

Abstract
Resistance to antibiotics is an increasingly dire threat to human health that warrants the development of new modes of treating infection. We recently identified 1 (CCG-2979) as an inhibitor of the expression of streptokinase, a critical virulence factor in Group A Streptococcus that endows blood-borne bacteria with fibrinolytic capabilities. In this report, we describe the synthesis and biological evaluation of a series of novel 5,6-dihydrobenzo[h]quinazolin-4(3H)-one analogs of 1 undertaken with the goal of improving the modest potency of the lead. In addition to achieving an over 35-fold increase in potency, we identified structural modifications that improve the solubility and metabolic stability of the scaffold. The efficacy of two new compounds 12c (CCG-203592) and 12k (CCG-205363) against biofilm formation in Staphylococcus aureus represents a promising additional mode of action for this novel class of compounds.
Keywords: Group A Streptococcus, virulence inhibitor, antibiotic, streptokinase, antibiotic resistance, metabolic oxidation, biofilm, antivirulence
1. Introduction
Bacterial infection remains one of the most critical threats to human health, especially in third world countries and in children. Streptococcus pyogenes, or Group A Streptococcus (GAS), represents one aspect of this global threat. GAS is a ubiquitous human pathogen responsible for up to 700 million infections per year worldwide.1 While most commonly associated with upper respiratory infection (“strep throat”) and superficial skin infections, systemic infections with GAS such as streptococcal toxic shock syndrome, rheumatoid fever, and necrotizing fasciitis are very often fatal. Cases of systemic GAS infection are thought to approach 500,000 cases annually, with a mortality rate of 15–35%.2,3
The rapid emergence of bacterial resistance to antibiotic treatment is an alarming aspect of systemic infection, which can rapidly progress in the absence of effective treatment. As dangerous resistant strains such as methicillin-resistant Staphylococcus aureus (MRSA) and vancomycin-resistant Enterococcus (VRE) become increasingly prevalent, especially in the hospital setting,4,5 the necessity for antibiotics with novel mechanisms of action grows. Virulence factors are among the targets currently being evaluated for antibiotic activity.6 By inhibiting the mechanisms bacteria use to spread and thrive in a host organism, virulence inhibitors are intended to retain highly infective pathogenic bacteria in a relatively benign state, allowing additional time for the host’s immune system or a course of antibiotics to clear the infection. Since inhibitors of virulence do not necessarily affect the reproduction of bacterial cells, they may represent an avenue of antibiotic treatment that does not encourage the development of resistance. A number of virulence mechanisms have been targeted for inhibition by small molecules in the literature, including bacterial toxins,7 adhesion factors,8 quorum-sensing systems,9,10 and virulence-inducing signaling pathways.11 Though no anti-virulence drugs have yet progressed to use in the clinic, inhibitors of anthrax toxin cell permeability,12 N-acyl homoserine lactone-mediated quorum sensing,13 and QseC histidine sensor kinase activation11 have demonstrated efficacy in murine models of infection.
Sun et al. previously identified the protein streptokinase (SK) as a potent virulence factor in GAS.14 SK is an activator of human plasminogen, which generates the fibrinolytic plasmin and allows the bacteria to circumvent the host’s defense mechanism of clotting around foci of infection. We recently reported a high-throughput screening (HTS) campaign that identified lead compound 1 (CCG-2979, Fig. 1) and analog 6b (CCG-102487) as capable of reducing the expression of streptokinase at the transcriptional level, confirmed by Western blot and RNA microarray analysis. This compound was also found to elicit a significant protective effect in a transgenic mouse model of GAS infection.15 These findings prompted the initiation of a structure-activity relationship (SAR) study based on 1 with the intent of increasing its modest potency (% of plasmin activity at 50 μM = 48 ± 28%) and improving the predicted pharmacokinetic properties of the scaffold. In particular, we were concerned about its high lipophilicity (cLogP = 7.1), which can be associated with poor solubility and metabolic stability. Ultimately we would also like to identify the macromolecular target for 1, which is currently unknown due to the phenotypic nature of the HTS and biological assay. To design affinity probes for this purpose, we needed to perform SAR in order to both improve potency and to identify areas on the molecule amenable to substitution without loss of activity.
Figure 1.
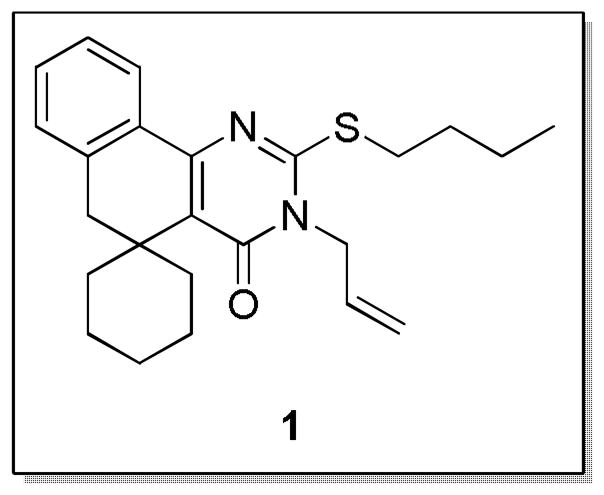
Structure of HTS lead compound 1.
2. Chemistry
Access to the core structure of 1 was accomplished with a known 5-step synthetic pathway (Scheme 1). β-aminoester 4 was synthesized from cyclohexanone via a Knoevenagel condensation with ethyl cyanoacetate followed by Michael addition of benzylmagnesium chloride, and finally acid-mediated cyclization.16 The 2-thioxo-dihydropyrimidinone ring could then be installed via nucleophilic addition of 4 to allyl isothiocyanate, followed by base-catalyzed cyclization.17 Finally, sulfur alkylation under basic conditions gave access to 1 and S-substituted analogs 6a–f. Additional analogs that replace the sulfide with ether (7a) and amine (7b) linkages were accessed via the S-methyl derivative 6f, either by direct nucleophilic substitution with sodium ethoxide, or further functionalization to the methyl sulfone and displacement with ethylamine under basic conditions.18
Scheme 1. Preparation of spirocyclohexyl analogs of 1.a.

aReagents and conditions: a) ethyl cyanoacetate, AcOH, NH4OAc, toluene, 150°C, 5h; b) BnMgCl, Et2O, RT, 72h; c) H2SO4, 0°C-RT, 7h; d) allyl isothiocyanate, EtOH, reflux, 10h; e) KOH, EtOH:H2O (1:1), reflux, 10h; f) R-X, base, EtOH or MEK, 0–70°C, 10min-16h; g) NaOEt, EtOH, 40°C, 48h; h) (i) mCPBA, DCM, RT, 16h, (ii) EtNH2, K2CO3, THF:DMF (1:1), RT, 5h.
Given the relative ease with which the original spirocyclohexyl-substituted analogs were synthesized, we were surprised to find that we were not able to generate gem-dimethyl analogs (e.g. 12, Scheme 2) using this scheme. While Michael addition of benzylmagnesium chloride into ethyl 2-cyano-3-methylbut-2-enoate proceeded as desired, no set of conditions was successful in generating the gem-dimethyl analog of 4. It was finally determined that the β-aminoesters 10a–d could be accessed in one step beginning from the corresponding o-tolunitriles 9a–d, available commercially or by SNAr addition of cyanide into the corresponding chlorotoluenes 8a,b19 (Scheme 2). In the presence of LDA, smooth addition of 9a–d to the β-position of ethyl 3,3-dimethyl acrylate afforded an enol ester intermediate that readily cyclized in the presence of ZnI2, providing amino esters 10a–d.20 Modified conditions for condensation with alkyl isothiocyanates were required to efficiently produce the 2-thioxo-dihydropyrimidinone intermediates 11a–f. Subsequent alkylation at sulfur ultimately allowed the synthesis of gem-dimethyl analogs 12a–n in a total of 3 to 4 steps (Scheme 2). Further elaboration of these compounds to generate ether and amino derivatives 13a,b was achieved in the same manner as for 7a,b. Chemoselective sulfur oxidation of 12a via treatment with 1.1 eq of Oxone®21 or an excess of mCPBA delivered sulfoxide and sulfone-modified analogs 14 and 15, respectively. No epoxidation of the allyl group was observed under these mild oxidation conditions.
Scheme 2. Synthesis of gem-dimethyl substituted analogs of 1.a.

aReagents and conditions: a) NiBr2, Zn(dust), PPh3, then KCN, THF, 50–60°C, 24h; b) LDA, diglyme, −78°C, 1h,, then ethyl 3,3-dimethyl acrylate, ZnI2, −78°C-RT, 2h; c) R4-NCS, AcOH, EtOH, 70°C, 16h; d) R5-X, Cs2CO3, MEK or DMF, 70°C, 16h; e) NaOEt, EtOH, 40°C, 48h; f) (i) mCPBA, DCM, 0°C-RT, 16h, (ii) EtNH2, K2CO3, THF:DMF (1:1), RT, 5h; g) 1.1 eq Oxone, THF:H2O (1:1), 0°C-RT, 16h; h) mCPBA, DCM, RT, 12h.
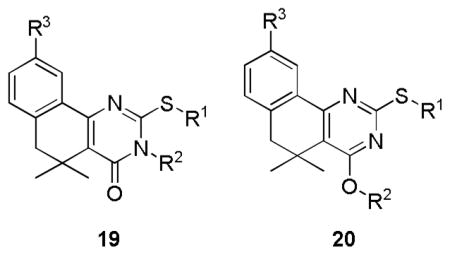
Small changes or additions to the core procedure allowed access to a number of other substitution patterns. Beginning from the methoxy-substituted aryl ethers 12a–c, treatment with BBr3 revealed phenols 16a–c, which could be elaborated into a diverse set of aryl ethers 17a–k (Scheme 3). Exploring diversity at the N- and O- positions of the pyrimidinone required assembling the unsubstituted thioxo-dihydropyrimidinone ring via treatment of 10a or 10d with benzoyl isothiocyanate, followed by ring closure and hydrolysis of the resulting N-benzoyl group with aqueous KOH to generate 18a and 18d22 (Scheme 4). Selective alkylation of the sulfur was achieved under neutral conditions, while subsequent base-catalyzed alkylation of the amide generally gave some mixture of N- and O- alkylated analogs 19a–b and 20a–c.
Scheme 3. Synthesis of aryl ethers 17a–k.a.

aReagents and conditions: a) BBr3, DCM, 0°C-RT, 16h; b) R-X, Cs2CO3, MEK or DMF, RT-70°C, 16h.
Scheme 4. Generation of N- and O-alkylated substitution pairs 19a–b and 20a–c.a.

aReagents and conditions: a) (i) benzoyl isothiocyanate, EtOH, 75°C, 5h, (ii) KOH, EtOH:H2O (2:1) 70°C, 2h; b) R2-X, NaHCO3, DMF, RT-70°C, 30min-16h; c) R3-X, base, EtOH or DMF, 70–80°C, 16–24h.
Finally, carboxylic acid derivatives at the 8-position (Scheme 5) were accessed via lithiation and carboxylation of 4-bromo-2-methylbenzonitrile 21, followed by cyclization and esterification to generate 23. Sensitivity to the standard acid-catalyzed cyclization with allyl isothiocyanate necessitated the use of the stepwise procedure seen for the conversion of 10 to 18 (Scheme 4). Subsequent alkylations at sulfur and nitrogen delivered the 8-methylcarboxylate analog 25, which could be saponified to acid 26. Amides 27a and 27b were accessed via amide coupling conditions using HOBt and EDC.
Scheme 5. Synthesis of carboxylic acid derivatives 24–26.a.

aReagents and conditions: a) nBuLi, then CO2, THF, −78°C-RT, 1h; b) (i) LDA, diglyme, −78°C, 20min, then ethyl 3,3-dimethyl acrylate, ZnI2, −78°C-RT, 3h, (ii) TMS-CHN2, MeOH:toluene (1:5), RT, 30min; c) benzoyl isothiocyanate, EtOH, reflux, 2.5h; d) NaOMe, MeOH, 70°C, 2h; e) 2-methoxyethyl-4-methylbenzenesulfonate, Cs2CO3, DMF, 40°C-RT, 16h; f) allyl bromide, NaOMe, MeOH, 65°C, 2h; g) NaOH, MeOH, RT, 16h; h) NH2R, HOBt, EDC, THF, RT, 16h.
3. Results and Discussion
3.1. SK Expression Assay
All new compounds were evaluated for their ability to suppress SK expression via a chromogenic assay of plasmin activity. Two identical cultures of GAS were grown in the presence of the desired concentration of test compound or DMSO and allowed to grow to OD600 ≈ 1.0. After centrifugation, an aliquot of supernatant was combined with human plasma and synthetic plasmin substrate S-2403 (see Section 5.1.1). Decreased expression of SK induced by the test compound lowers the amount of activated plasmin, in turn reducing the rate of cleavage of S-2403 to colored product p-nitroaniline. The concentration of p-nitroaniline was measured by absorbance at 405 nm (A405) and compared to the DMSO control. Activity data is reported as the ratio of A405(test) divided by A405(control) (T/C). To provide an approximate readout of potency, the assay was run initially at both 5 and 50 μM of test compound. An optical density reading at 600 nm (OD600) gave an estimate of cell density and thus overall bacterial growth inhibition; this data is also reported for the higher concentration of compound (50 μM) as a ratio of the OD600 of the test culture divided by the OD600 of the DMSO control culture. Selected compounds that inhibited SK activity by more than 50% at 50 μM were subjected to full dose-response titrations to determine approximate IC50 values using the same assay.
During the course of evaluating the new analogs, it was observed that there was a relatively high level of inter-assay variability in the activity assay. There are a number of likely contributing factors, most notably the variability inherent to working in a live bacterial system. For example, it has been noted that minimum inhibitory concentration (MIC) values for determining bacteriotoxicity tend to vary by up to three-fold from assay to assay.23 Furthermore, the currently unknown macromolecular target of these compounds likely affects a pathway including a transcription factor, which are known to be low-abundance proteins24 that exhibit significant temporal expression changes, potentially contributing to the variability. It was noted early into the SAR effort that 12h was both moderately active and particularly consistent from assay to assay. In an effort to reduce the number of anomalous data points, 12h was included in each assay as a positive control. Assays in which 12h exhibited activity deviating from the mean activity at 5 and 50 μM by more than one standard deviation (calculated from a total of 35 assays) were discarded.
3.2. SAR Development
We first examined a series of spirocycloalkyl substituted compounds with varying substitution patterns at the 2-position of the pyrimidinone ring system (Table 1). Aromatic substitution at R1 (6a–c) did not appreciably increase potency over 1, but methyl substitution (6f) conferred a modest increase in activity (IC50 = 19 μM vs. >50 μM). Interestingly, polar groups were tolerated as part of smaller alkyl groups at – XR1(e.g. 6d and 7a). The replacement of sulfur with nitrogen (7b) led to an unacceptable decrease in growth. Similarly, basic amine functionality appended to sulfur (6e) led to growth inhibition in some trials. We defined acceptable growth inhibition to be no greater than 15% at 50 μM of test compound. The favorable potency of commercially available spirocyclopentyl analog 28 relative to closely related spirocyclohexyl analog 6b led us to hypothesize that the size of the geminal substitution on the central ring could be reduced, allowing reduction of both the lipophilicity and the molecular weight of future analogs.
Table 1.
Inhibition of SK activity by spirocycloalkyl analogs.
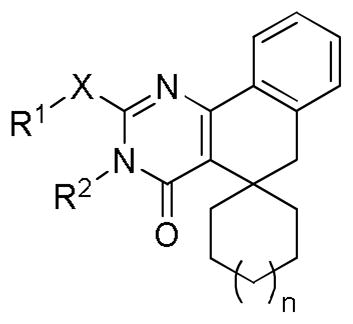
| ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| No. | R1 | R2 | X | n | 5 μM SK T/C a | 50 μM SK T/Ca | 50 μM Growth T/Cb | IC50 (μM) | MLM t1/2 (min)c | Aq. Sol. (μM)d |
| 1 | n-Bu | allyl | S | 1 | 0.84 ± 0.14 | 0.48 ± 0.28 | 0.95 ± 0.08 | >50 | 37.2 | 9 ± 2 |
| 6a | 4-MeOPhCH2 | allyl | S | 1 | 1.00 ± 0.33 | 0.82 ± 0.44 | 1.05 ± 0.06 | |||
| 6b | PhCH2 | allyl | S | 1 | 0.74 ± 0.16 | 0.57 ± 0.21 | 0.96 ± 0.06 | >50 | 0.6 | 9 ± 2 |
| 6c | 4-AcNHPhCH2 | allyl | S | 1 | 0.96 ± 0.46 | 0.74 ± 0.28 | 0.96 ± 0.14 | |||
| 6d | MeOCH2CH2 | allyl | S | 1 | 0.47 ± 0.28 | 0.29 ± 0.15 | 0.99 ± 0.10 | 6.4 | 9 ± 2 | |
| 6e | Me2NCH2CH2 | allyl | S | 1 | 0.72 ± 0.12 | 0.52 ± 0.37 | 0.88 ± 0.20 | |||
| 6f | Me | allyl | S | 1 | 0.60 ± 0.19 | 0.42 ± 0.03 | 0.98 ± 0.03 | 19 ± 6.0 | 13.5 | 22 ± 5 |
| 7a | Et | allyl | O | 1 | 0.53 ± 0.23 | 0.14 ± 0.05 | 1.02 ± 0.09 | 14.5 ± 3.5 | ||
| 7b | Et | allyl | NH | 1 | 0.78 ± 0.47 | 0.56 ± 0.70 | 0.32 ± 0.18 | 9 ± 2 | ||
| 28 | PhCH2 | Et | S | 0 | 0.44 ± 0.06 | 0.23 ± 0.16 | 1.07 ± 0.04 | |||
Ratio of A405 of SK-cleaved substrate in GAS culture treated with the indicated concentration of test compound divided by A405 of DMSO control (see Section 5.1.1). Values are the mean of at least three experiments ± standard deviation.
Ratio of OD (600 nm) for growth of GAS in the presence of test compound divided by DMSO control. Values are the mean of at least three experiments ± standard deviation.
Half-life of parent compound during incubation with mouse liver microsomes.
Kinetic solubility of compound in aqueous Todd Hewitt bacterial media.
Exploration of analogs with gem-dimethyl substitution of the central ring (Table 2) began with the assessment of commercially available analogs 29, 30, and 31, all of which showed an increase in potency in comparison to 1. Polar substitution at the R1 position was generally well-tolerated (12e, 12g, 12h, 31) except for primary amide 12d. Introducing an alcohol on the sidechain (12f) to improve solubility led to unacceptable bacteriotoxicity. This toxicity was also observed with heteroatomic analogs 13a and 13b versus sulfur analog 30, leading us to conclude that retention of the sulfur was necessary to avoid inhibition of bacterial growth. Altering the substitution of the N-position of the pyrimidinone was found to lead to bacterial toxicity (12i) or attenuation of activity (19a) compared to N-allyl (12h). Overall, the S-Et and S-CH2CH2OMe analogs 30 and 12h were the optimum compounds from this series (Table 2).
