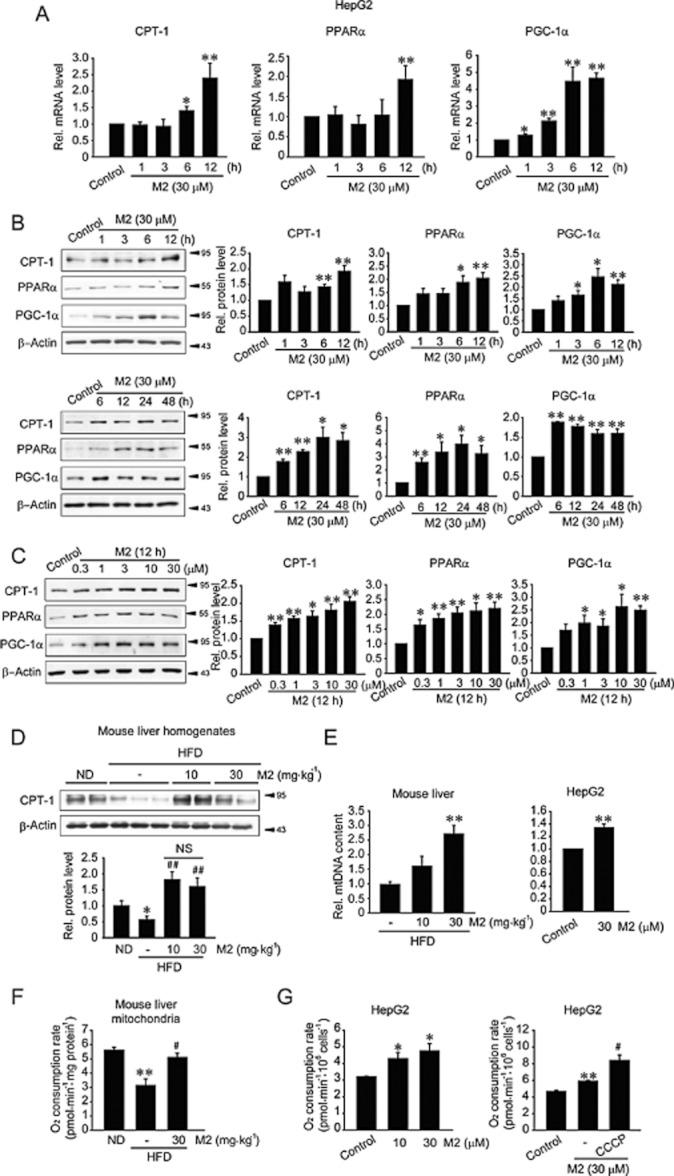
The effects of M2 on the expression of proteins associated with mitochondrial fuel oxidation and oxygen consumption. (A) qRT-PCR assays for CPT-1, PPARα and PGC-1α mRNA. HepG2 cells were treated with 30 μM M2 for the indicated times. (B) Time-course effects of M2 on CPT-1, PPARα and PGC-1α expression. Proteins of interest were immunoblotted in lysates of HepG2 cells treated with 30 μM M2 for the indicated times. (C) Concentration-dependent increases in CPT-1, PPARα and PGC-1α levels. HepG2 cells were treated with M2 for 12 h. For A–C, *P < 0.05,**P < 0.01, significantly different from vehicle. (D) Immunoblottings for CPT-1 in the liver homogenates. Mice were treated as described in the legend of Figure 1A. **P < 0.01, significantly different from ND alone; ##P < 0.01, significantly different from HFD alone. (n = 6–9 animals per group) (NS, not significant). (E) qRT-PCR assays for mtDNA. Total DNA was isolated from the liver tissue of mice fed a HFD with or without M2 treatment. For cell-based assays, HepG2 cells were treated with M2 for 6 h. The samples were subjected to qRT-PCR assays using primers for the mtDNA region COX II. Nuclear DNA-encoded gene RIP140 was amplified as a reference for data normalization. **P < 0.01, significantly different from HFD alone (left; n = 4–8 animals per group) or between M2 treatment and control (right). (F) Mitochondrial oxygen consumption rate in the liver. Mitochondria were isolated from the liver of mice fed HFD with or without M2. **P < 0.01, significantly different from ND alone; #P < 0.05, significantly different from HFD alone.(n = 3). (G) Oxygen consumption rate in HepG2 cells. HepG2 cells were treated with M2 alone (left), or in combination with CCCP (10 μM) for 12 h (right). *P < 0.05, **P < 0.01, significantly different from vehicle; #P < 0.05, significantly different from M2 alone.
M2 significantly increased the relative mtDNA content of the liver of mice fed a HFD or of HepG2 cells treated with M2 (Figure 3E), supporting the possibility that M2 may promote mitochondrial biogenesis. In order to link its anti-steatotic effect and enhancement of mitochondrial function, we determined oxygen consumption using in vivo and in vitro models. In an animal model, oxygen consumption was measured using the mitochondrial fraction prepared from mice fed either ND or HFD. HFD feeding decreased the oxygen consumption, an effect reversed by M2 treatment (Figure 3F). Similarly, oxygen consumption rate was increased in HepG2 cells exposed to M2 (Figure 3G, left). Treatment with CCCP, an uncoupler, significantly enhanced oxygen consumption rate in cells treated with M2 (Figure 3G, right), suggesting that the increase in mitochondrial oxygen consumption by M2 might not be due to uncoupling of mitochondrial respiration. These results indicate that M2 treatment may enhance the function and biogenesis of mitochondria in the liver.
