

Summary
Multiple sclerosis is the most common cause of non-traumatic neurological impairment in young adults. An energy deficient state has been implicated in the degeneration of axons, the pathological correlate of disease progression, in multiple sclerosis. Mitochondria are the most efficient producers of energy and play an important role in calcium homeostasis. We analysed the density and function of mitochondria using immunohistochemistry and histochemistry, respectively, in chronic active and inactive lesions in progressive multiple sclerosis. As shown before in acute pattern III and Balo’s lesions, the mitochondrial respiratory chain complex IV activity is reduced despite the presence of mitochondria in demyelinated axons with amyloid precursor protein accumulation, which are predominantly located at the active edge of chronic active lesions. Furthermore, the strong non-phosphorylated neurofilament (SMI32) reactivity was associated with a significant reduction in complex IV activity and mitochondria within demyelinated axons. The complex IV defect associated with axonal injury may be mediated by soluble products of innate immunity, as suggested by an inverse correlation between complex IV activity and macrophage/microglial density in chronic lesions. However, in inactive areas of chronic multiple sclerosis lesions the mitochondrial respiratory chain complex IV activity and mitochondrial mass, judged by porin immunoreactivity, are increased within approximately half of large (>2.5 μm diameter) chronically demyelinated axons compared with large myelinated axons in the brain and spinal cord. The axon-specific mitochondrial docking protein (syntaphilin) and phosphorylated neurofilament-H were increased in chronic lesions. The lack of complex IV activity in a proportion of Na+/K+ ATPase α-1 positive demyelinated axons supports axonal dysfunction as a contributor to neurological impairment and disease progression. Furthermore, in vitro studies show that inhibition of complex IV augments glutamate-mediated axonal injury (amyloid precursor protein and SMI32 reactivity). Our findings have important implications for both axonal degeneration and dysfunction during the progressive stage of multiple sclerosis.
Keywords: Mitochondria, axonal degeneration, multiple sclerosis
Introduction
Multiple sclerosis is the most common non-traumatic neurological disease among young adults in Europe and North America (Compston, 2005). Over two-thirds of patients develop progressive deterioration in neurological function. Whilst currently available immunomodulatory therapy is effective in reducing the rate of relapses in the disease, its progression remains unaltered by such agents (Ebers et al., 2008). The degeneration of demyelinated axons is considered as the pathological correlate of disease progression (Dutta and Trapp, 2007). The recently identified changes in electrogenic machinery in chronically demyelinated axons suggest that axonal dysfunction, a well recognized and positively reversible entity during relapsing remitting phase, may also contribute to the disability in the late stages of multiple sclerosis (Black et al., 2007; Waxman, 2008; Young et al., 2008).
Following demyelination and subsequent loss of saltatory conduction, the axonal membrane undergoes a number of changes including an increase in number of sodium channels within the demyelinated part of the axon (Craner et al., 2004; Waxman, 2006a; Black et al., 2007; Smith, 2007). The maintenance of intra-axonal ionic balance and resting membrane potential following the influx of sodium through the increased sodium channels relies on the largest consumer of energy in the central nervous system, Na+/K+ ATPase (Caldwell et al., 1960; Ames, 2000). In non-inflammatory environments this increase in energy demand of axons lacking a healthy myelin sheath is apparent by the changes in density and activity of energy producing organelles (mitochondria) in shiverer mice with a defect in the myelin basic protein (MBP) gene as well as unmyelinated segments of retinal ganglion cell axons in the lamina cribrosa of control optic nerves (Bristow et al., 2002; Barron et al., 2004; Andrews et al., 2006).
Mitochondria are the most efficient producers of ATP and play a role in apoptosis, reactive oxygen species generation and calcium buffering (DiMauro and Schon, 2003; Andreyev et al., 2005; Rizzuto and Pozzan, 2006). An imbalance of intra-axonal calcium appears to be involved in the degeneration of axons following a number of insults including ischaemia, trauma and possibly inflammation (Waxman et al., 1992; Coleman, 2005; Stys, 2005). The mitochondrial respiratory chain located in the inner mitochondrial membrane consists of four complexes (complexes I–IV) and complex V, which is directly involved in ATP synthesis (DiMauro and Schon, 2003; Taylor and Turnbull, 2005). The terminal complex of mitochondrial respiratory chain, complex IV or cytochrome c oxidase (COX), is where over 90% of oxygen is converted to water. Complex IV is targeted and can be irreversibly inhibited by nitric oxide, found in multiple sclerosis lesions, depending on its concentration and duration of exposure (Smith and Lassmann, 2002). In inflammatory environments, the activity of mitochondrial respiratory chain complexes may be impaired through reactive oxygen species-mediated post-transcriptional modification and nitration of the subunits as well as damage to the only non-nuclear DNA, or mitochondrial DNA, also to be present within axons (Lu et al., 2000; Wei et al., 2002; Qi et al., 2006). Defects of mitochondrial respiratory chain complexes and depletion of mitochondria not only cause an energy deficit but may increase the susceptibility of axons to excitotoxic injury through impaired calcium handling capacity (Rizzuto and Pozzan, 2006; von Kleist-Retzow et al., 2007).
