

Abstract
Lung cancer is the leading cause of cancer deaths in the United States. Current therapies are inadequate. Histone deacetylase inhibitors (HDACi) are a recently developed class of anticancer agents that cause increased acetylation of core histones and nonhistone proteins leading to modulation of gene expression and protein activity involved in cancer cell growth and survival pathways. We examined the efficacy of the HDACi panobinostat (LBH589) in a wide range of lung cancers and mesotheliomas. Panobinostat was cytotoxic in almost all 37 cancer cell lines tested. IC50 and LD50 values were in the low nmol/L range (4–470 nmol/L; median, 20 nmol/L). Small cell lung cancer (SCLC) cell lines were among the most sensitive lines, with LD50 values consistently <25 nmol/L. In lung cancer and mesothelioma animal models, panobinostat significantly decreased tumor growth by an average of 62% when compared with vehicle control. Panobinostat was equally effective in immunocompetent and severe combined immunodeficiency mice, indicating that the inhibition of tumor growth by panobinostat was not due to direct immunologic effects. Panobinostat was, however, particularly effective in SCLC xenografts, and the addition of the chemotherapy agent etoposide augmented antitumor effects. Protein analysis of treated tumor biopsies revealed elevated amounts of cell cycle regulators such as p21 and proapoptosis factors, such as caspase 3 and 7 and cleaved poly[ADP-ribose] polymerase, coupled with decreased levels of antiapoptotic factors such as Bcl-2 and Bcl-XL. These studies together suggest that panobinostat may be a useful adjunct in the treatment of thoracic malignancies, especially SCLC.
Introduction
Lung cancer has a worldwide incidence of 1.2 million cases. In the United States the annual death rate due to this disease for 2006 (162,460) approximated its annual incidence rate (174,470), making it the leading cause of cancer deaths in both men and women in the United States (17.8%). Mesothelioma represents 1.5% of these cases, and small cell lung cancer (SCLC) accounts for approximately 13%, with non–small cell lung cancer (NSCLC) being responsible for the rest (1). Given the overall poor survival rates despite treatment, and with only little to modest progress achieved to date in the development of effective treatments, there is a clear need for new therapeutic approaches.
Histone deacetylase inhibitors (HDACi) are a recently developed class of anticancer agents. These agents inhibit the deacetylation both of histones and nonhistone cellular proteins, inducing hyperacetylation and an “open chromatin” state leading to increased transcriptional activity. HDAC inhibition leads to modulation of gene expression leading to the reexpression of silenced tumor suppressor genes, cell cycle arrest, terminal differentiation, and apoptosis (2). In addition, HDAC inhibition can target nonhistone proteins involved in cancer cell growth and survival pathways. The antitumor effects of HDACi have been shown in a variety of cancer cell lines (NSCLC, prostate, colon, ovarian, breast, bladder, pancreas, leukemias, and lymphomas), in vivo tumor models, and in patients with both hematologic and solid tumors (2).
This study focuses on the use of a new pan-HDACi, panobinostat, a cinnamic hydroxamic acid analog (class II HDACi). This agent has been shown to exert antitumor effects on a variety of cancer cell lines (3-5) and is currently being investigated in clinical trials in patients with advanced solid tumors (such as hormone refractory prostate cancer, breast, clear renal cell, colorectal, bladder, and ovarian cancers) and hematologic malignancies (6, 7). A large clinical trial with another HDACi, suberoylanilide hydroxamic acid (SAHA), for treating mesothelioma is ongoing.
To date, the effects of panobinostat on cell lines derived from thoracic malignancies (mesothelioma, NSCLC, SCLC) have not been systematically characterized. Our goal was to examine the efficacy of panobinostat in a wide range of thoracic malignancies using both in vitro and in vivo models. We therefore screened a panel of 37 cell lines drawn from all three types of thoracic malignancies. Although the growth of most cell lines was inhibited in this pilot screen, we found that SCLC cell lines were especially sensitive to this compound. Animal studies confirmed these results, suggesting that panobinostat may be a useful adjunct in the treatment of thoracic malignancies, especially SCLC.
Materials and Methods
Cell Lines
Mesothelioma Cells Lines
The human mesothelioma cell lines OK-2 (SF151720), OK-4 (SF080125B), OK-5 (SF161815), and OK-6 (SF160520) were provided by the Stehlin Foundation (8). The human REN, LRK, and M30 cell lines were derived at the University of Pennsylvania from mesothelioma tumor tissues from individual patients. H513 cells were purchased from the American Type Culture Collection. The murine AB-1, AC29, AB12, and AE17-ova cell lines (provided by Drs. Bruce Robinson and Delia Nelson, University of Western Australia) were derived from mesothelioma tumors arising in mice that were first given i.p. administrations of asbestos (9, 10). OK-2, OK-4, OK-5, OK-6, REN, M30, and AE17 cells were cultured in RPMI 1640 (Gibco Invitrogen) supplemented with 10% fetal bovine serum (FBS; FBS Premium select, Atlanta Biologicals). H513 cells were cultured in RPMI 1640 supplemented with 5% FBS. LRK, AB-1, AC29, and AB12 cells were cultured in DMEM (Gibco) supplemented with 10% FBS.
NSCLC Cell Lines
The human NSCLC lines H1299, A549, H460, H1264, H322, and H661, and the murine NSCLC lines LLC and L1C2 were purchased from the American Type Culture Collection. L55 was derived at the University of Pennsylvania from the NSCLC of a patient. The murine TC-1 cell line (11) was provided by Dr. Yvonne Paterson (University of Pennsylvania), and the LKR cell line, derived from a tumor which developed in a transgenic mouse expressing an activated Kras gene (12), was provided by Dr. Joseph Friedberg (University of Pennsylvania). All cell lines were cultured in RPMI 1640 supplemented with 10% FBS with the exception of LLC, L1C2, and LKR cells, which were cultured in DMEM supplemented with 10% FBS.
SCLC Cell Lines
RG-1, LD-T, H209, GL-E, H209/CP, BK-T, LV-E, TY-E, HG-E, SH-A, AD-A, and SM-E cells were all derived from the individual tumors of patients with SCLC (13). H526 and H69 cells were purchased from the American Type Culture Collection. All cell lines were cultured in RPMI 1640 supplemented with 5% FBS.
All cell culture media formulations were supplemented with 2 mmol/L L-glutamine, 100 U/mL of penicillin, and 100 μg/mL of streptomycin (all from Gibco). All the cell lines were maintained at 37°C in a humidified atmosphere with 95% air: 5% CO2.
Reagents
Panobinostat (formerly called LBH589), a HDACi, was provided by Novartis Pharmaceuticals in suspension with 5% dextrose. The HDACi SAHA was kindly provided by Dr Xiaobo Cao, from M. D. Anderson. Etoposide (a topoisomerase II inhibitor) was purchased from Sigma-Aldrich.
Cell Proliferation MTS Assays
The 3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium (MTT) cell proliferation assays (Promega) were carried out for 37 cell lines (12 mesothelioma cell lines, 11 NSCLC cell lines, and 14 SCLC cell lines). Briefly, cells were plated in quadruplicate in 96-well plates at a confluence of approximately 70%. Cells were incubated overnight and panobinostat or SAHA was then added the following day. The cells remained exposed to each drug for the entire testing period; no new drug was subsequently added. Concentrations of drug tested ranged from 0 to 800 nmol/L for panobinostat and from 0 to 25 μmol/L for SAHA. The MTS reagents were added, and plates were read on a multiwell scanning spectrophotometer 48 and 72 h after drug exposure.
