

Abstract
Exposure to ethanol during the prenatal period contributes to increased alcohol consumption and preference in rodents and increased risk for alcoholism in humans. With studies in adult animals showing the orexigenic peptides, enkephalin (ENK), galanin (GAL) and orexin (OX), to stimulate ethanol consumption, the question addressed here is whether prenatal ethanol alters the development in utero of specific neurons that express these peptides. With reports describing suppressive effects of high doses of ethanol, we examined the offspring of dams gavaged from embryonic day 9 to parturition with a control solution or lower ethanol doses, 1 and 3 g/kg/day, known to promote ethanol consumption in the offspring. To understand underlying mechanisms, measurements were taken in postnatal offspring of the expression of ENK in the hypothalamic paraventricular nucleus (PVN) and nucleus accumbens (NAc), GAL in the PVN, and OX in the perifornical lateral hypothalamus (PFLH) using real-time qPCR and in situ hybridization, and also of the cell proliferation marker, 5-bromo-2-deoxyuridine (BrdU), and its double-labeling with either neuronal nuclei (NeuN), a marker of mature neurons, or the peptides. On postnatal day 15 (P15), after two weeks without ethanol, the offspring showed increased expression of ENK in the PVN and NAc core but not shell, GAL in the PVN, and OX in the PFLH. In these same areas, prenatal ethanol compared to control increased the density at birth (P0) of neurons expressing these peptides and at P0 and P15 of neurons double-labeling BrdU and NeuN, indicating increased neurogenesis. These BrdU-positive neurons were found to express ENK, GAL and OX, indicating that prenatal ethanol promotes neurogenesis in these specific peptide systems. There were no changes in gliogenesis or apoptosis. This increase in neurogenesis and density of peptide-expressing neurons suggests the involvement of these hypothalamic and accumbal peptide systems in mediating the increased alcohol consumption observed in prenatal ethanol-exposed offspring.
Keywords: prenatal ethanol, hypothalamus, nucleus accumbens, orexigenic peptides, neurogenesis
INTRODUCTION
In addition to being genetically determined, preference for ethanol is known to be affected by early environmental factors, including exposure to ethanol itself. Alcohol during the prenatal period significantly alters normal growth and development and has long-term physiological and behavioral consequences. In humans, maternal drinking during pregnancy increases risk for the development of alcohol use and abuse and also early onset of alcohol-related disorders (Alati et al., 2006). Laboratory animals with in utero exposure to alcohol also exhibit an increase in ethanol preference and consumption (Chotro et al., 2007), in addition to behavioral changes associated with drug abuse (Brocardo et al., 2011). While these studies provide clear evidence showing prenatal ethanol exposure to stimulate excess alcohol consumption in the offspring, the neurochemical mechanisms underlying this phenomenon still remain to be characterized.
Studies to date have focused primarily on the neurotransmitters, dopamine (DA), serotonin (5-HT), and gamma-aminobutyric acid (GABA), which in adult animals are known to be stimulated by ethanol and have a role in mediating ethanol acquisition, consumption, reward, reinforcement and tolerance (De Witte, 1996; Vengeliene et al., 2008; Tabakoff et al., 2009). The evidence obtained with prenatal exposure shows that ethanol, particularly at moderate to high doses, reduces the functioning of these neurochemical systems in postnatal and pubertal offspring, causing disturbances that may contribute to excessive ethanol consumption (Chotro et al., 2007). These effects of ethanol in utero depend greatly on blood ethanol concentration (BEC) in the dams, with higher concentrations that raise BEC to between 300 and 600 mg/dl tending to reduce cell density, cell proliferation, neurogenesis and brain volume (Miller, 1995a; Livy et al., 2003; Gil-Mohapel et al., 2010) and lower concentrations causing little change or, in one case, a stimulatory effect on cell proliferation at a BEC of 150 mg/dl (Maier and West, 2001; Mooney and Miller, 2010; Boehme et al., 2011). These effects may also be area dependent, with ethanol generally causing profound changes in the hippocampus, cortex and olfactory bulb but not the thalamus (Miller, 1995b; Livy et al., 2003; Gil-Mohapel et al., 2010). The few studies conducted in the hypothalamus and striatum found a BEC of 300 mg/dl to reduce brain volume in the preoptic area but have no effect on brain volume and cell number in the nucleus accumbens (NAc) (Ahmed et al., 1991; Lawrence et al., 2012). These findings in extra-hypothalamic areas underscore the dose-dependent as well as region-dependent nature of the changes produced by prenatal ethanol exposure in brain development.
In recent years, there has been a surge in research on orexigenic peptides in the hypothalamus and limbic system, which in addition to food intake are found to stimulate the drinking of ethanol (Morganstern et al., 2011). These peptides include enkephalin (ENK) expressed in the hypothalamic paraventricular nucleus (PVN) and NAc, galanin (GAL) also expressed in the PVN, and orexin (OX) expressed in the perifornical lateral hypothalamus (PFLH). Studies in adult animals show that the endogenous peptides measured in these areas are generally stimulated by the consumption and injection of ethanol (Oliva and Manzanares, 2007; Chang et al., 2010; Barson et al., 2011; Morganstern et al., 2011), while central injections of these peptides in turn significantly stimulate the drinking of ethanol, with their receptor antagonists producing the opposite effect (Barson et al., 2011). This evidence, suggesting the existence of a positive feedback loop in these brain sites (Morganstern et al., 2011), contrasts with that obtained in the arcuate nucleus (ARC), where GAL is unaffected by ethanol and neuropeptide Y (NPY) is negatively affected by ethanol (Barson et al., 2011). Whereas there are few studies of these peptide systems in offspring exposed in utero to ethanol, the evidence obtained so far shows that prenatal ethanol can stimulate the expression and levels of ENK in whole hypothalamus and NAc, as shown in preweanling and pubertal offspring (Druse et al., 1999; Lugo et al., 2006) but has little impact on NPY in whole hypothalamus (Dembele et al., 2006). These findings give some indication that the effects of prenatal ethanol on peptide expression in the offspring may be similar to those produced by ethanol intake in adult animals, raising the possibility that ethanol may alter in utero development of neurons expressing these orexigenic peptides known to stimulate ethanol drinking.
This report of prenatal ethanol exposure focused on the hypothalamus and NAc, structures known have important functions in controlling consummatory behavior. It examined the development in these areas of the specific peptide systems that promote ethanol consumption, and in light of evidence in extra-hypothalamic areas showing suppressive effects at higher doses, this investigation tested two lower doses of ethanol, 1 and 3 g/kg/day. From embryonic days 9–21, the dams were gavaged daily with one of these doses, and the offspring were examined at birth and postnatal day 15 (P15) for changes in the expression of ENK, GAL and OX and also the generation of new neurons that express these peptides. The specific hypothesis tested in this report is that prenatal ethanol exposure, at low concentrations that raise BEC to <100 mg/dl, increases neurogenesis in these peptide systems and that this phenomenon occurs specifically in the PVN, PFLH and NAc, where these peptides are known to have a role in stimulating the consumption of ethanol.
EXPERIMENTAL PROCEDURES
Animals
Time-pregnant, Sprague–Dawley rats (220–240 g) from Charles River Breeding Laboratories (Hartford, CT, USA) arrived on embryonic day 5 (E5) and were individually housed in plastic cages, in a fully accredited AAALAC facility (22 °C, 12:12-h light–dark cycle with lights off at 2 pm), according to institutionally approved protocols as specified in the NIH Guide to the Care and Use of Laboratory Animals and with approval of The Rockefeller University Animal Care and Use Committee. Rats were acclimated to laboratory conditions until E9, when they were given ethanol, either 1 g/kg/day (30% solution) or 3 g/kg/day (40% solution), or a control solution (maltose-dextrin made isocaloric to 35% ethanol solution). The daily dose of ethanol was split in half, with all rats gavaged two times daily, 4 h before the end of the light cycle and 4 h into the dark cycle, until E21. Blood ethanol concentration, measured 2 h after gavage, yielded 30 mg/dl in the 1 g/kg ethanol group and 90 mg/dl in the 3 g/kg ethanol group, similar to a previous report (Qiang et al., 2002). Rodent chow (LabDiet Rodent Chow 5001, St. Louis, MO, USA) and water were available ad libitum. Food intake was measured three times per week, and body weight was recorded weekly. There were no differences between control and ethanol-treated dams in their body weight (250–270 g) or chow intake (65–75 kcal/day). At birth, on P0, litters were culled to n = 8. Litters of the ethanol and control dams were similar in terms of size (from 9 to 15), body weight (5.7–6.8 g), and male/female ratio (1.1/1.0), with no spontaneous abortions observed in either group. Only male offspring were tested, with 1 male pup taken from each litter and the number of rats/group (n = 4–8) equal to the number of litters. Offspring were sacrificed by rapid decapitation, and whole brain or dissected brain tissue was collected for further analyses, all of which were performed by an evaluator blind to the experimental conditions.