There is increasing evidence implicating mitochondria in the pathogenesis of multiple sclerosis. Mitochondrial respiratory chain complex I and complex III activity is reduced in non-lesional motor cortex, where a number of nuclear DNA-encoded transcripts of mitochondrial proteins are decreased (Dutta et al., 2006). There is oxidative damage to mitochondrial DNA in chronic active white matter lesions, with an associated decrease in complex I activity (Lu et al., 2000). Interestingly, complex IV activity and mitochondrial DNA copy number are increased in chronic active lesion homogenates and within normal appearing grey matter neurons, respectively, possibly as a compensatory mechanism (Lu et al., 2000; Blokhin et al., 2008). We recently identified a defect in the main catalytic subunit of complex IV, COX-I, within acutely demyelinated axons in pattern III and Balo’s type MS lesions (Mahad et al., 2008). In this study we analysed mitochondrial density and complex IV activity in active and inactive lesions in chronic (progressive) multiple sclerosis. Our data suggest that the deficiency of complex IV activity may play a major role in axonal degeneration in active lesions in this condition. However, the mitochondrial mass and complex IV activity are increased in inactive demyelinated lesions, possibly reflecting a compensatory reaction to the increased energy demand of chronically demyelinated axons.
Methods
Autopsy tissue and classification of lesions
The brain and spinal cord tissue blocks, snap frozen in isopentane and stored at −70°C, from multiple sclerosis cases and controls were obtained from the UK multiple sclerosis tissue resource, London and Newcastle Brain Bank, UK (Table 1). Sixteen blocks with macroscopic lesions from posterior frontal lobe of nine multiple sclerosis cases and six blocks with macroscopic lesions from cervical and/or thoracic spinal cord of five multiple sclerosis cases were characterized based upon distribution of Luxol Fast Blue (LFB) and staining for proteolipidprotein and HLA-D expression in cryostat sections (10 μm thickness). The chronic active or slowly expanding and chronic inactive lesions were identified as previously described (van der Valk and De Groot, 2000). The cause of death for multiple sclerosis cases was pneumonia (n = 7), cardiac failure (n = 1) or sepsis (n = 2) and for controls was either cardiorespiratory failure/arrest or pneumonia. The Newcastle and North Tyneside Local Research Ethics Committee approved the study (205/Q0906/182).
Table 1.
Details of autopsy cases
| Case number |
Age (years)/sex |
Diagnosis | Duration of disease (years) |
PM interval (h) |
Number of spinal cord lesions |
Number of brain lesions |
|---|---|---|---|---|---|---|
| MS1 | 64/F | SP | 36 | 7 | NA | 3 |
| MS2 | 71/F | SP | 34 | 11 | NA | 1 |
| MS3 | 42/F | PP | 6 | 7 | 1 | 2 |
| MS4 | 57/M | SP | 33 | 8 | NA | 2 |
| MS5 | 81/F | SP | 41 | 10 | 1 | NA |
| MS6 | 46/M | SP | 8 | 8 | NA | 2 |
| MS7 | 73/M | SP | 52 | 10 | NA | 1 |
| MS8 | 53/M | SP | 11 | 12 | 1 | 1 |
| MS9 | 39/F | SP | 21 | 8 | 2 | 2 |
| MS10 | 40/M | SP | 9 | 10 | 1 | 2 |
| CON1 | 75/M | – | – | 6 | NAWM | NAWM |
| CON2 | 88/F | – | – | 12 | NAWM | NAWM |
| CON3 | 65/F | – | – | 10 | NAWM | NAWM |
| CON4 | 69/F | – | – | 7 | NAWM | NAWM |
| CON5 | 66/M | – | – | 8 | NA | NAWM |
| CON6 | 63/F | – | – | 11 | NA | N |
CON = control; MS = multiple sclerosis; NA = not applicable as either spinal cord or brain tissue was not available; NAWM = normal appearing white matter; SP = secondary progressive; PP = primary progressive.