For panobinostat, the IC50 and LD50 were determined and compared with that of SAHA in selected cell lines. IC50 was calculated as the concentration of the compound at which 50% of cells were growth-inhibited compared with vehicle treatment. LD50 was the concentration at which 50% of cells originally plated were killed. A standard curve with known numbers of control cells was included with every assay and assayed under identical conditions as the experimental groups. The assays were repeated to ensure consistency of results.
Fluorescent Activated Cell Sorting Analysis for Cell Cycle Status and Cellular Viability
Cell cycle status and cellular viability were determined via fluorescent activated cell sorting analysis of DNA content as previously described (14). Briefly, H69 cells were plated and mock-treated (control cells), or exposed to either etoposide (34 μmol/L) or panobinostat (20 nmol/L) alone, or in combination. The effects of the combination of etoposide and panobinostat added after 24 h and vice versa were also tested. During fluorescent activated cell sorting, no gating was done during data acquisition on the propidium iodide–stained nuclei in order to not exclude any cells from the analysis, including those with sub-G1 content representative of nuclear fragmentation. The aggregate results were then analyzed for the proportion of cells with DNA content corresponding to each respective cell cycle phase.
Cells were harvested at different time points (from 0 to 60 h).
Immunoblotting
Tumor lysates were prepared by mechanical tissue homogenization in lysis buffer [consisting of 50 mmol/L Tris-HCl (pH 8), 150 mmol/L NaCl, 1% NP-40, 0.5% sodium deoxycholate, and 0.1% SDS supplemented with a protease inhibitor cocktail (Mini Complete, Roche)]. The protein concentrations of the supernatants were quantified via the BCA Protein Assay Kit (Pierce). Twenty micrograms of lysate were mixed with SDS buffer (1× LDS sample buffer plus 1× sample reducing agent; Invitrogen) and were loaded into each lane of a 4% to 12% SDS-polyacrylamide gel. After electrophoresis, the proteins were transferred onto a polyvinylidene fluoride membrane (Perkin Elmer). The membranes were subsequently blocked with PBS containing 5% nonfat dry milk, and then probed with primary antibodies overnight at 4°C. The following primary antibodies and dilutions were used: anti-β-actin (1:10,000; Sigma-Aldrich), anti-Bcl-XL (1:1,000; Santa Cruz Biotechnology), anti-Bid (1:500; Becton Dickinson Pharmingen), anti-p21 (1:1,000; Santa Cruz), anti-Bcl-2 (1:100; Dako Cytomation), anti-acetylated histone H4 (AH4; 1:1,000; Upstate Biotechnology), anti-acetylated histone H3 (AH3; 1:5,000; Upstate Biotechnology), anti–cleaved caspase 3 (1:1,000; Cell Signaling), anti–cleaved caspase 7 (1:1,000; Cell Signaling), anti–poly[ADP-ribose] polymerase (PARP; 1:1,000; Cell Signaling), and anti-α-tubulin (1:5,000; Sigma-Aldrich). The membranes were washed repeatedly in PBS containing 0.5% milk and 0.2% Tween-20, and then incubated for 1 h with polyclonal horseradish peroxidase–conjugated secondary antibodies (1:10,000; donkey anti-rabbit or donkey anti-mouse antibody, from Jackson ImmunoResearch Laboratories). After further washes, enhanced chemiluminescence was done on the membranes with the Western Lightning Chemiluminescence Reagent Plus (Perkin Elmer), prior to exposure to film. Three separate tumor biopsies were analyzed from each treatment condition.
In vivo Experiments
The experiments done on the mice were in accord with a protocol reviewed and approved by the Institutional Animal Care and Usage Committee at the University of Pennsylvania, which was in compliance with the Guide for the Care and Use of Laboratory Animals. AE17 and TC-1 cancer cells (1 × 106 cells) were injected into the flanks of adult female C57Bl/6 mice (Taconic Labs) and severe combined immunodeficiency (SCID) mice (Jackson Laboratories). M30 (10 × 106 cells), A549 (5 × 106 cells), H69 (2.5 × 106 cells), BK-T (6.5 × 106), H526 (10 × 106), and RG1 (10 × 106) cells were also injected, but in the presence of matrigel (BD Biosciences), into the flanks of SCID mice. There were 5 to 10 mice in each treatment group. The experiments with the A549 and H69 cell lines were repeated to ensure the statistical consistency of the results. Experiments were terminated when the tumors in the control mice had grown to a size that threatened the quality of life of the mice.
When tumors reached 100 to 500 mm3, panobinostat was administered via i.p. injections (10–20 mg/kg) on a daily schedule (5-days-on, 2-days-off regimen) for the entire duration of the experiment. Control mice received i.p. injections with dextrose 5% in water (“vehicle treatment”). Every tumor was measured with a caliper at least twice weekly. For evaluation of the effects of combination therapy on SCLC-derived tumors, SCID mice with H69 tumors were administered panobinostat as described above. Three days after the initiation of panobinostat, and again 1 wk later, etoposide (40 mg/kg) was administered i.p.
Statistical Analysis
Statistical analyses were done using the Prism statistical package. Student's t-test or χ2 analyses were utilized to compare results between treatment groups. The Kruskal-Wallis one-way ANOVA by ranks with posthoc Dunn's testing was used for the experiments with more than two groups. P < 0.05 was considered statistically significant. Error bars in the graphs represent SE calculated with Microsoft Excel.
Results
Panobinostat Has Potent In vitro Growth Inhibitory and Cytotoxic Effects in Thoracic Cancer Cell Lines
The effects of panobinostat on the inhibition of cancer cell growth (IC50) were determined in a wide range of human and murine lung cancer (NSCLC and SCLC) and mesothelioma cell lines via MTS assays (Table 1). We also measured the ability to induce cell death (LD50) in many of the cell lines. Panobinostat exhibited potent antiproliferative activity and cytotoxicity in seven human and four murine NSCLC cell lines. For the human NSCLC cell lines, the IC50 ranged from 5 to 100 nmol/L, with a median value of 35 nmol/L (except for the cell line H661). The LD50 ranged from 50 to 400 nmol/L, with a median value of 105 nmol/L. For the murine NSCLC cell lines, the IC50 ranged from 20 to 310 nmol/L, with a median value of 87.5 nmol/L, and the LD50 ranged from 75 to 690 nmol/L, with a median value of 200 nmol/L.
Table 1.
IC50 of NSCLC, mesothelioma, and SCLC lines to panobinostat
| Species | Cell line | Panobinostat |
SAHA LD50 | ||
|---|---|---|---|---|---|
| IC50 (nmol/L) | LD50 (nmol/L) | ||||
| NSCLC | Human | H1299 | 5 | 120 | >25 μmol/L |
| Human | L55 | 11 | 80 | ||
| Human | A549 | 30 | 50 | ||
| Human | H460 | 40 | 90 | ||
| Human | H1264 | 100 | 250 | ||
| Human | H322 | 100 | 400 | ||
| Human | H661 | >800 | |||
| Mouse | TCI | 20 | 75 | >800 nmol/L | |
| Mouse | LLC | 85 | 200 | ||
| Mouse | L1C2 | 90 | |||
| Mouse | LKR | 310 | 690 | ||
| MESO | Human | OK-6 | 5 | 5 | 12 μmol/L |
| Human | OK-5 | 7 | 7 | 1.5 μmol/L | |
| Human | LRK | <10 | 30 | >800 nmol/L | |
| Human | OK-4 | 25 | 60 | ||
| Human | M30 | 30 | 100 | ||
| Human | REN | 30 | |||
| Human | H513 | 35 | |||
| Human | OK-2 | 50 | 620 | 14 μmol/L | |
| Mouse | AB-1 | 20 | |||
| Mouse | AE17-ova | 25 | 80 | ||
| Mouse | AC29 | 40 | |||
| Mouse | AB12 | 470 | |||
| SCLC | Human | RG-1 | 4 | 6 | |
| Human | LD-T | 5 | 11 | ||
| Human | GLE | 8 | 8 | 800 nmol/L | |
| Human | H209 | 6 | 10 | ||
| Human | H209/CP | 8 | 20 | ||
| Human | BKT | 9 | 13 | ||
| Human | LVE | 9 | |||
| Human | TY-E | 10 | 19 | 3 μmol/L | |
| Human | H526 | 10 | 12 | ||
| Human | HG-E | 10 | 12 | ||
| Human | SH-A | 12 | |||
| Human | H69 | 20 | 25 | ||
| Human | ADA | 25 | |||
| Human | SM-E | 175 | |||
NOTE: Comparison of IC50 and LD50 of panobinostat for 37 lung cancer cell lines: 11 NSCLC cell lines (7 human, 4 mouse), 12 mesothelioma (MESO) cell lines (8 human, 4 mouse), and 14 SCLC cell lines (all human). Selected cell lines were compared with SAHA. Note that most of the IC50 values are in the low nmol/L range.