Inhibition of LXRα-mediated lipogenesis
Liver steatosis may occur as a result of an increase in lipogenesis as well as reduction in energy expenditure. In subsequent experiments, we determined the effect of M2 treatment on LXRα-mediated lipogenesis both in vitro and in vivo. In HepG2 cells, CYP7A1-LXRE-luciferase reporter assays revealed that treatment with T090 (a synthetic agonist of LXRα) increased the transcriptional activity of LXRα, which was antagonized by M2 treatment (Figure 4A, upper). In parallel, M2 suppressed an increase in LXRα mRNA level by T090 (Figure 4A, lower). Likewise, it prevented T090-mediated increases in SREBP-1c, fatty acid synthase (FAS), ACC and stearoyl CoA desaturase-1 (SCD-1) mRNAs, confirming LXRα inhibition by M2 (Figure 4B). Treatment with M2 inhibited the induction of SREBP-1c by either T090 or GW3965 (another specific agonist of LXRα): both premature and mature forms of SREBP-1c levels in lysates or nuclear fractions were repressed (Figure 4C,D; Supporting Information Figure S2). To verify the anti-lipogenic effect of M2 in vitro, intracellular TG was measured in HepG2 cells incubated with palmitate or palmitate plus M2. M2 inhibited TG accumulation promoted by palmitate (Figure 4E). In the liver of HFD-fed mice, M2 treatment prevented increases in the mRNA levels of LXRα, SREBP-1c, FAS and ACC (downstream targets of LXRα) (Figure 4F). Moreover, the induction of LXRα and SREBP-1c proteins was diminished (Figure 4G). Collectively, our results demonstrated that M2 may inhibit LXRα-mediated lipogenesis in hepatocytes.
Figure 4.

The anti-lipogenic effect of M2. (A) LXRE reporter activity and qRT-PCR assays for LXRα mRNA. After LXRE transfection, HepG2 cells were treated with vehicle or M2 for 1 h, and continuously incubated with 10 μM T090 for 12 h. LXRα mRNA levels were measured in HepG2 cells similarly treated without transfection. (B) qRT-PCR assays for lipogenic gene transcripts. HepG2 cells were treated with vehicle or M2 for 1 h, and continuously incubated with 10 μM T090 for 12 h. (C) Immunoblotting for SREBP-1c in HepG2 cells. Premature and mature forms of SREBP-1c were immunoblotted in whole cell lysates or nuclear fraction of HepG2 cells treated as described in the legend of Figure 4A. WCL, whole cell lysate; NF, nuclear fraction. (D) Immunoblotting for SREBP-1c in HepG2 cells. HepG2 cells were treated with vehicle, M2, GW3965 (10 μM), or M2 + GW3965. For (A)–(D), **P < 0.01, significantly different from vehicle; #P < 0.05, ##P < 0.01, significantly different from T090/GW3965 alone. (E) Relative intracellular TG levels. TG levels were measured in HepG2 cells treated with 0.5 mM palmitate or palmitate + M2 for 24 h. **P < 0.01, significantly different from BSA; ##P < 0.01, significantly different from palmitate alone. (F) qRT-PCR assays for lipogenic gene transcripts in the liver. **P < 0.01, significantly different from ND alone; #P < 0.05, significantly different from HFD alone (n = 3–8 animals per group). (G) Immunoblotting for LXRα or SREBP-1c in the liver homogenates.
AMPK activation
To understand the mechanisms governing the beneficial effect of M2 on lipid homeostasis, we determined the role of AMPK in the regulation of mitochondrial fuel oxidation and lipogenesis. Treatment of HepG2 cells with M2 increased the phosphorylation of AMPK in a time-dependent manner (Figure 5A), with a maximum at 30 min and decreasing thereafter, although AMPK phosphorylation was maintained for up to 12 h. M2 treatment also increased the phosphorylation of ACC, a downstream substrate of AMPK, confirming the activation of AMPK. Inhibition of AMPK activity by Compound C (a chemical inhibitor of AMPK) completely prevented M2 from enhancing CPT-1, PPARα and PGC-1α expression in HepG2 cells (Figure 5B). Moreover, AMPK inhibition by either over-expression of dominant negative (DN)-AMPK or Compound C treatment reversed the inhibitory effect of M2 on SREBP-1c induction (Figure 5C). Our results provide evidence that AMPK activation by M2 contributes to not only the induction of the genes encoding for proteins associated with fuel oxidation, but the repression of SREBP-1c (i.e. anti-lipogenic effect).
Figure 5.

The role of AMPK activation by M2 in the induction of proteins associated with fuel oxidation or in the inhibition of lipogenesis. (A) The activation of AMPK by M2. Phosphorylated and total forms of AMPK or ACC were immunoblotted in lysates of HepG2 cells treated with M2 for the indicated times. *P < 0.05, **P < 0.01, significantly different from vehicle. (B) The effect of Compound C treatment on lipogenic gene transcript levels. qRT-PCR assays were done on HepG2 cells treated with 3 μM Compound C for 1 h and continuously incubated M2 for 12 h. *P < 0.05, **P < 0.01, significantly different from vehicle; #P < 0.05, ##P < 0.01, significantly different from M2 alone. (C) The effect of AMPK inhibition on SREBP-1c repression by M2. After DN-AMPK transfection (24 h) or Compound C treatment (1 h), HepG2 cells were treated with vehicle or M2 for 1 h, and continuously incubated with 10 μM T090 for 12 h. SREBP-1c was immunoblotted on the whole cell lysates. *P < 0.05, **P < 0.01, significantly different from vehicle; #P < 0.05, significantly different from T090 alone. NS, not significant.