Table 2.
Inhibition of SK activity by gem-dimethyl analogs.
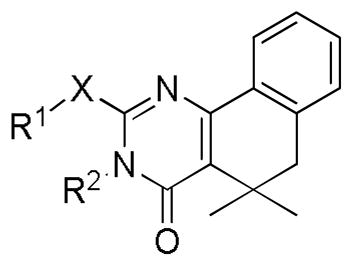
| |||||||||
|---|---|---|---|---|---|---|---|---|---|
| No. | R1 | R2 | X | 5 μM SK T/Ca | 50 μM SK T/Ca | 50 μM Growth T/Cb | IC50 (μM) | MLM t1/2 (min)c | Aq. Sol. (μM)d |
| 12d | H2NCOCH2 | Allyl | S | 0.79 ± 0.17 | 0.64 ± 0.16 | 1.00 ± 0.02 | <2 | ||
| 12e | 1-pyridylCH2 | Allyl | S | 0.40 ± 0.09 | 0.13 ± 0.07 | 1.05 ± 0.02 | |||
| 12f | HOCH2CH2 | Allyl | S | 0.48 ± 0.17 | - | 0.06 ± 0.03 | |||
| 12g | NCCH2 | Allyl | S | 0.37 ± 0.17 | 0.23 ± 0.06 | 1.00 ± 0.03 | 20.6 | 34.5 ± 7.5 | |
| 12h | MeOCH2CH2 | Allyl | S | 0.35 ± 0.11 | 0.13 ± 0.07 | 0.86 ± 0.10 | 3.2 ± 0.6 | 1.7 | 22.5 ± 4.5 |
| 12i | MeOCH2CH2 | Et | S | 0.41 ± 0.23 | - | 0.39 ± 0.15 | 0.8 | ||
| 13a | Et | Allyl | O | 0.34 ± 0.08 | 0.23 ± 0.10 | 0.50 ± 0.23 | 0.9 | ||
| 13b | Et | Allyl | NH | 0.48 ± 0.19 | - | 0.06 ± 0.03 | 1.1 | ||
| 19a | MeOCH2CH2 | 4-MeOPhCH2CH2 | S | 0.71 ± 0.40 | 0.61 ± 0.32 | 0.97 ± 0.07 | 9.4 | ||
| 29 | PhCH2 | Allyl | S | 0.65 ± 0.25 | 0.35 ± 0.14 | 1.03 ± 0.03 | 10.8 ± 3.0 | 9 ± 2 | |
| 30 | Et | Allyl | S | 0.24 ± 0.07 | 0.17 ± 0.08 | 0.89 ± 0.05 | 2.0 ± 0.5 | ||
| 31 | EtO2CCH2 | Allyl | S | 0.54 ± 0.10 | 0.27 ± 0.12 | 1.01 ± 0.02 | |||
Ratio of A405 of SK-cleaved substrate in GAS culture treated with the indicated concentration of test compound divided by A405 of DMSO control (see Section 5.1.1). Values are the mean of at least three experiments ± standard deviation.
Ratio of OD (600 nm) for growth of GAS in the presence of test compound divided by DMSO control. Values are the mean of at least three experiments ± standard deviation.
Half-life of parent compound during incubation with mouse liver microsomes.
Kinetic solubility of compound in aqueous Todd Hewitt bacterial media.
Our attention then turned to exploring substitution of the aromatic ring (Table 3) at 3 different positions. Assessment of methyl ether analogs 12a–c revealed that substitution at the 8- and 9- positions conferred a moderate increase in potency over 30, while substitution of the 7-position did not. The installation of larger and/or more polar ethers (17a–d, 17h–k) generally led to the attenuation of these gains, though a few compounds retained activity similar to 30 (17f, g). Incorporation of a basic amine (17e) introduced significant growth inhibition. Benzoic acid-derived analogs 25–27 also displayed reduced potency compared to 30. Ultimately, 9-methoxy analog 12a was found to be the optimal compound from this series, exhibiting a 90% reduction in streptokinase activity at 5 μM and an IC50 of 1.3 μM, representing a greater than 35-fold improvement over 1, although some modest inhibition of bacterial growth at high concentrations was introduced.
Table 3.
Inhibition of SK activity by analogs with 7-, 8-, and 9-position substitution.

| ||||||||
|---|---|---|---|---|---|---|---|---|
| No. | R1 | R3 | 5 μM SK T/Ca | 50 μM SK T/Ca | 50 μM Growth T/Cb | IC50 (μM) | MLM t1/2 (min)c | Aq. Sol. (μM)d |
| 12a | Et | 9-OMe | 0.10 ± 0.06 | 0.07 ± 0.02 | 0.73 ± 0.07 | 1.3 ± 0.6 | 2.3 | 9 ± 2 |
| 12b | Et | 7-OMe | 0.26 ± 0.07 | 0.29 ± 0.18 | 0.28 ± 0.26 | |||
| 12c | Et | 8-OMe | 0.18 ± 0.09 | 0.07 ± 0.01 | 0.87 ± 0.19 | 3.1 ± 0.7 | 0.8 | 14.5 ± 3.5 |
| 12j | MeOCH2CH2 | 8-OMe | 0.47 ± 0.25 | 0.25 ± 0.18 | 0.89 ± 0.16 | |||
| 12k | Allyl | 8-OMe | 0.33 ± 0.15 | 0.28 ± 0.16 | 0.92 ± 0.16 | 6.9 ± 0.3 | 0.6 | 14.5 ± 3.5 |
| 17a | Et | 9-MeOCH2CH2O | 0.58 ± 0.24 | 0.43 ± 0.15 | 1.02 ± 0.10 | |||
| 17b | Et | 7-MeOCH2CH2O | 0.84 ± 0.23 | 0.40 ± 0.21 | 0.94 ± 0.08 | |||
| 17c | Et | 8-MeOCH2CH2O | 0.46 ± 0.35 | 0.36 ± 0.41 | 0.85 ± 0.22 | |||
| 17d | Et | 8-EtO2CCH2 | 0.64 ± 0.67 | 0.47 ± 0.48 | 1.02 ± 0.10 | |||
| 17e | Et | 8-
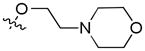
|
0.53e | 0.08e | 0.41e | |||
| 17f | Et | 8-O-iPr | 0.29 ± 0.19 | 0.13 ± 0.12 | 0.87 ± 0.23 | 4.1 ± 0.7 | 14.5 ±3.5 | |
| 17g | Et | 8-NCCH2O | 0.45 ± 0.25 | 0.20 ± 0.17 | 0.94 ± 0.15 | |||
| 17h | Et | 8-n-BuO | 0.48 ± 0.26 | 0.35 ± 0.29 | 0.87 ± 0.10 | |||
| 17i | Et | 8-PhCH2CH2O | 0.68 ± 0.34 | 0.59 ± 0.55 | 1.01 ± 0.03 | |||
| 17j | Et | 8-CH3(CH2)6O | 1.03 ± 0.18 | 0.87 ± 0.12 | 1.05 ± 0.03 | |||
| 17k | Et | 8-HO2CCH2O | 0.96 ± 0.21 | 0.44 ± 0.12 | 0.86 ± 0.07 | |||
| 25 | MeOCH2CH2 | 8-CO2Me | 0.71 ± 0.30 | 0.58 ± 0.19 | 1.01 ± 0.04 | |||
| 26 | MeOCH2CH2 | 8-CO2H | 0.75 ± 0.36 | 0.70 ± 0.52 | 0.86 ± 0.11 | |||
| 27a | MeOCH2CH2 | 8-CONHBn | 1.14 ± 0.24 | 0.91 ± 0.18 | 1.07 ± 0.02 | |||
| 27b | MeOCH2CH2 | 8-
|
0.84 ± 0.10 | 0.77 ± 0.27 | 0.99 ± 0.02 | |||
Ratio of A405 of SK-cleaved substrate in GAS culture treated with the indicated concentration of test compound divided by A405 of DMSO control (see Section 5.1.1). Values are the mean of at least three experiments ± standard deviation.
Ratio of OD (600 nm) for growth of GAS in the presence of test compound divided by DMSO control. Values are the mean of at least three experiments ± standard deviation.
Half-life of parent compound during incubation with mouse liver microsomes.
Kinetic solubility of compound in aqueous Todd Hewitt bacterial media.
Values derived from only 1 experiment due to toxicity.
The final analogs for the SAR effort were designed to assess the effect of alkylation at the pyrimidinone oxygen in comparison to identical substitution at nitrogen (Table 4). No general efficacy advantage for N- or O- alkylation was observed; O-alkylated 20a was somewhat more potent than 19a, while 12a remained much more potent than its O-alkylated counterpart 20c. Compounds 19b and 20b displayed similarly weak activity.
Table 4.
Effect of N-vs. O-alkylation on GAS-SK inhibition and metabolic stability.
| ||||||||
|---|---|---|---|---|---|---|---|---|
| No. | R1 | R2 | R3 | 5 μM SK T/Ca | 50 μM SK T/Ca | 50 μM Growth T/Cb | IC50 (μM) | MLM t1/2 (min)c |
| 19a | MeOCH2CH2 | 4-MeOPhCH2CH2 | H | 0.71 ± 0.40 | 0.61 ± 0.32 | 0.97 ± 0.07 | 9.4 | |
| 20a | MeOCH2CH2 | 4-MeOPhCH2CH2 | H | 0.46 ± 0.37 | 0.24 ± 0.15 | 1.00 ± 0.09 | 5.5 ± 0.7 | 45.1 |
| 19b | Et | CF3CH2 | MeO | 0.78 ± 0.36 | 0.49 ± 0.17 | 1.01 ± 0.04 | 3.0 | |
| 20b | Et | CF3CH2 | MeO | 0.51 ± 0.20 | 0.38 ± 0.23 | 0.89 ± 0.02 | 45.5 | |
| 12a | Et | Allyl | MeO | 0.10 ± 0.06 | 0.07 ± 0.02 | 0.73 ± 0.07 | 1.3 ± 0.6 | 2.3 |
| 20c | Et | Allyl | MeO | 0.89 ± 0.33 | 0.68 ± 0.39 | 0.96 ± 0.02 | 8.3 | |
Ratio of A405 of SK-cleaved substrate in GAS culture treated with the indicated concentration of test compound divided by A405 of DMSO control (see Section 5.1.1). Values are the mean of at least three experiments ± standard deviation.
Ratio of OD (600 nm) for growth of GAS in the presence of test compound divided by DMSO control. Values are the mean of at least three experiments ± standard deviation.
Half-life of parent compound during incubation with mouse liver microsomes.
3.3. Microsomal Stability and Metabolite Identification
In our previous publication identifying 1 and 6b as inhibitors of SK expression, it was determined that 1 displayed efficacy in mouse models of GAS infection while 6b did not, despite having similar activity in bacterial assays.15 We hypothesized that a difference in susceptibility to oxidative metabolism might be playing a role in the differential activity in mammalian systems. Incubation of each of these compounds in mouse liver microsomal extract (MLM) supported this hypothesis, with 1 displaying more than 60-fold higher stability to oxidation than 6b (t1/2 = 37.2 min vs. 0.6 min, respectively). A survey of several more compounds in the series (Tables 1–3) revealed that most were highly unstable (t1/2 < 5 minutes), despite several structural modifications that would potentially reduce their propensity to oxidation, including lowered lipophilicity, fewer unsubstituted aliphatic/olefinic/aryl carbons, and replacement of sulfur with oxygen or nitrogen. Interestingly, we also noted that the spirocyclohexyl compounds were generally more stable than the corresponding gem-dimethyl compounds (e.g. 6d t1/2 = 6.4 min vs. 12h t1/2 = 1.7 min), despite their higher lipophilicity.
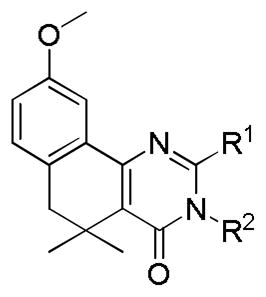
A metabolite ID study was performed on compound 6d to elucidate the most metabolically labile sites of the scaffold. Possible metabolites were identified by LC-MS analysis, then fragmented via collision-induced dissociation (CID). Subsequent MS analysis of the resulting metabolite fragments allowed us to deduce the structure of each metabolite. This study indicated that the most metabolically labile portions of the molecule were the substitutions on the pyrimidinone ring (Figure 2). No oxidation was indicated on the spirocyclohexane ring or any of the four unsubstituted aromatic carbons. Based on the findings of the metabolite ID study, we prepared five analogs of our optimum compound 12a that deactivated this portion of the scaffold to oxidation (Table 5); however, these were also found to be quickly metabolized. Interestingly, metabolic assessment of the O-alkylated compounds 20a–c (Table 4) showed that they were consistently more stable (4–15 fold) than their N-alkylated counterparts. This observation, paired with the generally greater stability of the hindered spirocyclohexyl analogs vs. the corresponding gem-dimethyl analogs, suggests that hydrolysis of the pyrimidinone amide may play a key role in the metabolic breakdown of these compounds. Compound 20a, despite being four-fold less potent than 12a (IC50 = 5.5 μM vs. 1.3 μM), demonstrates the potential viability of O-alkylated analogs as more metabolically stable alternatives to the N-alkylated compounds.
Figure 2.
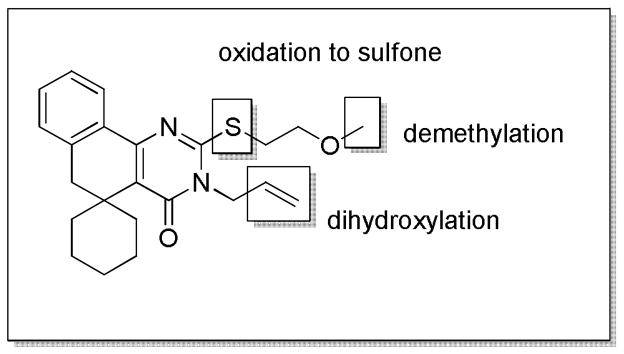
Potential routes of metabolism identified by MLM incubation and MS analysis. No oxidation of the left-hand side of the molecule was observed.
Table 5.
Analogs of 12a with potentially reduced metabolic liability.
| ||||||
|---|---|---|---|---|---|---|
| No. | R1 | R2 | 5 μM SK T/Ca | 50 μM SK T/C SDa | 50 μM Growth T/Cb | MLM t1/2 (min)c |
| 12a | S-Et | Allyl | 0.10 ± 0.06 | 0.07 ± 0.02 | 0.73 ± 0.07 | 2.3 |
| 12l | S-CH2CF3 | Allyl | 0.45 ± 0.19 | 0.22 ± 0.08 | 0.86 ± 0.07 | 4.8 |
| 12m | S-Et | Me | 0.66 ± 0.20 | 0.20 ± 0.14 | 0.83 ± 0.15 | 1.7 |
| 14 | (SO)Et | Allyl | 1.00 ± 0.13 | 0.51 ± 0.31 | 1.01 ± 0.05 | 2.8 |
| 15 | (SO2)Et | Allyl | 0.83 ± 0.31 | 0.70 ± 0.13 | 1.03 ± 0.03 | 3.8 |
| 19b | S-Et | CF3CH2 | 0.78 ± 0.36 | 0.49 ± 0.17 | 1.01 ± 0.04 | 3.0 |
Ratio of A405 of SK-cleaved substrate in GAS culture treated with the indicated concentration of test compound divided by A405 of DMSO control (see Section 5.1.1). Values are the mean of at least three experiments ± standard deviation.
Ratio of OD (600 nm) for growth of GAS in the presence of test compound divided by DMSO control. Values are the mean of at least three experiments ± standard deviation.
Half-life of parent compound during incubation with mouse liver microsomes.
3.4. Solubility
In addition to microsomal stability, the aqueous solubility of small molecules has been shown to be a critical physicochemical property for predicting the efficacy of compounds in living systems. Lipinski suggests that to achieve oral bioavailability at a dose of 1 mg/kg, a compound minimally needs to achieve an aqueous solubility of 52 μg/mL,25 corresponding to a solubility of ~150 μM for small analogs such as 12a. Therefore, several key compounds from the SAR library were assessed for kinetic solubility in Todd Hewitt (THY) bacterial media. The solubility of all compounds tested (Tables 1–3) was equal to or less than 25 μM, with the exception of nitrile 12g (aq. sol. = 34.5 ± 7.5 μM). It was found that the solubility of these compounds was considerably lower in phosphate-buffered saline (PBS) than in THY media (eg. 2 ± 1 vs. 9 ± 2 μM [PBS:THY] for 1, 3 ± 1 vs. 34.5 ± 7.5 μM [PBS:THY] for 12g), suggesting that protein binding may increase the effective aqueous solubility of this compound class. Preliminary activity studies that included human serum albumin (HSA) in the reaction mixture also suggested some compounds in this series may bind to protein.a The measured solubility has a weak negative correlation to the cLogP (R2 = 0.533), but the most soluble compound 12g does indeed have the lowest cLogP (3.55). Further decreases in cLogP should logically result in greater aqueous solubility; however, nearly all efforts to significantly decrease lipophilicity, including replacement of the fused phenyl ring with pyridyl and replacement of the central ring with lactones or lactams (unpublished data), have thus far led to the reduction or loss of activity.
3.5. Mammalian Cytotoxicity
A representative subset of compounds (1, 12a, 12c, 12h, 19a, 20a, 29) was assessed for toxic effect in HeLa cells using a standard tetrazolium-formazan assay of cell viability. It was found that none of the compounds inhibited growth by more than 35% up to 100 μM, the highest concentration tested.