The antiproliferative and cytotoxic effects of panobinostat were then studied in eight human and four murine mesothelioma cell lines. As shown in Table 1, mesothelioma (Meso) cell lines tended to be more sensitive than NSCLC lines to panobinostat. For the human Meso cell lines, the IC50 ranged from 5 to 50 nmol/L, with a median value of 30 nmol/L, and the LD50 ranged from 5 to 620 nmol/L, with a median value of 45 nmol/L. For the murine Meso cell lines, the IC50 ranged from 20 to 470 nmol/L, with a median value of 32.5 nmol/L.
SCLC cell lines showed the highest sensitivity to panobinostat. As shown in Table 1, the IC50 for all but one of the 14 human SCLC cell lines was <25 nmol/L, with a median of 9.5 nmol/L; in fact, 10 of 14 SCLC lines showed an IC50 of ≤10 nmol/L. The LD50 ranged from 6 to 25 nmol/L, with a median value of 12 nmol/L. The SCLC line H209/CP (a variant of H209 cells that has developed cisplatinum resistance) showed very similar IC50 and LD50 values when compared with the parental cisplatinum-sensitive H209 cell line, results suggesting that panobinostat may usefully overcome chemotherapy resistance in SCLC cell lines.
Finally, in selected cell lines, we compared panobinostat with SAHA, another HDACi currently in clinical trials. The LD50 of SAHA were 10- to 1,000-fold higher than those seen with panobinostat (Table 1).
To further explore the increased sensitivity of SCLC lines to panobinostat, we assessed the time course of cell cycle effects invoked by exposure to panobinostat. H69 cells exposed to panobinostat showed accumulation of cells with G2-M DNA content, which was initially coincident with but eventually was superseded by the proportion of nonviable cells (marked by sub-G1 DNA content; Fig. 1A). With sustained exposure to panobinostat, e.g. 60 hours of exposure, >70% of the cells had become nonviable.
Figure 1.
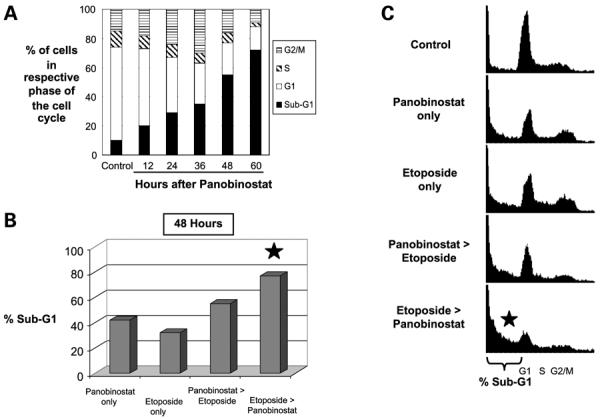
The in vitro effects of panobinostat on H69 SCLC cells: time-dependent cell cycle changes and cytotoxicity. A, time-dependent cell cycle and cytotoxic effects of panobinostat. Bar graphs, percentage of H69 cells in the respective phases of cell cycle after different exposure times to panobinostat, as determined by DNA flourescent activated cell sorting (FACS). The proportion of cells with Sub-G1 DNA content progressively increased with increasing exposure times to panobinostat. B and C, cell death is increased with combined panobinostat and etoposide. Bar graphs, respective percentages of H69 cells with Sub-G1 DNA content after exposure to panobinostat alone, etoposide alone, or combination of both agents. All cells were harvested after 48 h of drug exposure. Panobinostat > Etoposide, cells were treated with panobinostat for 12 h before etoposide was added, and then exposed to both drugs for an additional 36 h. Etoposide > Panobinostat, cells were treated with etoposide for 12 h before panobinostat was added, and then exposed to both drugs for an additional 36 h. Cells treated with panobinostat only or etoposide only were treated continuously for 48 h. Either sequence of combination treatment showed significantly greater proportions of nonviable cells than each of the drugs alone. Error bars are not available because these experiments did not involve parallel plates processed in triplicate. Statistical significance of differences in the proportion of cells was evaluated by χ2 analysis. We repeated this experiment three times, with all three experiments resulting in similar findings. *, statistically significant differences when compared with panobinostat only or etoposide only (P < 0.01); C, DNA FACS histograms derived from the experiment described in Fig. 1B, showing the respective proportions of H69 cells in each of the cell cycle phases, after the indicated drug treatment (mock-treated controls, panobinostat alone, etoposide alone, or combination of both agents). Bracket, proportion of nonviable cells with Sub-G1 DNA content.
The rate of cell death induced by panobinostat could also be accelerated by combined treatment with etoposide, a standard chemotherapeutic agent for SCLC. Whereas near complete eradication of H69 cells with panobinostat alone required ≥60 hours of continuous exposure, a comparable degree of cell death with only 36 hours of panobinostat exposure was achieved if the cells were pretreated with etoposide for 12 hours (compare the last bar graph in Fig. 1A with last bar graph in Fig. 1B). The proportion of cells killed by the combination treatment was also significantly greater than with either etoposide or panobinostat alone for the full 48 hours of the experiment (Fig. 1B and C). Interestingly, pretreatment with etoposide before the addition of panobinostat seemed to be more effective at rapidly killing cells than panobinostat followed by etoposide.
Panobinostat Significantly Slows the In vivo Growth of Tumors Derived from Meso and NSCLC Cells
We next tested the effects of doses between 10 and 20 mg/kg of panobinostat (given i.p., 5 days/week) on selected cell lines grown as flank tumors in mice, including the human Meso cell line M30 (grown in SCID mice; Fig. 2A), the mouse Meso cell line AE17 (grown in syngeneic C57/B6 mice; Fig. 2B), the human NSCLC line A549 (grown in SCID mice; Fig. 2C), and the mouse NSCLC line TC1 (grown in C57/B6 mice; Fig. 2D). Although tumor regression was not observed, panobinostat significantly slowed tumor growth when compared with vehicle control for all four different in vivo models tested, and by an average of almost 60% (P < 0.05). Interestingly, there seemed to be no statistically significant differences among the effects of the 10, 15, and 20 mg/kg doses for most of the tumors, with the exception of the tumors grown from the A549 cells, for which the 20 mg/kg dose was clearly more efficacious than the 10 mg/kg dose. Finally, there was no increased mortality or obvious toxicity noted with the panobinostat treatment during the course of the experiments, except for mild weight loss (<10%), and fur changes in the M30 cell study.
Figure 2.