Increases in LKB1 activity and AMP/ATP ratio
To find the upstream regulator activating AMPK, we determined whether M2 activated LKB1, a tumour suppressor that functions as a major kinase for AMPK (Shaw et al.,2004), in HepG2 cells (Figure 6A). M2 treatment notably increased LKB1 phosphorylation, which began from as early as 5 min post-treatment and was maintained at least up to 12 h. A deficiency of LKB1 using siLKB1 failed to inhibit the induction of CPT-1, PPARα and PGC-1α or the repression of SREBP-1c by M2 (Figure 6B,C). Knockdown of LKB1 still allowed the phosphorylation of AMPK or ACC in response to M2 (Figure 6D). In addition, treatment with STO-609 (a specific inhibitor of CaMKKβ) failed to inhibit the activation of AMPK (data not shown). These results raised the possibility that the ability of M2 to activate AMPK may not depend solely on LKB1.
Figure 6.
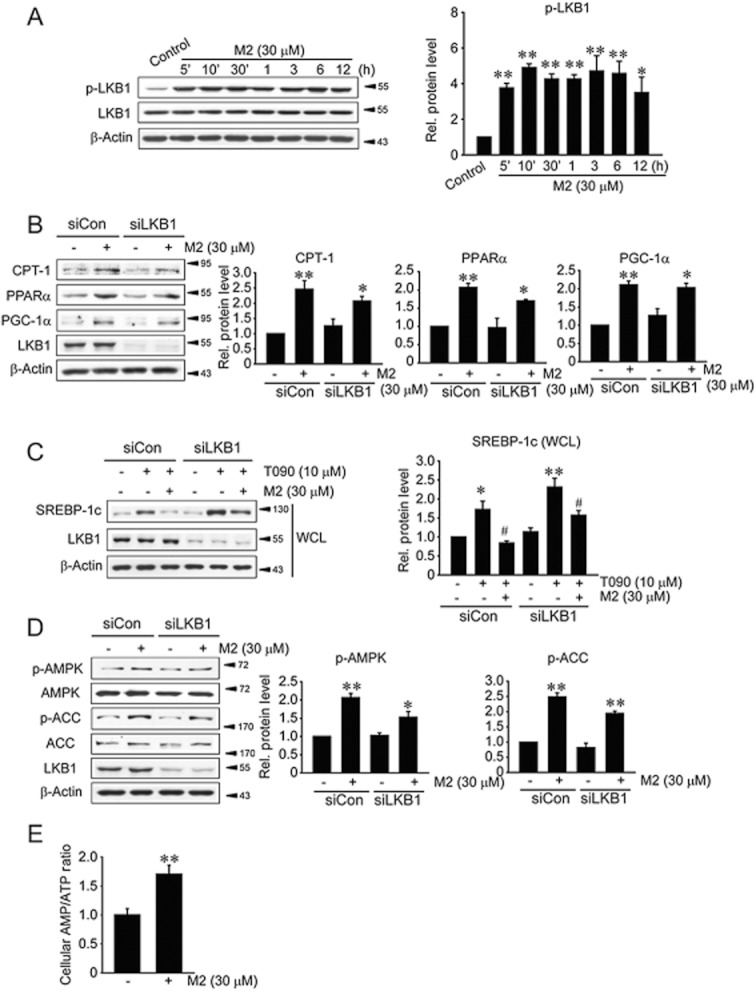
LKB1 activation and increased AMP/ATP ratio induced by M2. (A) The activation of LKB1 by M2. Phosphorylated or total LKB1 was immunoblotted in lysates of HepG2 cells treated with M2 for the indicated times. *P < 0.05, **P < 0.01, significantly different from vehicle. (B) The effect of LKB1 knockdown on CPT-1, PPARα and PGC-1α induction by M2. HepG2 cells were treated with M2 for 12 h following control knockdown (siCon) or LKB1 knockdown (siLKB1). (C) The effect of LKB1 knockdown on SREBP-1c repression by M2. Cells were treated with vehicle or M2 for 1 h, and continuously incubated with 10 μM T090 for 12 h following LKB1 knockdown. *P < 0.05, **P < 0.01, significantly different from vehicle; #P < 0.05, significantly different from T090 alone. WCL, whole cell lysate. (D) The effect of LKB1 knockdown on AMPK activation by M2. For B and D, *P < 0.05, **P < 0.01, significantly different from vehicle. (E) Increase in cellular AMP/ATP ratio by M2. AMP and ATP contents were measured using HPLC assays in HepG2 cells treated with M2 for 1 h. **P < 0.01, M2 treatment significantly different from control.
AMPK senses and is activated by an increase in AMP level coupled with a decrease in ATP (Lage et al.,2008; Zhang et al.,2009). To further define the regulatory mechanism governing AMPK activation by M2, we measured the intracellular AMP/ATP ratio. HPLC analyses revealed that M2 treatment increased the ratio of AMP to ATP in HepG2 cells (Figure 6E). Thus, it is highly likely that both the activation of LKB1 and the increase in AMP/ATP contribute to activating AMPK. Our findings demonstrate that M2 inhibits liver fat accumulation by enhancing both mitochondrial fuel oxidation and anti-lipogenic effect, and which results from AMPK activated by enhanced activity of LKB1 and increase in cellular AMP/ATP ratio (Figure 7).