Immunohistochemistry
The cryostat sections (10 μm thickness) from snap frozen tissue blocks were air dried for 60 min. The sections were briefly (5 min) fixed in 2% paraformaldehyde prior to antigen retrieval using EDTA for 1 min, incubation in 2% hydrogen peroxide, blocking in normal goat serum and immunostaining using avidin-biotin-horseradish peroxidase complex (ABC) and 3,3-diaminobenzidine as previously described (Table 2).
Table 2.
Details of antibodies used for immunohistochemistry and Western blots
| Antigen | Target | Antibody type | Source |
|---|---|---|---|
| PLP | Proteolipid protein | Mouse IgG | Serotec |
| HLA-DP, DQ, DR |
Human leukocyte antigen | Mouse IgG1 | Dako |
| SMI31 | Phosphorylated neurofilaments | Mouse IgG1 | Vector Laboratories |
| SMI32 | Non-phosphorylated neurofilaments |
Mouse IgG1 | Vector Laboratories |
| TNF | Total neurofilaments | Rabbit polyclonal | Millipore |
| β-APP | Amyloid precursor protein | Mouse IgG1/rabbit polyclonal |
Millipore |
| Syntaphilin | Syntaphilin | Rabbit polyclonal | Santa Cruz Biotechnology |
| Porin | Mitochondrial transmembrane protein |
Mouse IgG2b | Molecular probes, Invitrogen |
| SDHA II | SDH 70 kDa subunit | Mouse IgG1 | Molecular probes, Invitrogen |
| β-Tubulin | Class III β-tubulin | Rabbit Polyclonal | Covance |
| β-Actin | β-Actin | Mouse IgG1 | Sigma |
| Na+/K+ATPase | Na+/K+ATPase α-1 | Mouse IgG1 | Upstate biotechnology |
For double and triple immunofluorescent labelling, the snap frozen sections were prepared as for immunohistochemistry, without hydrogen peroxide, and subclass specific secondary antibodies directly conjugated with fluorochromes (Alexa fluor 488, 568 or 633) were used from molecular probes (Invitrogen). The appropriate controls were performed to exclude cross-reactivity and non-specific binding in double- and triple-labelling experiments.
COX and SDH histochemistry
Blocks of brain and spinal cord, snap frozen in isopentane and stored at −70°C, were mounted for cryostat sectioning as for immunohistochemistry and the serial sections (10 μm thickness) were air dried for 30 min and incubated in COX medium (100 mM cytochrome c, 4 mM diaminobenzidine tetrahydrochloride and 20 mg/ml catalase in 0.2 M phosphate buffer, pH 7.0) at 37°C for 55 min (Clark et al., 1997; Mahad et al., 2008). Following COX histochemistry, axons and mitochondria were identified using double immunofluorescent histochemistry, as described above, in the same section. To detect complex II activity serial cryostat sections were incubated in SDH medium (130 mM sodium succinate, 200 μM phenazine methosulphate, 1 mM sodium azide, 1.5 mM nitroblue tetrazolium in 0.2 M phosphate buffer, pH 7.0) at 37°C for 40 min, once the sections were washed following incubation in COX medium for 50 min. For the identification of complex IV activity within axons, the sections were immunofluorescently labelled using antibodies against neurofilaments and amyloid precursor protein, as described above (Table 2), following completion of COX histochemistry. The stained sections were dehydrated in 70%, 90% and 100% ethanol prior to mounting in aqueous medium.
COX electron microscopy
Immediately following tissue harvest, small pieces of tissue with ~1 mm thickness were dissected from the spinal cord lesions and normal appearing white matter and fixed in 4% glutaraldehyde for 15 min. To determine complex IV activity the pieces of tissue were then washed in phosphate buffer prior to incubation in COX medium (100 mM cytochrome c, 4 mM diaminobenzidine tetrahydro-chloride and 20 mg/ml catalase in 0.2 M phosphate buffer, pH 7.0) at 37°C for 90 min. The pieces of tissue were washed in phosphate buffer, fixed with 4% glutaraldehyde overnight at 4°C and then post-fixed with 1% osmium tetroxide in 0.1 M phosphate overnight (Kirkinezos et al., 2005). The samples were dehydrated in a series of acetone prior to embedding in Epoxy resin. Ultrathin sections without any further staining were examined with a Philips CM100 EM (Biomedical EM unit, Newcastle University).