3.6. Biofilm inhibition
As reported previously, a gene expression microarray assay of GAS mRNA levels after incubation with 6b confirmed the down-regulation of several virulence genes, including streptokinase, antiphagocytic factors, and cytolytic toxins. Reduced transcription of genes related to cell adhesion and biofilm formation, such as laminin- and fibronectin-binding proteins and collagen-like surface protein, was also observed.15 Biofilm formation is critical for bacterial colonization of solid substrates in the body and serves to mechanically sequester bacteria from the effects of the immune system and antibiotics.26 Diminishing the ability of bacteria to form biofilms represents a new mode of action by which compounds in this series can exert anti-virulence activity, and more importantly, expands the range of susceptible bacterial species to clinically important strains such as S. aureus. The compounds produced for the SK effort were assessed for their ability to inhibit biofilm formation in this strain using a previously developed plate-based OD assay.27 These assays identified compounds 12c (CCG-203592) and 12k (CCG-205363) as potent S. aureus biofilm inhibitors; these results are reported in greater detail elsewhere.28
4. Conclusions
We report the preparation and biological evaluation of 45 analogs of 1, resulting in the achievement of a greater than 35-fold improvement in activity (IC50 >50 μM to 1.3 μM) over compound 1 with optimum analog 12a. During the optimization process, we observed a general instability of new analogs to oxidative metabolism. Through careful structure metabolism relationship analysis, we have developed a hypothesis that a primary route of metabolism for this class of compounds is through hydrolysis of the pyrimidinone amide, which should guide the design of new analogs with improved potential for activity in vivo. Murine infection studies are in fact currently planned for the most stable active analogs from this work (6f, 12g, 20a, 20b) and will be reported in due course. Concurrently, a more focused effort toward optimizing the biofilm inhibition activity of this class of compounds is underway.
5. Experimental Section
5.1. Biological/Physicochemical Characterization
5.1.1. SK Expression Assay
The GAS strain UMAA2616 used in this study was derived from the GAS M type 1 strain MGAS166.29 UMAA2616 was originally designated as UMAA2392 and contains mutations in the CovR/S system to generate increased expression of SK and other virulence factors.30
Extracellular GAS-SK activity was measured by a previously described chromogenic assay.15 Briefly, a single colony of UMAA2616 was inoculated into Todd-Hewitt broth containing 0.2% yeast extract (THY) (Difco, Detroit, MI) supplemented with 100 μg/mL streptomycin and grown overnight at 37 °C. Vials of THY medium (4 mL) containing different concentrations of test compound in a final concentration of 0.1% DMSO were inoculated with 4 μl of the UMAA2616 overnight culture. The cultures were grown in triplicate at 37 °C to an OD600 ≈ 1.0. Each sample was then centrifuged at 11,000g for 8 minutes. An aliquot (20 μL) of supernatant was mixed with 100 μl PBS, 10 μl human plasma (Innovative Research, Novi, MI), and 10μl of 1 mg/ml S-2403 solution (Figure 3; Diapharma Group Inc., West Chester, OH), then incubated at 37 °C for 2 hours. Activity was reported as the ratio of SK activity as measured by absorbance at 405 nm of the test cultures compared to that of a culture treated with only DMSO. A GAS strain deficient in SK activity, UMAA2641,31 was used as a blank. The SK activity was calculated based on a standard curve made with serial dilutions of control UMAA2616 cultures grown under the same conditions with 0.1% DMSO. The numbers were then corrected for inhibition of growth by each test compound vs. DMSO control, as measured by OD600. The experiments were performed in triplicate to obtain mean and standard deviation values for each test concentration.
Figure 3.
Structure of chromogenic plasmin substrate S-2403 and microsomal stability assay internal standard 32.
5.1.2. Aqueous Solubility Assay
Compounds were dissolved in DMSO to a stock concentration of 10 mM. 100 μL of each stock was diluted with DMSO through a series of twelve 35% dilutions, resulting in working solutions with concentrations ranging from 10 mM to 0.0875 mM. 2 μL of each working solution was added to one well of a clear plastic, round-bottom 96-well plate containing 200 μL of standard-strength Todd-Hewitt broth or phosphate-buffered saline solution warmed to 37°C, resulting in final testing concentrations of 0.87 μM to 99 μM. After addition of compound to all test wells, the plate was loaded into a Molecular Devices SpectraMax Plus UV/Vis spectrophotometer, shaken for 15 seconds, and incubated for 5 minutes at 37°C. Measurement of the OD600 resulted in a series of curves. The concentration at which the average (n = 3 for each concentration and test compound) OD600 reading rose above background (OD600 ≥ 0.005 AU) was noted, and the aqueous solubility is reported as the mean concentration between this point and the previous point (± the concentration difference between the two points * 0.5).
5.1.3 Microsomal Stability Assay
Stock solutions of test compounds were generated via the dilution of 100mM DMSO stocks 1000-fold with phosphate-buffered saline containing up to 10% MeOH as a co-solvent. To 366 μL of 100 mM phosphate buffer containing 3.3 mM MgCl2 was added 10 μL of 20 mg/mL mouse liver microsomal extract (XenoTech, Lenexa, KS) and 4 μL of test compound solution (100 μM). Enzymatic oxidation was initiated by adding 15 μL of 16.7 mg/mL NADPH in 100 mM phosphate buffer containing 3.3 mM MgCl2 (final concentrations of mouse liver microsomes and test compound = 0.5 mg/mL and 1 μM, respectively). The reactions were carried out at 37 °C for 60 minutes. At each time point (0, 1, 3, 5, 10, 30, and 60 min), an aliquot of the reaction mixture (30 μL) was removed and added to 90 μL of cold acetonitrile containing a known concentration of internal standard compound (32, Figure 3) to quench the reaction. The quenched sample mixtures were centrifuged at 16,000g for 10 minutes. The supernatant was then analyzed via an LC-MS/MS system equipped with a reverse-phase column.
LC-MS/MS analysis of test compounds was performed on an LC-MS/MS-3200 system (AB Sciex, Framingham, MA) equipped with an electrospray ionization (ESI) source. The AB Sciex LC-MS/MS 2800 system consisting of Agilent 1200 series (Agilent, Santa Clara, CA) with a Zorbax Extend C-18 column (5 μm, 50 × 2.1 mm) was used for the separation and the effluent from the column was directly fed into the ionization source. The system was controlled by Analyst software (version 1.4.2) to collect and process data. The mobile phase consisted of water containing 0.1% formic acid (Solvent A) and acetonitrile containing 0.1% formic acid (Solvent B) for all test compounds with the solvent B gradient changing from 10–95% during a 15 minute run. The LC-MS/MS was operated at a flow rate of 0.4 mL/min. Metabolic half-lives were derived from the rate of disappearance of the parent compound MS/MS readout as determined by comparison to the internal standard.
5.1.4. Metabolite Identification Study
Metabolic oxidation and HPLC separation were performed in the same manner as in the metabolic half-life determination assays. Samples from the metabolic oxidation of 6d were injected into the HPLC and subjected to enhanced product ion (EPI), MS/MS (MS2), and MS3 scanning to obtain MS2 and MS3 fragmentation spectra of each potential metabolite derived from the M+H ion of 6d. Samples of 6d incubated with inactivated (boiled) MLM or omitting the addition of NADPH to the reaction mixture served as negative controls. Potential metabolites were identified using multiple reaction monitoring (MRM), enhanced mass spectrometry (EMS) full scan, and precursor scanning detection modes. Only the ions detected in the test sample and absent in both control samples were regarded as possible metabolites. Based on the MS2 and MS3 spectra of the possible metabolites and the proposed fragmentation pathways of 6d, the likely structures of the metabolites were deduced.
5.1.5. Mammalian Cytotoxicity Assay
HeLa cells (ATCC, Manassas, VA) were cultured in Dulbecco’s modified Eagle’s medium (DMEM) (Life Technologies, Grand Island, NY) supplemented with 10% fetal bovine serum, 1% penicillin-streptomycin and 1% L-glutamine at 37 °C under 5% CO2 atmosphere. Cell suspensions (200 μL, 2.5×104 cells/well) with different concentrations (0.39 μM to 100 μM) of test compounds or DMSO were plated in 96-well plates and cultured for 24 hours in triplicate. CellTiter 96 Aqueous One Solution reagent (Promega, Madison, WI) (20 μL) was added to each sample and incubated for 2 hours. Viability was reported as the ratio of absorbance at 490 nm of the test compound wells in comparison to DMSO control. The experiment was repeated three times to obtain mean and standard error values.
5.2. Chemistry Procedures
General information
Chemical names follow CAS nomenclature. Starting materials were purchased from Fisher, Sigma-Aldrich Lancaster, Fluka or TCI-America and were used as supplied unless otherwise indicated. All reaction solvents were purchased from Fisher and used as received. Reactions were monitored by TLC using precoated silica gel 60 F254 plates. All anhydrous reactions were run under an atmosphere of dry nitrogen. Silica gel chromatography was performed with silica gel (220–240 mesh) obtained from Silicycle. Solvent abbreviations used: CDCl3, deutero-chloroform; DCM, dichloromethane; DMSO, dimethyl sulfoxide; EtOH, ethanol; MEK, methyl ethyl ketone; THF, tetrahydrofuran; DMF, N,N-dimethylformamide; EtOAc, ethyl acetate; hex, hexanes. Reagent abbreviations used: Cs2CO3, cesium carbonate; Na2SO4, sodium sulfate; MgSO4, magnesium sulfate; mCPBA, meta-chloro peroxybenzoic acid; KOH, potassium hydroxide; LDA, lithium diisopropylamide; NaHCO3, sodium bicarbonate; NaOMe, sodium methoxide; TMS, trimethylsilyl; HOBt 1-hydroxybenzotriazole; EDC, 1-(3-dimethylaminopropyl)-3-ethylcarbodiimide; ZnI2, zinc iodide.
NMR spectra were recorded on a Bruker 400 MHz, Bruker 500 MHz, Varian 400 MHz, or Varian 500 MHz spectrometer. Chemical shifts are reported in δ (parts per million), by reference to the hydrogen residues of deuterated solvent as internal standard CDCl3: δ = 7.28 (1H NMR), or in reference to the hydrogen peaks of tetramethylsilane, δ = 0.00 (1H NMR). Mass spectra were recorded on a Micromass LCT time-of-flight instrument utilizing electrospray ionization operating in positive-ion (ESI+) or negative-ion (ESI-) modes where indicated. Melting points were measured on a MEL-TEMP melting point apparatus and are uncorrected. The purity of the compounds was assessed via analytical rpHPLC with one of three gradient methods. “Method A”: 10% B to 90% B over 6 minutes, hold at 90% B for 7 additional minutes; “Method B”: 50% B to 90% B, hold at 90% B for 7 additional minutes; “Method C”: 90% B over 12 minutes (solvent A H2O, solvent B acetonitrile, C18 column, 3.5 um, 4.6×100mm, 254 nm μ).
5.2.1. Ethyl 2-cyano-2-cyclohexylideneacetate (3)
To a solution of cyclohexanone (1.50 ml, 14.47 mmol) in toluene (24.12 ml) was added ethyl cyanoacetate (1.556 ml, 14.62 mmol), acetic acid (0.166 ml, 2.89 mmol), and ammonium acetate (0.112 g, 1.447 mmol). The mixture was heated to reflux at 150°C in a Dean-Stark apparatus. After 5 hours, the reaction was cooled and washed with water and saturated NaHCO3 solution. The organics were dried over Na2SO4, filtered, and concentrated. The resulting residue (2.48g, 89% yield) was used without further purification. 1H NMR (500 MHz, CDCl3) δ (ppm) 4.29 (q, J = 7.2 Hz, 2H), 3.00 (t, J = 6.0 Hz, 2H), 2.68 (t, J = 6.1 Hz, 2H), 1.82(p, J = 6.0 Hz, 2H), 1.75 (p, J = 6.1 Hz, 2H), 1.70-1.65 (m, 2H), 1.37 (t, J = 7.2 Hz, 3H).
5.2.2. Ethyl 4′-amino-1′H-spiro[cyclohexane-1,2-naphthalene]-3′-carboxylate (4)
1M benzylmagnesium chloride in diethyl ether (18.92 ml) was added dropwise to a solution of 3 (1.828 g, 9.46 mmol) in diethyl ether (6.31 ml) at room temperature. The mixture was stirred at room temperature for 3 days, then 10% hydrochloric acid (7.83 ml, 255 mmol) was added dropwise at 0°C while stirring. The organic layer was separated, washed with water, dried over Na2SO4, filtered, and concentrated to dryness in vacuo. Concentrated H2SO4 (4.59 ml) was added dropwise to the crude solid at 0°C. The mixture was stirred at room temperature for 7 hours, then ice water was added to the mixture resulting in a precipitate. The precipitate was dissolved in Et2O and washed with 28% NH3 solution. The organic layer was washed with water, dried over Na2SO4, filtered, and concentrated to an orange oil (1.66g, 68% yield). 1H NMR (500 MHz, CDCl3) δ (ppm) 7.42 (d, J = 6.8 Hz, 1H), 7.36-7.25 (m, 2H), 7.22 (d, J = 6.8 Hz, 1H), 6.04 (bs, 2H), 4.25 (q, J = 7.0 Hz, 2H), 2.87 (s, 2H), 2.24-2.05 (m, 2H), 1.76-1.20 (m, 9H).
5.2.3. 3-allyl-2-thioxo-2,3-dihydro-1H-spiro[benzo[h]quinazoline-5,1′-cyclohexan]-4(6H)-one (5)
Intermediate 4 (1.66 g, 5.82 mmol) and allyl isothiocyanate (0.600 ml, 6.13 mmol) were dissolved in ethanol (9.69 ml) and refluxed at 85°C. for 10 hours. A solution of KOH (0.653 g, 11.63 mmol) in water (9.69 ml) was then added and the reaction mixture was refluxed for 3 hours. The cooled reaction mixture was acidified to pH 3.0–3.5 resulting in precipitation. The precipitate was collected via vacuum filtration, washed with water, and recrystallized from butanol. Recovered as pale yellow crystals (587 mg, 29.8% yield). TLC Rf = 0.25 (10% EtOAc:hex). 1H NMR (500 MHz, CDCl3) δ (ppm) 9.28 (s, 1H), 7.50–7.35 (m, 3H), 7.32 (d, J = 7.4 Hz, 1H), 6.00 (ddt, J = 16.2, 11.3, 5.8 Hz, 1H), 5.37 (dd, 1H), 5.27 (dd, 1H), 5.06 (d, J = 5.8 Hz, 2H), 3.03 (s, 2H), 2.48 (td, J = 13.3, 4.4 Hz, 2H), 1.72 (d, J = 13.3 Hz, 1H), 1.62–1.44 (m, 4H), 1.33 (d, J = 14.3 Hz, 3H).
5.2.4. General Method A for generating compounds 1, 6a–e
Compound 5 (100 mg, 0.295 mmol) was combined with base (0.443 mmol) and alkylating agent (0.325 mmol) in ethanol or MEK (1.75 mL). The suspension was warmed to 70°C and allowed to stir for 16 hours. The suspension was subsequently diluted with EtOAc and washed with water and brine. The isolated organic layer was then dried over MgSO4, filtered, and concentrated in vacuo. Further purification via flash chromatography (0 to 10% EtOAc:hex) delivered the desired compounds in 72–83% yield.
5.2.5. 3-allyl-2-(butylthio)-3H-spiro[benzo[h]quinazoline-5,1′-cyclohexan]-4(6H)-one (CCG-2979) (1)
Prepared from 5 according to General Method A, using 1-iodobutane as the alkylating agent, Cs2CO3 as the base, and MEK as the solvent. Isolated after flash chromatography (0–5% EtOAc:hex) as white crystals (87 mg, 74% yield). 1H NMR (400 MHz, CDCl3) δ (ppm) 8.08 (d, J = 6.7 Hz, 1H), 7.38–7.28 (m, 2H), 7.20 (d, J = 6.8 Hz, 1H), 5.99–5.87 (m, 1H), 5.33–5.22 (m, 2H), 4.68 (d, J = 5.2 Hz, 2H), 3.31 (t, J = 7.3 Hz, 2H), 3.03 (s, 2H), 2.67–2.52 (m, 2H), 1.80 (p, J = 7.4 Hz, 2H), 1.71 (d, J = 11.3 Hz, 1H), 1.63–1.45 (m, 6H), 1.44–1.31 (m, 3H), 0.98 (t, J = 7.4 Hz, 3H). ESI+MS m/z = 395.2 (M + H+), 417.2 (M + Na+). HPLC (Method C, tR = 7.44 min), purity >95%.
5.2.6. 3-allyl-2-((4-methoxybenzyl)thio)-3H-spiro[benzo[h]quinazoline-5,1′-cyclohexan]-4(6H)-one (6a)
Prepared from 5 according to General Method A, using 4-methoxybenzyl bromide as the alkylating agent, KOH as the base, and ethanol as the solvent. Isolated as a clear oil (97 mg, 72% yield). 1H NMR (500 MHz, CDCl3) δ (ppm) 8.17 (d, J = 7.6 Hz, 1H), 7.41–7.36 (m, 3H), 7.34 (t, J = 6.8 Hz, 1H), 7.24 (d, J = 7.2 Hz, 1H), 6.89 (d, J = 8.6 Hz, 2H), 5.94 (ddt, J = 17.1, 10.7, 5.6 Hz, 1H), 5.37–5.18 (m, 2H), 4.67 (d, J = 5.6 Hz, 2H), 4.57 (s, 2H), 3.82 (s, 3H), 3.07 (s, 2H), 2.72–2.51 (m, 2H), 1.74 (d, J = 12.8 Hz, 1H), 1.66–1.50 (m, 4H), 1.49–1.32 (m, 3H). ESI+MS m/z = 481.2 (M + Na+). HPLC (Method C, tR = 5.11 min), purity >95%.
5.2.7. 3-allyl-2-(benzylthio)-3H-spiro[benzo[h]quinazoline-5,1′-cyclohexan]-4(6H)-one (CCG-102487) (6b)
Prepared from 5 according to General Method A, using benzyl bromide as the alkylating agent, KOH as the base, and ethanol as the solvent. Isolated as a clear oil (95 mg, 75% yield). 1H NMR (400 MHz, CDCl3) δ (ppm) 8.11 (d, J = 7.4 Hz, 1H), 7.44 (d, J = 7.2 Hz, 2H), 7.39–7.27 (m, 5H), 7.21 (d, J = 7.2 Hz, 1H), 5.92 (ddt, J = 15.8, 10.7, 5.5 Hz, 1H), 5.32–5.21 (m, 2H), 4.66 (d, J = 5.5 Hz, 2H), 4.59 (s, 2H), 3.04 (s, 2H), 2.65–2.53 (m, 2H), 1.71 (d, J = 12.7 Hz, 1H), 1.61–1.52 (m, 4H), 1.43-1.32 (m, 3H). ESI+MS m/z = 429.2 (M + H+), 451.2 (M + Na+). HPLC (Method C, tR = 4.68 min), purity >95%.