Figure 7.
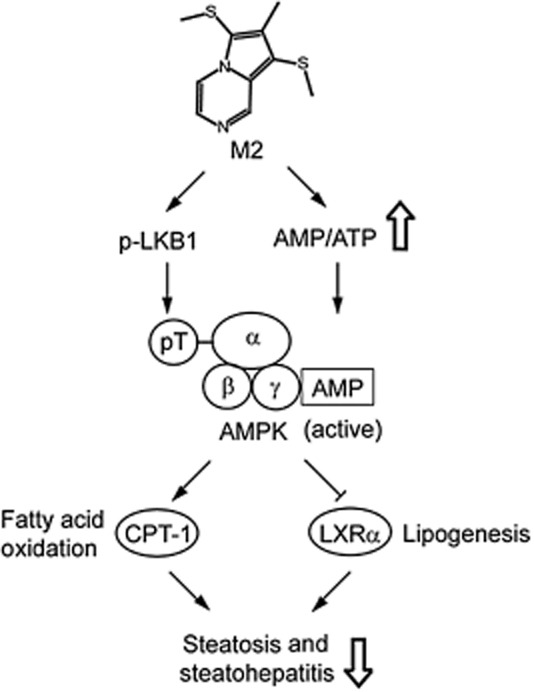
A schematic diagram illustrating the proposed mechanism by which M2 inhibits liver steatosis and steatohepatitis. Inhibition of liver steatosis and steatohepatitis by M2 may result from the activation of AMPK elicited by increases in LKB1 activity and the AMP/ATP ratio.
Discussion
Inducing an increase in FFA oxidation capacity and an inhibition of de novo lipogenesis may be a promising strategy for NAFLD treatment. Oltipraz has outstanding properties as an anti-oxidative agent (Kang et al.,2002; 2003; Bae et al.,2007; Hwahng et al.,2009); it prevents chemical carcinogenesis by inducing antioxidant and phase II enzymes, which may account for detoxification of carcinogens and other toxicants (Kang et al.,2003). We also reported that oltipraz attenuates liver cirrhosis in animals (Kang et al.,2002). Moreover, oltipraz was capable of inhibiting liver steatosis (Hwahng et al.,2009). Here we have shown that M2, a major metabolite of oltipraz, had an anti-steatotic effect. Consistent with this, M2 treatment repressed abnormal accumulation of liver fat in mice fed on HFD and reduced the body weight gain.
Oltipraz, when administered i.v. or p.o. to mammalian species, is being metabolized to yield two major metabolites: 4-methyl-5-(pyrazin-2-yl)-3H-1,2-dithiol-3-one (M1) and M2 (Bae et al.,2003; Kim et al.,2010). The formation of M1 results from oxidative desulfuration of the thione group of oltipraz, whereas M2 is produced from oltipraz by a series of chemical reactions and molecular rearrangement (Bae et al.,2003; Ko et al.,2006; Kwon et al.,2009). This was also confirmed in our clinical trial that when oltipraz was orally administered to patients with liver fibrosis/cirrhosis as a single dose or multiple doses for 24 weeks. A considerable concentration of M2 was observed in the plasma through 24 weeks of treatment, at a higher level than that of oltipraz (Kim et al.,2010). This pharmacokinetic profile implies that M2 may be pharmacologically active and the amount of M2 produced from oltipraz is apparently safe and tolerable (Kim et al.,2010).
The pathogenesis of NAFLD due to fat accumulation seems multifactorial. For example, fatty liver, which is defined by accumulated fat in the liver tissue exceeding ∼5% of total liver weight, results from not only impaired fatty acid utilization within mitochondria but also increased de novo fatty acid synthesis and subsequent TG accumulation. As excessive energy intake into the liver drives mitochondria to oxidize the increased amount of FFAs beyond the basal capacity, there is robust production of ROS/RNS in mitochondria. Thus excess deposition of fat within the hepatocytes not only elicits FFA oxidation, but also provokes oxidative stress. Hence, hepatic steatosis may progress to a variety of metabolic diseases accompanying inflammation and hepatocyte injury (Day and James,1998; Berk,2008; Zhang et al.,2009; Fabbrini et al.,2010). In the present study, we used two different approaches, (i) an in vivo model involving mice fed a HFD to assess the pharmacological effect of M2 on the inhibition of liver fat accumulation and (ii) an in vitro model using HepG2 cell model to study the underlying molecular mechanisms. Our findings demonstrated that M2 markedly inhibited hepatic inflammation, oxidative stress and hepatocyte injury caused by HFD feeding, and enabled the liver to restore GSH content. Previously, it has been shown that M2 protected mitochondria from injury against the insult of arachidonic acid and iron, and exhibited a cytoprotective activity (Kwon et al.,2009). M2 also activated Nrf2 and induced antioxidant and phase II enzymes (Ko et al.,2006). Thus, our results shown here in this study parallel the previous finding that the effects of M2 may reflect its antioxidant activity.