Western blotting
To prepare homogenates, white matter from CON (n = 6), normal appearing white matter (n = 6) and chronic inactive multiple sclerosis lesions (n = 6) from posterior frontal cortex was carefully removed from cryosectioned slides (20 μm thickness) using a scalpel and homogenized in buffer containing 250 mM sucrose, 2 mM HEPES and 0.1 mM EDTA. Samples were diluted in sample buffer (0.5 M Tris–HCl buffer, pH 6.8, 20% Glycerol, 4%-SDS, 0.1% Bromophenol blue) containing 2% β-mercaptoethanol, incubated for 30 min at 37°C and separated on a 15% SDS polyacrylamide gel at 150 V. Proteins were transferred electrophoretically to 0.45 μm polyvinylidene fluoride membranes (Millipore Corporation, USA) for 3 h at 300 mA in CAPS buffer (2.2 g CAPS, 900 ml of 10% methanol, pH 11.0) at 4°C. The membranes were blocked overnight in 5% milk-TTBS and incubated for 90 min with primary antibodies against β-actin, porin, phosphorylated as well as total neurofilaments, syntaphilin and complex II 70 kDa subunit (Table 2). After three washes with TTBS, the membranes were incubated for 90 min with horseradish peroxidise-conjugated polyclonal rabbit anti-mouse antibody (DAKO, Denmark) diluted in 5% milk-TTBS. The secondary antibody was detected using ECL Plus Western Blotting detection system (GE Healthcare, UK). The signal was visualized using STORM 860 scanner (GE Healthcare, UK) and the peak heights of bands were determined using Image Quant TL software (GE Healthcare, UK).
Neuron culture
We used neurons differentiated from a wild-type mouse embryonic stem cell line CC9.3.1 (gift of Dr Allan Bradley, Sanger Centre, Cambridge, UK). Undifferentiated mouse embryonic stem cells were maintained on 0.1% gelatine coated flasks (Greiner Bio-One) in Glasgow modified Eagle’s medium (Invitrogen), 10% foetal calf serum (Biosera) supplemented with 2 mM L-glutamine (Invitrogen), 1 mM sodium pyruvate, 1% non-essential amino acids (Invitrogen) and 0.1 mM 2-mercaptoethanol (Invitrogen). Embryoid bodies were generated by transferring mouse embryonic stem cells onto Petri dishes in mouse embryonic stem medium, which was changed every 2 days (Day 2, Day 4, Day 6 and Day 8). A total of 1 μMtrans-retinoic acid (Sigma) was added at Day 4 and Day 6 of embryoid body formation. At Day 8, embryoid bodies were trypsinized and cells were plated on 6-well tissue culture plates containing coverslips coated with 10 μg/ml poly-D-lysine (Sigma) and 2 μg/ml laminin (Sigma) at a density of 8 × 105 cells per well. The cells were cultured in Neurobasal medium + bFGF (Invitrogen), Dulbecco’s modified Eagle’s medium (Sigma), 1.6% B27 (Invitrogen), 1 mM sodium pyruvate, 2 mM L-glutamine (Invitrogen), 1% non-essential amino acids (Invitrogen), 0.2% N2 (Invitrogen) and 10 ng/ml of bFGF (Peprotech) for 2 days. On Day 3, the medium was replaced by Neurobasal medium without bFGF and cell cultured for further 4 days with media changed every 2 days. The experiments were performed on 15 days old mature neurons. Less than 5% of the population consisted of GFAP-positive (astrocytes) or OX-42 positive (microglia) cells.
The mature neurons were exposed to a range of concentrations of sodium azide (1, 10 and 100 μM), a specific inhibitor of complex IV, alone or glutamate alone (1, 10 and 100 μM) for 1, 2, 4, 8 and 16 h. Furthermore, in parallel experiments glutamate in varying concentrations (1, 10 and 100 μM) was added to mature neurons for the last 1 h of sodium azide exposure at each time point. Using ethidium homodimer exclusion method, previously described, we determined a sublethal concentration of sodium azide and glutamate, either in isolation or together, as 10 μM of sodium azide alone for 2 h, 100 μM of glutamate alone for 1 h and the addition of 100 μM glutamate to the second 1 h of the 2 h 10 μM sodium azide exposure. The experiments were repeated on four separate occasions for each time point and the cover slips were immediately fixed in 2% paraformaldehyde for 10 min prior to immunofluorescent double labelling with β-tubulin and APP or non-phosphorylated NF (SMI32). The number of ethidium homodimer positive cells, judged by fluorescent microscopy, was <1% and not significantly different at these sublethal concentrations and exposure times. The exposure of neuron cultures to 10 mM sodium azide inhibited complex IV activity by 36% as determined using spectrophotometry and when corrected for citrate synthase activity (Taylor and Turnbull, 1997).