Brain dissections
Immediately after sacrifice, the brain was placed in a matrix with the ventral surface facing up, and six 0.5 mm coronal sections were made, yielding three slices used for microdissection. Using the middle optic chiasm as a main boundary, a slice was made at the level of A 3.8–3.5 mm for microdissection of the PVN and at A 2.9–2.3 mm for the PFLH and ARC using the stereotaxic atlas of a 10-day-old rat brain for guidance (Sherwood and Timiras, 1970) as previously described (Chang et al., 2008). A final slice was made at A 6.5–5.6 mm for microdissection of the nucleus accumbens core (NAcC) and nucleus accumbens shell (NAcSh). The NAcC was dissected bilaterally as an oval, lateral to the lateral ventricle and medial to the lateral stripe of the striatum. The NAcSh was dissected bilaterally in a moon shape, with the dorsal tip beginning at the lateral ventricle, the medial aspect at the semilunar nucleus, and the ventral edge along the ventral pallidum.
Quantitative real-time PCR
Total RNA from each microdissected sample was purified and cDNA reverse transcribed as previously described (Barson et al., 2012). Resulting A260/A280 ratios of total RNA from all the animals ranged between 2.03 and 2.12. qRT-PCR was then performed as described (Barson et al., 2012). Levels of target gene expression were quantified relative to the level of cyclophilin, using the relative quantification method, and the average Ct values for the endogenous control vs. target genes differed by no more than four Ct. The primers, designed with ABI Primer Express V.1.5a software from published sequences, were: (1) cyclophilin: 5′-GTGTTCTTCGACATCACGGCT-3′ (forward) and 5′-CTGTCTTTGGAACTTTGTCTGCA-3′ (reverse); (2) ENK: 5′-GGACTGCGCTAAATGCAGCTA-3′ (forward) and 5′-GTGTGCA TGCCAGGAAGTTG-3′ (reverse); (3) GAL: 5'-TTCCCACCACTG CTCAAGATG-3' (forward) and 5'-TGGCTGACAGGGTTGCAA-3' (reverse); and (4) OX: 5′-AGATACCATCTCTCCGGATTGC-3′ (forward) and 5′-CCAGGGAACCTTTGTAGAAGGA-3′ (reverse). The concentration of the ENK, GAL and OX primers was 100 nM and of the CYC primer was 200 nM. These measurements of mRNA levels were performed in the specific areas where the peptide-expressing neurons are most concentrated, namely, GAL in the PVN, ENK in the PVN and NAc, and OX in the PFLH.
Radiolabeled in situ hybridization histochemistry
The mRNA levels of ENK, GAL and OX were measured, as previously described (Chang et al., 2008, 2010; Morganstern et al., 2010b), using radiolabeled in situ hybridization histochemistry (RISH) in the P15 offspring exposed in utero to the control solution or 1 g/kg or 3 g/kg dose of ethanol (n = 6–8/group). Briefly, immediately after sacrifice, whole brains were removed, fixed in 4% paraformaldehyde phosphate-buffer (0.1 M, pH 7.2) at 4 °C for 2 days, and then cryoprotected in 20% sucrose at 4 °C for 3 days. Thirty-micrometer alternate free-floating coronal sections were used for RISH, as previously described (Chang et al., 2010). Gene expression level, expressed as optical density, was determined with a computer-assisted microdensitometry of autoradiographic images on the MCID image analysis system (Image Research, Inc., St. Catherines, ON, Canada) as described (Reagan et al., 2004; Chang et al., 2008). Gray-level/optical density calibrations were performed with a calibrated film-strip ladder (Imaging Research Inc.) for optical density. This was plotted as a function of microscale calibration values. All subsequent optical density values of digitized autoradiographic images fell within the linear range of the function. The values obtained represent the average of measurements taken from 10 sections per animal. Within each section, the optical density for the nucleus was recorded, from which the background optical density from a same-size area in the corpus callosum was subtracted. Mean value of the optical density for the PVN, PFLH and NAc, in control, 1 g/kg ethanol, and 3 g/kg ethanol groups in each experiment, was compared and reported as percentage of the control group as previously described (Chang et al., 2008).
Digoxigenin-labeled in situ hybridization histochemistry
For better visualization, in situ hydridization histochemistry with a digoxigenin-labeled probe (digoxigenin-labeled in situ hybridization; DIG-ISH) was performed to reveal the anatomical distribution at birth (P0) of changes in peptide expression in the control, 1 g/kg ethanol and 3 g/kg dose ethanol groups (n = 6–8/group) as described (Chang et al., 2008, 2010). Briefly, digoxigenin-labeled antisense RNA probes and 30 μm free-floating cryostat coronal sections were used for in situ hybridization histochemistry. AP-conjugated sheep anti-digoxigenin Fab fragments (1:1000; Roche Diagnostics, Indianapolis, IN, USA) and NBT/BCIP (Roche Diagnostics) were used to visualize the signal. Sections were viewed on a Leitz microscope (10× objective). The images were captured with a Nikon DXM 1200 digital camera (Nikon, Tokyo, Japan) and analyzed using Image-Pro Plus software (Version 4.5, Media Cybernetics Inc., Silver Spring, MD, USA) on a gray-value scale from 1 to 255. In each animal, 8–10 sections at the same level were used to examine each nucleus of interest, and the entire nucleus was outlined and analyzed. The population density was used to determine the cell density in these areas. Before measurements, a threshold for each nucleus was established. Using the selected sections, this threshold was set by matching the number of cells counted by the software in a defined area with the number of cells counted manually in that same area. This method, which was the same for all experiments and brain areas, yielded different threshold values (average of thresholds obtained within the same area in the ten sections) for the different brain areas, experiments and measurements of cells. This semi-quantitative procedure allowed us to count the number of neurons in a specific area, which were then expressed as the cell density (number of cells/μm2), reflecting the density of mRNA containing cells. In all analyses, the cell number was counted only on one plane in each section, and only those cells containing a nucleus in the plane (>10 μm2) were counted, thereby excluding fractions of cells. The average cell density for the different groups was then compared and statistically analyzed.
5-Bromo-2-deoxyuridine (BrdU) immunofluorescence histochemistry
To label proliferating cells in the hypothalamus and NAc of the embryo, the control, 1 g/kg ethanol and 3 g/kg ethanol dams were given intraperitoneal (i.p.) injections of BrdU (20 mg/kg, Sigma, St. Louis, MO, USA) in 0.9% NaCl and 0.007 N NaOH every 8 h from E11–E14, the period of peak cell birth in the rat hypothalamus (Altman and Bayer, 1978), and from E17–E20, the period of peak cell birth in the NAc (Bayer et al., 1993). The offspring (n = 10/age/group) were sacrificed at different postnatal ages (P0, P15, P25), and their brains were processed for revealing BrdU by immunofluorescence histochemistry as previously reported (Chang et al., 2008).