NAFLD results from not only impaired FFA utilization, but increased de novo synthesis of fat and subsequent accumulation (Day and James,1998). Mitochondria contribute to maintaining overall energy balance within the cell by generating ATP as a major energy source (Bonnefont et al.,2004; Orellana-Gavaldà et al.,2012). NAFLD progression is strongly associated with mitochondrial dysfunction due to excess influx of fatty acids into the liver (Bonnefont et al.,2004; Orellana-Gavaldà et al.,2012). Thus, increased mitochondrial capacity for the metabolism of fatty acids may be of help in treating NAFLD. A series of proteins are involved in this mitochondrial function. One of them, CPT-1, is localized in the mitochondrial outer membrane and enables FFAs to enter the mitochondria for β-oxidation (Kerner and Hoppel,2000). Over-expression of CPT-1A or its active form promotes FFA oxidation and improves metabolic profiles, such as steatosis, fasting blood glucose and inflammation (Orellana-Gavaldà et al.,2012). Our results demonstrate induction of CPT-1 by M2 and M2 prevented CPT-1 repression in mice fed a HFD, implying that this agent may facilitate the influx of FFAs into the mitochondria.
PPARα, mainly expressed in the liver and brown adipose tissue, controls lipid metabolism; as it is a transcription factor regulating the expression of genes responsible for mitochondrial and peroxisomal β-oxidation, FFA uptake, lipoprotein assembly and transport (Yoon,2009; Pyper et al.,2010). In our cell model, PPARα was up-regulated by M2. As a deficiency of PPARα causes drastic fat accumulation in the liver and depresses FFA oxidation and uptake (Kersten et al.,1999), the ability of M2 to induce PPARα would help to facilitate fuel oxidation in the mitochondria. Another important protein is PGC-1α, which is a master regulator of mitochondrial biogenesis (Aharoni-Simon et al.,2011). Our finding that M2 treatment up-regulated PGC-1α is compatible with the increased mitochondrial function. As PGC-1α interacts with PPARα and nuclear respiration factor 1/2 for energy utilization (Arany et al.,2005), the anti-steatotic effect of M2 may be explained in part by increased capacity for mitochondrial FFA oxidation and biogenesis. PGC-1α regulates transcription, replication and the amount of mtDNA (Arany et al.,2005; Aquilano et al.,2010). Thus, induction of PGC-1α represents mitochondrial biogenesis, consistent with our observation that M2 increased mtDNA content and oxygen consumption rate.
LXRα transcriptionally regulates a series of genes involved in lipid synthesis and is activated in the liver of mice fed a HFD or genetically obese mice. LXRα binds to LXRE upon endogenous ligand binding in the promoter region of the LXRα gene itself and SREBP-1c, and induces SREBP-1c (Laffitte et al.,2001; Zelcer and Tontonoz,2005). As SREBP-1c transcriptionally activates the genes involved in fatty acid biogenesis, such as FAS, ACC and SCD-1, LXRα stimulates fat accumulation under NAFLD conditions and contributes to steatosis. Previously, we reported the anti-steatotic effect of oltipraz, which results from the inhibition of LXRα via (i) threonine phosphorylation of LXRα by AMPK, an energy sensor governing diverse signaling pathways, and (ii) suppression of LXRα phosphorylation at serine residues by S6K1 (Hwahng et al.,2009). In the present study, M2 also antagonized LXRα activation in vitro and in vivo, which depended on the activation of AMPK. Hence, both oltipraz and its metabolite M2 have the ability to activate AMPK, which accounts for LXRα inhibition.
The activity of AMPK is regulated by two major mechanisms: (i) post-transcriptional modification by LKB1, CaMKKβ or TAK1, and (ii) sensing and allosterical activation by AMP, a condition affected by hypoxia, glucose deprivation, oxidative stress and cytokines (Lage et al.,2008; Zhang et al.,2009). Previously, we showed that oltipraz protected cells from mitochondrial impairment and injury against oxidative stress (Shin and Kim,2009). Moreover, it inhibits insulin resistance caused by HFD feeding (Bae et al.,2007). All of these effects of oltipraz relied on AMPK activation without activating LKB1 (Bae et al.,2007). By contrast, M2 strongly increased the activating phosphorylation of LKB1, which is distinct from the effect of oltipraz (Bae et al.,2007; Hwahng et al.,2009). This may result from the differences between M2 and oltipraz in chemico-biological properties (e.g. backbone structure, pro-oxidant effect or Nrf2-activating effect).
Our present findings demonstrated that both mitochondrial fuel oxidation and repression of SREBP-1c-mediated lipogenesis depended on AMPK. Nevertheless, up-regulation of CPT-1, PPARα and PGC-1α by M2 was only weakly reversed by LKB1 knockdown, implying that there exists another mechanism in this event. As an increase in AMP/ATP ratio plays a key role in the allosteric regulation of AMPK (Lage et al.,2008; Zhang et al.,2009), AMPK activation by M2 may be explained by an increase in the AMP/ATP ratio as well as LKB1 activation (Figure 7). Taken together, our results show that M2 inhibited steatosis and steatohepatitis, which may be associated with AMPK-dependent enhanced fuel oxidation in the mitochondria and repression of LXRα-dependent lipogenesis. As oltipraz is being studied for metabolic liver disease, our finding that M2 inhibits hepatic fat accumulation and injury provides key information on the effect of oltipraz in vivo, and the potential use of these agents for the treatment of liver diseases.
Acknowledgments
This work was supported by the National Research Foundation of Korea grant funded by the government of Korea (MEST) (No. 2012-0000843) and by the Leading Industry Development for Economic Region of the Chungcheong Leading Industry Office (CCLIO), Korea Institute for Advancement of Technology (KIAT) and Ministry of Knowledge Economy (MKE) (2010-C-2-B-YO-A-25).