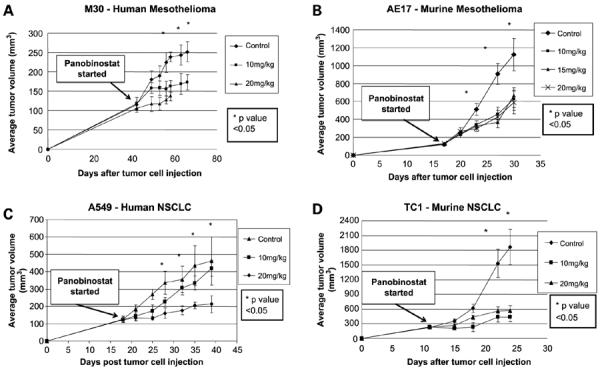
The in vivo effects of panobinostat on mesothelioma and NSCLC cells growing in mice. Tumor cells were injected in the flank of appropriate mice on day 0. Panobinostat treatment was started when tumors reached approximately 100 to 300 mm3. A, M30 mesothelioma xenograft model. B, AE17 mesothelioma murine syngeneic model. C, A549 NSCLC murine xenograft model. D, TC1 NSCLC murine syngeneic model. *, P < 0.05, between control and panobinostat treatment groups, except for C where the statistical difference is present between control and 20 mg/kg dose treatment group only. Arrow from “Panobinostat started” box indicates when panobinostat treatment was started.
The experiments with the AE17 and TC-1 cells were done in syngeneic animals with intact immune systems. To test whether the therapeutic responses we observed might be related to immune effects, we repeated the experiments in immunodeficient SCID mice. The slowing of tumor growth by panobinostat in the SCID mice was virtually identical to that previously noted in immunocompetent C57/B6 mice (Supplementary Fig. S1), therefore indicating that the inhibition of tumor growth caused by panobinostat is not likely to be due to T-cell- or B-cell-mediated immunologic effects.
Panobinostat Dramatically Slows the In vivo Growth of Tumors Derived from SCLC Cells and Induces Remissions
We next tested the effect of panobinostat on tumors derived from four human SCLC cell lines grown as xenografts in SCID mice. Panobinostat had a dramatic effect on all the SCLC-derived tumors that was significant compared with control animals with identically derived tumors but mock-treated. Daily panobinostat, given i.p. at 20 mg/kg for 5 days per week, resulted in an average decrease in growth of 70% at the end of all the experiments. Compared with the corresponding control tumors, panobinostat resulted in a 53% decrease for H526-derived tumors (Fig. 3A), a 81% decrease for BK-T–derived tumors (Fig. 3B), a 76% decrease for RG-1–derived tumors (Fig. 3C), and a 70% decrease for H69-derived tumors (Fig. 3D). In contrast to the lack of tumor regression noted in NSCLC and Meso-derived xenografted tumors that were treated under identical conditions and doses, panobinostat resulted in dramatic tumor regression in two of the SCLC-derived tumors, with the size of the BK-T–derived tumors reduced from an average of 296 mm3 at the beginning of the treatment course to 116 mm3 at the end of the experiment, and RG-1–derived tumors decreased from an average of 185 mm3 at the beginning of the treatment course to 86 mm3 at the end of the experiment.
Figure 3.
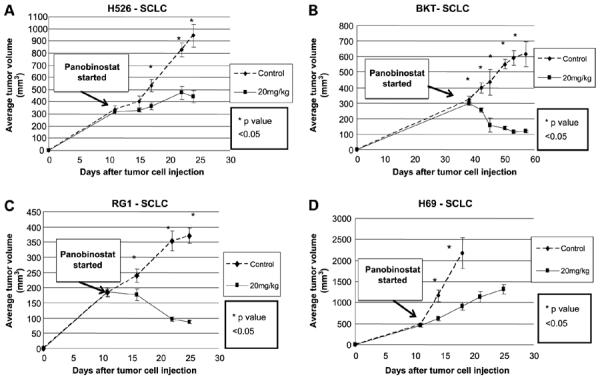
The in vivo effects of panobinostat on SCLC cells growing in mice. SCLC tumor cells were injected in the flanks of SCID mice (murine xenograft model) on day 0. Panobinostat treatment was started when tumors reached approximately 200 to 500 mm3. Cell lines H526 (A), BKT (B), RG1 (C), and H69 (D). *, P < 0.05 between control and panobinostat treatment groups. Arrow from “Panobinostat stated” box indicates when panobinostat treatment was started.
Panobinostat Combines with Etoposide to Maximally Inhibit the In vivo Growth of Tumors Derived from SCLC Cells
Patients with SCLC are commonly treated with chemotherapy that includes the drug etoposide (VP16). Studies have reported that HDACi exposure promotes histone hyperacetylation, resulting in a more accessible chromatin structure that may be more susceptible to DNA-damaging agents such as etoposide (15, 16), a mechanism that may underlie the enhanced in vitro efficacy of combining panobinostat with etoposide that we noted for SCLC cell lines (Fig. 1). We therefore studied the in vivo effects of combining these two agents to treat tumors derived from SCLC cells. As shown in Fig. 4, etoposide alone (40 mg/kg given i.p. weekly) did not slow tumor growth in all animals to a degree significantly different than that in mock-treated animals. Panobinostat at 20 mg/kg (given i.p. on a daily basis) by itself significantly reduced tumor growth, as we had previously found in the experiments described in Fig. 3. However, the combination of etoposide and panobinostat resulted in the maximal antitumor effects that were significantly better than either drug alone (P < 0.05).
Figure 4.
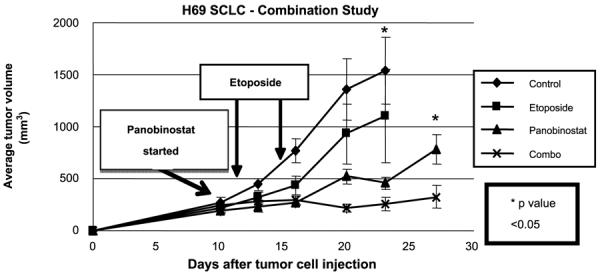
The combination of panobinostat and etoposide has enhanced in vivo effects on SCLC cells growing in the mice. Graph shows in vivo experiment where H69 SCLC tumor cells were injected in the flank of SCID mice (murine xenograft model) on day 0. Panobinostat treatment was started when tumors reached approximately 200 mm3. Groups were as follows: control, no treatment provided (vehicle only); panobinostat, 20 mg/kg panobinostat administered 5 d weekly i.p.; etoposide, 40 mg/kg administered i.p. once weekly × 2; combination, etoposide 40 mg/kg plus panobinostat 20 mg/kg administered i.p. *, P < 0.05 between control and panobinostat 20 mg/kg and combo (by ANOVA on day 23) and between panobinostat 20 mg/kg and combo (day 27). Arrows from “Etoposide” box point to days when Etoposide treatment was administered. Arrow from “Panobinostat started” box indicates when panobinostat treatment was started.
Effects of Panobinostat on Histones and Nonhistone Proteins; Modulation of Apoptosis-Related Components
In order to characterize the mechanisms that may underlie the antitumor effects of panobinostat, xenografts were established with H69 human SCLC cells in SCID mice and were subsequently treated as in the experiments described in Fig. 4. As before, mice were injected with vehicle only (control group), etoposide alone, panobinostat alone, or etoposide together with panobinostat (“combo” group). These tumors were harvested from the mice at the end of the experiment, 6 hours after the last dose of panobinostat and 24 hours after a dose of etoposide had been given and processed for immunoblot analyses. As shown in the respective columns of Fig. 5, acetylation of histone H3 and H4 was markedly increased by panobinostat treatment, indicating HDAC inhibition. Panobinostat was also associated with marked up-regulation of the proapoptotic markers t-Bid, cleaved PARP, cleaved caspase 3, and cleaved caspase 7; a marked decrease of the antiapoptosis factor Bcl-XL; and a small increase in the cyclin-dependent kinase inhibitor p21. Interestingly, compared with mock-treated controls, etoposide alone also led to a small increase of histone H4 acetylation and cleaved PARP. However, unlike panobinostat alone or in combination, etoposide alone did not discernibly increase histone H3 acetylation, or increase the apoptosis-related t-Bid, caspase 3, or caspase 7.