5.2.8. N-(4-(((3-allyl-4-oxo-4,6-dihydro-3H-spiro[benzo[h]quinazoline-5,1′-cyclohexan]-2-yl)thio)methyl)phenyl)acetamide (6c)
Prepared from 5 according to General Method A, using N-(4-(chloromethyl)phenyl)acetamide as the alkylating agent, Cs2CO3 as the base, and MEK as the solvent. Isolated as a white solid (71 mg, 83% yield). 1H NMR (500 MHz, CDCl3) δ (ppm) 8.11 (d, J = 7.2 Hz, 1H), 7.46 (d, J = 8.5 Hz, 2H), 7.39 (d, J = 8.5 Hz, 2H), 7.35 (t, J = 7.3 Hz, 1H), 7.31 (t, J = 6.9 Hz, 1H), 7.22 (d, J = 7.3 Hz, 1H), 7.16 (s, 1H), 5.91 (ddt, J = 15.9, 10.5, 5.6 Hz, 1H), 5.35–5.11 (m, 2H), 4.65 (d, J = 5.6 Hz, 2H), 4.55 (s, 2H), 3.49 (d, J = 5.3 Hz, 1H), 3.04 (s, 2H), 2.65–2.51 (m, 2H), 2.18 (s, 3H), 1.71 (d, J = 13.4 Hz, 1H), 1.60–1.51 (m, 4H), 1.45–1.32 (m, 3H). ESI+MS m/z = 508.2 (M + Na+). HPLC (Method C, tR = 2.75 min), purity >95%.
5.2.9. 3-allyl-2-((2-methoxyethyl)thio)-3H-spiro[benzo[h]quinazoline-5,1′-cyclohexan]-4(6H)-one (6d)
Prepared from 5 according to General Method A, using 2-methoxyethyl p-toluenesulfonate as the alkylating agent, KOH as the base, and ethanol as the solvent. Isolated as a yellow oil (88 mg, 75% yield). 1H NMR (500 MHz, CDCl3) δ (ppm) 8.08 (d, J = 7.5 Hz, 1H), 7.38 (t, J = 7.3 Hz, 1H), 7.33 (t, J = 6.8 Hz, 1H), 7.24 (d, J = 7.2 Hz, 1H), 5.96 (ddt, J = 15.9, 11.0, 5.6 Hz, 1H), 5.35–5.26 (m, 2H), 4.71 (d, J = 5.6 Hz, 2H), 3.79 (t, J = 6.2 Hz, 2H), 3.56 (t, J = 6.2 Hz, 2H), 3.44 (s, 3H), 3.06 (s, 2H), 2.66–2.56 (m, 2H), 1.73 (d, J = 13.0 Hz, 1H), 1.62–1.54 (m, 4H), 1.44–1.35 (m, 3H). ESI+MS m/z = 397.2 (M + H+), 419.2 (M + Na+). HPLC (Method C, tR = 3.54 min), purity >95%.
5.2.10. 3-allyl-2-((2-(dimethylamino)ethyl)thio)-3H-spiro[benzo[h]quinazoline-5,1′-cyclohexan]-4(6H)-one (6e)
Prepared from 5 according to General Method A, using β-dimethylaminoethyl bromide hydrobromide as the alkylating agent, KOH as the base, and MEK as the solvent. Isolated as white crystals (88 mg, 75% yield). 1H NMR (500 MHz, CDCl3) δ (ppm) 8.10 (d, J = 7.6 Hz, 1H), 7.35 (t, J = 7.3 Hz, 1H), 7.30 (t, J = 7.4 Hz, 1H), 7.21 (d, J = 7.1 Hz, 1H), 5.93 (ddt, J = 15.9, 10.4, 5.5 Hz, 1H), 5.32–5.23 (m, 2H), 4.69 (d, J = 5.5 Hz, 2H), 3.46 (t, J = 7.0 Hz, 2H), 3.04 (s, 2H), 2.73 (t, J = 7.0 Hz, 2H), 2.64–2.53 (m, 2H), 2.34 (s, 6H), 1.71 (d, J = 12.6 Hz, 1H), 1.59–1.51 (m, 4H), 1.42–1.33 (m, 3H). ESI+MS m/z = 410.3 (M + H+). HPLC (Method A, tR = 6.29 min), purity >95%.
5.2.11. 3-allyl-2-(methylthio)-3H-spiro[benzo[h]quinazoline-5,1′-cyclohexan]-4(6H)-one (6f)
Compound 5 (150 mg, 0.443 mmol) was dissolved in absolute EtOH at 0°C (2.61 mL) to which KOH (37 mg, 0.665 mmol) and methyl iodide (33 μL, 0.532 mmol) were added. The solution was allowed to stir for 10 minutes, resulting in the precipitation of white crystals. The suspension was diluted with H2O and vacuum filtered to collect the precipitate. The precipitate was washed with water and dried in vacuo. Recovered 150 mg (95% yield). 1H NMR (500 MHz, CDCl3) δ (ppm) 8.14 (d, J = 7.4 Hz, 1H), 7.35 (t, J = 7.3 Hz, 1H), 7.31 (t, J = 6.8 Hz, 1H), 7.21 (d, J = 7.1 Hz, 1H), 5.94 (ddt, J = 15.9, 10.8, 5.6 Hz, 1H), 5.32–5.24 (m, 2H), 4.68 (d, J = 5.6 Hz, 2H), 3.04 (s, 2H), 2.68 (s, 3H), 2.64–2.54 (m, 2H), 1.71 (d, J = 12.8 Hz, 1H), 1.60–1.51 (m, 4H), 1.42–1.33 (m, 3H). ESI+MS m/z = 353.2 (M + H+), 375.2 (M + Na+). HPLC (Method C, tR = 4.19 min), purity >95%.
5.2.12. General Method B for generating 7a and 13a
Metallic sodium (78 mg, 3.40 mmol) was added to absolute ethanol (1.67 mL) and allowed to stir at room temperature for 30 min. Once all solid had dissolved, the corresponding 2-methylthio 5,6-dihydrobenzo[h]quinazolin-4(3H)-one (6f or 12n, 0.284 mmol) was added. The reaction was warmed to 40°C and allowed to stir for 48 hours. After the completion of the reaction, the solution was diluted with H2O and extracted with 3 portions of ethyl acetate. The organic layers were combined, washed with water and brine, then isolated, dried over MgSO4, vacuum filtered, and concentrated to a yellow solid that was further purified via silica flash chromatography (15% EtOAc:hex), 69–73% yield.
5.2.13. 3-allyl-2-ethoxy-3H-spiro[benzo[h]quinazoline-5,1′-cyclohexan]-4(6H)-one (7a)
Prepared according to General Method B from 6f. Isolated as a light yellow crystalline solid (73 mg, 73% yield). 1H NMR (400 MHz, CDCl3) δ (ppm) 8.07 (d, J = 8.0 Hz, 1H), 7.38–7.25 (m, 2H), 7.20 (d, J = 6.8 Hz, 1H), 5.99–5.85 (m, 1H), 5.27–5.15 (m, 2H), 4.61 (d, J = 5.8 Hz, 3H), 4.56 (q, J = 7.2 Hz, 2H), 3.03 (s, 2H), 2.66–2.53 (m, 2H), 1.70 (d, J = 12.1 Hz, 1H), 1.60–1.50 (m, 4H), 1.45 (t, J = 7.2 Hz, 3H), 1.41–1.33 (m, 3H). ESI+MS m/z = 351.2 (M + H+), 373.2 (M + Na+) HPLC (Method B, tR = 9.75 min), purity >95%.
5.2.14. General Method C for generating 7b and 13b
Compound 6f or 12n (1.28 mmol) was dissolved in DCM (19.3 mL), then mCPBA (70 wt%, 787 mg, 3.19 mmol) was added and the reaction mixture allowed to stir over the course of 16 hours at room temperature. At this time the reaction mixture was diluted with DCM, washed with saturated aqueous NaHCO3 solution, water, and brine. The organic layer was dried over MgSO4, vacuum filtered, and concentrated in vacuo. Purification via flash chromatography isolated the sulfone intermediate which was then dissolved in a 1:1 mixture of THF:DMF (2 mL). Potassium carbonate (165 mg, 1.19 mmol) and 2M ethylamine solution in THF (1.00 mL, 2.00 mmol) were added, then the reaction vessel was tightly capped and allowed to stir 5 hours at RT. The reaction mixture was diluted with diethyl ether, then washed with 3 portions of H2O followed by brine. The organic layer was isolated, dried over MgSO4, vacuum filtered, and concentrated in vacuo. The residue was further purified by flash chromatography (10–33% EtOAc:hex) and isolated in 41–53% yield over 2 steps.
5.2.15. 3-allyl-2-(ethylamino)-3H-spiro[benzo[h]quinazoline-5,1′-cyclohexan]-4(6H)-one (7b)
Prepared from 6f according to General Method C. Isolated as a white crystalline solid, 53% yield over 2 steps. 1H NMR (500 MHz, CDCl3) δ (ppm) 8.13 (d, J = 7.4 Hz, 1H), 7.34–7.27 (m, 2H), 7.18 (d, J = 6.6 Hz, 1H), 5.92 (ddt, J = 17.3, 10.5, 5.3 Hz, 1H), 5.36–5.26 (m, 2H), 4.66 (d, J = 5.2 Hz, 2H), 4.56 (d, J = 5.0 Hz, 1H), 3.57 (qd, J = 7.2, 5.0 Hz, 2H), 3.01 (s, 2H), 2.64–2.54 (m, 2H), 1.70 (d, J = 12.6 Hz, 1H), 1.62–1.48 (m, 5H), 1.38 (d, J = 12.8 Hz, 3H), 1.28 (t, J = 7.2 Hz, 3H). ESI+MS m/z = 350.2 (M + H+), 372.2 (M + Na+). HPLC (Method B, tR = 7.18 min), purity = 94%.
5.2.16. General method D for generating 9a and 9b
To anhydrous THF (6.4 mL) in a dry round-bottom flask was added nickel(II)bromide (279 mg, 1.28 mmol), zinc powder (250 mg, 3.83 mmol), and triphenylphosphine (1.68 g, 6.39 mmol). The mixture was heated to 50°C and stirred for 30 minutes. The selected o-chlorotoluene (8a or 8b, 12.78 mmol) was added, the temperature raised to 60°C, and the reaction was tightly capped and allowed to stir 30 minutes, then potassium cyanide (1.66g, 25.5 mmol) was added over the course of 5 hours in 2 equal portions. The mixture was allowed to stir an additional 16 hours. Water was added to quench the reaction, and the suspension extracted 3x with ether. The resulting organic layer was washed with H2O and brine, dried over MgSO4, filtered, and concentrated to a heterogeneous mixture of white crystals and clear oil. The residue was diluted with 10 mL of toluene, then methyl iodide (997 mg, 7.02 mmol) was added and allowed to stir 16 hours at room temperature. The resulting white crystals were removed via vacuum filtration. The filtrate was concentrated in vacuo to a clear oil. Flash chromatography with 2% EtOAc:hex delivered the pure product in 68–90% yield.
5.2.17. 5-methoxy-2-methylbenzonitrile (9a)
Synthesized from 8a according to General Method D. Isolated in 90% yield. 1H NMR (400 MHz, CDCl3) δ (ppm) 7.21 (d, J = 8.5 Hz, 1H), 7.08 (d, J = 2.8 Hz, 1H), 7.03 (dd, J = 8.5, 2.8 Hz, 1H), 3.81 (s, 3H), 2.47 (s, 3H).
5.2.18. 3-methoxy-2-methylbenzonitrile (9b)
Synthesized from 8b according to General Method D. Isolated as a clear oil, 68% yield. 1H NMR (500 MHz, CDCl3) δ (ppm) 7.24 (t, J = 7.9 Hz, 1H), 7.19 (d, J = 7.7 Hz, 1H), 7.03 (d, J = 8.1 Hz, 1H), 3.86 (s, 3H), 2.42 (s, 3H).
5.2.19. General Method E for synthesizing 10a–10d, 23
A dry round bottom flask was charged with anhydrous diglyme (24 mL) and diisopropylamine (2.91 mL, 20.4 mmol), then cooled to −78°C in a dry ice/acetone bath. To this solution was added n-butyllithium (2.5M in hexanes, 8.15 mL, 20.4 mmol). The reaction was removed from the dry ice/acetone bath for 10 minutes, then re-cooled to −78°C. The desired o-tolunitrile 9a–d (6.79 mmol), dissolved in anhydrous diglyme (2 mL), was then added slowly dropwise then allowed to stir at −78°C for 45 minutes.
A separate dry flask was charged with anhydrous diglyme (8.0 mL) and zinc powder (1.11 g, 17.0 mmol). Molecular iodine (3.45 g, 13.6 mmol) was added portion wise over the course of 10 minutes. The suspension was subsequently heated via heat gun in 30-second intervals until a silver precipitate of ZnI2 had formed and all iodine color had disappeared (caution: exothermic).
Ethyl-3,3-dimethyl acrylate (1.42 mL, 10.2 mmol) was added to the first flask dropwise over 10 minutes. The flask containing the ZnI2 suspension was then added to the reaction vessel and the resulting suspension allowed to stir for an additional 2 hours, slowly warming to room temperature. The reaction was quenched by the addition of saturated ammonium chloride solution and the resulting biphasic suspension was extracted with diethyl ether (3 × 30 mL), then washed with water (4 × 50 mL) and brine (1 × 50 mL). The organic extract was dried over MgSO4, vacuum filtered, and concentrated in vacuo. Further purification via flash chromatography (silica gel, 5% EtOAc:hex) delivered the desired 10a–d in 33–59% yields.
5.2.20. Ethyl 1-amino-7-methoxy-3,3-dimethyl-3,4-dihydronaphthalene-2-carboxylate (10a)
Prepared according to General Method E from 9a. Isolated as a pale yellow oil (623 mg, 33% yield). 1H NMR (500 MHz, CDCl3) δ (ppm) 7.11 (d, J = 8.1 Hz, 1H), 6.96 (s, 1H), 6.88 (d, J = 8.1 Hz, 1H), 6.27 (s, 2H), 4.27 (q, J = 7.1 Hz, 2H), 3.85 (s, 3H), 2.60 (s, 2H), 1.37 (t, J = 7.1 Hz, 3H), 1.20 (s, 6H).
5.2.21. Ethyl 1-amino-5-methoxy-3,3-dimethyl-3,4-dihydronaphthalene-2-carboxylate (10b)
Prepared according to General Method E from 9b. Isolated as a yellow oily solid (636 mg, 35% yield). 1H NMR (500 MHz, CDCl3) δ (ppm) 7.24 (t, J = 8.1 Hz, 2H), 7.04 (d, J = 8.1 Hz, 1H), 6.92 (d, J = 8.1 Hz, 1H), 6.31 (s, 2H), 4.25 (q, J = 7.1 Hz, 2H), 3.85 (s, 3H), 2.66 (s, 2H), 1.34 (t, J = 7.1 Hz, 3H), 1.20 (s, 6H).
5.2.22. Ethyl 1-amino-6-methoxy-3,3-dimethyl-3,4-dihydronaphthalene-2-carboxylate (10c)
Prepared according to General Method E from 9c. Isolated as pale yellow crystals (1.108 g, 59% yield). TLC Rf = 0.14 (10% EtOAc:hex). 1H NMR (500 MHz, CDCl3) δ (ppm) 7.35 (d, J = 8.5 Hz, 1H), 6.81 (dd, J = 8.5, 2.4 Hz, 1H), 6.73 (d, J = 2.4 Hz, 1H), 6.37 (s, 1H), 4.26 (q, J = 7.1 Hz, 2H), 3.86 (s, 3H), 2.64 (s, 2H), 1.36 (t, J = 7.1 Hz, 3H), 1.22 (s, 6H).
5.2.23. Ethyl 1-amino-3,3-dimethyl-3,4-dihydronaphthalene-2-carboxylate (10d)
Prepared according to General Method E from 9d. Isolated as a pale yellow oil (860 mg, 41% yield). TLC Rf = 0.30 (10% EtOAc:hex). 1H NMR (500 MHz, CDCl3) δ (ppm) 7.42 (d, J = 8.6 Hz, 1H), 7.36–7.29 (m, 2H), 7.20 (d, J = 7.3 Hz, 1H), 6.35 (s, 1H), 4.28 (q, J = 7.1 Hz, 2H), 2.68 (s, 2H), 1.37 (t, J = 7.1 Hz, 3H), 1.22 (s, 6H).
5.2.24. General Method F for synthesizing compounds 11a–f
Glacial acetic acid (147 μL, 2.56 mmol) and an alkyl isothiocyanate (2.56 mmol) were combined with the selected intermediate 10a–d (1.28 mmol) in absolute ethanol (1.7 mL). The solution was allowed to stir at reflux under nitrogen atmosphere for 1 hour. Additional allyl isothiocyanate (373 μL, 3.84 mmol) was added in equal portions over the course of 3 hours. The reaction was allowed to stir at reflux 16 additional hours, then diluted with ethyl acetate. The organic mixture was washed with water and brine, dried over MgSO4, filtered, and concentrated in vacuo. Trituration or flash chromatography delivered compounds 11a–f in 21–59% yield.
5.2.25. 3-allyl-9-methoxy-5,5-dimethyl-2-thioxo-2,3,5,6-tetrahydrobenzo[h]quinazolin-4(1H)-one (11a)
Prepared according to general method F from 10a and allyl isothiocyanate. Purified via trituration with hexanes and diethyl ether (125 mg, 21% yield). 1H NMR (400 MHz, CDCl3) δ (ppm) 9.29 (s, 1H), 7.35 (d, J = 8.0 Hz, 1H), 7.06 (d, J = 8.0 Hz, 1H), 7.00 (s, 1H), 6.08 (ddt, J = 15.8, 10.9, 5.5 Hz, 1H), 5.45–5.30 (m, 2H), 5.18 (d, J = 5.5 Hz, 2H), 3.85 (s, 3H), 2.73 (s, 2H), 1.34 (s, 6H).
5.2.26. 3-allyl-7-methoxy-5,5-dimethyl-2-thioxo-2,3,5,6-tetrahydrobenzo[h]quinazolin-4(1H)-one (11b)
Prepared according to general method F from 10c and allyl isothiocyanate. Purified via trituration of the crude organic isolate with hexanes and diethyl ether (133 mg, 24% yield). 1H NMR (500 MHz, CDCl3) δ (ppm) 9.29 (s, 1H), 7.35 (t, J = 8.0 Hz, 1H), 7.08–7.01 (m, 2H), 6.00 (ddt, J = 16.8, 10.8, 5.8 Hz, 1H), 5.37 (d, J = 16.8 Hz, 1H), 5.27 (d, J = 10.8 Hz, 1H), 5.06 (d, J = 5.8 Hz, 2H), 3.89 (s, 3H), 2.79 (s, 2H), 1.34 (s, 6H).