Glossary
- ACC
acetyl CoA carboxylase
- ALT
alanine aminotransferase
- AMPK
AMP-activated protein kinase
- CCCP
carbonyl cyanide 3-chlorophenylhydrazone
- CPT-1
carnitine palmitoyltransferase-1
- DN-AMPK
dominant negative AMPK
- FAS
fatty acid synthase
- FFA
free fatty acid
- HFD
high-fat diet
- LKB1
liver kinase B1
- LXRα
liver X receptor-α
- LXRE
LXRα response element
- mtCOX II
mitochondrial cytochrome c oxidase subunit II
- NAFLD
non-alcoholic fatty liver disease
- ND
normal diet
- Nrf2
nuclear factor (erythroid-derived 2)-like 2
- PGC-1α
peroxisome proliferator-activated receptor gamma co-activator 1α
- RIP140
receptor-interacting protein 140
- ROS
reactive oxygen species
- S6K1
p70 S6 kinase 1
- SCD-1
stearoyl-CoA desaturase-1
- SREBP-1c
sterol regulatory element binding protein-1c
- TG
triglyceride
- T090
T0901317
Conflict of interest
The authors disclose no conflicts.
Supporting information
Additional Supporting Information may be found in the online version of this article at the publisher's web-site:
Figure S1 The effects of M2 on PPARα and PGC-1α levels in the liver. (A) Immunoblottings for PPARα and PGC-1α. Proteins of interest in liver homogenates were immunoblotted. (B) qRT-PCR assays for hepatic PPARα or PGC-1α mRNA. Mice were treated as described in the legend of Figure 1A. **P < 0.01, significantly different from ND alone; #P < 0.05, ##P < 0.01, significantly different from HFD alone.
Figure S2 Immunoblotting for SREBP-1c in HepG2 cells. Premature form of SREBP-1c was immunoblotted on the lysates of HepG2 cells treated as described in the legend to Figure 4C. **P < 0.01, significantly different from vehicle; #P < 0.05, ##P < 0.01, significantly different from T090 alone.
Table S1 The sequences of primer pairs and Tm for qRT-PCR assays.
References
- Aharoni-Simon M, Hann-Obercyger M, Pen S, Madar Z, Tirosh O. Fatty liver is associated with impaired activity of PPARγ-coactivator 1α (PGC-1α) and mitochondrial biogenesis in mice. Lab Invest. 2011;91:1018–1028. doi: 10.1038/labinvest.2011.55. [DOI] [PubMed] [Google Scholar]
- Aquilano K, Vigilanza P, Baldelli S, Pagliei B, Rotilio G, Ciriolo MR. Peroxisome proliferator-activated receptor gamma co-activator 1alpha (PGC-1alpha) and sirtuin 1 (SIRT1) reside in mitochondria: possible direct function in mitochondrial biogenesis. J Biol Chem. 2010;285:21590–21599. doi: 10.1074/jbc.M109.070169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arany Z, He H, Lin J, Hoyer K, Handschin C, Toka O, et al. Transcriptional coactivator PGC-1 alpha controls the energy state and contractile function of cardiac muscle. Cell Metab. 2005;1:259–271. doi: 10.1016/j.cmet.2005.03.002. [DOI] [PubMed] [Google Scholar]
- Bae EJ, Yang YM, Kim JW, Kim SG. Identification of a novel class of dithiolethiones that prevent hepatic insulin resistance via the adenosine monophosphate-activated protein kinase-p70 ribosomal S6 kinase-1 pathway. Hepatology. 2007;46:730–739. doi: 10.1002/hep.21769. [DOI] [PubMed] [Google Scholar]
- Bae SK, Kim EJ, Chung SJ, Kim SG, Lee MG. Pharmacokinetic interaction between oltipraz and dimethyl-4,4′-dimethoxy-5,6,5′,6′-dimethylene dioxybiphenyl-2,2′-dicarboxylate (DDB) after single intravenous and oral administration to rats. J Pharm Pharmacol. 2003;55:1241–1249. doi: 10.1211/0022357021648. [DOI] [PubMed] [Google Scholar]
- Berk PD. Regulatable fatty acid transport mechanisms are central to the pathophysiology of obesity, fatty liver, and metabolic syndrome. Hepatology. 2008;48:1362–1376. doi: 10.1002/hep.22632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolton MG, Muñoz A, Jacobson LP, Groopman JD, Maxuitenko YY, Roebuck BD, et al. Transient intervention with oltipraz protects against aflatoxin-induced hepatic tumorigenesis. Cancer Res. 1993;53:3499–3504. [PubMed] [Google Scholar]
- Bonnefont JP, Djouadi F, Prip-Buus C, Gobin S, Munnich A, Bastin J. Carnitine palmitoyl transferases 1 and 2: biochemical, molecular and medical aspects. Mol Aspects Med. 2004;25:495–520. doi: 10.1016/j.mam.2004.06.004. [DOI] [PubMed] [Google Scholar]
- Bugianesi E, Gentilcore E, Manini R, Natale S, Vanni E, Villanova N, et al. A randomized controlled trial of metformin versus vitamin E or prescriptive diet in nonalcoholic fatty liver disease. Am J Gastroenterol. 2005;100:1082–1090. doi: 10.1111/j.1572-0241.2005.41583.x. [DOI] [PubMed] [Google Scholar]