Figure 5.
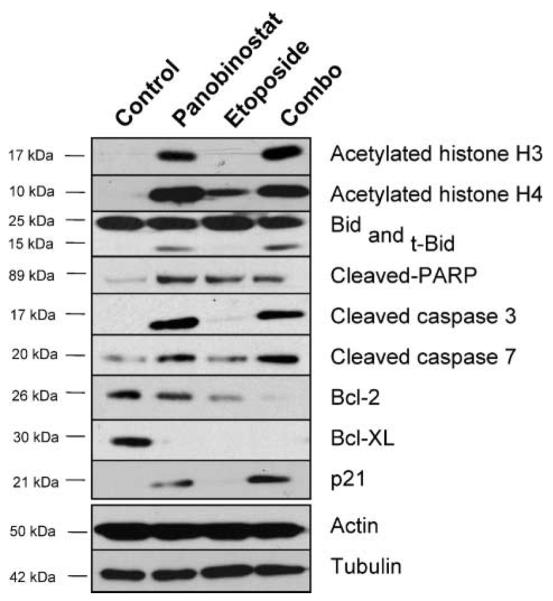
Effects of panobinostat, etoposide, or combined treatment on key proteins in SCLC cells growing in the mouse: In vivo effects on core histone acetylation, cell-cycle and apoptosis-related component. Analysis of H69 tumors from in vivo combination therapy experiment shown in Fig. 4. Tumor lysates from control (control) mice or mice treated with panobinostat (panobinostat), etoposide (etoposide), or the combination of panobinostat and etoposide (combo) were analyzed by immunoblot with indicated antibodies. Protein concentrations were normalized according to actin and tubulin levels.
We were especially interested in the relative effects of panobinostat combined with etoposide (i.e. combo treatment), compared with controls and either panobinostat or etoposide alone, given that the antitumor efficacy with combo treatment was more effective than either treatment alone. We noted that combo treatment led to changes in proteins similar to panobinostat alone, but with important differences. In particular, combo treatment led to near complete eradication of the antiapoptotic factor Bcl-2 and maximal increase in p21. Panobinostat alone had no effect on Bcl-2 and only a small increase in p21.
Discussion
The unsatisfactory rate of cure with standard treatments for disseminated thoracic malignancies has encouraged the development of novel treatment strategies such as those including histone deacetylase inhibitors. In vitro studies on select NSCLC, SCLC, and mesothelioma cell lines have thus been conducted with HDACi such as SAHA (17–20), MGCD0103 (21), trichostatin-A (19, 22–27), MS-275 (28), depsipeptide/FR901228 (29), trapoxin A (30), sodium butyrate (31), and valproic acid (32). A common finding has been that HDACi inhibit cell growth and induce apoptosis in these cell lines studied with varying degrees of efficacy. In vitro mechanisms of cell death induced by these HDACi have included activation of caspases, down-regulation of antiapoptotic factors such as Bcl-2 and Bcl-XL, and up-regulation of p21 (17, 18, 21, 22, 24, 27, 29, 31, 33–35). A number of other studies have also suggested that HDACi may affect antitumor immune responses (18, 36–38).
The wealth of published in vitro data has been accompanied by relatively few in vivo studies of these agents in animal models of tumors derived from thoracic malignancies (21, 33). There have also been early reports incorporating HDACi in clinical trials, such as for NSCLC with Pivanex (39) and Romidepsin (depsipeptide; ref. 40). A large randomized trial of SAHA for mesothelioma (41) is ongoing, and it is based on responses seen in an earlier phase I trial (42).
Panobinostat (formerly known as LBH589) is a cinnamic hydroxamic acid analog (class II HDACi) that has shown activity for hematologic malignancies and cutaneous lymphomas (6, 7). However, studies involving panobinostat for thoracic malignancies are still relatively sparse (33, 43). We therefore sought to compare the effects of panobinostat in a large panel of thoracic tumor cell lines, to test its efficacy in xenograft and syngeneic animal models, to study whether combining panobinostat with chemotherapy may result in superior efficacy, and to analyze the in vivo modulation of histone and nonhistone proteins as potential mechanisms of the antitumor efficacy of panobinostat alone or in combination with chemotherapy.
We found that panobinostat was highly effective in the vast majority of NSCLC, SCLC, and Meso cell lines, of both murine and human origin. Panobinostat at nanomolar concentrations inhibited the growth of all but one cancer cell line. This efficacy seemed superior to SAHA, which generally required micromolar concentrations for similar effects (Table 1).
Of particular interest was the high sensitivity of SCLC lines to panobinostat. The median LD50 was 120 nmol/L for NSCLC cell lines and 60 nmol/L for mesothelioma cell lines, but was only 12 nmol/L for SCLC cell lines. Although our findings of HDACi efficacy in NSCLC cell lines were generally consistent with those previously reported for panobinostat (33, 43) and MGCD0103 (21), we are not aware of previous analyses of panobinostat efficacy in a panel of SCLC cell lines. The in vitro susceptibility of SCLC cell lines to panobinostat we found (Table 1) was confirmed when the in vivo susceptibility to panobinostat of tumors derived from all four different SCLC cell lines tested was shown. In contrast to only slowing tumor growth seen after panobinostat treatment of animals with NSCLC and mesothelioma tumors, we noted substantial tumor regressions or stabilization in three of the four human SCLC xenografts and a substantial effect in the tumors from the remaining H69 SCLC cell line (Fig. 3). In addition, the in vivo efficacy of panobinostat may be even more enhanced when given with chemotherapy. Although this report may be the first to show in vivo data of HDACi efficacy against SCLC, we note that others previously found susceptibility of a small number of SCLC cell lines to other HDACi such as trichostatin A (22, 26), FR901228 (27, 29), or valproic acid (32).
Our in vivo experiments also included analyses of protein status and expression in tumors derived from SCLC cells that were grown in mice that were then separated into treatment groups (consisting of mice that were mock-treated and mice that were treated with panobinostat alone, etoposide alone, or the two drugs in combination). These analyses found evidence of anti-HDAC enzyme activity, with increased acetylation of histones H3 and H4 after treatment of the animals with panobinostat. However, we also found that panobinostat resulted in increased levels of the proapoptotic factors t-Bid, cleaved PARP, cleaved caspase 3, and cleaved caspase 7, accompanied by the disappearance of Bcl-XL and an increase in levels of the cyclin-dependent kinase (cdk) inhibitor p21. Our results may supply the first in vivo confirmation of modulation of these proteins, which has been described only previously in cells grown and treated under in vitro conditions (16–18, 21, 22, 27, 29, 31, 34, 35, 43, 44). Thus, the proapoptotic effects of HDACi that have been observed for cells in culture are also likely to be operative in animal models of SCLC. It should be noted that our analysis was not exhaustive and that other pathways of cell death or growth inhibition are also likely involved (32). Nonetheless, our observations suggest that a subset of these proteins that are modulated by panobinostat may merit testing as potentially useful biomarkers of HDACi efficacy in clinical trials.