Immunofluorescence histochemistry
Single- and double-labeling of immunofluorescence were used to examine BrdU, glial fibrillary acidic protein (GFAP), and coexistence of BrdU with NeuN in the PVN and NAc and of BrdU with OX in the PFLH in P15 offspring as previously described (Chang et al., 2008). Density of immunofluorescent cells was quantified with Image-Pro Plus software (Version 4.5, Media Cybernetics Inc., Silver Spring, MD, USA) as described below and reported as cell density (objects/μm2). Double-labeled cells were counted and reported as percentage of total single-labeled cells.
Double-labeling of digoxigenin in situ hybridization of peptide with BrdU immunofluorescence histochemistry
To determine whether BrdU-labeled cells express the peptides, GAL and ENK, DIG-ISH of these peptides combined with BrdU immunofluorescence histochemistry was performed as previously described (Chang et al., 2008). Double-labeled cells were counted and expressed as percentage of total single-labeled cells (Chang et al., 2008).
Antibodies
Antibodies used were: rat anti-BrdU monoclonal antibody (1:200, Novus Biologicals, Inc., CO, USA); mouse anti-NeuN and mouse anti-GFAP monoclonal antibody (1:50, Chemicon-Millipore, Temecula, CA, USA); goat anti-OX polyclonal antibody (1:200, Santa Cruz, CA, USA); and fluorescence-conjugated secondary antibodies (1:200, Jackson Immunoresearch Laboratories, Inc., PA, USA).
Terminal uridine nucleotide end labeling (TUNEL)
To determine whether maternal exposure to ethanol leads to apoptosis, TUNEL was used to label DNA strand breaks for detection of apoptotic cells in the offspring at P15, by which time hypothalamic apoptosis is generally completed. Using an In Situ Cell Death Detection Kit (Fluorescein, Roche Applied Science, Indianapolis, IN, USA), TUNEL, with positive and negative controls, was performed with 30 μm free-floating coronal sections following the manufacturer's protocol.
Semiquantification of digoxigenin-labeled in situ hybridization and immunofluorescence
The density of ENK-, GAL- and OX-expressing neurons and of BrdU, GFAP, NeuN and OX-immunoreactive cells was measured by semiquantification, with Image-Pro Plus (Version 4.5, Cybernetics Inc., Silver Spring, MD, USA) as described (Chang et al., 2008) and it is expressed in terms of density (number of objects/μm2).
Statistical analysis
Comparisons between the three groups, control, 1 g/kg ethanol and 3 g/kg ethanol, were made using a univariate analysis of variance, followed-up by Tukey's post-hoc tests when appropriate. The criterion for significance was p < 0.05, and all significance values greater than this are reported as ns (not significant). Data are reported as mean ± standard error of the mean (SEM).
RESULTS
Experiment 1: Effect of prenatal ethanol on peptide expression in P15 offspring as measured using qRT-PCR
This experiment examined the effect of in utero ethanol exposure on the expression of the orexigenic peptides, ENK, GAL and OX, in specific brain areas of the P15 offspring, with time-pregnant dams gavaged daily from E9 to E21 with either ethanol, at 1 g/kg or 3 g/kg, or the isocaloric control solution. Measurements of peptide gene expression in the hypothalamus and NAc revealed a significant difference between the groups (n = 8/group) (Fig. 1). This was observed for ENK (F(2,24) = 33.42, p < 0.001) and GAL (F(2,24) = 12.66, p < 0.001) in the PVN, for OX in the PFLH (F(2,24) = 18.5, p < 0.001), and for ENK in the NAcC (F(2,24) = 15.7, p < 0.001). Post-hoc analyses showed that the ethanol-exposed offspring, as compared to control, had significantly higher expression of these peptides in the PVN, PFLH and NAcC (p < 0.05 for both ethanol groups vs. control) (Fig. 1), with this increase ranging from 28% to 48% and occurring with similar magnitude at the two doses (ns for 1 vs. 3 g/kg). There was no change evident in the ARC, with measurements of either ENK (F(2,24) = 0.82, ns) or GAL (F(2,24) = 0.35, ns), or in the NAcSh, with measurements of ENK (F(2,24) = 0.18, ns). This provides the first evidence that ethanol during pregnancy, at low to moderate doses, has a stimulatory effect on the expression of these three orexigenic peptides and that this effect is site-specific in nature and long lasting, persisting for at least 15 days postnatally in the absence of ethanol.
Fig. 1.
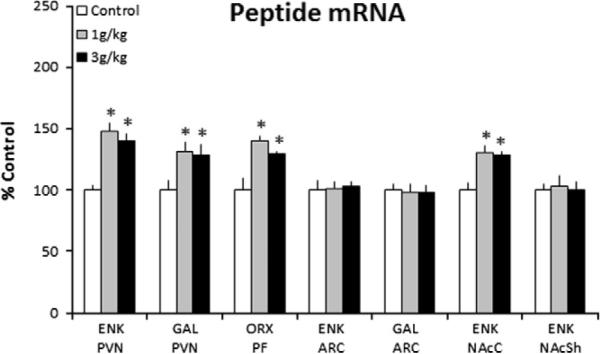
Prenatal ethanol increases peptide mRNA in the P15 offspring prenatally exposed to 1 g/kg or 3 g/kg ethanol relative to control (n = 8/group), as assessed by quantitative real-time PCR. Peptide mRNA in ethanol vs. control groups expressed as percent change relative to control. Data are mean ± SEM,*p < 0.05 vs. control. Abbreviations used in the figures: aca, anterior commissure; ARC, arcuate nucleus; ENK, enkephalin; F, fornix; GAL, galanin; NAcC, nucleus accumbens core; NAcSh, nucleus accumbens shell; OX, orexin; PVN, hypothalamic paraventricular nucleus; PFLH, perifornical lateral hypothalamus.
Experiment 2: Effect of prenatal ethanol on peptide expression in P15 offspring as measured using ISH
This experiment, with similar test procedures as Experiment 1, measured peptide gene expression using ISH with radiolabeled probes in different brain areas of P15 offspring exposed to ethanol (1 g/kg or 3 g/kg) or control solution. Measurements of peptide gene expression in the hypothalamus and NAc revealed a significant difference between the groups (n = 6–8/group) (Fig. 2A), as illustrated in the photomicrographs (Fig. 2B). This was observed for ENK (F(2,21) = 12.32, p < 0.001) and GAL (F(2,21) = 21.51, p < 0.001) in the PVN, for OX in the PFLH (F(2,21) = 7.07, p < 0.001), and for ENK in the NAcC (F(2,20) = 9.81, p < 0.001). Post-hoc analyses showed that the ethanol-exposed offspring, as compared to control, had significantly higher expression of these peptides in the PVN, PFLH and NAcC (p < 0.05 for both ethanol groups vs. control) (Fig. 2A), with this increase ranging from 25% to 56% and occurring with similar magnitude at the two doses (ns for 1 vs. 3 g/kg). There was no change evident in the ARC, with measurements of either ENK (F(2,20) = 0.01, ns) or GAL (F(2,20) = 0.10, ns), or in the NAcSh, with measurements of ENK (F(2,19) = 1.21, ns). These data confirm our findings in Experiment 1 and underscore the anatomical specificity and long-lasting nature of the effect of in utero ethanol exposure.
Fig. 2.

Prenatal ethanol increases peptide mRNA in the P15 offspring prenatally exposed to 1 g/kg or 3 g/kg ethanol relative to control (n = 6–8/group), as assessed by radiolabeled in situ hybridization. (A) Peptide mRNA in ethanol vs. control groups expressed as percent change relative to control. Data are mean ± SEM, *p < 0.05 vs. control. (B) Photomicrographs illustrating this effect of 1 g/kg ethanol vs. control group on ENK, GAL and OX expression. See legend to Fig. 1 for abbreviations.