- Crunkhorn S, Dearie F, Mantzoros C, Gami H, da Silva WS, Espinoza D, et al. Peroxisome proliferator activator receptor gamma coactivator-1 expression is reduced in obesity: potential pathogenic role of saturated fatty acids and p38 mitogen-activated protein kinase activation. J Biol Chem. 2007;282:15439–15450. doi: 10.1074/jbc.M611214200. [DOI] [PubMed] [Google Scholar]
- Day CP, James OF. Steatohepatitis: a tale of two ‘hits’? Gastroenterology. 1998;114:842–845. doi: 10.1016/s0016-5085(98)70599-2. [DOI] [PubMed] [Google Scholar]
- Fabbrini E, Sullivan S, Klein S. Obesity and nonalcoholic fatty liver disease: biochemical, metabolic, and clinical implications. Hepatology. 2010;51:679–689. doi: 10.1002/hep.23280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frezza C, Cipolat S, Scorrano L. Organelle isolation: functional mitochondria from mouse liver, muscle and cultured fibroblasts. Nat Protoc. 2007;2:287–295. doi: 10.1038/nprot.2006.478. [DOI] [PubMed] [Google Scholar]
- Hahn-Windgassen A, Nogueira V, Chen CC, Skeen JE, Sonenberg N, Hay N. Akt activates the mammalian target of rapamycin by regulating cellular ATP level and AMPK activity. J Biol Chem. 2005;280:32081–32089. doi: 10.1074/jbc.M502876200. [DOI] [PubMed] [Google Scholar]
- Han CY, Ki SH, Kim YW, Noh K, Lee Y, Kang B, et al. Ajoene, a stable garlic by-product, inhibits high-fat diet-induced hepatic steatosis and oxidative injury through LKB1-dependent AMPK activation. Antioxid Redox Signal. 2011;14:187–202. doi: 10.1089/ars.2010.3190. [DOI] [PubMed] [Google Scholar]
- Hancock CR, Han DH, Chen M, Terada S, Yasuda T, Wright DC, et al. High-fat diets cause insulin resistance despite an increase in muscle mitochondria. Proc Natl Acad Sci U S A. 2008;105:7815–7820. doi: 10.1073/pnas.0802057105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hwahng SH, Ki SH, Bae EJ, Kim HE, Kim SG. Role of adenosine monophosphate-activated protein kinase-p70 ribosomal S6 kinase-1 pathway in repression of liver X receptor-alpha-dependent lipogenic gene induction and hepatic steatosis by a novel class of dithiolethiones. Hepatology. 2009;49:1913–1925. doi: 10.1002/hep.22887. [DOI] [PubMed] [Google Scholar]
- Kang KW, Kim YG, Cho MK, Bae SK, Kim CW, Lee MG, et al. Oltipraz regenerates cirrhotic liver through CCAAT/enhancer binding protein-mediated stellate cell inactivation. FASEB J. 2002;16:1988–1990. doi: 10.1096/fj.02-0406fje. [DOI] [PubMed] [Google Scholar]
- Kang KW, Cho IJ, Lee CH, Kim SG. Essential role of phosphatidylinositol 3-kinase-dependent CCAAT/enhancer binding protein beta activation in the induction of glutathione S-transferase by oltipraz. J Natl Cancer Inst. 2003;95:53–66. doi: 10.1093/jnci/95.1.53. [DOI] [PubMed] [Google Scholar]
- Kang SG, Lee WH, Lee YH, Lee YS, Kim SG. Hypoxia-inducible factor-1α inhibition by a pyrrolopyrazine metabolite of oltipraz as a consequence of microRNAs 199a-5p and 20a induction. Carcinogenesis. 2012;33:661–669. doi: 10.1093/carcin/bgr320. [DOI] [PubMed] [Google Scholar]
- Karaskov E, Scott C, Zhang L, Teodoro T, Ravazzola M, Volchuk A. Chronic palmitate but not oleate exposure induces endoplasmic reticulum stress, which may contribute to INS-1 pancreatic beta-cell apoptosis. Endocrinology. 2006;147:3398–3407. doi: 10.1210/en.2005-1494. [DOI] [PubMed] [Google Scholar]
- Kerner J, Hoppel C. Fatty acid import into mitochondria. Biochim Biophys Acta. 2000;1486:1–17. doi: 10.1016/s1388-1981(00)00044-5. [DOI] [PubMed] [Google Scholar]
- Kersten S, Seydoux J, Peters JM, Gonzalez FJ, Desvergne B, Wahli W. Peroxisome proliferator-activated receptor alpha mediates the adaptive response to fasting. J Clin Invest. 1999;103:1489–1498. doi: 10.1172/JCI6223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kilkenny C, Browne W, Cuthill IC, Emerson M, Altman DG. NC3Rs Reporting Guidelines Working Group. Br J Pharmacol. 2010;160:1577–1579. doi: 10.1111/j.1476-5381.2010.00872.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim SG, Kim YM, Choi YH, Lee MG, Choi JY, Han JY, et al. Pharmacokinetics of oltipraz and its major metabolite (RM) in patients with liver fibrosis or cirrhosis: relationship with suppression of circulating TGF-beta1. Clin Pharmacol Ther. 2010;88:360–368. doi: 10.1038/clpt.2010.89. [DOI] [PubMed] [Google Scholar]
- Ko MS, Lee SJ, Kim JW, Lim JW, Kim SG. Differential effects of the oxidized metabolites of oltipraz on the activation of CCAAT/enhancer binding protein-beta and NF-E2-related factor-2 for GSTA2 gene induction. Drug Metab Dispos. 2006;34:1353–1360. doi: 10.1124/dmd.106.009514. [DOI] [PubMed] [Google Scholar]