Microscopy
For the analysis of complex IV activity within axons, firstly, bright field grey scale images of COX histochemistry were obtained using a 100× oil lens and, secondly, immunofluorescent double labelling of axons (SMI31, SMI32 or APP) was captured in exactly the same field and plane of focus (Zeiss Axioplan) in each area. In brain lesions, all large axons (>2.5 μm in diameter and at least 50 μm in length) identified by immunofluorescent labelling were captured whereas in the spinal cord three adjacent 100× images perpendicular to the longitudinal axis of axons were captured in each area due to the relative mass of longitudinally sectioned phosphorylated neurofilament (SMI31) positive axons compared with the posterior frontal lobe sections. For the analysis of complex IV activity to correlate with microglial/macrophage density, four bright field grey scale images of COX histochemical and HLA immunohistochemical staining per area were obtained at 20× magnification in serial sections. The exposure time of grey scale images were kept constant between areas as well as sections. The myelinated and demyelinated areas were located based on the lesion classification in serial sections.
For the analysis of mitochondrial mass within axons in tissue sections, triple-labelled sections with porin, syntaphilin and axonal marker (SMI31, SMI32 or APP) were imaged using a 63× oil lens and Leica laser scanning microscope (Heidelberg, Germany). One micrometre thick individual optical sections were taken through the tissue sections (10 μm). The minimum number of optical sections was combined to track longitudinally sectioned axons. FITC, TRIC and Cy5 channels were imaged sequentially and the offset and gain were kept constant during imaging of all sections. A x–z optical section was taken through each large axon (>2.5 μm in diameter and at least 50 μm in length) to ensure the axonal localization of porin and syntaphilin reactive elements. All large axons in brain sections and three adjacent sections perpendicular to the longitudinal axis of axons in spinal cord were captured for each area.
In neuron cultures, axons were identified with immunofluorescently labelled β-tubulin (Rhodamine) and imaged using a 63 × oil lens (Zeiss Axiovert microscope). Fifty β-tubulin positive axons (>100 μm in length) were imaged in adjacent fields per cover slip with the SMI32 (FITC) or amyloid precursor protein (FITC) immunofluorescent labelling related to β-tubulin positive axons captured by keeping the exposure time for FITC images constant.
Quantitation of axonal complex IV activity and mitochondrial mass
To identify the complex IV active elements within large axons, the outline of the immunofluorescently labelled axons (SMI31, SMI32 or APP) was superimposed on the bright field image of COX histochemistry. The area occupied by complex IV active elements was determined as a percentage of the total area of the large (>2.5 μm in diameter and at least 50 μm in length) morphologically intact (without terminal ovoids or beading) axons. The complex IV active elements within axons were subjected to further analysis with respect to their intensity using densitometry and Zeiss Axiovision version 4.6. Densitometric analysis of COX histochemical product is a valid means of assessing complex IV activity in tissue sections (Hevner et al., 1995; Mahad et al., 2008). For the correlation of complex IV activity with microglial/macrophage density, the densitometric value of entire 20× grey scale images of COX histochemical staining was determined with the densitometric value of normal appearing white matter, as an internal control, subtracted from the corresponding value of lesion.
In confocal images, the area of porin reactive elements within x–y images of large axons, as a percentage of axonal area, was determined. The axonal location of porin elements was confirmed in x–z images. The myelinated and demyelinated axons were identified based on the lesion classification in serial sections. The selection of long (50 μm or more) axonal segments were based on establishing morphological integrity as well as minimizing the identification of false positive segments lacking mitochondrial activity or elements because of the naturally discontinuous distribution of mitochondrial elements and variability in inter-mitochondrial distance within axons.