Experiment 3: Impact of prenatal ethanol on peptide expression in P0 offspring
The next question to address is whether these effects of prenatal ethanol on peptide expression are detected immediately at birth (P0), possibly reflecting events occurring in utero. Pregnant dams were gavaged with ethanol or control solution, and their offspring's brains (n = 6–8/group) were analyzed using DIG-ISH. Analyses in the PVN revealed significant differences between groups for ENK (F(2,20) = 4.95, p < 0.05) and GAL (F(2,20) = 5.43, p < 0.05). While the control offspring had very few detectable ENK+ or GAL+ neurons in the PVN, the ethanol-exposed offspring at birth showed a significantly greater density of neurons expressing these peptides, specifically in the medial parvocellular region of the PVN (p < 0.05 for both ethanol groups vs. control) (Fig. 3A and B). There were also differences in OX expression in the PFLH (F(2,20) = 7.08, p < 0.05) and ENK in the NAc (F(2,11) = 7.98, p < 0.01), with prenatal ethanol at both doses increasing the density of these neurons compared to control (p < 0.05) (Fig. 3A and B). This stimulatory effect of ethanol, a 15–48% increase, was similar in magnitude at both doses for all peptides (ns for 1 vs. 3 g/kg). This increase in neuronal density at P0 suggests that prenatal ethanol affects in utero development of these peptide systems in the hypothalamus and mesolimbic region.
Fig. 3.

Prenatal ethanol increases the density of peptide-expressing neurons in P0 offspring prenatally exposed to 1 g/kg or 3 g/kg ethanol relative to control (n = 6–8/group), as assessed by in situ hybridization histochemistry with digoxigenin-labeled probes. (A) Density of peptide-expressing neurons. Data are mean ± SEM, *p < 0.05, vs. control. (B) Photomicrographs illustrating this effect of 1 g/kg ethanol vs. control group on ENK, GAL and OX (10× magnification). Scale bar in left panel = 200 μm.
Experiment 4: Effect of in utero ethanol on cell generation in P0 and P15 offspring
To determine whether prenatal ethanol stimulates the generation of new cells during pregnancy, the offspring (n = 4–6/group) of dams gavaged daily with ethanol or control solution were injected with the cell proliferation marker, BrdU, for analyses of the hypothalamus or NAc. They were examined at two different ages, P0 when many cells are still undergoing apoptosis and P15 when postnatal development in the hypothalamic PVN and PFLH has primarily ceased (Ifft, 1972). The P0 offspring exhibited significant differences in the density of BrdU+ cells in the PVN (F(2,11) = 20.50, p < 0.0001), PFLH (F(2,11) = 6.43, p < 0.05), and NAc (F(2,11) = 76.68, p < 0.0001) (Fig. 4A), as illustrated in the photomicrographs (Fig. 4B), but no difference in the ARC (F(2,11) = 0.002, ns). Prenatal ethanol significantly increased, by 25%, the density of BrdU+ cells in these areas compared to control (p < 0.05), with the two doses having a similar magnitude of effect (ns for 1 vs. 3 g/kg). This stimulatory effect of ethanol was similarly seen in the P15 offspring (Table 1), with a significant difference in the density of BrdU+ cells evident in the PVN (F(2,17) = 70.92, p < 0.0001), PFLH (F(2,17) = 60.29, p < 0.001), and NAcC (F(2,17) = 33.58, p < 0.0001), reflecting a significant increase in each area compared to control (p < 0.05) that was similar in magnitude at the two ethanol doses (ns for 1 vs. 3 g/kg). These changes were not evident in the ARC (F(2,17) = 0.002, ns) or NAcSh (F(2,17) = 0.98, ns), nor were they detected in another extra-hypothalamic area examined, the hippocampus (F(2,17) = 0.79, ns). These results underscore the robustness and anatomical specificity of the stimulatory effect of prenatal ethanol on the generation of new cells and show once again its persistent nature, with many of the newly generated cells at birth still evident two weeks later in the absence of ethanol.
Fig. 4.

Prenatal ethanol stimulates cell proliferation in the hypothalamic and limbic areas of P0 offspring prenatally exposed to 1 g/kg or 3 g/kg ethanol relative to control (n = 4–6/group). (A) Prenatal ethanol increases the density of BrdU+ cells. Data are mean ± SEM, *p < 0.05. (B) Photomicrographs illustrating this effect of 1 g/kg ethanol vs. control group in the PVN, PFLH and NAc (10× magnification). Scale bar in left panel = 200 μm. See legend to Figs. 1 and 2 for abbreviations.
Table 1.
Prenatal ethanol stimulates cell proliferation in the PVN, PFLH and NAc of P15 offspring prenatally exposed to 1 g/kg or 3 g/kg ethanol relative to control (n = 6–8/group), as indicated by a significant increase in BrdU+ cells in ethanol vs. control offspring.
| Brain areas | BrdU+ cell density (objects/μm2 × 10−4) |
||
|---|---|---|---|
| Control | 1 g/kg | 3 g/kg | |
| PVN | 12.83 ± 2.50 | 18.37 ± 0.39* | 15.57 ± 0.21* |
| PFLH | 12.72 ± 0.30 | 18.95 ± 0.55* | 16.57 ± 0.23* |
| ARC | 4.81 ± 0.62 | 4.79 ± 0.25 | 4.71 ± 0.28 |
| NAcC | 2.93 ± 0.07 | 3.69 ± 0.62* | 3.87 ± 0.07* |
| NAcSh | 0.50 ± 0.01 | 0.52 ± 0.01 | 0.52 ± 0.01 |
| Hippocampus | 3.69 ± 0.19 | 3.76 ± 0.16 | 3.81 ± 0.20 |
Experiment 5: Phenotype of BrdU+ cells stimulated by ethanol exposure in P15 offspring
To determine the phenotype of ethanol-stimulated BrdU+ cells in the hypothalamus and NAc, this experiment performed single- or double-labeling immunofluorescence with antibodies against NeuN (marker for mature neurons), GFAP (marker for astrocytes), and TUNEL (marker for apoptotic cell death). The P15 offspring (n = 4–5/group) of dams gavaged with ethanol compared to the control solution exhibited a marked increase in the density of BrdU+ cells in the PVN, PFLH and NAc that double-labeled NeuN. This was indicated by the increased percentage of double-labeled cells relative to total number of single-labeled cells immunoreactive for BrdU or NeuN (Fig. 5A), as illustrated in the photomicrographs (Fig. 5B). The number of BrdU+/NeuN+ cells was significantly different between groups in the PVN relative to BrdU (F(2,11) = 126.07, p < 0.05) and NeuN (F(2,11) = 29.82, p < 0.05), the PFLH relative to BrdU (F(2,11) = 50.02, p < 0.05) and NeuN (F(2,11) = 38.12, p < 0.05), and the NAcC relative to BrdU (F(2,11) = 7.11, p < 0.05) and NeuN (F(2,11) = 11.53, p < 0.05). There was no difference in the NAcSh relative to BrdU (F(2,11) = 2.33, ns) or NeuN (F(2,11) = 2.02, ns). The two doses of ethanol were both effective in significantly increasing, by 18–36%, the number of double-labeled cells (p < 0.05) in these areas, with a dose-dependent change evident only in the PFLH where the 1 g/kg dose produced a significantly greater increase than the 3 g/kg dose in density of BrdU+/NeuN+ cells relative to BrdU (p < 0.0001) and NeuN (p < 0.05). In contrast to these changes in NeuN+ cells in P15 offspring, prenatal ethanol produced no change in the density of GFAP cells in the PVN (F(2,11) = 0.02, ns), PFLH (F(2,11) = 0.62, ns), or NAc (F(2,11) = 0.02, ns) (Table 2). It also had no effect on TUNEL+ cells, with analyses in the control P15 offspring revealing no immunoreactive cells in the PVN or PFLH and a few faint TUNEL+ cells in the NAc, and with no significant differences between ethanol-exposed and control offspring in the PVN (F(2,11) = 0.01, ns), PFLH (F(2,11) = 0.06, ns), or NAc (F(2,11) = 0.02, ns). Together, these data demonstrate that in utero ethanol exposure, at low to moderate doses, has a significant, stimulatory effect on the generation and development of new neurons in the hypothalamus and accumbens but has little impact on gliogenesis or apoptosis in these areas.
Fig. 5.