The striking effects of panobinostat on the Bcl-2 pathway and related components may help explain why SCLC cell lines are especially sensitive to this agent. The literature supports the idea that SCLC, more than NSCLC or mesotheliomas, have high levels of expression of Bcl-2 and seem especially “addicted” to this pathway (45). For example, the highly potent Bcl-2 inhibitors ABT-737 and ABT-263 exhibited single-agent activity in in vitro and animal models of SCLC and hematologic malignancies, but not in the majority of other tumor types (46, 47). This Bcl-2 sensitivity has resulted in SCLC being the primary target for a recent clinical trial using a Bcl-2 antisense oligonucleotide (48).
Given the potentially different mechanisms of action and toxicities, the proven value of multitargeted approaches in cancer, the experimental data showing the interactions of histone deacetylase with DNA topoisomerase II (15, 16), and a report showing the combinatorial action of the HDACi trichostatin A with etoposide (23), we examined the combination of panobinostat with the topoisomerase inhibitor and SCLC chemotherapy agent etoposide (VP16). The combination of the two agents increased the ability to induce apoptosis in H69 cells in vitro (Fig. 1B). The combination seemed to work in an additive fashion in the in vivo H69 animal xenograft model (Fig. 4). Analysis of the tumor biopsies using immunoblotting that allowed comparison of the effects with panobinostat alone, etoposide alone, or the combination, showed what also seemed to be “additive” effects, in that in the combination there was enhanced loss of Bcl-2 and an augmented increase in p21 compared with either treatment alone (Fig. 5).
The combination of panobinostat and etoposide increased the ability to induce apoptosis in H69 cells in culture and as a tumor xenograft in the mouse. In the immunoblotting studies, however, the combination of these two agents did not increase the expression of cleaved caspase 3, caspase 7, PARP, and acetylated H4 compared with panobinostat treatment only. There may be two potential explanations for this seeming contradiction. First, there may be differences between in vitro and in vivo effects. In vivo, panobinostat may inhibit angiogenesis or other microenvironment-related mechanisms that contribute to tumor growth and treatment resistance, thereby sensitizing the tumor to etoposide-induced mitotic death and inhibition of tumor growth, as shown in Fig. 4. These mechanisms would not be in play for in vitro experiments such as shown in Fig. 1. Second, the combination of panobinostat and etoposide led to levels of p21 that were substantially increased over either drug alone, as shown in Fig. 5. The combination of the drugs also led to levels of Bcl-2 that were maximally decreased to almost undetectable levels, in contrast to levels with either drug alone. Consequently, it is also possible that panobinostat modulates intracellular components beyond caspase activation, to sensitize cancer cells to killing by etoposide.
Our ability to study murine cell lines grown in syngeneic mice allowed us to examine the question of whether panobinostat exerted any of its antitumor efficacy via immune effects mediated by T or B cells. The effect of HDACi on antitumor immunity is complex. On one hand, HDACi have been reported to up-regulate MHC class I and class II molecules (36), MHC class I–related chain A and B (MICA/B) molecules (38), and TRAIL (49–51), all of which could potentially enhance antitumor immune effects. On the other hand, these agents have been reported to inhibit dendritic cell differentiation and immunogenicity (52) and can stimulate the production of T-regulatory CD4 cells (53), all of which would inhibit antitumor immune effects and promote tumor growth. In the TC-1 lung cancer model, in which we have previously shown some endogenous antitumor immune response (54), and in the AE17-ova mesothelioma model, panobinostat had virtually identical antitumor efficacy in normal compared with immune-deficient mice (Supplementary Fig. S1), suggesting no acquired immune response component to the activity seen. We did not study the effect of panobinostat with active immunotherapy, however. This might be interesting, because at least one report suggests that combining an HDACi with immunotherapy (i.e., interleukin-2) could be helpful (37).
In summary, panobinostat in nanomolar concentrations seems to have activity against cell lines and tumors derived from human NSCLC, SCLC, and mesothelioma. Cell lines and tumors derived from SCLC seem to be especially susceptible to the antigrowth effects of panobinostat, and maximal efficacy was noted when combined with etoposide. Animal studies suggest that this agent causes histone acetylation within tumors and results in nonimmunologically induced tumor cell apoptosis with acceptable toxicity. SCLC remains a deadly form of lung cancer (1). Given the high relapse rate after chemotherapy and the relative dearth of second line treatments, these data support clinical trials incorporating panobinostat for treatment of SCLC.
Acknowledgments
We acknowledge Peter Atadja and Wenlin Shao of the Novartis Institutes of Biomedical Research (Cambridge, MA) for their helpful comments and suggestions.
Grant support: Novartis Pharmaceuticals.
Footnotes
Supplementary material for this article is available at Molecular Cancer Therapeutics Online (http://mct.aacrjournals.org/).
Disclosure of Potential Conflicts of Interest The authors received a research grant from Novartis Pharmaceuticals.
References
- 1.Govindan R, Page N, Morgensztern D, et al. Changing epidemiology of small cell lung cancer in the United States over the last 30 years: analysis of the surveillance, epidemiologic, and end results database. J Clin Oncol. 2006;24:4539–44. doi: 10.1200/JCO.2005.04.4859. [DOI] [PubMed] [Google Scholar]
- 2.Minucci S, Pelicci PG. Histone deacetylase inhibitors and the promise of epigenetic (and more) treatments for cancer. Nat Rev Cancer. 2006;6:38–50. doi: 10.1038/nrc1779. [DOI] [PubMed] [Google Scholar]
- 3.Verheul HM, Salumbides B, Van Erp K, et al. Combination strategy targeting the hypoxia inducible factor-1 alpha with mammalian target of rapamycin and histone deacetylase inhibitors. Clin Cancer Res. 2008;14:3589–97. doi: 10.1158/1078-0432.CCR-07-4306. [DOI] [PubMed] [Google Scholar]