In neuron culture, the fluorescence intensity of FITC-labelled amyloid precursor protein (APP) or SMI32 related to the β-tubulin positive unmyelinated axons (Rhodamine) was densitometrically determined in 50 axons per coverslip for each of four separate experiments. The mean densitometric value over the outline of 100 μm or longer β-tubulin positive axonal segments was used to quantitate SMI32 and amyloid precursor protein immunofluorescent intensity. The background densitometric value in bright field images for complex IV activity and immunofluorescent images for amyloid precursor protein and SMI32 reactivity was determined and subtracted from the axonal densitometric values in order to correct for variation in the background intensity between sections.
Statistics
Parametric tests and SPSS version 14 were used for comparison of complex IV activity and porin immunoreactivity between demyelinated and myelinated axons as well as unmyelinated axons exposed to sodium azide and glutamate, in vitro.
Results
As mitochondrial defects are considered important in the pathogenesis of multiple sclerosis lesions and axonal degeneration (Lu et al., 2000; Dutta et al., 2006) we studied mitochondrial activity and mass in chronic active and inactive lesions. We then investigated the implications of mitochondrial dysfunction, in particular complex IV, for nerve impulse transmission and axonal viability.
A complex IV defect is present in amyloid precursor protein and SMI32 immunoreactive injured demyelinated axons
We studied mitochondrial respiratory chain complex IV activity in relation to axonal injury. As previously described we detected a number of amyloid precursor protein or SMI32 reactive injured axons in active and, to a lesser extent, inactive areas of chronic lesions (Fig. 1) (Ferguson et al., 1997; Trapp et al., 1998; Kornek et al., 2000). The linear segments of amyloid precursor protein and SMI32 positive demyelinated axons without signs of acute transection or terminal ovoids in the brain and spinal cord multiple sclerosis tissue are deficient in complex IV activity, which is not due to the absence of mitochondria (Fig. 1F–I). The complex IV defect in injured axons in chronic lesions was confirmed by the quantitation of the area occupied by as well as the intensity of complex IV active elements (Fig. 2 and Table 3). Within amyloid precursor protein positive axons complex IV activity was significantly lower compared with SMI31 positive myelinated and demyelinated axons in control white matter and inactive areas of chronic lesions, respectively. Furthermore, there was a significant reduction in complex IV activity in SMI32 compared with SMI31 positive chronically demyelinated axons in inactive areas of chronic lesions (Fig. 2 and Table 3). While axonal mitochondrial mass determined by porin reactivity was unchanged in amyloid precursor protein positive axons, SMI32 reactive chronically demyelinated axons revealed a significant decrease in mitochondrial content (Table 3). These data are consistent with a complex IV defect in amyloid precursor protein positive demyelinated axons, whereas the lack of complex IV activity in SMI32 relative to SMI31 reactive chronically demyelinated axons appears to be partly due to mitochondrial depletion.
Figure 1.

Complex IV activity within injured demyelinated axons. (A–E) In the active rim of chronic active multiple sclerosis lesions, identified by the loss of Luxol Fast Blue (A) and presence of MHC class II positive cells (B) in serial sections, the complex IV activity is at an intermediate level compared with normal appearing white matter and relatively inactive lesion centre (C). Surprisingly complex IV activity is increased in the inactive area of chronic lesions. There are a number of ovoid shaped structures containing intense complex IV activity in the active rim (C and insert). When the same serial section used for COX histochemistry (C) is immunofluorescently labelled for amyloid precursor protein (APP) (D), the complex IV active ovoid structures are APP-positive (D, E). However, not all APP reactive ovoids show complex IV activity (red, E). (F–I) The APP-positive linear segments of demyelinated axons without terminal ovoids (red in F and G) in chronic lesions are devoid of complex IV activity, brown punctate elements apparent at 100× magnification (F, arrows). The lack of complex IV activity in APP-positive demyelinated axons is not due to the lack of mitochondria as shown by the presence of porin reactive elements (green) in x–y and x–z confocal images (G arrows and inserts). In a separate chronic lesion, the immunofluorescent labelling of non-phosphorylated neurofilaments (SMI32) identifies an injured demyelinated axon (red in H and I) which also lacks complex IV active elements (H, arrowheads). The SMI32 (red) reactive axon lacking complex IV active elements contain numerous porin (green) reactive elements (I, arrowheads). x– y and x–z; confocal images of APP and porin immunoreactivity in x–y and x–z planes. Asterisk indicates inactive area of chronic multiple sclerosis lesion.
Figure 2.