Prenatal ethanol stimulates neurogenesis in the hypothalamic and limbic areas in P15 offspring prenatally exposed to 1 g/kg or 3 g/kg ethanol relative to control (n = 4–5/group). (A, top) Prenatal ethanol increases the percentage of BrdU+/NeuN+ cells relative to total number of single-labeled BrdU+ cells in the PVN, PFLH and NAcC of the offspring from dams receiving i.p. injection of BrdU from E11–E14 or E17–E20. (A, bottom) Prenatal ethanol increases the percentage of BrdU+/NeuN+ cells relative to total number of single-labeled NeuN+ cells. Data are mean ± SEM, *p < 0.05. (B) Photomicrographs demonstrate this effect for 1 g/kg ethanol vs. control group, illustrating BrdU+ cells (red), NeuN+ cells (green) and BrdU+/NeuN+ cells (yellow) as indicated with arrowheads for double-labeled cells in the PVN, PFLH and NAc (20× magnification). Insets are double-labeled BrdU+/NeuN+cells marked with dashed square in main images (40× magnification). Scale bar in left panel = 100 μm. See legends to Figs. 1 and 2 for abbreviations.
Table 2.
Prenatal ethanol has no effect on gliogenesis in the PVN, PFLH or NAc of P15 offspring prenatally exposed to 1 g /kg or 3 g/kg ethanol relative to control (n = 4–6/diet group), as indicated by no change in GFAP+ cells, a marker for astrocytes.
| Brain areas | GFAP+ cell density (objects/μm2 × 10−4) |
||
|---|---|---|---|
| Control | 1 g/kg | 3 g/kg | |
| PVN | 1.51 ± 0.12 | 1.52 ± 0.17 | 1.53 ± 0.07 |
| PFLH | 0.91 ± 0.08 | 0.91 ± 0.06 | 0.94 ± 0.04 |
| NAc | 2.35 ± 0.19 | 2.39 ± 0.16 | 2.40 ± 0.16 |
Data are mean ± -SEM. See legend to Fig. 1 for abbreviations
Experiment 6: Impact of in utero ethanol exposure on genesis of peptide-expressing neurons in P15 or P25 offspring
To determine whether the BrdU+ neurons stimulated by prenatal ethanol exposure can express or synthesize the orexigenic peptides that are also stimulated, DIG-ISH combined with BrdU immunofluorescence or double-labeling immunofluorescence was used to examine the offspring (n = 4–5/group). As in Experiment 3, the dams were gavaged daily with ethanol or control solution and were also injected with BrdU, for analyses of the hypothalamus or NAc. Whereas the control offspring showed almost no colocalization of peptides with BrdU, the prenatal ethanol-exposed offspring exhibited a significant increase in the percentage of double-labeled cells relative to single-labeled peptide+ or BrdU+ cells. As illustrated in Figs. 6 and 7, there were significant group differences in the PVN, with measurements of ENK+/BrdU+ cells relative to single-labeled ENK+ (F(2,11) = 11.53, p < 0.05) and BrdU+ (F(2,11) = 9.73, p < 0.05)cells and of GAL+/BrdU+ cells relative to single-labeled GAL+ (F(2,11) = 36.65, p < 0.005) and BrdU+ (F(2,11) = 6.79, p < 0.05) cells in P15 offspring. Significant differences were also evident in the PFLH, with measurements of OX+/BrdU+ cells relative to single-labeled OX+ (F(2,11) = 54.64, p < 0.05) and BrdU+ (F(2,11) = 48.47, p < 0.05) cells in P25 offspring. While not detected in the NAcSh, the double-labeled ENK+/BrdU+ cells were evident in the NAcC of P15 offspring, where there were significant differences in the number of double-labeled cells relative to single-labeled ENK+ (F(2,14) = 34.12, p < 0.05) and BrdU+ (F(2,14) = 18.65, p < 0.05) cells (Fig. 6). Whereas both doses of ethanol had a similar, stimulatory effect on GAL+/BrdU+ neurons in the PVN (p < 0.05 vs. control; ns for 1 vs. 3 g/kg) and ENK+/BrdU+ neurons in the PVN and NAcC (p < 0.05 vs. control; ns for 1 vs. 3 g/kg), the increase in OX+/BrdU+ neurons in the PFLH (p < 0.05 vs. control) was significantly greater with the 1 g/kg ethanol dose compared to the 3 g/kg ethanol dose (p < 0.05). Together, these results demonstrate that in utero ethanol at these low to moderate doses can stimulate the generation of new neurons in the PVN, PFLH and NAc that, as shown in postnatal offspring in the absence of ethanol, have the specific phenotype of expressing the orexigenic peptides known to be stimulated by ethanol in adult animals.
Fig. 6.

Prenatal ethanol stimulates neurogenesis of peptide-expressing systems in hypothalamic and limbic areas in P15 offspring prenatally exposed to 1 g/kg or 3 g/kg ethanol relative to control (n = 4–5/group). This is demonstrated by a significant increase in the percentage of double-labeled, BrdU+/peptide+ neurons relative to total number of single-labeled BrdU+ neurons (left) or peptide+ (right) neurons. Data are mean ± SEM, *p < 0.05, vs. control. See legend to Fig. 1 for abbreviations.
Fig. 7.

Photomicrographs illustrate the stimulatory effect of 1 g/kg prenatal ethanol on neurogenesis of peptide-expressing neurons in the hypothalamus. (A) Photomicrographs of ethanol vs. control P15 offspring showing cells in the PVN that co-label BrdU (red) and ENK (black) mRNA, as indicated by the arrowheads. Insets are double-labeled BrdU+/ENK+ neurons marked with dashed square in main images (20× magnification). (B) Photomicrographs of ethanol vs. control P25 offspring showing cells in the PFLH that co-label BrdU (red) and OX (green), with BrdU+/OX+ cells stained yellow, as indicated by the arrowheads. Insets are double-labeled BrdU+/OX+ neurons marked with dashed square in main images (40× magnification). Scale bar in left panel = 200 μm.
DISCUSSION
This study provides the first evidence that prenatal exposure to ethanol, at 1 or 3 g/kg/day, stimulates expression of ENK specifically in the PVN and NAcC, but not the NAcSh, of postnatal offspring, consistent with studies showing a higher dose of ethanol (4.5 g/kg) to increase ENK levels in whole hypothalamus and accumbens of adolescent or adult offspring (Druse et al., 1999; Lugo et al., 2006). It additionally shows a similar increase in mRNA levels of GAL in the PVN and OX in the PFLH and reveals these effects both at birth and P15. This stimulation of all three peptides in these areas observed using two different techniques, qRT-PCR and ISH, underscores the broader scope of the changes produced by in utero ethanol exposure and demonstrates how its effects are the same as those observed with oral ethanol in adult rats (Morganstern et al., 2010a; Barson et al., 2011). In fact, they occur in precisely the same areas within the PVN and PFLH, with ethanol in newborn offspring as in adults increasing the number and density of neurons expressing GAL and ENK in the medial parvocellular region of the PVN and OX in both the medial and lateral regions of the PFLH. Whereas PVN GAL neurons like ENK responded similarly to both doses of ethanol, the PFLH OX neurons were differentially responsive, with the 1 g/kg dose consistently producing a stronger stimulation than the 3 g/kg dose. This may be related to the finding in adult rats, that OX expression is stimulated by acute ethanol more strongly at 0.75 g/kg than at 2.5 g/kg (Morganstern et al., 2010a), possibly reflecting a greater inhibitory control of these OX neurons by other neurotransmitters, such as DA and GABA (Li and van den Pol, 2005; Xie et al., 2006), which are stimulated by ethanol (De Witte, 1996; Vengeliene et al., 2008). This increase in peptide-expressing neurons at birth, and its persistence in the absence of ethanol in P15 offspring, indicates that ethanol exposure during gestation affects in utero development of these peptide systems and produces neuronal changes that are long-lasting.