- 4.Haefner M, Bluethner T, Niederhagen M, et al. Experimental treatment of pancreatic cancer with two novel histone deacetylase inhibitors. World J Gastroenterol. 2008;14:3681–92. doi: 10.3748/wjg.14.3681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zhou Q, Agoston AT, Atadja P, Nelson WG, Davidson NE. Inhibition of histone deacetylases promotes ubiquitin-dependent proteasomal degradation of DNA methyltransferase 1 in human breast cancer cells. Mol Cancer Res. 2008;6:873–83. doi: 10.1158/1541-7786.MCR-07-0330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Giles F, Fischer T, Cortes J, et al. A phase I study of intravenous LBH589, a novel cinnamic hydroxamic acid analogue histone deacetylase inhibitor, in patients with refractory hematologic malignancies. Clin Cancer Res. 2006;12:4628–35. doi: 10.1158/1078-0432.CCR-06-0511. [DOI] [PubMed] [Google Scholar]
- 7.Ellis L, Pan Y, Smyth GK, et al. Histone deacetylase inhibitor panobinostat induces clinical responses with associated alterations in gene expression profiles in cutaneous T-cell lymphoma. Clin Cancer Res. 2008;14:4500–10. doi: 10.1158/1078-0432.CCR-07-4262. [DOI] [PubMed] [Google Scholar]
- 8.Li Q, Verschraegen CF, Mendoza J, Hassan R. Cytotoxic activity of the recombinant anti-mesothelin immunotoxin, SS1(dsFv)PE38, towards tumor cell lines established from ascites of patients with peritoneal mesotheliomas. Anticancer Res. 2004;24:1327–35. [PubMed] [Google Scholar]
- 9.Jackaman C, Bundell CS, Kinnear BF, et al. IL-2 intratumoral immunotherapy enhances CD8+ T cells that mediate destruction of tumor cells and tumor-associated vasculature: a novel mechanism for IL-2. J Immunol. 2003;171:5051–63. doi: 10.4049/jimmunol.171.10.5051. [DOI] [PubMed] [Google Scholar]
- 10.Davis MR, Manning LS, Whataker D, Garlepp MJ, Robinson BWS. Establishment of a murine model of malignant mesotheoioma. Int J Cancer. 1992;52:881–86. doi: 10.1002/ijc.2910520609. [DOI] [PubMed] [Google Scholar]
- 11.Lin KY, Guarnieri FG, Staveley-O'Carroll KF, et al. Treatment of established tumors with a novel vaccine that enhances major histocompatibility class II presentation of tumor antigens. Cancer Res. 1996;56:21–26. [PubMed] [Google Scholar]
- 12.Wilderman M, Sun J, Khan M, et al. Intrapulmonary interferon-β gene therapy using an adenoviral vector is highly effective in a murine orthotopic model of lung adenocarcinoma via a combination of direct toxicity, NK cell, and CD8 T-cell mediated effects. Cancer Res. 2005;65:8379–87. doi: 10.1158/0008-5472.CAN-05-0920. [DOI] [PubMed] [Google Scholar]
- 13.Campling BG, Haworth AC, Baker HM, et al. Establishment and characterization of a panel of human lung cancer cell lines. Cancer. 1992;69:2064–74. doi: 10.1002/1097-0142(19920415)69:8<2064::aid-cncr2820690811>3.0.co;2-#. [DOI] [PubMed] [Google Scholar]
- 14.Kao GD, McKenna WG, Guenther MG, Muschel RJ, Lazar MA, Yen TJ. Histone deacetylase 4 interacts with 53BP1 to mediate the DNA damage response. J Cell Biol. 2003;160:1017–27. doi: 10.1083/jcb.200209065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tsai SC, Valkov N, Yang WM, Gump J, Sullivan D, Seto E. Histone deacetylase interacts directly with DNA topoisomerase II. Nat Genet. 2000;26:349–53. doi: 10.1038/81671. [DOI] [PubMed] [Google Scholar]
- 16.Kim MS, Blake M, Baek JH, Kohlhagen G, Pommier Y, Carrier F. Inhibition of histone deacetylase increases cytotoxicity to anticancer drugs targeting DNA. Cancer Res. 2003;63:7291–300. [PubMed] [Google Scholar]
- 17.Rundall BK, Denlinger CE, Jones DR. Suberoylanilide hydroxamic acid combined with gemcitabine enhances apoptosis in non-small cell lung cancer. Surgery. 2005;138:360–7. doi: 10.1016/j.surg.2005.06.016. [DOI] [PubMed] [Google Scholar]
- 18.Neuzil J, Swettenham E, Gellert N. Sensitization of mesothelioma to TRAIL apoptosis by inhibition of histone deacetylase: role of Bcl-xL down-regulation. Biochem Biochem Res Commun. 2004;314:186–91. doi: 10.1016/j.bbrc.2003.12.074. [DOI] [PubMed] [Google Scholar]
- 19.Miyanaga A, Gemma A, Noro R, et al. Antitumor activity of histone deacetylase inhibitors in non-small cell lung cancer cells: development of a molecular predictive model. Mol Cancer Ther. 2008;7:1923–30. doi: 10.1158/1535-7163.MCT-07-2140. [DOI] [PubMed] [Google Scholar]
- 20.Komatsu N, Kawamata N, Takeuchi S, et al. SAHA, a HDAC inhibitor, has profound anti-growth activity against non-small cell lung cancer cells. Oncol Rep. 2006;15:187–91. [PubMed] [Google Scholar]
- 21.Fournel M, Bonfils C, Hou Y, et al. MGCD0103, a novel isotype-selective histone deacetylase inhibitor, has broad spectrum antitumor activity in vitro and in vivo. Mol Cancer Ther. 2008;7:759–68. doi: 10.1158/1535-7163.MCT-07-2026. [DOI] [PubMed] [Google Scholar]
- 22.Platta CS, Greenblatt DY, Kunnimalaiyaan M, Chen H. The HDAC inhibitor trichostatin A inhibits growth of small cell lung cancer cells. JSR. 2007;142:219–26. doi: 10.1016/j.jss.2006.12.555. [DOI] [PubMed] [Google Scholar]
- 23.Hajji N, Wallenborg K, Vlachos P, Nyman U, Hermanson O, Joseph B. Combinatorial action of the HDAC inhibitor trichostatin A and etoposide induces caspase-mediated AIF-dependent apoptotic cell death in non-small cell lung carcinoma cells. Oncogene. 2008;27:3134–44. doi: 10.1038/sj.onc.1210976. [DOI] [PubMed] [Google Scholar]
- 24.Kim HR, Kim EJ, Yang SH, et al. Trichostatin A induces apoptosis in lung cancer cells via simultaneous activation of the death receptor-mediated and mitochondrial pathway? Exp Mol Med. 2006;38:616–24. doi: 10.1038/emm.2006.73. [DOI] [PubMed] [Google Scholar]
- 25.Choi YH. Induction of apoptosis by trichostatin A, a histone deacetylase inhibitor, is associated with inhibition of cyclooxygenase-2 activity in human non-small cell lung cancer cells. Int J Oncol. 2005;27:473–9. [PubMed] [Google Scholar]
- 26.El-Khoury V, Gomez D, Liautaud-Roger F, Trussardi-Régnier A, Dufer J. Effects of the histone deacetylase inhibitor trichostatin A on nuclear texture and c-jun gene expression in drug-sensitive and drug-resistant human H69 lung carcinoma cells. Cytometry A. 2004;62:109–17. doi: 10.1002/cyto.a.20088. [DOI] [PubMed] [Google Scholar]
- 27.Doi S, Soda H, Oka M, et al. The histone deacetylase inhibitor FR901228 induces caspase-dependent apoptosis via the mitochondrial pathway in small cell lung cancer cells. Mol Cancer Ther. 2004;3:1397–402. [PubMed] [Google Scholar]
- 28.Witta SE, Gemmill RM, Hirsch FR, et al. Restoring E-cadherin expression increases sensitivity to epidermal growth factor receptor inhibitors in lung cancer cell lines. Cancer Res. 2006;66:944–50. doi: 10.1158/0008-5472.CAN-05-1988. [DOI] [PubMed] [Google Scholar]
- 29.Tsurutani J, Soda H, Oka M, et al. Antiproliferative effects of the histone deacetylase inhibitor FR901228 on small-cell lung cancer lines and drug-resistant sublines. Int J Cancer. 2003;104:238–42. doi: 10.1002/ijc.10921. [DOI] [PubMed] [Google Scholar]
- 30.Wang S, Yan-Neale Y, Fischer D, et al. Histone deacetylase 1 represses the small GTPase RhoB expression in human nonsmall lung carcinoma cell line. Oncogene. 2003;22:6204–13. doi: 10.1038/sj.onc.1206653. [DOI] [PubMed] [Google Scholar]