5.2.27. 3-allyl-8-methoxy-5,5-dimethyl-2-thioxo-2,3,5,6-tetrahydrobenzo[h]quinazolin-4(1H)-one (11c)
Prepared according to general method F from 10c and allyl isothiocyanate. Purified via trituration of the crude organic isolate with hexanes and diethyl ether; isolated as tan crystals (252 mg, 59% yield). TLC Rf = 0.11 (10% EtOAc:hex). 1H NMR (400 MHz, CDCl3) δ (ppm) 9.27 (s, 1H), 7.38 (d, J = 8.6 Hz, 1H), 6.88 (dd, J = 8.6, 2.5 Hz, 1H), 6.80 (d, J = 2.5 Hz, 1H), 5.99 (ddt, J = 16.6, 10.4, 5.7 Hz, 1H), 5.36 (dd, J = 16.6, 1.3 Hz, 2H), 5.26 (dd, J = 10.4, 1.3 Hz, 1H), 5.06 (d, J = 5.7 Hz, 2H), 3.87 (s, 3H), 2.74 (s, 2H), 1.33 (s, 6H). ESI-MS m/z = 327 (M−H+).
5.2.28. 3-allyl-5,5-dimethyl-2-thioxo-2,3,5,6-tetrahydrobenzo[h]quinazolin-4(1H)-one (11d)
Prepared according to general method F from 10d and allyl isothiocyanate. Purified via trituration of the crude organic isolate with hexanes and diethyl ether; recovered in 24% yield. Rf = 0.34 (10% EtOAc:hex). 1H NMR (500 MHz, CDCl3) δ (ppm) 9.28 (s, 1H), 7.50–7.37 (m, 3H), 7.29 (d, J = 7.4 Hz, 1H), 6.00 (ddt, J = 17.1, 10.3, 5.8 Hz, 1H), 5.37 (d, J = 17.1 Hz, 1H), 5.27 (d, J = 10.3 Hz, 1H), 5.07 (d, J = 5.8 Hz, 2H), 2.79 (s, 2H), 1.34 (s, 6H).
5.2.29. 3-ethyl-5,5-dimethyl-2-thioxo-2,3,5,6-tetrahydrobenzo[h]quinazolin-4(1H)-one (11e)
Prepared according to general method F from 10d and ethyl isothiocyanate. Purified via flash chromatography (0 to 10% EtOAc:hex); recovered in 31% yield. TLC Rf = 0.20 (10% EtOAc:hex). 1H NMR (500 MHz, CDCl3) δ (ppm) 9.24 (s, 1H), 7.49–7.35 (m, 3H), 7.29 (d, J = 7.4 Hz, 1H), 4.49 (q, J = 7.0 Hz, 2H), 2.79 (s, 2H), 1.35 (t, J = 7.0 Hz, 3H), 1.34 (s, 6H).
5.2.30. 9-methoxy-3,5,5-trimethyl-2-thioxo-2,3,5,6-tetrahydrobenzo[h]quinazolin-4(1H)-one (11f)
Prepared according to general method F from ethyl 10a and methyl isothiocyanate. Purified via flash chromatography (0–10% EtOAc:hex) and recovered in 27% yield. 1H NMR (400 MHz, CDCl3) δ (ppm) 9.35 (s, 1H), 7.20 (d, J = 8.3 Hz, 1H), 6.99 (dd, J = 8.3, 2.3 Hz, 1H), 6.94 (d, J = 2.3 Hz, 1H), 3.87 (s, 3H), 3.74 (s, 3H), 2.71 (s, 2H), 1.33 (s, 6H).
5.2.31. General method G for the generation of compounds 12a–m
The selected intermediate 11a–f (0.101 mmol) was dissolved in MEK (0.591 mL), to which Cs2CO3 (66 mg, 0.201 mmol) and alkylating agent (0.151 mmol) were added. The reaction was heated to 70°C and allowed to stir 16 hours. The reaction mixture was diluted with H2O and extracted 2x with EtOAc. The combined organic layers were washed with water and brine, then isolated, dried over MgSO4, vacuum filtered, and concentrated in vacuo. Further purification via flash chromatography in an appropriate solvent system delivered the desired S-alkylated compounds in 24–88% yield.
5.2.32. 3-allyl-2-(ethylthio)-9-methoxy-5,5-dimethyl-2,3,5,6-tetrahydrobenzo[h]quinazolin-4(1H)-one (12a)
Prepared according to General Method G from intermediate 11a, using iodoethane as the alkylating agent. Isolated via flash chromatography as a white solid (74 mg, 71% yield). 1H NMR (400 MHz, CDCl3) δ (ppm) 7.70 (s, 1H), 7.10 (d, J = 8.2 Hz, 1H), 6.91 (d, J = 8.2 Hz, 1H), 5.93 (ddt, J = 15.8, 10.9, 5.5 Hz, 1H), 5.33–5.21 (m, 2H), 4.68 (d, J = 5.5 Hz, 2H), 3.85 (s, 3H), 3.31 (q, J = 7.3 Hz, 2H), 2.72 (s, 2H), 1.49 (t, J = 7.3 Hz, 3H), 1.37 (s, 6H). ESI+MS m/z = 357.2 (M + H+), 379.2 (M + Na+). HPLC (Method B, tR = 8.53 min) purity >95%.
5.2.33. 3-allyl-2-(ethylthio)-7-methoxy-5,5-dimethyl-5,6-dihydrobenzo[h]quinazolin-4(3H)-one (12b)
Prepared according to General Method G from intermediate 11b, using iodoethane as the alkylating agent. Isolated via flash chromatography (75 mg, 69% yield). 1H NMR (500 MHz, CDCl3) δ (ppm) 7.80 (d, J = 7.8 Hz, 1H), 7.30 (t, J = 8.0 Hz, 1H), 6.97 (d, J = 8.1 Hz, 1H), 5.95 (ddt, J = 16.0, 10.6, 5.6 Hz, 1H), 5.34–5.25 (m, 2H), 4.70 (d, J = 5.5 Hz, 2H), 3.90 (s, 3H), 3.33 (q, J = 7.3 Hz, 2H), 2.82 (s, 2H), 1.49 (t, J = 7.3 Hz, 3H), 1.41 (s, 6H). ESI+MS m/z = 357.2 (M + H+), 379.1 (M + Na+). HPLC (Method B, tR = 8.52 min), purity >95%.
5.2.34. 3-allyl-2-(ethylthio)-8-methoxy-5,5-dimethyl-5,6-dihydrobenzo[h]quinazolin-4(3H)-one (CCG-203592) (12c)
Prepared according to General Method G from intermediate 11c, using iodoethane as the alkylating agent. Isolated as white crystals via flash chromatography (105 mg, 88% yield). 1H NMR (500 MHz, CDCl3) δ (ppm) 8.05 (d, J = 8.6 Hz, 1H), 6.84 (dd, J = 8.6, 2.4 Hz, 1H), 6.71 (d, J = 2.1 Hz, 1H), 5.93 (ddt, J = 15.8, 10.8, 5.5 Hz, 1H), 5.30–5.22 (m, 2H), 4.67 (d, J = 5.4 Hz, 2H), 3.86 (s, 3H), 3.30 (q, J = 7.3 Hz, 2H), 2.75 (s, 2H), 1.47 (t, J = 7.3 Hz, 3H), 1.38 (s, 6H). ESI+MS m/z = 357.2 (M + H+), 379.2 (M + Na+). HPLC (Method B, tR = 8.26 min), purity >95%.
5.2.35. 2-((3-allyl-5,5-dimethyl-4-oxo-3,4,5,6-tetrahydrobenzo[h]quinazolin-2-yl)thio)acetamide (12d)
Prepared according to General Method G from intermediate 11d, using 2-bromoacetamide as the alkylating agent. Further purification via flash chromatography (2:1 EtOAc:hex) delivered the desired product as a white crystalline solid (26 mg, 74% yield). 1H NMR (500 MHz, CDCl3) δ (ppm) 8.07 (d, J = 7.1 Hz, 1H), 7.41–7.31 (m, 2H), 7.20 (d, J = 7.9 Hz, 1H), 6.83 (s, 1H), 5.93 (ddt, J = 17.3, 10.1, 5.6 Hz, 1H), 5.39 (s, 1H), 5.34–5.27 (m, 2H), 4.70 (d, J = 5.6 Hz, 2H), 3.96 (s, 2H), 2.80 (s, 2H), 1.38 (s, 6H). ESI+MS m/z = 378.1 (M + Na+). HPLC (Method B, tR = 2.77 min), purity >95%.
5.2.36. 3-allyl-5,5-dimethyl-2-((pyridin-2-ylmethyl)thio)-5,6-dihydrobenzo[h]quinazolin-4(3H)-one (12e)
Prepared according to General Method G from intermediate 11d, using 2-(bromomethyl)pyridine hydrobromide as the alkylating agent, as well as an additional equivalent of Cs2CO3 to neutralize the acid. Isolated via flash chromatography (2:1 EtOAc:hex) as a white crystalline solid (34 mg, 87% yield). 1H NMR (500 MHz, CDCl3) δ (ppm) 8.62 (d, J = 5.5 Hz, 1H), 8.11 (d, J = 7.7 Hz, 1H), 7.66 (t, J = 7.7 Hz, 1H), 7.52 (d, J = 7.8 Hz, 1H), 7.41–7.30 (m, 2H), 7.26–7.18 (m, 2H), 5.96 (ddt, J = 16.0, 10.5, 5.6 Hz, 1H), 5.35–5.26 (m, 2H), 4.76 (s, 2H), 4.73 (d, J = 5.6 Hz, 2H), 2.81 (s, 2H), 1.40 (s, 6H). ESI+MS m/z = 390.1 (M + H+), 412.1 (M + Na+). HPLC (Method B, tR = 2.36 min), purity >95%.
5.2.37. 3-allyl-2-((2-hydroxyethyl)thio)-5,5-dimethyl-5,6-dihydrobenzo[h]quinazolin-4(3H)-one (12f)
Prepared according to General Method G from intermediate 11d, using 2-bromoethanol as the alkylating agent. Isolated via flash chromatography (5% EtOAc:hex) as a white crystalline solid (27 mg, 78% yield). 1H NMR (500 MHz, CDCl3) δ (ppm) 8.04 (d, J = 7.2 Hz, 1H), 7.39–7.29 (m, 2H), 7.19 (d, J = 6.7 Hz, 1H), 5.94 (ddt, J = 15.9, 10.7, 5.6 Hz, 1H), 5.34–5.26 (m, 2H), 4.72 (d, J = 5.6 Hz, 2H), 4.04 (t, J = 5.3 Hz, 2H), 3.55 (t, J = 5.6 Hz, 2H), 2.93 (s, 1H), 2.78 (s, 2H), 1.37 (s, 6H). ESI+MS m/z = 343.1 (M + H+), 365.1 (M + Na+). HPLC (Method B, tR = 4.71 min), purity >95%.
5.2.38. 2-((3-allyl-5,5-dimethyl-4-oxo-3,4,5,6-tetrahydrobenzo[h]quinazolin-2-yl)thio)acetonitrile (12g)
Prepared according to General Method G from intermediate 11d, using α-chloroacetonitrile as the alkylating agent. Isolated via flash chromatography (5% EtOAc:hex) as a white crystalline solid (92 mg, 81% yield). 1H NMR (500 MHz, CDCl3) δ (ppm) 8.16 (d, J = 7.0 Hz, 1H), 7.41–7.32 (m, 2H), 7.19 (d, J = 6.3 Hz, 1H), 5.91 (ddt, J = 16.1, 10.6, 5.6 Hz, 1H), 5.34–5.26 (m, 2H), 4.65 (d, J = 5.6 Hz, 2H), 4.06 (s, 2H), 2.80 (s, 2H), 1.39 (s, 6H). ESI+MS m/z = 338.1 (M + H+), 360.1 (M + Na+). HPLC (Method B, tR = 5.57 min), purity >95%.
5.2.39. 3-allyl-2-((2-methoxyethyl)thio)-5,5-dimethyl-5,6-dihydrobenzo[h]quinazolin-4(3H)-one (12h)
Prepared according to General Method G from intermediate 11d, using 2-methoxyethyl p-toluenesulfonate as the alkylating agent. Isolated via flash chromatography (5% EtOAc:hex) as a white crystalline solid (86 mg, 72% yield). 1H NMR (500 MHz, CDCl3) δ (ppm) 8.08 (d, J = 7.4 Hz, 1H), 7.35 (t, J = 7.3 Hz, 1H), 7.31 (t, J = 7.3 Hz, 1H), 7.19 (d, J = 7.1 Hz, 1H), 5.93 (ddt, J = 15.9, 10.8, 5.6 Hz, 1H), 5.32–5.24 (m, 2H), 4.70 (d, J = 5.6 Hz, 2H), 3.76 (t, J = 6.2 Hz, 2H), 3.54 (t, J = 6.2 Hz, 2H), 3.42 (s, 3H), 2.79 (s, 2H), 1.38 (s, 6H). ESI+MS m/z = 357.1 (M + H+), 379.1 (M + Na+). HPLC (Method B, tR = 7.47 min), purity = 94%.
5.2.40. 3-ethyl-2-((2-methoxyethyl)thio)-5,5-dimethyl-5,6-dihydrobenzo[h]quinazolin-4(3H)-one (12i)
Prepared according to General Method G from intermediate 11e, using 2-methoxyethyl p-toluenesulfonate as the alkylating agent. Isolated as a clear oil after flash chromatography (66 mg, 71% yield). 1H NMR (500 MHz, CDCl3) δ (ppm) 8.09 (dd, J = 7.5, 1.3 Hz, 1H), 7.37 (td, J = 7.3, 1.6 Hz, 1H), 7.33 (td, J = 7.5, 1.4 Hz, 1H), 7.21 (d, J = 6.9 Hz, 1H), 4.14 (q, J = 7.1 Hz, 2H), 3.80 (t, J = 6.2 Hz, 2H), 3.57 (t, J = 6.2 Hz, 2H), 3.46 (s, 3H), 2.81 (s, 2H), 1.43–1.36 (m, 9H). ESI+MS m/z = 345.2 (M + H+), 367.2 (M + Na+). HPLC (Method B, tR = 7.53 min), purity >95%.
5.2.41. 3-allyl-8-methoxy-2-((2-methoxyethyl)thio)-5,5-dimethyl-5,6-dihydrobenzo[h]quinazolin-4(3H)-one (12j)
Prepared according to General Method G from intermediate 11c, using 2-methoxyethyl p-toluenesulfonate as the alkylating agent. Isolated as a crystalline white solid after flash chromatography (67 mg, 76% yield). 1H NMR (500 MHz, CDCl3) δ (ppm) 8.01 (d, J = 8.6 Hz, 1H), 6.83 (d, J = 8.6 Hz, 1H), 6.71 (s, 1H), 5.92 (ddt, J = 16.1, 10.6, 5.5 Hz, 1H), 5.31–5.23 (m, 2H), 4.69 (d, J = 5.5 Hz, 2H), 3.86 (s, 3H), 3.75 (t, J = 6.3 Hz, 2H), 3.52 (t, J = 6.3 Hz, 2H), 3.42 (s, 3H), 2.75 (s, 2H), 1.38 (s, 6H). ESI+MS m/z = 387.2 (M + H+), 409.2 (M + Na+). HPLC (Method B, tR = 7.05 min), purity >95%.
5.2.42. 3-Allyl-2-(allylthio)-8-methoxy-5,5-dimethyl-5,6-dihydrobenzo[h]quinazolin-4(3H)-one (CCG-205363) (12k)
Prepared using a method similar to General Method G from intermediate 11c, using allyl bromide as the alkylating agent, DMF as the solvent, and stirring for 18h at room temperature. Flash chromatography (hexanes/diethyl ether 3:2) afforded the product (22 mg, 78% yield) as a white solid; mp 75–78°C. 1H NMR (400 MHz, dmso) δ (ppm) 8.03 (d, J = 8.6, 1H), 6.92 (dd, J = 8.6, 2.5, 1H), 6.86 (d, J = 2.3, 1H), 6.01 (dd, J = 16.9, 10.1, 1H), 5.94–5.78 (m, 1H), 5.39 (d, J = 17.0, 1H), 5.27–5.07 (m, 3H), 4.60 (d, J = 5.1,2H), 4.00 (d, J = 6.8, 2H), 3.82 (s, 3H), 2.75 (s, 2H), 1.29 (s, 3H), 1.24 (s, 3H). ESI+MS m/z = 369 (M + H+), 391 (M + Na+). HPLC (Method A tR = 9.40 min), purity = 96%.
5.2.43. 3-allyl-9-methoxy-5,5-dimethyl-2-((2,2,2-trifluoroethyl)thio)-5,6-dihydrobenzo[h]quinazolin-4(3H)-one (12l)
Prepared according to General Method G from intermediate 11a, using 2,2,2-trifluoro-1-iodoethane as the alkylating agent. Reaction was heated to 50°C to avoid evaporation of the alkylating agent. Flash chromatography (5–10% EtOAc:hex) delivered the product (7 mg, 24% yield). 1H NMR (400 MHz, CDCl3) δ (ppm) 7.61 (s, 1H), 7.10 (d, J = 8.1 Hz, 1H), 6.93 (d, J = 8.1 Hz, 1H), 5.93 (ddt, J = 16.5, 11.1, 5.6 Hz, 1H), 5.34–5.23 (m, 2H), 4.71 (d, J = 5.6 Hz, 2H), 4.16 (q, J = 9.7 Hz, 2H), 3.84 (s, 3H), 2.73 (s, 2H), 1.37 (s, 6H). ESI+MS m/z = 411.2 (M + H+), HPLC (Method B, tR = 7.98 min), purity = 88%.
5.2.44. 2-(ethylthio)-9-methoxy-3,5,5-trimethyl-5,6-dihydrobenzo[h]quinazolin-4(3H)-one (12m)
Prepared according to General Method G from intermediate 11f, using iodoethane as the alkylating agent. Isolated as a colorless oil (20 mg, 55% yield) after flash chromatography. 1H NMR (400 MHz, CDCl3) δ (ppm) 7.70 (s, 1H), 7.10 (d, J = 8.2 Hz, 1H), 6.90 (d, J = 8.2 Hz, 1H), 3.85 (s, 3H), 3.50 (s, 3H), 3.32 (q, J = 7.3 Hz, 2H), 2.72 (s, 2H), 1.50 (t, J = 7.3 Hz, 3H), 1.38 (s, 6H). ESI+MS m/z = 331.2 (M + H+), 353.2 (M + Na+). HPLC (Method A, tR = 9.25 min), purity >95%.