The results indicate further that the mechanism underlying the prenatal ethanol-induced change in peptides involves an increased generation of new cells, specifically neurons in the PVN, PFLH and NAcC. As revealed by a significant increase in the number of single-labeled BrdU+ cells and of BrdU+ cells that double-label NeuN, this effect that was evident at both doses (1 and 3 g/kg) and both ages (P0 and P15) tested. In contrast, prenatal ethanol had no effect on the number of GFAP+ cells in these areas, indicating no change in gliogenesis, consistent with some studies in other brain areas (Ramos et al., 2002; Uban et al., 2010), or on TUNEL+ cells, indicating no effect on apoptosis as previously found in hippocampus and cortex (Fakoya and Caxton-Martins, 2006; Rubert et al., 2006). These newly generated neurons stimulated by in utero ethanol exposure expressed the peptides, as demonstrated by an increased density of BrdU+ cells that co-labeled ENK in the PVN and NAcC, GAL in the PVN, and OX in the PFLH, and they were evident in precisely the same areas where peptide expression is stimulated by ethanol intake in adults (Barson et al., 2011; Morganstern et al., 2011). Whereas studies with moderate to high doses of ethanol have sometimes described a reduction or no change in the volume of these brain areas (Rudeen, 1986; Miller, 1995b; McGivern et al., 1998; Livy et al., 2001), it will be interesting to determine with such measurements whether the stimulatory effect of lower ethanol doses on neurogenesis is associated with an increase in the volume of these structures, as previously shown in hippocampus (Gonzalez-Burgos et al., 2006). A particularly intriguing finding in this study is the absence of effects of prenatal ethanol on the ENK- and GAL-expressing neurons in the ARC. This clearly demonstrates anatomical specificity of ethanol's action, as similarly shown in adult rats (de Gortari et al., 2000; Barson et al., 2011), and it may help to elucidate the mechanism through which low to moderate ethanol stimulates neurogenesis. For example, the PVN and ARC respond very differently to dietary fat and circulating lipids that are elevated by ethanol similar to fat (Barson et al., 2011), with ENK and GAL in the PVN but not ARC increased by consumption of a high-fat diet and positively related to the lipids (Chang et al., 2007; Barson et al., 2010). Fatty acids are found to stimulate neuronal proliferation and differentiation (Maekawa et al., 2009) and increase ENK transcription in PC12 cells (Parab et al., 2007). The PVN and ARC are also dissimilar in their responsiveness to female steroids (Barson et al., 2010), which are elevated by ethanol and dietary fat (Emanuele et al., 2001; Barson et al., 2010) and can stimulate neurogenesis (Fowler et al., 2008), and also to neurotrophins (Steinberg et al., 2006), which are affected by ethanol (Heaton et al., 2003) and promote neurogenesis (Schaffer and Gage, 2004). These studies provide examples of mechanisms, involving fatty acids, steroids and neurotrophins, which may be involved in mediating the effects of prenatal ethanol on neuronal development.
The increased consumption of and preference for alcohol in the offspring produced by prenatal ethanol exposure, particularly at low to moderate doses, has been attributed to changes in the dopaminergic system and HPA axis (Alati et al., 2006; Chotro et al., 2007). The present results, revealing a stimulation of neurogenesis in the peptide systems and a persistent increase in the density of peptide-expressing neurons, lead us to propose a novel mechanism that may also mediate this phenomenon. In addition to central injection studies showing ENK, GAL and OX to each stimulate ethanol drinking in adult rats (see Introduction), analyses and manipulations of the endogenous peptide systems provide further support for their role in promoting drinking in ethanol-exposed offspring. They show that GAL knockout compared to wildtype mice drink less ethanol while GAL overexpressors drink more and that GAL haplotypes and GAL receptor alleles in humans are linked to alcoholism (Barson et al., 2010). Further, endogenous OX has been related to ethanol-seeking (Bayerlein et al., 2011), and ENK is significantly elevated in the NAc of ethanol-preferring mice (Jamensky and Gianoulakis, 1999) and the cortex of ethanol-preferring rats (Marinelli et al., 2000). The anatomically specific effect of prenatal ethanol on ENK neurons, in the NAcC but not the NAcSh, suggests that the functional consequences of increased alcohol intake in the offspring may be more closely related to enhanced ethanol-seeking due to strengthened instrumental performance mediated by the NAcC, than to enhanced ethanol reward due to greater incentive salience mediated by the NAcSh (Di Chiara, 2002). Direct support for a function of endogenous ENK in these effects of in utero ethanol exposure comes from evidence showing administration of naltrexone a few days before birth to reverse the stimulatory effect of prenatal ethanol on alcohol drinking in adolescent offspring (Arias and Chotro, 2005). The present findings focus attention on the genesis of peptide-expressing neurons specifically in the hypothalamus and NAcC and provide support for their role in promoting ethanol consumption in offspring exposed prenatally to ethanol. Further studies in these offspring with agents that block peptide expression or function could provide a direct test of this hypothesis.
CONCLUSIONS
The findings of this report demonstrate that in utero exposure to ethanol, at lower doses that raise BEC to < 100 mg/dl, significantly stimulates neurogenesis in the peptide systems of the hypothalamus and NAc known to promote ethanol consumption. It shows that the ENK-, GAL- and OX-expressing neurons during in utero development respond in a similar manner to that described in adult animals consuming excess ethanol and that the stimulatory effect of prenatal ethanol on neurogenesis of these peptide neurons occurs in some but not all brain areas, possibly attributed to their differential responsiveness to circulating nutrients, hormones and neurotrophins affected by ethanol. This stimulatory effect of ethanol is specifically on neurogenesis, accompanied by little change in gliogenesis and apoptosis. The marked increase in density of neurons expressing the orexigenic peptides helps to explain why maternal drinking during pregnancy increases the risk of alcohol use disorders in the offspring.
Acknowledgements
This research was supported by USPHS Grant AA12882.
Abbreviations
- 5-HT
serotonin
- ARC
arcuate nucleus
- BEC
blood ethanol concentration
- BrdU
5-bromo-2-deoxyuridine
- DA
dopamine
- DIG-ISH
digoxigenin-labeled in situ hybridization
- E
embryonic day
- ENK
enkephalin
- GABA
gamma-aminobutyric acid
- GAL
galanin
- GFAP
glial fibrillary acidic protein
- ISH
in situ hybridization
- NAc
nucleus accumbens
- NAcC
nucleus accumbens core
- NAcSh
nucleus accumbens shell
- NeuN
neuronal nuclei
- NPY
neuropeptide Y
- ns
not significant
- P
postnatal day
- PFLH
perifornical lateral hypothalamus
- PVN
paraventricular nucleus
- OX
orexin
- TUNEL
terminal uridine nucleotide end labeling
REFERENCES
- Ahmed II, Shryne JE, Gorski RA, Branch BJ, Taylor AN. Prenatal ethanol and the prepubertal sexually dimorphic nucleus of the preoptic area. Physiol Behav. 1991;49:427–432. doi: 10.1016/0031-9384(91)90260-u. [DOI] [PubMed] [Google Scholar]