- 31.Cao XX, Mohuiddin I, Ece F, McConkey DJ, Smythe WR. Histone deacetylase inhibitor downregulation of bcl-xl gene expression leads to apoptotic cell death in mesothelioma. Am J Respir Cell Mol Biol. 2001;25:562–8. doi: 10.1165/ajrcmb.25.5.4539. [DOI] [PubMed] [Google Scholar]
- 32.Platta CS, Greenblatt DY, Kunnimalaiyaan M, Chen H. Valproic acid induces Notch1 signaling in small cell lung cancer cells. J Surg Res. 2008;148:31–7. doi: 10.1016/j.jss.2008.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Geng L, Cuneo KC, Fu A, Tu T, Atadja PW, Hallahan DE. Histone deacetylase (HDAC) inhibitor LBH589 increases duration of gamma-H2AX foci and confines HDAC4 to the cytoplasm in irradiated non-small cell lung cancer. Cancer Res. 2006;66:11298–304. doi: 10.1158/0008-5472.CAN-06-0049. [DOI] [PubMed] [Google Scholar]
- 34.Ruefli AA, Ausserlechner MJ, Bernhard D, et al. The histone deacetylase inhibitor and chemotherapeutic agent suberoylanilide hydroxamic acid (SAHA) induces a cell-death pathway characterized by cleavage of Bid and production of reactive oxygen species. Proc Natl Acad Sci U S A. 2001;98:10833–8. doi: 10.1073/pnas.191208598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Shao Y, Gao Z, Marks PA, Jiang X. Apoptotic and autophagic cell death induced by histone deacetylase inhibitors. Proc Natl Acad Sci U S A. 2004;101:18030–5. doi: 10.1073/pnas.0408345102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Magner WJ, Kazim AL, Stewart C, et al. Activation of MHC class I, II, and CD40 gene expression by histone deacetylase inhibitors. J Immunol. 2000;165:7017–24. doi: 10.4049/jimmunol.165.12.7017. [DOI] [PubMed] [Google Scholar]
- 37.Kato Y, Yoshimura K, Shin T, et al. Synergistic in vivo antitumor effect of the histone deacetylase inhibitor MS-275 in combination with interleukin 2 in a murine model of renal cell carcinoma. Clin Cancer Res. 2007;13:4538–46. doi: 10.1158/1078-0432.CCR-07-0014. [DOI] [PubMed] [Google Scholar]
- 38.Skov S, Pedersen MT, Andresen L, Straten PT, Woetmann A, Odum N. Cancer cells become susceptible to natural killer cell killing after exposure to histone deacetylase inhibitors due to glycogen synthase kinase-3-dependent expression of MHC class I-related chain A and B. Cancer Res. 2005;65:11136–45. doi: 10.1158/0008-5472.CAN-05-0599. [DOI] [PubMed] [Google Scholar]
- 39.Reid T, Valone F, Lipera W, et al. Phase II trial of the histone deacetylase inhibitor pivaloyloxymethyl butyrate (Pivanex, AN-9) in advanced non-small cell lung cancer. Lung Cancer. 2004;45:381–6. doi: 10.1016/j.lungcan.2004.03.002. www.clinical-trials.gov NCT00073385. [DOI] [PubMed] [Google Scholar]
- 40.Schrump DS, Fischette MR, Nguyen DM, et al. Clinical and molecular responses in lung cancer patients receiving Romidepsin. Clin Cancer Res. 2008;14:188–98. doi: 10.1158/1078-0432.CCR-07-0135. [DOI] [PubMed] [Google Scholar]
- 41.Krug LM, Curley T, Schwartz L, et al. Potential role of histone deacetylase inhibitors in mesothelioma: clinical experience with suberoylanilide hydroxamic acid. Clin Lung Cancer. 2006;7:257–61. doi: 10.3816/CLC.2006.n.003. [DOI] [PubMed] [Google Scholar]
- 42.Kelly WK, O'Connor OA, Krug LM, et al. Phase I study of an oral histone deacetylase inhibitor, suberoylanilide hydroxamic acid, in patients with advanced cancer. J Clin Oncol. 2005;23:3923–31. doi: 10.1200/JCO.2005.14.167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Edwards A, Li J, Atadja P, Bhalla K, Haura EB. Effect of the histone deacetylase inhibitor LBH589 against epidermal growth factor receptor-dependent human lung cancer cells. Mol Caner Ther. 2007;6:2515–24. doi: 10.1158/1535-7163.MCT-06-0761. [DOI] [PubMed] [Google Scholar]
- 44.Maiso P, Carvajal-Vergara X, Ocio EM, et al. The histone deacetylase inhibitor LBH589 is a potent antimyeloma agent that overcomes drug resistance. Cancer Res. 2006;66:5781–9. doi: 10.1158/0008-5472.CAN-05-4186. [DOI] [PubMed] [Google Scholar]
- 45.Ziegler A, Luedke GH, Fabbro D, Altmann KH, Stahel RA, Zanegemeister-Wittke U. Induction of apoptosis in small-cell lung cancer cells by an antisense oligonucleotide targeting the Bcl-2 coding sequence. J Natl Cancer Inst. 1997;89:1027–36. doi: 10.1093/jnci/89.14.1027. [DOI] [PubMed] [Google Scholar]
- 46.Oltersdorf T, Elmore SW, Shoemaker AR, et al. An inhibitor of Bcl-2 family proteins induces regression of solid tumours. Nature. 2005;435:677–81. doi: 10.1038/nature03579. [DOI] [PubMed] [Google Scholar]
- 47.Tse C, Shoemaker AR, Adickes J, et al. ABT-263: a potent and orally bioavailable Bcl-2 family inhibitor. Cancer Res. 2008;68:3421–8. doi: 10.1158/0008-5472.CAN-07-5836. [DOI] [PubMed] [Google Scholar]
- 48.Rudin CM, Salgia R, Wang X, et al. Randomized phase II study of carboplatin and etoposide with or without the bcl-2 antisense oligonucleotide oblimersen for extensive-stage small-cell lung cancer: CALGB 30103. J Clin Oncol. 2008;26:870–6. doi: 10.1200/JCO.2007.14.3461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Nakata S, Yoshida T, Horinaka M, Shiraishi T, Wakada M, Sakai T. Histone deacetylase inhibitors upregulate death receptor 5/TRAIL-R2 and sensitize apoptosis induced by TRAIL/APO2-L in human malignant tumor cells. Oncogene. 2004;23:6261–71. doi: 10.1038/sj.onc.1207830. [DOI] [PubMed] [Google Scholar]
- 50.Guo F, Sigua C, Tao J, et al. Cotreatment with histone deacetylase inhibitor LAQ824 enhances Apo-2L/tumor necrosis factor-related apoptosis inducing ligand-induced death inducing signaling complex activity and apoptosis of human acute leukemia cells. Cancer Res. 2004;64:2580–9. doi: 10.1158/0008-5472.can-03-2629. [DOI] [PubMed] [Google Scholar]
- 51.Lundqvist A, Abrams SI, Schrump DS, et al. Bortezomib and depsipeptide sensitize tumors to tumor necrosis factor-related apoptosis-inducing ligand: a novel method to potentiate natural killer cell tumor cytotoxicity. Cancer Res. 2006;66:7317–25. doi: 10.1158/0008-5472.CAN-06-0680. [DOI] [PubMed] [Google Scholar]
- 52.Nencioni A, Beck J, Werth D, et al. Histone deacetylase inhibitors affect dendritic cell differentiation and immunogenicity. Clin Cancer Res. 2007;13:3933–41. doi: 10.1158/1078-0432.CCR-06-2903. [DOI] [PubMed] [Google Scholar]
- 53.Tao R, de Zoeten EF, Ozkaynak E, et al. Deacetylase inhibition promotes the generation and function of regulatory T cells. Nat Med. 2007;13:1299–307. doi: 10.1038/nm1652. [DOI] [PubMed] [Google Scholar]
- 54.Tanaka T, Delong P, Amin K, et al. Treatment of lung cancer using clinically relevant oral doses of the cyclooxygenase-2 inhibitor rofecoxib: potential value as adjuvant therapy after surgery. Ann Surg. 2005;241:168–78. doi: 10.1097/01.sla.0000149427.84712.d9. [DOI] [PMC free article] [PubMed] [Google Scholar]