5.2.45. 3-allyl-5,5-dimethyl-2-(methylthio)-5,6-dihydrobenzo[h]quinazolin-4(3H)-one (12n)
Intermediate 11d (116 mg, 0.39 mmol) was dissolved in EtOH (2.3 mL), to which KOH (38 mg, 0.58 mmol) and methyl iodide (27 μL, 0.43 mmol) were added. The solution was stirred for 30 minutes, precipitating a crystalline white solid. The solution was diluted with ethyl acetate and water. The aqueous layer was extracted with additional EtOAc, then the combined organic layers were washed with water and brine. The organic layer was isolated, dried over MgSO4, vacuum filtered, and concentrated in vacuo. The recovered crystalline material (116 mg, 96% yield) was found to be pure by NMR and used without further purification. 1H NMR (400 MHz, CDCl3) δ 8.15 (d, J = 7.3, 1H), 7.38-7.28 (m, 2H), 7.18 (d, J = 6.9, 1H), 6.01-5.75 (m, 1H), 5.33–5.23 (m, 2H), 4.69 (d, J = 5.5, 2H), 2.79 (s, 2H), 2.69 (s, 3H), 1.39 (s, 6H). HPLC (Method B, tR = 7.57 min), purity >95%.
5.2.46. 3-allyl-2-ethoxy-5,5-dimethyl-5,6-dihydrobenzo[h]quinazolin-4(3H)-one (13a)
Prepared according to General Method B from intermediate 12n. Isolated as a light yellow solid (22 mg, 69% yield). 1H NMR (500 MHz, CDCl3) δ (ppm) 8.09 (d, J = 7.3 Hz, 1H), 7.37–7.27 (m, 2H), 7.18 (d, J = 7.5 Hz, 1H), 5.92 (ddt, J = 16.0, 10.3, 5.8 Hz, 1H), 5.25–5.16 (m, 2H), 4.62 (d, J = 5.8 Hz, 2H), 4.57 (q, J = 7.1 Hz, 2H), 2.78 (s, 2H), 1.45 (t, J = 7.1 Hz, 3H), 1.37 (s, 6H). ESI+MS m/z = 311.1 (M + H+), 333.1 (M + Na+). HPLC (Method B, tR = 7.94 min), purity >95%.
5.2.47. 3-allyl-2-(ethylamino)-5,5-dimethyl-5,6-dihydrobenzo[h]quinazolin-4(3H)-one (13b)
Prepared according to General Method C from intermediate 12n. Isolated as a white crystalline solid (23 mg, 41% yield over 2 steps). 1H NMR (500 MHz, CDCl3) δ (ppm) 8.15 (d, J = 7.3 Hz, 1H), 7.36–7.26 (m, 2H), 7.16 (d, J = 7.4 Hz, 1H), 5.92 (ddt, J = 17.3, 10.5, 5.3 Hz, 1H), 5.36–5.26 (m, 2H), 4.68 (d, J = 5.2 Hz, 2H), 4.58 (t, J = 4.9 Hz, 1H), 3.57 (qd, J = 7.2, 5.2 Hz, 2H), 2.77 (s, 2H), 1.37 (s, 6H), 1.28 (t, 3H). ESI+MS m/z = 310.1 (M + H+), 332.1 (M + Na+). HPLC (Method B, tR = 5.47 min), purity >95%.
5.2.48. 3-allyl-2-(ethylsulfinyl)-9-methoxy-5,5-dimethyl-5,6-dihydrobenzo[h]quinazolin-4(3H)-one (14)
Compound 12a (20 mg, 0.056 mmol) was added to a 3.5:1 THF:water mixture (0.617 mL), and the reaction then cooled to 0°C. A solution of Oxone (34 mg, 0.056 mmol) in water (0.411 mL) was added slowly dropwise, and the reaction was allowed to stir 3.5 hours at 0°C, then allowed to warm to room temperature over 16 hours. At this point the reaction was partitioned between EtOAc and water. The organic layer was washed with water and brine, then isolated, dried over MgSO4, vacuum filtered, and concentrated under reduced pressure. The resulting residue was purified via flash chromatography (10–33% EtOAc:hex) to deliver the product as light yellow crystals (13 mg, 62% yield). 1H NMR (400 MHz, CDCl3) δ (ppm) 7.66 (d, J = 2.7 Hz, 1H), 7.12 (d, J = 8.2 Hz, 1H), 6.93 (dd, J = 8.2, 2.7 Hz, 1H), 6.56–5.61 (m, 1H), 5.30 (d, J = 10.3 Hz, 1H), 5.24 (d, J = 17.2 Hz, 1H), 5.13 (ab with 5.00; ddt, J = 1.6, 5.0, 15.8 Hz, 1H), 5.00 (ab with 5.13; dd, J = 5.0, 15.8 Hz, 1H), 3.86 (s, 3H), 3.46–3.27 (m, 2H), 2.75 (s, 2H), 1.43 (t, J = 7.5 Hz, 3H), 1.39 (s, 3H), 1.38 (s, 3H). ESI+MS m/z = 395.2 (M + H+). HPLC (Method A, tR = 7.39 min), purity >95%.
5.2.49. 3-allyl-2-(ethylsulfonyl)-9-methoxy-5,5-dimethyl-5,6-dihydrobenzo[h]quinazolin-4(3H)-one (15)
Compound 12a (28 mg, 0.79 mmol) and mCPBA (70 wt%, 48 mg, 0.196 mmol) were combined in DCM (1.2mL) at 0°C. The reaction mixture was kept at 0°C for 1 hour, then allowed to warm to RT over the course of 16 hours. The reaction mixture was then diluted with more DCM and washed with saturated sodium bicarbonate solution, water, and brine. The organic layer was isolated, dried over MgSO4, vacuum filtered, and concentrated in vacuo. Further purification via flash chromatography (5–10% EtOAc:hex) delivered the desired product as a light yellow solid (19 mg, 62% yield). 1H NMR (400 MHz, CDCl3) δ (ppm) 7.43 (s, 1H), 7.14 (d, J = 8.2 Hz, 1H), 6.94 (d, J = 8.2 Hz, 1H), 6.04 (ddt, J = 16.8, 10.3, 5.5 Hz, 1H), 5.40 (d, J = 16.8 Hz, 1H), 5.30 (d, J = 10.3 Hz, 1H), 5.02 (d, J = 5.5 Hz, 2H), 3.84 (s, 3H), 3.81 (q, J = 7.3 Hz, 2H), 2.75 (s, 2H), 1.62 (t, J = 7.3 Hz, 3H), 1.38 (s, 6H). ESI+MS m/z = 389.2 (M + H+), 411.2 (M + Na+). HPLC (Method A, tR = 8.64 min), purity >95%.
5.2.50. General Method H for preparation of phenols 16a–c
Boron tribromide solution (1M in dichloromethane, 4.42 mL) was added gradually to a stirred solution of the selected intermediate 12a–c (2.1 mmol) in dichloromethane (14 mL) at room temperature under N2. The mixture was heated to reflux for 6 hours, then cooled to room temperature. Water (10 mL) was then added dropwise and the mixture was partitioned between DCM and water. The layers were separated and the organic layer washed with water, saturated aqueous NaHCO3, and saturated brine, then dried over MgSO4. The solvent was removed under reduced pressure and the residue was triturated in ethyl acetate, filtered, and dried under high vacuum. The resulting crystalline phenols 16a–c (43–62% isolated yield) were used without further purification.
5.2.51. 3-allyl-2-(ethylthio)-9-hydroxy-5,5-dimethyl-5,6-dihydrobenzo[h]quinazolin-4(3H)-one (16a)
Prepared according to General Method H from compound 12a. Isolated as a tan solid (60 mg, 62% yield). 1H NMR (500 MHz, CDCl3) δ (ppm) 7.91 (s, 1H), 7.05 (d, J = 8.2 Hz, 1H), 6.98 (d, J = 8.2 Hz, 1H), 5.90 (ddt, J = 16.3, 10.7, 5.6 Hz, 1H), 5.44–5.36 (m, 2H), 4.83 (d, J = 5.6 Hz, 2H), 3.95 (q, J = 6.8 Hz, 2H), 2.75 (s, 2H), 1.52 (t, J = 6.8 Hz, 3H), 1.36 (s, 6H).
5.2.52. 3-allyl-2-(ethylthio)-7-hydroxy-5,5-dimethyl-5,6-dihydrobenzo[h]quinazolin-4(3H)-one (16b)
Prepared according to General Method H from compound 12b. Isolated as a tan solid (43 mg, 45% yield). 1H NMR (500 MHz, CDCl3) δ (ppm) 7.76 (d, J = 7.8 Hz, 1H), 7.18 (t, J = 7.8 Hz, 1H), 6.89 (d, J = 7.8 Hz, 1H), 5.99–5.87 (m, 1H), 5.31–5.24 (m, 2H), 5.18 (s, 1H), 4.68 (d, J = 5.5 Hz, 2H), 3.31 (q, J = 7.4 Hz, 2H), 2.78 (s, 2H), 1.47 (t, J = 7.4 Hz, 3H), 1.41 (s, 6H).
5.2.53. 3-allyl-2-(ethylthio)-8-hydroxy-5,5-dimethyl-5,6-dihydrobenzo[h]quinazolin-4(3H)-one (16c)
Prepared according to General Method H from compound 12c. Isolated as tan crystals (310 mg, 43% yield). 1H NMR (500 MHz, CDCl3) δ (ppm) 7.62 (d, J = 8.2 Hz, 1H), 6.85–6.79 (m, 2H), 5.88 (ddt, J = 16.3, 10.8, 5.9 Hz, 1H), 5.48–5.40 (m, 2H), 4.86 (d, J = 5.9 Hz, 2H), 4.22 (q, J = 7.3 Hz, 2H), 2.74 (s, 2H), 1.59 (t, J = 7.3 Hz, 3H), 1.31 (s, 6H).
5.2.54. General Method I to generate compounds 17a–j
The selected phenol intermediate 12a–c (0.622 mmol) was dissolved in DMF (3.66 mL), to which Cs2CO3 (304 mg, 0.933 mmol) and an alkylating agent (0.716 mmol) were added. The resulting suspension was stirred at 25°C to 70°C for 30 minutes to 16 hours until the completion of the reaction. Compounds were purified as indicated in 35–92% yield.
5.2.55. 3-allyl-2-(ethylthio)-9-(2-methoxyethoxy)-5,5-dimethyl-5,6-dihydrobenzo[h]quinazolin-4(3H)-one (17a)
Prepared using general method I from 12a and 2-methoxyethyl p-toluenesulfonic ester, heated to 70°C for 16 hours. Reaction mixture was partitioned between ethyl acetate and water, then the organic layer washed with water and brine. The organic layer was isolated, dried over MgSO4, vacuum filtered, and concentrated in vacuo. Further purification via flash chromatography (0–10% EtOAc:hex) delivered the desired product (38 mg, 65% yield). 1H NMR (500 MHz, CDCl3) δ (ppm) 7.71 (s, 1H), 7.09 (d, J = 8.2 Hz, 1H), 6.94 (d, J = 8.2 Hz, 1H), 5.93 (ddt, J = 16.0, 10.7, 5.6 Hz, 1H), 5.31–5.23 (m, 2H), 4.68 (d, J = 5.5 Hz, 2H), 3.81–3.75 (m, 2H), 3.61–3.55 (m, 2H), 3.47 (s, 3H), 3.31 (q, J = 7.3 Hz, 2H), 2.72 (s, 2H), 1.48 (t, J = 7.3 Hz, 3H), 1.37 (s, 6H). ESI+MS m/z = 401.2 (M + H+), 423.2 (M + Na+). HPLC (Method B, tR = 7.83 min), purity >95%.
5.2.56. 3-allyl-2-(ethylthio)-7-(2-methoxyethoxy)-5,5-dimethyl-5,6-dihydrobenzo[h]quinazolin-4(3H)-one (17b)
Prepared using general method I from phenol intermediate 12b and 2-methoxyethyl p-toluenesulfonic ester, heated to 70°C for 16 hours. Reaction mixture was partitioned between ethyl acetate and water, then the organic layer washed with water and brine. The organic layer was isolated, dried over MgSO4, vacuum filtered, and concentrated in vacuo. Further purification via flash chromatography (0–10% EtOAc:hex) delivered the desired product (36 mg, 68% yield). 1H NMR (500 MHz, CDCl3) δ (ppm) 7.78 (d, J = 7.8 Hz, 1H), 7.25 (t, J = 8.0 Hz, 1H), 6.96 (d, J = 8.1 Hz, 1H), 5.99–5.87 (m, 1H), 5.31–5.23 (m, 2H), 4.68 (d, J = 5.5 Hz, 2H), 4.17 (dd, J = 5.5, 4.2 Hz, 2H), 3.80 (dd, J = 5.5, 4.2 Hz, 2H), 3.48 (s, 3H), 3.30 (q, J = 7.4 Hz, 2H), 2.83 (s, 2H), 1.47 (t, J = 7.4 Hz, 3H), 1.39 (s, 6H). ESI+MS m/z = 401.2 (M + H+), 423.2 (M + Na+). HPLC (Method B, tR = 7.91 min), purity >95%.
5.2.57. 3-allyl-2-(ethylthio)-8-(2-methoxyethoxy)-5,5-dimethyl-5,6-dihydrobenzo[h]quinazolin-4(3H)-one (17c)
Prepared using General Method I from phenol intermediate 12c and 2-methoxyethyl p-toluenesulfonic ester, heated to 70°C for 16 hours. Reaction mixture was partitioned between ethyl acetate and water, then the organic layer washed with water and brine. The organic layer was isolated, dried over MgSO4, vacuum filtered, and concentrated in vacuo. Further purification via flash chromatography (0–10% EtOAc:hex) delivered the desired product (39 mg, 71% yield). 1H NMR (500 MHz, CDCl3) δ (ppm) 8.04 (d, J = 8.6 Hz, 1H), 6.86 (dd, J = 8.6, 2.4 Hz, 1H), 6.74 (d, J = 2.2 Hz, 1H), 5.92 (ddt, J = 15.9, 10.6, 5.4 Hz, 1H), 5.30–5.22 (m, 2H), 4.67 (d, J = 5.5 Hz, 2H), 4.18 (t, J = 5.0 Hz, 2H), 3.78 (t, J = 5.0 Hz, 2H), 3.47 (s, 3H), 3.30 (q, J = 7.4 Hz, 2H), 2.74 (s, 2H), 1.47 (t, J = 7.3 Hz, 3H), 1.37 (s, 6H). ESI+MS m/z = 401.1 (M + H+), 423.1 (M + Na+). HPLC (Method B, tR = 7.56 min), purity >95%.
5.2.58. Ethyl 2-((3-allyl-2-(ethylthio)-5,5-dimethyl-4-oxo-3,4,5,6-tetrahydro-benzo[h]-quinazolin-8-yl)oxy)acetate (17d)
Prepared using General Method I from phenol intermediate 12c and ethyl bromoacetate, stirred at room temperature for 2.5 hours. The mixture was poured into 40 mL of water, then the precipitate was filtered off, rinsed with water 3 times and dried to afford the product (54 mg, 86% yield) as a white powder. 1H NMR (400 MHz, dmso) δ (ppm) 8.01 (d, J = 8.6, 1H), 6.90 (dd, 1H), 6.86 (s, 1H), 5.94–5.83 (m, 1H), 5.22 (d, J = 10.2, 1H), 5.13 (d, J = 17.2, 1H), 4.85 (s, 2H), 4.59 (d, J = 4.3, 2H), 4.19 (q, 2H), 3.29 (q, 2H), 2.75 (s, 2H), 1.39 (t, J = 7.2, 3H), 1.29 (s, 6H), 1.23 (t, J = 7.1, 3H). ESI+MS m/z = 429 (M + H+), 451 (M + Na+). HPLC (Method A, tR = 10.21 min), purity = 92%.
5.2.59. 3-Allyl-2-(ethylthio)-5,5-dimethyl-8-(2-morpholinoethoxy)-5,6-dihydrobenzo[h]-quinazolin-4(3H)-one (17e)
Prepared using General Method I from phenol intermediate 12c and 4-(2-chloroethyl)-morpholine hydrochloride, stirred at 80°C for 4 hours. The mixture was poured into water and extracted with ethyl acetate three times, then the combined extracts were washed with water and saturated brine, dried over MgSO4, and concentrated in vacuo to a tan solid (0.035g, 69% yield), mp 91–92°C. 1H NMR (400 MHz, dmso) δ (ppm) 8.06 (d, 1H), 6.98 (dd, 1H), 6.92 (d, 1H), 5.93 (ddt, 1H), 5.23 (d, 1H), 5.19 (d, 1H), 4.64 (d, 2H), 4.21 (t, 2H), 3.65 (m, 4H), 3.34 (q, 2H), 2.80 (m, 4H), 2.53 (m, 2H), 1.44 (t, 3H), 1.35 (s, 6H). ESI+MS m/z = 456 (M+H+). HPLC (Method A, tR = 10.23 min), purity = 91%.
5.2.60. 3-Allyl-2-(ethylthio)-8-isopropoxy-5,5-dimethyl-5,6-dihydrobenzo[h]quinazolin-4(3H)-one (17f)
Prepared using General Method I from phenol intermediate 12c, using 2-bromopropane as the alkylating agent and stirring at room temperature for 4 hours. The reaction mixture was poured into water, then extracted into ethyl acetate. The organic layer was washed with water and brine, then isolated, dried over MgSO4, vacuum filtered, and concentrated in vacuo delivering the final product as a sticky yellow oil (34 mg, 92% yield).. 1H NMR (400 MHz, dmso) δ (ppm) 8.04 (d, 1H), 6.94 (dd, 1H), 6.87 (d, 1H), 5.92 (m, 1H), 5.27 (dd, 1H), 5.18 (m, 1H), 4.82–4.69 (m, 1H), 4.64 (d, 2H), 3.31–3.39 (m, 2H), 2.79 (s, 2H), 1.44(t, 3H), 1.35 (m, 12H). ESI+MS m/z = 385 (M + H+), 407 M+Na+). HPLC (Method A, tR = 9.25 min), purity = 93%.
5.2.61. 2-((3-Allyl-2-(ethylthio)-5,5-dimethyl-4-oxo-3,4,5,6-tetrahydrobenzo[h]quinazolin-8-yl)oxy)-acetonitrile (17g)
Prepared using General Method I from phenol intermediate 12c using bromoacetonitrile as the alkylating agent and stirring at room temperature for 4 hours. The reaction mixture was partitioned between ethyl acetate and water, then the organic layer was washed with water and brine. After drying over MgSO4, filtration, and concentration in vacuo, recrystallization of the residue from isopropanol gave the pure product (13 mg, 39% yield); mp 130–131°C. 1H NMR (400 MHz, dmso) δ (ppm) 8.03 (d, 1H), 7.01 (dd, 1H),6.95 (d, 1H), 5.91–5.74 (m, 1H), 5.19 (m, 3H), 5.09 (d, 1H), 4.55 (d, 2H), 3.26 (q, 2H), 2.74 (s, 2H), 1.35 (t, 3H), 1.26 (s, 6H). ESI+MS m/z = 382 (M + H+), 404 (M +Na+). HPLC (Method A, tR = 8.69 min), purity = 89%.