- Alati R, Al Mamun A, Williams GM, O'Callaghan M, Najman JM, Bor W. In utero alcohol exposure and prediction of alcohol disorders in early adulthood: a birth cohort study. Arch Gen Psychiatry. 2006;63:1009–1016. doi: 10.1001/archpsyc.63.9.1009. [DOI] [PubMed] [Google Scholar]
- Altman J, Bayer SA. Development of the diencephalon in the rat. I. Autoradiographic study of the time of origin and settling patterns of neurons of the hypothalamus. J Comp Neurol. 1978;182:945–971. doi: 10.1002/cne.901820511. [DOI] [PubMed] [Google Scholar]
- Arias C, Chotro MG. Increased palatability of ethanol after prenatal ethanol exposure is mediated by the opioid system. Pharmacol Biochem Behav. 2005;82:434–442. doi: 10.1016/j.pbb.2005.09.015. [DOI] [PubMed] [Google Scholar]
- Barson JR, Fagan SE, Chang GQ, Leibowitz SF. Neurochemical heterogeneity of rats predicted by different measures to be high ethanol consumers. Alcohol Clin Exp Res. 2012 doi: 10.1111/j.1530-0277.2012.01858.x. Epub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barson JR, Morganstern I, Leibowitz SF. Galanin and consummatory behavior: special relationship with dietary fat, alcohol and circulating lipids. EXS. 2010;102:87–111. doi: 10.1007/978-3-0346-0228-0_8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barson JR, Morganstern I, Leibowitz SF. Similarities in hypothalamic and mesocorticolimbic circuits regulating the overconsumption of food and alcohol. Physiol Behav. 2011;104:128–137. doi: 10.1016/j.physbeh.2011.04.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bayer SA, Altman J, Russo RJ, Zhang X. Timetables of neurogenesis in the human brain based on experimentally determined patterns in the rat. Neurotoxicology. 1993;14:83–144. [PubMed] [Google Scholar]
- Bayerlein K, Kraus T, Leinonen I, Pilniok D, Rotter A, Hofner B, Schwitulla J, Sperling W, Kornhuber J, Biermann T. Orexin A expression and promoter methylation in patients with alcohol dependence comparing acute and protracted withdrawal. Alcohol (Fayetteville, NY) 2011;45:541–547. doi: 10.1016/j.alcohol.2011.02.306. [DOI] [PubMed] [Google Scholar]
- Boehme F, Gil-Mohapel J, Cox A, Patten A, Giles E, Brocardo PS, Christie BR. Voluntary exercise induces adult hippocampal neurogeneis and BDNF expression in a rodent model of fetal alcohol spectrum disorders. Eur J Neurosci. 2011;33:1799–1811. doi: 10.1111/j.1460-9568.2011.07676.x. [DOI] [PubMed] [Google Scholar]
- Brocardo PS, Boehme F, Patten A, Cox A, Gil-Mohapel J, Christie BR. Anxiety- and depression-like behaviors are accompanied by an increase in oxidative stress in a rat model of fetal alcohol spectrum disorders: protective effects of voluntary physical exercise. Neuropharmacology. 2011;62:1607–1618. doi: 10.1016/j.neuropharm.2011.10.006. [DOI] [PubMed] [Google Scholar]
- Chang GQ, Barson JR, Karatayev O, Chang SY, Chen YW, Leibowitz SF. Effect of chronic ethanol on enkephalin in the hypothalamus and extra-hypothalamic areas. Alcohol Clin Exp Res. 2010;34:761–770. doi: 10.1111/j.1530-0277.2010.01148.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang GQ, Gaysinskaya V, Karatayev O, Leibowitz SF. Maternal high-fat diet and fetal programming: increased proliferation of hypothalamic peptide-producing neurons that increase risk for overeating and obesity. J Neurosci. 2008;28:12107–12119. doi: 10.1523/JNEUROSCI.2642-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang GQ, Karatayev O, Ahsan R, Gaysinskaya V, Marwil Z, Leibowitz SF. Dietary fat stimulates endogenous enkephalin and dynorphin in the paraventricular nucleus: role of circulating triglycerides. Am J Physiol Endocrinol Metab. 2007;292:E561–E570. doi: 10.1152/ajpendo.00087.2006. [DOI] [PubMed] [Google Scholar]
- Chotro MG, Arias C, Laviola G. Increased ethanol intake after prenatal ethanol exposure: studies with animals. Neurosci Biobehav Rev. 2007;31:181–191. doi: 10.1016/j.neubiorev.2006.06.021. [DOI] [PubMed] [Google Scholar]
- de Gortari P, Mendez M, Rodriguez-Keller I, Perez-Martinez L, Joseph-Bravob P. Acute ethanol administration induces changes in TRH and proenkephalin expression in hypothalamic and limbic regions of rat brain. Neurochem Int. 2000;37:483–496. doi: 10.1016/s0197-0186(00)00059-0. [DOI] [PubMed] [Google Scholar]
- De Witte P. The role of neurotransmitters in alcohol dependence. animal research. Alcohol Alcohol. 1996;1:13–16. [PubMed] [Google Scholar]
- Dembele K, Yao XH, Chen L, Nyomba BL. Intrauterine ethanol exposure results in hypothalamic oxidative stress and neuroendocrine alterations in adult rat offspring. Am J Physiol. 2006;291:R796–R802. doi: 10.1152/ajpregu.00633.2005. [DOI] [PubMed] [Google Scholar]
- Di Chiara G. Nucleus accumbens shell and core dopamine: differential role in behavior and addiction. Behav Brain Res. 2002;137:75–114. doi: 10.1016/s0166-4328(02)00286-3. [DOI] [PubMed] [Google Scholar]
- Druse MJ, Hao HL, Eriksen JL. In utero ethanol exposure increases proenkephalin, a precursor of a neuropeptide that is inhibitory to neuronal growth. Alcohol Clin Exp Res. 1999;23:1519–1527. [PubMed] [Google Scholar]
- Emanuele NV, LaPaglia N, Steiner J, Kirsteins L, Emanuele MA. Effect of chronic ethanol exposure on female rat reproductive cyclicity and hormone secretion. Alcohol Clin Exp Res. 2001;25:1025–1029. [PubMed] [Google Scholar]
- Fakoya FA, Caxton-Martins EA. Neocortical neurodegeneration in young adult Wistar rats prenatally exposed to ethanol. Neurotoxicol Teratol. 2006;28:229–237. doi: 10.1016/j.ntt.2005.11.001. [DOI] [PubMed] [Google Scholar]
- Fowler CD, Liu Y, Wang Z. Estrogen and adult neurogenesis in the amygdala and hypothalamus. Brain Res Rev. 2008;57:342–351. doi: 10.1016/j.brainresrev.2007.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gil-Mohapel J, Boehme F, Kainer L, Christie BR. Hippocampal cell loss and neurogenesis after fetal alcohol exposure: insights from different rodent models. Brain Res Rev. 2010;64:283–303. doi: 10.1016/j.brainresrev.2010.04.011. [DOI] [PubMed] [Google Scholar]
- Gonzalez-Burgos I, Alejandre-Gomez M, Olvera-Cortes ME, Perez-Vega MI, Evans S, Feria-Velasco A. Prenatal-through-postnatal exposure to moderate levels of ethanol leads to damage on the hippocampal CA1 field of juvenile rats: a stereology and Golgi study. Neurosci Res. 2006;56:400–408. doi: 10.1016/j.neures.2006.08.007. [DOI] [PubMed] [Google Scholar]
- Heaton MB, Paiva M, Madorsky I, Mayer J, Moore DB. Effects of ethanol on neurotrophic factors, apoptosis-related proteins, endogenous antioxidants, and reactive oxygen species in neonatal striatum: relationship to periods of vulnerability. Brain Res. 2003;140:237–252. doi: 10.1016/s0165-3806(02)00610-7. [DOI] [PubMed] [Google Scholar]
- Ifft JD. An autoradiographic study of the time of final division of neurons in rat hypothalamic nuclei. J Comp Neurol. 1972;144:193–204. doi: 10.1002/cne.901440204. [DOI] [PubMed] [Google Scholar]
- Jamensky NT, Gianoulakis C. Comparison of the proopiomelanocortin and proenkephalin opioid peptide systems in brain regions of the alcohol-preferring C57BL/6 and alcohol-avoiding DBA/2 mice. Alcohol (Fayetteville, NY) 1999;18:177–187. doi: 10.1016/s0741-8329(99)00002-6. [DOI] [PubMed] [Google Scholar]