- Kwon YN, Shin SM, Cho IJ, Kim SG. Oxidized metabolites of oltipraz exert cytoprotective effects against arachidonic acid through AMP-activated protein kinase-dependent cellular antioxidant effect and mitochondrial protection. Drug Metab Dispos. 2009;37:1187–1197. doi: 10.1124/dmd.108.025908. [DOI] [PubMed] [Google Scholar]
- Laffitte BA, Joseph SB, Walczak R, Pei L, Wilpitz DC, Collins JL, et al. Autoregulation of the human liver X receptor alpha promoter. Mol Cell Biol. 2001;21:7558–7568. doi: 10.1128/MCB.21.22.7558-7568.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lage R, Dieguez C, Vidal-Puig A, Lopez M. AMPK: a metabolic gauge regulating whole-body energy homeostasis. Trends Mol Med. 2008;14:539–549. doi: 10.1016/j.molmed.2008.09.007. [DOI] [PubMed] [Google Scholar]
- Lee Y, Wang MY, Kakuma T, Wang ZW, Babcock E, McCorkle K, et al. Liporegulation in diet-induced obesity. The antisteatotic role of hyperleptinemia. J Biol Chem. 2001;276:5629–5635. doi: 10.1074/jbc.M008553200. [DOI] [PubMed] [Google Scholar]
- McGrath J, Drummond G, McLachlan E, Kilkenny C, Wainwright C. Guidelines for reporting experiments involving animals: the ARRIVE guidelines. Br J Pharmacol. 2010;160:1573–1576. doi: 10.1111/j.1476-5381.2010.00873.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahady SE, Webster AC, Walker S, Sanyal A, George J. The role of thiazolidinediones in non-alcoholic steatohepatitis – a systematic review and meta analysis. J Hepatol. 2011;55:1383–1390. doi: 10.1016/j.jhep.2011.03.016. [DOI] [PubMed] [Google Scholar]
- Matthews DR, Hosker JP, Rudenski AS, Naylor BA, Treacher DF, Turner RC. Homeostasis model assessment: insulin resistance and beta-cell function from fasting plasma glucose and insulin concentrations in man. Diabetologia. 1985;28:412–419. doi: 10.1007/BF00280883. [DOI] [PubMed] [Google Scholar]
- Orellana-Gavaldà JM, Herrero L, Malandrino MI, Pañeda A, Sol Rodríguez-Peña M, Petry H, et al. Molecular therapy for obesity and diabetes based on a long-term increase in hepatic fatty-acid oxidation. Hepatology. 2012;53:821–832. doi: 10.1002/hep.24140. [DOI] [PubMed] [Google Scholar]
- Patsouris D, Reddy JK, Müller M, Kersten S. Peroxisome proliferator-activated receptor alpha mediates the effects of high-fat diet on hepatic gene expression. Endocrinology. 2006;147:1508–1516. doi: 10.1210/en.2005-1132. [DOI] [PubMed] [Google Scholar]
- Pyper SR, Viswakarma N, Yu S, Reddy JK. PPARα: energy combustion, hypolipidemia, inflammation and cancer. Nucl Recept Signal. 2010;8:e002. doi: 10.1621/nrs.08002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ratziu V, de Ledinghen V, Oberti F, Mathurin P, Wartelle-Bladou C, Renou C, et al. A randomized controlled trial of high-dose ursodesoxycholic acid for nonalcoholic steatohepatitis. J Hepatol. 2011;54:1011–1019. doi: 10.1016/j.jhep.2010.08.030. [DOI] [PubMed] [Google Scholar]
- Shaw RJ, Kosmatka M, Bardeesy N, Hurley RL, Witters LA, DePinho RA, et al. The tumor suppressor LKB1 kinase directly activates AMP-activated kinase and regulates apoptosis in response to energy stress. Proc Natl Acad Sci U S A. 2004;101:3329–3335. doi: 10.1073/pnas.0308061100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin SM, Kim SG. Inhibition of arachidonic acid and iron-induced mitochondrial dysfunction and apoptosis by oltipraz and novel 1,2-dithiole-3-thione congeners. Mol Pharmacol. 2009;75:242–253. doi: 10.1124/mol.108.051128. [DOI] [PubMed] [Google Scholar]
- Velayutham M, Muthukumaran RB, Sostaric JZ, McCraken J, Fishbein JC, Zweier JL. Interactions of the major metabolite of the cancer chemopreventive drug oltipraz with cytochrome c: a novel pathway for cancer chemoprevention. Free Radic Biol Med. 2007;43:1076–1085. doi: 10.1016/j.freeradbiomed.2007.06.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoon MC. The role of PPARalpha in lipid metabolism and obesity: focusing on the effects of estrogen on PPARalpha actions. Pharmacol Res Rev. 2009;60:151–159. doi: 10.1016/j.phrs.2009.02.004. [DOI] [PubMed] [Google Scholar]
- Zelcer N, Tontonoz P. Liver X receptors as integrators of metabolic and inflammatory signaling. J Clin Invest. 2005;116:607–614. doi: 10.1172/JCI27883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang BB, Zhou G, Li C. AMPK: an emerging drug target for diabetes and the metabolic syndrome. Cell Metab. 2009;9:407–416. doi: 10.1016/j.cmet.2009.03.012. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.