Quantitation of complex IV activity within axons in brain and spinal cord. (A, B) The percentage area of complex IV active elements within large (>2.5 μm) axons indicates a significant decrease in complex IV activity within acutely injured APP positive axons in brain (A) and spinal cord (B) multiple sclerosis lesions compared with SMI31 positive myelinated axons. Furthermore, SMI32 positive injured demyelinated axons from inactive areas of chronic lesions contain significantly less complex IV activity compared with SMI31 positive demyelinated axons from the same area. The complex IV activity within SMI31 positive chronically demyelinated axons are significantly greater compared with myelinated axons in normal appearing white matter and control white matter of brain and spinal cord. The SMI31 positive myelinated axons in normal appearing white matter contain significantly greater complex IV activity compared with SMI31 positive myelinated axons in control white matter in brain (A) but not spinal cord (B). The dotted line indicates the upper range of 95% confidence interval based on complex IV activity in myelinated axons from control white matter. The number of large axons analysed is indicated within parentheses. The values are means ± SD of percentage area of complex IV active elements within large axons. *P < 0.001.
Table 3.
Quantitation of the intensity of complex IV active elements and mitochondrial mass within axons
|
|
|||||
|---|---|---|---|---|---|
| Control (SMI31) | NAWM (SMI31) |
Lesion (APP) |
Lesion (SMI32) |
Lesion (SMI31) |
|
|
|
|||||
| Complex IV (intensity) brain |
17.93 ± 5.89 (76) |
16.14 ± 6.54 (76) |
9.85 ± 6.94# (37) |
10.26 ± 5.43# (58) |
20.10 ± 7.38†,‡ (98) |
| Complex IV (intensity) spinal cord |
16.99 ± 15.49 (105) |
16.87 ± 12.44 (87) |
9.01 ± 11.16# (38) |
15.93 ± 13.01 (49) |
23.04 ± 11.07* (89) |
| Porin (percentage area) brain |
6.74 ± 2.82 (64) |
7.35 ± 4.30 (73) |
7.93 ± 4.50 (32) |
4.19 ± 3.95† (43) |
11.15 ± 5.23* (92) |
| Porin (percentage area) spinal cord |
5.93 ± 4.38 (138) |
3.46 ± 3.92 (135) |
8.26 ± 7.40 (45) |
1.64 ± 1.39# (55) |
13.97 ± 7.23* (132) |
The intensity of complex IV active elements represents the difference in densitometric value between background and complex IV active elements in inverted grey scale 100× brightfield images of cytochrome c oxidase or COX histochemistry. The percentage area of porin reactive elements within axons was calculated based on the total area of axonal porin reactive elements in triple labelled (porin, syntaphilin and axonal marker) and area of axons in confocal images.
P = 0.001 (versus NAWM) and
P = 0.002 (versus CON).
P < 0.001 (versus SMI31 in CON, NAWM and lesion).
P < 0.001 (versus SMI31 in NAWM and CON as well as SMI32 in lesion and APP in lesion).
Mitochondrial respiratory chain complex IV activity is increased in chronic inactive areas of multiple sclerosis lesions
To our surprise, histochemistry revealed the activity of mitochondrial respiratory chain complex II and complex IV to be increased particularly in the inactive centre of chronic active and in inactive lesions (Figs 1A–C and 3C–D). The punctate complex IV active elements, typical of mitochondria, were more prominent in terms of density and intensity in chronically demyelinated areas (Fig. 3E and G) compared with normal appearing white matter (Fig. 3F and H). When the complex IV active elements within large axons (diameter >2.5 μm) were identified using histochemistry and immunofluorescent labelling of SMI31, a proportion of demyelinated axons were abundant in mitochondrial elements with intense complex IV activity (Fig. 3I–N). Amyloid precursor protein or strong SMI32 reactivity was not detected in demyelinated axons with abundant complex IV active elements. Furthermore, COX electron microscopy showed mitochondria with greater complex IV activity in demyelinated axons in relatively inactive chronic lesions compared with myelinated axons in the normal appearing white matter (Fig. 4).
Figure 3.