- Lawrence RC, Otero NK, Kelly SJ. Selective effects of perinatal ethanol exposure in medial prefrontal cortex and nucleus accumbens. Neurotoxicol Teratol. 2012;34:128–135. doi: 10.1016/j.ntt.2011.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, van den Pol AN. Direct and indirect inhibition by catecholamines of hypocretin/orexin neurons. J Neurosci. 2005;25:173–183. doi: 10.1523/JNEUROSCI.4015-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Livy DJ, Maier Se, West JR. Fetal alcohol exposure and temporal vulnerability: effects of binge-like alcohol exposure on the ventrolateral nucleus of the thalamus. Alcohol Clin Exp Res. 2001;25:774–780. [PubMed] [Google Scholar]
- Livy DJ, Miller EK, Maier SE, West JR. Fetal alcohol exposure and temporal vulnerability: effects of binge-like alcohol exposure on the developing rat hippocampus. Neurotoxicol Teratol. 2003;25:447–458. doi: 10.1016/s0892-0362(03)00030-8. [DOI] [PubMed] [Google Scholar]
- Lugo JN, Jr, Wilson MA, Kelly SJ. Perinatal ethanol exposure alters met-enkephalin levels of male and female rats. Neurotoxicol Teratol. 2006;28:238–244. doi: 10.1016/j.ntt.2005.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maekawa M, Takashima N, Matsumata M, Ikegami S, Kontani M, Hara Y, Kawashima H, Owada Y, Kiso Y, Yoshikawa T, Inokuchi K, Osumi N. Arachidonic acid drives postnatal neurogenesis and elicits a beneficial effect on prepulse inhibition, a biological trait of psychiatric illnesses. PLoS One. 2009;4:e5085. doi: 10.1371/journal.pone.0005085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maier SE, West JR. Regional differences in cell loss associated with binge-like alcohol exposure during the first two trimesters equivalent in the rat. Alcohol (Fayetteville, NY) 2001;23:49–57. doi: 10.1016/s0741-8329(00)00133-6. [DOI] [PubMed] [Google Scholar]
- Marinelli PW, Kiianmaa K, Gianoulakis C. Opioid propeptide mRNA content and receptor density in the brains of AA and ANA rats. Life Sci. 2000;66:1915–1927. doi: 10.1016/s0024-3205(00)00517-8. [DOI] [PubMed] [Google Scholar]
- McGivern RF, Ervin MG, McGeary J, Somes C, Handa RJ. Prenatal ethanol exposure induces a sexually dimorphic effect on daily water consumption in prepubertal and adult rats. Alcohol Clin Exp Res. 1998;22:868–875. [PubMed] [Google Scholar]
- Miller MW. Effect of pre- or postnatal exposure to ethanol on the total number of neurons in the principal sensory nucleus of the trigeminal nerve: cell proliferation and neuronal death. Alcohol Clin Exp Res. 1995a;19:1359–1363. doi: 10.1111/j.1530-0277.1995.tb01625.x. [DOI] [PubMed] [Google Scholar]
- Miller MW. Generation of neurons in the rat dentate gyrus and hippocampus: effects of prenatal and postnatal treatment with ethanol. Alcohol Clin Exp Res. 1995b;19:1500–1509. doi: 10.1111/j.1530-0277.1995.tb01014.x. [DOI] [PubMed] [Google Scholar]
- Mooney SM, Miller MW. Prenatal exposure to ethanol affects postnatal neurogenesis in thalamus. Exp Neurol. 2010;223:566–573. doi: 10.1016/j.expneurol.2010.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morganstern I, Barson JR, Leibowitz SF. Regulation of drug and palatable food overconsumption by similar Peptide systems. Curr Drug Abuse Rev. 2011;4:163–173. doi: 10.2174/1874473711104030163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morganstern I, Chang GQ, Barson JR, Ye Z, Karatayev O, Leibowitz SF. Differential effects of acute and chronic ethanol exposure on orexin expression in the perifornical lateral hypothalamus. Alcohol Clin Exp Res. 2010a;34:886–896. doi: 10.1111/j.1530-0277.2010.01161.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morganstern I, Chang GQ, Karatayev O, Leibowitz SF. Increased orexin and melanin-concentrating hormone expression in the perifornical lateral hypothalamus of rats prone to overconsuming a fat-rich diet. Pharmacol Biochem Behav. 2010b;96:413–422. doi: 10.1016/j.pbb.2010.06.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oliva JM, Manzanares J. Gene transcription alterations associated with decrease of ethanol intake induced by naltrexone in the brain of Wistar rats. Neuropsychopharmacology. 2007;32:1358–1369. doi: 10.1038/sj.npp.1301237. [DOI] [PubMed] [Google Scholar]
- Parab S, Nankova BB, La Gamma EF. Differential regulation of the tyrosine hydroxylase and enkephalin neuropeptide transmitter genes in rat PC12 cells by short chain fatty acids: concentration-dependent effects on transcription and RNA stability. Brain Res. 2007;1132:42–50. doi: 10.1016/j.brainres.2006.11.013. [DOI] [PubMed] [Google Scholar]
- Qiang M, Wang MW, Elberger AJ. Second trimester prenatal alcohol exposure alters development of rat corpus callosum. Neurotoxicol Teratol. 2002;24:719–732. doi: 10.1016/s0892-0362(02)00267-2. [DOI] [PubMed] [Google Scholar]
- Ramos AJ, Evrard SG, Tagliaferro P, Tricarico MV, Brusco A. Effects of chronic maternal ethanol exposure on hippocampal and striatal morphology in offspring. Ann N Y Acad Sci. 2002;965:343–353. doi: 10.1111/j.1749-6632.2002.tb04176.x. [DOI] [PubMed] [Google Scholar]
- Reagan LP, Rosell DR, Wood GE, Spedding M, Munoz C, Rothstein J, McEwen BS. Chronic restraint stress up-regulates GLT-1 mRNA and protein expression in the rat hippocampus: reversal by tianeptine. Proc Natl Acad Sci U S A. 2004;101:2179–2184. doi: 10.1073/pnas.0307294101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubert G, Minana R, Pascual M, Guerri C. Ethanol exposure during embryogenesis decreases the radial glial progenitorpool and affects the generation of neurons and astrocytes. J Neurosci Res. 2006;84:483–496. doi: 10.1002/jnr.20963. [DOI] [PubMed] [Google Scholar]
- Rudeen PK. Reduction of the volume of the sexually dimorphic nucleus of the preoptic area by in utero ethanol exposure in male rats. Neurosci Lett. 1986;72:363–368. doi: 10.1016/0304-3940(86)90542-2. [DOI] [PubMed] [Google Scholar]
- Schaffer DV, Gage FH. Neurogenesis and neuroadaptation. NeuroMol Med. 2004;5:1–9. doi: 10.1385/NMM:5:1:001. [DOI] [PubMed] [Google Scholar]
- Sherwood NM, Timiras PS. A sterotaxic atlas of the developing rat brain. University of California; Los Angeles: 1970. [Google Scholar]
- Steinberg GR, Watt MJ, Fam BC, Proietto J, Andrikopoulos S, Allen AM, Febbraio MA, Kemp BE. Ciliary neurotrophic factor suppresses hypothalamic AMP-kinase signaling in leptin-resistant obese mice. Endocrinology. 2006;147:3906–3914. doi: 10.1210/en.2005-1587. [DOI] [PubMed] [Google Scholar]
- Tabakoff B, Saba L, Printz M, Flodman P, Hodgkinson C, Goldman D, Koob G, Richardson HN, Kechris K, Bell RL, Hubner N, Heinig M, Pravenec M, Mangion J, Legault L, Dongier M, Conigrave KM, Whitfield JB, Saunders J, Grant B, Hoffman PL, State WISo. Trait Markers of A Genetical genomic determinants of alcohol consumption in rats and humans. BMC Biol. 2009;7:70. doi: 10.1186/1741-7007-7-70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uban KA, Sliwowska JH, Lieblich S, Ellis LA, Yu WK, Weinberg J, Galea LA. Prenatal alcohol exposure reduces the proportion of newly produced neurons and glia in the dentate gyrus of the hippocampus in female rats. Horm Behav. 2010;58:835–843. doi: 10.1016/j.yhbeh.2010.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vengeliene V, Bilbao A, Molander A, Spanagel R. Neuropharmacology of alcohol addiction. Br J Pharmacol. 2008;154:299–315. doi: 10.1038/bjp.2008.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie X, Crowder TL, Yamanaka A, Morairty SR, Lewinter RD, Sakurai T, Kilduff TS. GABA(B) receptor-mediated modulation of hypocretin/orexin neurones in mouse hypothalamus. J Physiol. 2006;574:399–414. doi: 10.1113/jphysiol.2006.108266. [DOI] [PMC free article] [PubMed] [Google Scholar]