5.2.62. 3-allyl-8-butoxy-2-(ethylthio)-5,5-dimethyl-5,6-dihydrobenzo[h]quinazolin-4(3H)-one (17h)
Prepared according to General Method I from intermediate 12c and 1-bromopentane. Isolated in 75% yield. 1H NMR (500MHz, DMSO) δ 8.00 (d, 1H), 6.90 (d, 1H), 6.84 (s, 1H), 5.94-5.77 (m, 1H), 5.21 (d, 1H), 5.13 (d, 1H), 4.59 (d, 2H), 4.03 (t, 2H), 3.28 (q, 2H), 2.74 (s, 2H), 1.71 (m, 2H), 1.48 (m, 2H), 1.41 (t, 3H), 1.29 (s, 6H), 0.95 (t, 3H). ESI+MS m/z = 399 (M + H+), 421 (M +Na+). HPLC (Method C, tR = 5.45 min), purity = 94%.
5.2.63. 3-allyl-2-(ethylthio)-5,5-dimethyl-8-phenethoxy-5,6-dihydrobenzo[h]quinazolin-4(3H)-one (17i)
Prepared according to General Method I from intermediate 12c and (2-bromoethyl)benzene. Isolated in 77% yield. 1H NMR (500MHz, DMSO) δ 8.05 (d, 1H), 7.5–7.29 (m, 5H), 6.97 (d, 1H), 6.91 (s, 1H), 6.09-5.81 (m, 1H), 5.27 (d, 1H), 5.18 (d, 1H), 4.64 (d, 2H), 4.31 (t, 2H), 3.33 (q, 2H), 3.12 (t, 2H), 2.79 (s, 2H), 1.45 (t, 3H), 1.33 (s, 6H). ESI+MS m/z = 447 (M + H+), 469 (M +Na+). HPLC (Method A, tR = 8.89 min), purity = 93%.
5.2.64. 3-allyl-2-(ethylthio)-8-(heptyloxy)-5,5-dimethyl-5,6-dihydrobenzo[h]quinazolin-4(3H)-one (17j)
Prepared according to General Method I from intermediate 12c and 1-bromoheptane. Isolated in 35% yield. 1H NMR (500MHz, DMSO) δ 7.95 (d, 1H), 6.85 (d, 1H), 6.80 (s, 1H), 5.89-5.78 (m, 1H), 5.18 (d, 1H), 5.09 (d, 1H), 4.54 (d, 2H), 3.98 (t, 2H), 3.23 (q, 2H), 2.70 (s, 2H), 1.69 (m, 2H), 1.45-1.12 (m, 17H), 0.84 (t, 3H). ESI+MS m/z = 441 (M + H+), 463 (M +Na+). HPLC (Method C, tR = 11.67 min), purity >95%.
5.2.65. 2-((3-Allyl-2-(ethylthio)-5,5-dimethyl-4-oxo-3,4,5,6-tetrahydrobenzo[h]quinazolin-8-yl)oxy)acetic acid (17k)
Compound 17d (40 mg, 0.93 mmol) was added to MeOH (0.5 mL), and KOH solution (1M, 0.75 mL) was added. The mixture was stirred and heated at 50°C for 90 minutes then cooled, poured into 20 mL of water, stirred and acidified with 2N HCl. After 2 hours the mixture was extracted twice with ethyl acetate. The combined extracts were washed with water then saturated brine and dried over MgSO4. The solvent was removed under reduced pressure and the residue recrystallized from acetonitrile to afford the product (10 mg, 27% yield) as a white solid; mp 167–168°C. 1H NMR (400 MHz, dmso) δ 13.03 (s, 1H), 7.97 (d, 1H), 6.85 (dd, 1H), 6.82 (d, 1H), 5.91–5.74 (m, 1H), 5.19 (dd, 1H), 5.09 (d, 1H), 4.70 (d, 2H), 4.54 (d, 2H), 3.26–3.19 (m, 2H), 2.71 (s, 2H), 1.34 (t, 3H), 1.25 (s, 6H). ESI+MS m/z = 401 (M + H+), 423 (M + Na+); ESI-MS m/z = 399 (M−H+). HPLC (Method A tR = 7.69 min), purity = 93%.
5.2.66. General Method J for generating unsubstituted 2-thioxopyrimidinone intermediates 18a, 18d, 24
To a solution of the selected β-aminoester intermediate (10a, 10d, 23)(12.64 mmol) in absolute EtOH (15 mL) was added benzoyl isothiocyanate (1.7 mL, 12.64 mmol), and the reaction mixture was warmed to reflux for 30 minutes. Additional benzoyl isothiocyanate (400 μL, 2.7 mmol) was added in two equal portions over the course of 1 hour, then the reaction was allowed to cool. The thiourea adduct was isolated via crystallization from ethanol or via flash chromatography, then added to a solution of KOH (1.21 g, 21.57 mmol) in ethanol:water (2:1, 30 mL). The reaction mixture was warmed to reflux and allowed to stir for 1.5 hours. The reaction mixture was cooled, then acidified to pH 5–6 with 1N HCl. The resulting precipitate was isolated via vacuum filtration, washed with EtOH and water and dried under high vacuum. Isolated in 61–83% yield.
5.2.67. 9-methoxy-5,5-dimethyl-2-thioxo-2,3,5,6-tetrahydrobenzo[h]quinazolin-4(1H)-one (18a)
Generated using General Method J from β-aminoester 10a. Isolated the desired product as a white solid (650 mg, 61% yield over 2 steps). 1H NMR (400 MHz, CDCl3) δ (ppm) 9.32 (s, 2H), 7.20 (d, J = 8.2 Hz, 1H), 7.00 (dd, J = 8.2, 2.4 Hz, 1H), 6.97 (d, J = 2.4 Hz, 1H), 3.87 (s, 3H), 2.71 (s, 2H), 1.32 (s, 6H).
5.2.68. 5,5-Dimethyl-2-thioxo-2,3,5,6-tetrahydrobenzo[h]quinazolin-4(1H)-one (18d)
Generated using General Method J from β-aminoester 10d. Isolated the desired product as a white solid (2.7g, 83% yield over 2 steps); Rf: 0.56 (2:3, EtOAc/hexanes); 1H NMR (400 MHz, CDCl3): δ (ppm) 9.95 (s, 1H, NH), 9.75 (s, 1H, NH), 7.57 (d, 1H, J = 7.7 Hz), 7.46 (t, 1H, J = 7.4 Hz), 7.39 (t, 1H, J = 7.5 Hz), 7.28 (d, 1H, J = 7.5 Hz), 2.78 (s, 2H), 1.33 (s, 6H).
5.2.69. 2-((2-Methoxyethyl)thio)-3-(4-methoxyphenethyl)-5,5-dimethyl-5,6-dihydrobenzo[h]quin-azolin-4(3H)-one, (19a) and 2-((2-Methoxyethyl)thio)-4-(4-methoxyphenethoxy)-5,5-dimethyl-5,6-dihydrobenzo[h]quinazoline, (20a)
A solution of compound 18d (2.7 g, 10.45 mmole) and KOH (590 mg, 10.45 mmole) in absolute EtOH (60 mL) was refluxed for 30 minutes. Then 2-methoxyethyl toluenesulfonic ester (2.41 g, 10.45 mmole) was added in EtOH (3 mL). The reaction was allowed to reflux for 15 hours, then allowed to cool to room temperature. The crystallized solid was filtered, washed with EtOH (3 mL) and water (75 mL), dried under suction and then under high vacuum. A portion of the resulting S-alkylated compound (25 mg, 0.079 mmol) in DMF (1 mL) was treated with lithium carbonate (17 mg, 0.23 mmol) and 1-(2-bromoethyl)-4-methoxybenzene (18 mg, 0.084 mmol). The reaction was warmed to 80°C and allowed to stir 24 hours, at which point additional lithium carbonate (17 mg, 0.23 mmol) and 1-(2-bromoethyl)-4-methoxybenzene (13 mg, 0.061 mmol) were added. After 24 additional hours at reflux, the solvent was removed in vacuo and the resulting mixture of N and O-alkylated products were separated via flash chromatography (4% EtOAc:hex).
5.2.69.1. 2-((2-Methoxyethyl)thio)-3-(4-methoxyphenethyl)-5,5-dimethyl-5,6-dihydrobenzo[h]quin-azolin-4(3H)-one, (19a)
Yield: 5 mg (11% over 2 steps); Rf: 0.30 (1:9, EtOAc/hexanes); 1H NMR (400 MHz, CDCl3) δ (ppm) 8.08 (dd, 1H, J = 1.6 & 7.1 Hz), 7.26–7.37 (m, 4H), 7.19 (d, 1H, J = 7.2 Hz), 6.86 (d, 2H, J = 8.6 Hz), 4.19 (t, 2H, J = 8.3 Hz), 3.80 (s, 3H), 3.77 (t, 2H, J = 6.2 Hz), 3.56 (t, 2H, J = 6.2 Hz), 3.44 (s, 3H), 3.00 (t, 2H, J = 8.3 Hz), 2.79 (s, 2H) & 1.39 (s, 6H); ESI+MS m/z = 451 (M + H+), 473 (M + Na+). HPLC (Method B, tR = 9.9 min), purity = 98.8%.
5.2.69.2. 2-((2-Methoxyethyl)thio)-4-(4-methoxyphenethoxy)-5,5-dimethyl-5,6-dihydrobenzo[h]quinazoline, (20a)
Yield: 24 mg (54% over 2 steps); Rf: 0.38 (1:9, EtOAc/hexanes); 1H NMR (400 MHz, CDCl3) δ (ppm) 8.20 (dd, 1H, J = 1.7 & 7.2 Hz), 7.29–7.36 (m, 2H), 7.14–7.21 (m, 3H), 6.85 (m, 2H), 4.60 (t, 2H, J = 7.0 Hz), 3.79 (s, 3H), 3.74 (t, 2H, J = 6.9 Hz), 3.41 (m, 5H), 3.06 (t, 2H, J = 7.0 Hz), 2.77 (s, 2H) & 1.25 (s, 6H); ESI+MS m/z = 451 (M + H+), 473 (M + Na+). HPLC (Method B, tR = 10.1 min), purity = 97.3%.
5.2. 70. 2-(ethylthio)-9-methoxy-5,5-dimethyl-3-(2,2,2-trifluoroethyl)-5,6-dihydrobenzo[h]quinazolin-4(3H)-one (19b) and 2-(ethylthio)-9-methoxy-5,5-dimethyl-4-(2,2,2-trifluoroethoxy)-5,6-dihydrobenzo[h]quinazoline (20b)
The 2-thioxo intermediate 18a (118 mg, 0.409 mmol) was dissolved in EtOH (2.4 mL), to which KOH (23 mg, 0.409 mmol) and iodoethane (64 mg, 0.409 mmol) were added. The reaction was warmed to 75°C and allowed to stir for 3 hours. The reaction mixture was diluted with 1N HCl and the resulting precipitate was collected via sintered glass funnel, washed with water, and dried under high vacuum. A portion of the S-alkylated adduct (27 mg, 0.085 mmol) was combined with 1,1,1-trifluoro-2-iodoethane (27 mg, 0.128 mmol) and Cs2CO3 (33 mg, 0.102 mmol) in DMF (500 μL). The reaction was tightly capped and heated to 70°C for 16 hours. The reaction mixture was partitioned between ethyl acetate and water, then the organic layer was washed with water and brine. The organic layer was concentrated in vacuo and purified via flash chromatography (0–3% EtOAc:hex) to yield compounds 19b (6 mg, 13% yield over 2 steps) and 20b (17 mg, 36% yield over 2 steps) as crystalline white solids.
5.2.70.1. 2-(ethylthio)-9-methoxy-5,5-dimethyl-3-(2,2,2-trifluoroethyl)-5,6-dihydrobenzo[h]quinazolin-4(3H)-one (19b)
1H NMR (400 MHz, CDCl3) δ (ppm) 7.68 (d, J = 2.8 Hz, 1H), 7.11 (d, J = 8.2 Hz, 1H), 6.93 (dd, J = 8.2, 2.8 Hz, 1H), 4.78 (q, J = 8.0 Hz, 2H), 3.85 (s, 3H), 3.35 (q, J = 7.3 Hz, 2H), 2.74 (s, 2H), 1.50 (t, J = 7.3 Hz, 3H), 1.36 (s, 6H). ESI+MS m/z = 399.2 (M + H+), 421.2 (M + Na+). HPLC (Method B, tR = 8.49 min), purity >95%.
5.2.70.2. 2-(ethylthio)-9-methoxy-5,5-dimethyl-4-(2,2,2-trifluoroethoxy)-5,6-dihydrobenzo[h]quinazoline (20b)
1H NMR (400 MHz, CDCl3) δ (ppm) 7.81 (d, J = 2.7 Hz, 1H), 7.10 (d, J = 8.2 Hz, 1H), 6.94 (dd, J = 8.2, 2.7 Hz, 1H), 4.80 (q, J = 8.5 Hz, 2H), 3.87 (s, 3H), 3.21 (q, J = 7.3 Hz, 2H), 2.76 (s, 2H), 1.47 (t, J = 7.3 Hz, 3H), 1.33 (s, 6H). ESI+MS m/z = 399.2 (M + H+). HPLC (Method A, tR = 9.84 min), purity >95%.
5.2.71. 4-(allyloxy)-2-(ethylthio)-9-methoxy-5,5-dimethyl-5,6-dihydrobenzo[h]quinazoline (20c)
The 2-thioxo intermediate 18a (118 mg, 0.409 mmol) was dissolved in EtOH (2.4 mL), to which KOH (23 mg, 0.409 mmol) and iodoethane (64 mg, 0.409 mmol) were added. The reaction was warmed to 75°C and allowed to stir for 3 hours. The reaction mixture was diluted with 1N HCl, and the precipitate (92 mg, 71% yield) was collected via sintered glass funnel, washed with water, and dried under high vacuum. A portion of the S-alkylated adduct (30 mg, 0.095 mmol) was combined with Cs2CO3 (37 mg, 0.114 mmol) and allyl bromide (17 mg, 0.142 mmol) in DMF (558 μL). The reaction mixture was tightly capped and warmed to 75°C, and allowed to stir for 16 hours. The reaction mixture was partitioned between ethyl acetate and water, then the organic layer was washed with water and brine. The organic layer was concentrated in vacuo, and the residue was purified via flash chromatography (0–5% EtOAc:hex) to yield 19e (14 mg, 30% yield over 2 steps). 1H NMR (400 MHz, CDCl3) δ (ppm) 7.81 (d, J = 2.5 Hz, 1H), 7.08 (d, J = 8.2 Hz, 1H), 6.91 (dd, J = 8.2, 2.5 Hz, 1H), 6.09 (ddt, J = 16.4, 10.6, 5.5 Hz, 1H), 5.40 (d, J = 16.4 Hz, 1H), 5.27 (d, J = 10.6 Hz, 1H), 4.94 (d, J = 5.5 Hz, 2H), 3.87 (s, 3H), 3.19 (q, J = 7.3 Hz, 2H), 2.74 (s, 2H), 1.47 (t, J = 7.3 Hz, 3H), 1.33 (s, 6H). ESI+MS m/z = 357.2 (M + H+), 379.2 (M + Na+). HPLC (Method B, tR = 10.41 min), purity >95%. N-allyl compound 12a was also isolated (10 mg, 21% yield over 2 steps).
5.2.72. 4-cyano-3-methylbenzoic acid (22)
A solution of 2.5M n-butyllithium in hexanes(6.7 mL, 16.8 mmol) was added to anhydrous THF (15 mL) at −78°C. A solution of 4-bromo-2-methylbenzonitrile 21 (3.0 g, 15.3 mmol) in THF (15 mL) was then added dropwise and allowed to stir for 30 min. Dry ice (solid CO2) was added and the solution was allowed to stir 1 hour while warming to room temperature. The reaction was then concentrated in vacuo and the resulting residue was triturated in ether and filtered. The solid was dissolved in EtOAc and washed with 2M HCl and brine. The organic layer was isolated, dried over MgSO4, concentrated in vacuo, then triturated in ether/hexane and dried under high vacuum. Obtained 4-cyano-3-methylbenzoic acid 22 (1.5 g, 60.8% yield) as a white solid. 1H NMR (400 MHz, CDCl3) δ (ppm) 8.18 (s, 1H), 8.02–8.09 (m, 1H), 7.75–7.81 (m, 1H), 2.61 (s, 3H).
5.2.73. 2-ethyl 6-methyl 1-amino-3,3-dimethyl-3,4-dihydronaphthalene-2,6-dicarboxylate (23)
Prepared according to General Method E from 4-cyano-3-methylbenzoic acid 22 (1.0 g, 6.2 mmol). The impure residue resulting from concentrating the crude organic extract was dissolved in 20% methanolic toluene (20 ml) and treated with trimethylsilyldiazomethane (2.7 ml, 5.4 mmol). The mixture was stirred 30 minutes at room temperature, then concentrated in vacuo. Purification via flash chromatography (5% to 10% to 20% EtOAc:hex) yielded the desired compound 23 as a yellow solid (0.78 g, 41.5% yield). 1H NMR (400 MHz, CDCl3) δ (ppm) 7.98–8.10 (m, 1H), 7.82–7.88 (m, 1H), 7.45–7.51 (m, 1H), 6.24 (br s, 2H), 4.31–4.39 (q, 2H), 3.93 (s, 3H), 2.68 (br s, 2H), 1.38–1.41 (t, 3H), 1.28 (s, 6H). HPLC (Method A, tR = 8.02 min), purity = 84%.
5.2.74. Methyl 5,5-dimethyl-4-oxo-2-thioxo-1,2,3,4,5,6-hexahydrobenzo[h]quinazoline-8-carboxylate (24)
Prepared according to General Method J from β-aminoester 23. Isolated as a white solid (550 mg, 61% yield over 2 steps) used without further purification. 1H NMR (400 MHz, CDCl3) δ 8.11–8.15 (m, 1H), 7.87–7.89 (m, 1H), 7.78–7.81 (m, 1H), 3.89 (s, 3H), 2.82 (br s, 2H), 1.27 (s, 6H). HPLC (Method A, tR = 6.26 min), purity = 82%.
5.2.75. Methyl 3-allyl-2-((2-methoxyethyl)thio)-5,5-dimethyl-4-oxo-3,4,5,6-tetrahydrobenzo[h]quinazoline-8-carboxylate (25)
To a solution of methyl 5,5-dimethyl-4-oxo-2-thioxo-1,2,3,4,5,6-hexahydrobenzo[h]quinazoline-8-carboxylate 24 (850 mg, 2.7 mmol) in DMF (10 mL) was added Cs2CO3 (1.8 g, 5.4 mmol) followed by 2-methoxyethyl 4-methylbenzenesulfonate (0.74 g, 3.2 mmol). The resulting mixture was stirred at 40°C for 3 hours, then allowed to cool to room temperature and stir an additional 16 hours. The reaction mixture was diluted with EtOAc, then washed with water, saturated NaHCO3 solution, and brine, then isolated and dried over MgSO4. The organic extract was concentrated in vacuo and purified by flash chromatography, (5 to 30% EtOAc:hex) to obtain the S-2-methoxyethyl adduct as a white solid. A portion of the resulting solid (40 mg, 0.11 mmol) was dissolved in anhydrous methanol (1.5 mL) along with sodium methoxide (10 mg, 0.21 mmol) and allyl bromide (39 mg, 0.32 mmol). The reaction was heated to 65°C and stirred for 30 minutes. Additional sodium methoxide (20 mg, 0.42 mmol) and allyl bromide (78 mg, 0.64 mmol) were added in two equal portions over the course of 1 hour and then allowed to stir an additional hour at 65°C. The mixture was then cooled and concentrated in vacuo. The resulting residue was partitioned between 2M HCl and EtOAc. The aqueous layer was extracted with additional EtOAc, then the organic extracts were combined, dried over MgSO4, vacuum filtered, and concentrated in vacuo. Purification by flash chromatogaphy (5–20% EtOAc:hex) provided methyl 3-allyl-2-((2-methoxyethyl)thio)-5,5-dimethyl-4-oxo-3,4,5,6-tetrahydrobenzo[h]quinazoline-8-carboxylate 25 (20 mg, 0.05 mmol, 30% yield over 2 steps). 1H NMR (400 MHz, CDCl3) δ (ppm) 8.14–8.16 (m, 1H), 7.98–8.00 (m, 1H), 7.88 (s, 1H), 5.92–5.97 (m, 1H), 5.28–5.33(m, 2H), 3.82 (s, 3H), 3.78–3.82 (t, 2H), 3.55–3.56 (m, 2H), 3.44 (s, 3H), 2.86 (s, 2H), 1.46 (s, 6H). ESI+MS m/z = 415.1 (M + H +). HPLC (Method A, tR = 9.06 min), purity = 95%.
5.2.76. 3-allyl-2-((2-methoxyethyl)thio)-5,5-dimethyl-4-oxo-3,4,5,6-tetrahydrobenzo[h]quinazoline-8-carboxylic acid (26)
To a solution of sodium hydroxide (3.1 mg, 0.055 mmol) in methanol (0.5 mL) was added methyl 3-allyl-2-((2-methoxyethyl)thio)-5,5-dimethyl-4-oxo-3,4,5,6-tetrahydrobenzo[h]quinazoline-8-carboxylate 25 (20 mg, 0.06 mmol). The resulting mixture was stirred overnight at room temperature, then concentrated in vacuo and partitioned between 2M HCl and EtOAc. The aqueous phase was extracted with additional EtOAc, then the organic extracts were combined and washed with brine. The organic layer was isolated, dried over MgSO4, vacuum filtered, and concentrated in vacuo to a white powder (19 mg, 0.05 mmol, 86% yield). 1 H NMR (400 MHz, CDCl3) δ (ppm) 8.18–8.19 (m, 1H), 8.01–8.05 (m, 1H), 7.95 (br s, 1H), 6.20–6.23 (m, 1H), 5.89–5.94 (m, 1H), 5.26–5.31 (m, 2H), 4.58–4.63 (m, 2H), 3.72–3.78 (m, 4H), 3.49–3.52 (m, 2H), 3.42 (s, 3H), 2.94–2.95 (m, 2H), 2.80 (s, 2H), 1.37 (s, 6H). ESI+MS m/z = 400.49 (M + H+). HPLC (Method A, tR = 7.5 min), purity = 95%.
5.2.77. 3-allyl-N-benzyl-2-((2-methoxyethyl)thio)-5,5-dimethyl-4-oxo-3,4,5,6-tetrahydrobenzo[h]quinazoline-8-carboxamide (27a)
To a solution of 3-allyl-2-((2-methoxyethyl)thio)-5,5-dimethyl-4-oxo-3,4,5,6-tetrahydrobenzo[h]quinazoline-8-carboxylic acid 26 (100 mg, 0.25 mmol), EDC (60 mg, 0.30 mmol), and HOBt (50 mg, 0.30 mmol) in dry THF (10 mL) was added benzylamine (30 mg, 0.30 mmol). The reaction was allowed to stir at room temperature for 16 hours, then diluted with diethyl ether and water. The organic layer was washed with saturated Na2CO3 solution, 2M HCl, water, and brine, then isolated, dried over MgSO4, vacuum filtered, and concentrated in vacuo. Further purification via flash chromatography (5–20% EtOAc:hex) delivered the desired compound (35 mg, 29% yield). 1H NMR (400 MHz, CDCl3) δ (ppm) 8.12 (d, J = 8Hz, 1H), 7.70 (d, J = 8Hz, 1H), 7.62 (br s, 1H), 7.32–7.38 (m, 4H), 6.39–4.41 (m, 1H), 5.89–5.93 (m, 1H), 5.26–5.31 (m, 2H), 4.67–4.73 (4H, M), 3.75 (t, J = 4Hz, 2H), 3.53 (t, J = 4Hz, 2H), 3.42 (s, 3H), 2.82 (s, 2H), 1.56 (s, 6H). ESI+MS m/z = 490.3 (M + H+). HPLC (Method A, tR = 8.33 min), purity = 98.6%.
5.2.78. N-(2-(1H-imidazol-4-yl)ethyl)-3-allyl-2-((2-methoxyethyl)thio)-5,5-dimethyl-4-oxo-3,4,5,6-tetrahydrobenzo[h]quinazoline-8-carboxamide (27b)
To a solution of 3-allyl-2-((2-methoxyethyl)thio)-5,5-dimethyl-4-oxo-3,4,5,6-tetrahydrobenzo[h]quinazoline-8-carboxylic acid 26 (50 mg, 0.13 mmol), EDC (30 mg, 0.15 mmol), and HOBt (20 mg, 0.15 mmol) in dry THF (5 mL) was added 2-(1H-imidazol-4-yl)ethanamine hydrochloride (20 mg, 0.15 mmol). The reaction was allowed to stir at room temperature for 16 hours, then diluted with diethyl ether and water. The organic layer was washed with saturated Na2CO3 solution, water, and brine, then isolated, dried over MgSO4, vacuum filtered, and concentrated in vacuo. Further purification via flash chromatography (5–30% EtOAc:hex) delivered the desired compound (13 mg, 21% yield). 1H NMR (400 MHz, CDCl3) δ (ppm) 8.11 (d, J = 8Hz, 1H), 7.73 (d, J = 8Hz, 1H), 7.64 (br s, 2H), 7.22–7.24 (br s, 1H), 7.01 (br s, 1H), 5.87–5.95 (m, 1H), 5.26–5.28 (m, 2H), 4.69–4.70 (2H, M), 4.60–4.61 (2H, m), 3.73 (t, J = 4Hz, 2H), 3.2 (t, J = 4Hz, 2H), 3.41 (s, 3H), 2.82 (s, 2H), 1.38 (s, 6H). (ESI+MS m/z = 494.2 (M+H+). HPLC (Method A, tR = 5.29 min), purity = 93%.
Supplementary Material
Acknowledgments
This work was supported by NIH Grant P01HL573461 to HS, a University of Michigan Life Sciences Institute Innovation Partnership grant to HS and SDL, and NIH Pharmacological Sciences Training Program fellowships (Grant T32 GM007767) for BDY and MEB. We thank Dr. David Ginsburg for his advice regarding optimization of the SK activity assay.
Abbreviations used
- GAS
Group A Streptococcus (Streptococcus pyogenes)
- SK
streptokinase
- MRSA
methicillin-resistant Staphylococcus aureus
- VRE
vancomycin-resistant Enterococcus
- HTS
high-throughput screening
- SAR
structure-activity relationship
- MLM
mouse liver microsomal extract
- CID
collision-induced dissociation
- ESI
electrospray ionization
- EPI
enhanced product ion scan
- MRM
multiple reaction monitoring
- EMS
enhanced mass spectrometry
- DMEM
Dulbecco’s modified Eagle’s medium
- THY
Todd-Hewitt media with 0.2% yeast extract
Footnotes
Compound 1 showed attenuated efficacy in the presence of 10% HSA compared to the standard assay conditions. This effect was less pronounced for the more hydrophilic compound 12g (data not shown).
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Carapetis JR, Steer AC, Mulholland EK, Weber M. The global burden of group A streptococcal diseases. Lancet Infect Dis. 2005;5(11):685–694. doi: 10.1016/S1473-3099(05)70267-X. [DOI] [PubMed] [Google Scholar]
- 2.World Health Organization. World Health Organization Discussion Papers on Child Health. 2005. The current evidence for the burden of group A streptococcal diseases. [Google Scholar]
- 3.O’Loughlin RE, Roberson A, Cieslak PR, Lynfield R, Gershman K, Craig A, Albanese BA, Farley MM, Barrett NL, Spina NL, Beall B, Harrison LH, Reingold A, Beneden CV, Team ABCS. The Epidemiology of Invasive Group A Streptococcal Infection and Potential Vaccine Implications: United States, 2000–2004. Clin Infect Dis. 2007;45(7):853–862. doi: 10.1086/521264. [DOI] [PubMed] [Google Scholar]
- 4.D’Agata EM. Rapidly rising prevalence of nosocomial multidrug-resistant, Gram-negative bacilli: a 9-year surveillance study. Infect Control Hosp Epidemiol. 2004;25(10):842–6. doi: 10.1086/502306. [DOI] [PubMed] [Google Scholar]
- 5.Archibald L, Phillips L, Monnet D, McGowan JE, Jr, Tenover F, Gaynes R. Antimicrobial resistance in isolates from inpatients and outpatients in the United States: increasing importance of the intensive care unit. Clin Infect Dis. 1997;24(2):211–5. doi: 10.1093/clinids/24.2.211. [DOI] [PubMed] [Google Scholar]
- 6.Rasko DA, Sperandio V. Anti-virulence strategies to combat bacteria-mediated disease. Nat Rev Drug Discov. 2010;9(2):117–128. doi: 10.1038/nrd3013. [DOI] [PubMed] [Google Scholar]
- 7.Karginov VA, Nestorovich EM, Yohannes A, Robinson TM, Fahmi NE, Schmidtmann F, Hecht SM, Bezrukov SM. Search for cyclodextrin-based inhibitors of anthrax toxins: Synthesis, structural features, and relative activities. Antimicrob Agents Ch. 2006;50(11):3740–3753. doi: 10.1128/AAC.00693-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chorell E, Pinkner JS, Bengtsson C, Banchelin TSL, Edvinsson S, Linusson A, Hultgren SJ, Almqvist F. Mapping pilicide anti-virulence effect in Escherichia coli, a comprehensive structure-activity study. Bioorg Med Chem. 2012;20(9):3128–3142. doi: 10.1016/j.bmc.2012.01.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Borlee BR, Geske GD, Blackwell HE, Handelsman J. Identification of Synthetic Inducers and Inhibitors of the Quorum-Sensing Regulator LasR in Pseudomonas aeruginosa by High-Throughput Screening. Appl Environ Microb. 2010;76(24):8255–8258. doi: 10.1128/AEM.00499-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Marsden DM, Nicholson RL, Skindersoe ME, Galloway WRJD, Sore HF, Givskov M, Salmond GPC, Ladlow M, Welchd M, Spring DR. Discovery of a quorum sensing modulator pharmacophore by 3D small-molecule microarray screening. Org Biomol Chem. 2010;8(23):5313–5323. doi: 10.1039/c0ob00300j. [DOI] [PubMed] [Google Scholar]
- 11.Rasko DA, Moreira CG, Li DR, Reading NC, Ritchie JM, Waldor MK, Williams N, Taussig R, Wei SG, Roth M, Hughes DT, Huntley JF, Fina MW, Falck JR, Sperandio V. Targeting QseC signaling and virulence for antibiotic development. Science. 2008;321(5892):1078–1080. doi: 10.1126/science.1160354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Moayeri M, Robinson TA, Leppla SH, Karginov VA. In vivo efficacy of beta-cyclodextrin derivatives against anthrax lethal toxin. Antimicrob Agents Ch. 2008;52(6):2239–2241. doi: 10.1128/AAC.00009-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wu H, Song Z, Hentzer M, Andersen JB, Molin S, Givskov M, Hoiby N. Synthetic furanones inhibit quorum-sensing and enhance bacterial clearance in Pseudomonas aeruginosa lung infection in mice. J Antimicrob Chemoth. 2004;53(6):1054–1061. doi: 10.1093/jac/dkh223. [DOI] [PubMed] [Google Scholar]
- 14.Sun H, Ringdahl U, Homeister JW, Fay WP, Engleberg NC, Yang AY, Rozek LS, Wang X, Sjobring U, Ginsburg D. Plasminogen is a critical host pathogenicity factor for group A streptococcal infection. Science. 2004;305(5688):1283–6. doi: 10.1126/science.1101245. [DOI] [PubMed] [Google Scholar]
- 15.Sun H, Xu Y, Sitkiewicz I, Ma Y, Wang X, Yestrepsky BD, Huang Y, Lapadatescu MC, Larsen MJ, Larsen SD, Musser JM, Ginsburg D. Inhibitor of streptokinase gene expression improves survival after group A streptococcus infection in mice. Proc Natl Acad Sci U S A. 2012;109(9):3469–74. doi: 10.1073/pnas.1201031109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Markosyan AI, Kuroyan RA, Lilanyan SV. Synthesis and transformation of 4-amino-3-ethoxycarbonyl-1,2-dihydrospiro(naphthalene-2,1′-cyclopentane) Khim Geterotsikl. 1998;(6):820–823. [Google Scholar]
- 17.Markosyan AI, Kuroyan RA, Karapetyan KV. Synthesis of 2-alkylthio-substituted 3-p-tolyl-4-oxo-3,4,5,6-tetrahydrospiro(benzo [h]quinazoline-5,1′-cycloalkanes) Khim Geterotsikl. 1999;(12):1655–1658. [Google Scholar]
- 18.Boehm TL, Christie LC, Devadas B, Madsen HM, Marrufo L, Selness S. World Intellectual Property Organization Patent WO2006040666. Substituted N-alkylpyrimidinones. 2006 Apr 20;
- 19.Chong JA, Abdesaken F, Wu CC. US Patent 5,965,766. Process for synthesizing benzoic acids. 1999 Oct 12;
- 20.Kobayashi K, Uneda T, Takada K, Tanaka H, Kitamura T, Morikawa O, Konishi H. Efficient synthesis of 1-amino-2-naphthalenecarboxylic acid derivatives via a sequential Michael addition enolate-nitrile coupling route and its application to facile preparation of 9-amino analogues of arylnaphthofuranone lignans. J Org Chem. 1997;62(3):664–668. doi: 10.1021/jo961744r. [DOI] [PubMed] [Google Scholar]
- 21.Peifer C, Abadleh M, Bischof J, Hauser D, Schattel V, Hirner H, Knippschild U, Laufer S. 3,4-Diaryl-isoxazoles and -imidazoles as Potent Dual Inhibitors of p38 alpha Mitogen Activated Protein Kinase and Casein Kinase 1 delta. J Med Chem. 2009;52(23):7618–7630. doi: 10.1021/jm9005127. [DOI] [PubMed] [Google Scholar]
- 22.Markosyan AI, Kuroyan RA, Dilanyan SV, Aleksanyan MS, Karapetyan AA, Struchkov YT. Synthesis and structure of 2-methyl-6-oxo-7,8-dihydrospiro(benzo[h]triazolo[3,4-b]quinazoline-7,1′-cyclopentane) Khim Geterotsikl. 2000;(5):658–662. [Google Scholar]
- 23.Craig BA. Modeling approach to diameter breakpoint determination. Diagn Micr Infec Dis. 2000;36(3):193–202. doi: 10.1016/s0732-8893(99)00130-3. [DOI] [PubMed] [Google Scholar]
- 24.Ghaemmaghami S, Huh W, Bower K, Howson RW, Belle A, Dephoure N, O’Shea EK, Weissman JS. Global analysis of protein expression in yeast. Nature. 2003;425(6959):737–741. doi: 10.1038/nature02046. [DOI] [PubMed] [Google Scholar]
- 25.Lipinski CA. Drug-like properties and the causes of poor solubility and poor permeability. J Pharmacol Toxicol. 2000;44(1):235–249. doi: 10.1016/s1056-8719(00)00107-6. [DOI] [PubMed] [Google Scholar]
- 26.Anwar H, Strap JL, Costerton JW. Establishment of Aging Biofilms - Possible Mechanism of Bacterial-Resistance to Antimicrobial Therapy. Antimicrob Agents Ch. 1992;36(7):1347–1351. doi: 10.1128/aac.36.7.1347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Christensen GD, Simpson WA, Younger JJ, Baddour LM, Barrett FF, Melton DM, Beachey EH. Adherence of Coagulase-Negative Staphylococci to Plastic Tissue-Culture Plates - a Quantitative Model for the Adherence of Staphylococci to Medical Devices. J Clin Microbiol. 1985;22(6):996–1006. doi: 10.1128/jcm.22.6.996-1006.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ma Y, Xu Y, Yestrepsky BD, Sorenson RJ, Chen M, Larsen SD, Sun H. Novel inhibitors of Staphylococcus aureus virulence gene expression and biofilm formation. PloS One. 2012;7(10):e47225. doi: 10.1371/journal.pone.0047255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Musser JM, Hauser AR, Kim MH, Schlievert PM, Nelson K, Selander RK. Streptococcus-Pyogenes Causing Toxic-Shock-Like Syndrome and Other Invasive Diseases - Clonal Diversity and Pyrogenic Exotoxin Expression. Proc Natl Acad Sci USA. 1991;88(7):2668–2672. doi: 10.1073/pnas.88.7.2668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Engleberg NC, Heath A, Miller A, Rivera C, DiRita VJ. Spontaneous mutations in the CsrRS two-component regulatory system of Streptococcus pyogenes result in enhanced virulence in a murine model of skin and soft tissue infection. J Infect Dis. 2001;183(7):1043–1054. doi: 10.1086/319291. [DOI] [PubMed] [Google Scholar]
- 31.Khil J, Im M, Heath A, Ringdahl U, Mundada L, Engleberg NC, Fay WP. Plasminogen enhances virulence of group A streptococci by streptokinase-dependent and streptokinase-independent mechanisms. J Infect Dis. 2003;188(4):497–505. doi: 10.1086/377100. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.