Mitochondrial respiratory chain complex IV and complex II activity in chronic multiple sclerosis lesions. (A–D) Serial sections of a posterior frontal tissue block containing a chronic active multiple sclerosis lesion (A, arrow), identified by the loss of Luxol Fast Blue (LFB, A) and a rim of inflammatory activity (HLA reactivity, B), shows increased activity of mitochondrial respiratory chain complex IV (C) or cytochrome c oxidase (COX) and complex II (D) or SDH throughout the lesion compared with normal appearing white matter. The complexes IV and II active mitochondria are enriched in the cortex (ctx). (E–H) The complex IV active mitochondrial elements are prominent within axons as well as glia in relatively inactive areas of chronic lesions in the brain (E) as well as spinal cord (G) lesions compared with adjacent normal appearing white matter (F and H). (I–N) The chronically demyelinated large diameter (>2.5 μm) axons in brain (outlined in E) and spinal cord (outlined in G), identified by SMI31 reactivity (J and M) within chronic lesions, contain intense complex IV active elements (I and L) as evident in the superimposed images (K and N). Most complex IV active elements are punctate (I and L), typical of mitochondria, but others are elongated aligning the longitudinal axis of axons (I).
Figure 4.

COX electronmicroscopy. (A,B) Electronmicroscopy of chronic multiple sclerosis lesions following COX histochemistry shows mitochondria with dark cristae and membranes abundant in complex IV activity (A, arrows) within demyelinated axons compared with mitochondria (B, arrowheads) in myelinated axons in normal appearing white matter.
The quantitation of the area occupied by the complex IV active elements as a percentage of the total area of large axons confirmed the apparent increase in complex IV activity within SMI31 positive chronically demyelinated axons as significantly different from myelinated axons (Fig. 2). Furthermore, the intensity of complex IV active elements, when determined using densitometry, showed greater complex IV activity per mitochondrial element in chronically demyelinated compared with myelinated SMI31 positive axons (Table 3). Based on the 95% confidence intervals of complex IV activity in myelinated axons in control white matter, 55 and 59% of chronically demyelinated axons in multiple sclerosis brain and spinal cord, respectively, contained the increased complex IV activity (Fig. 2). The increase in complex IV activity in multiple sclerosis tissue appears also to involve the myelinated segments of axons in normal appearing white matter of brain but not spinal cord (Fig. 2). The mean diameter of demyelinated (4.47 μm) and myelinated (4.21 μm) axons analysed was not significantly different.
Mitochondrial mass, syntaphilin and phosphorylated neurofilaments are increased within morphologically intact demyelinated axons in chronic inactive areas of multiple sclerosis lesions
Given the changes in complex IV activity in relation to acute axonal injury and chronic demyelination, we determined the mitochondrial mass in lesions and within axons using western blotting and triple-labelled immunofluorescent histochemistry with monoclonal antibodies against porin, a voltage gated anion channel expressed on the outer membrane of all mitochondria (Dutta et al., 2006; Mahad et al., 2008), syntaphilin—a mitochondrial docking protein expressed specifically within axons—(Kang et al., 2008) and SMI31, respectively (Table 2 and Fig. 5). In chronic lesions, western blotting identified a 1.6-fold increase (n = 5 and P = 0.006) in porin and 3.2-fold increase (n = 4 and P = 0.002) in syntaphilin compared with control white matter from the same area (Fig. 5A–C).
Figure 5.

Mass and docking of axonal mitochondria and phosphorylation status of neurofilaments in chronic multiple sclerosis lesions. (A–C) Mitochondrial mass, judged by porin immunoreactivity, is increased throughout chronic multiple sclerosis lesions, as apparent in a serial section of the lesion in Fig. 1. Western blots of porin, a voltage gated anion channel expressed on all mitochondria, and a subunit of complex II (SDH70 kDa or SDHA), which is entirely encoded by nuclear DNA, shows a 1.6-fold increase in mitochondrial mass in chronic lesions compared with control white matter (n = 5, P = 0.006). (B). Western blots of syntaphilin, an axon specific mitochondrial docking protein, shows a 3.2- and 1.8-fold increase (n = 4 P = 0.002) in chronic lesions compared with normal appearing white matter and controls, respectively (C). (D–I) Porin (green) and syntaphilin (red) reactive elements in triple immunofluorescently labelled confocal images are abundant in chronic lesions (D and E) compared with normal appearing white matter (G and H). The majority of syntaphilin reactive mitochondrial elements are located within SMI31 positive (blue) demyelinated axons (F), whereas myelinated SMI31 positive axons in the normal appearing white matter contain strikingly less porin and syntaphilin reactive elements (I). Interestingly syntaphilin immunoreactivity within demyelinated axons does not always co-localize with porin immunoreactive elements (F). (J, K) Western blots of phosphorylated and total neurofilaments (NF) in chronic lesions show an increase in phosphorylated neurofilament-H in lesions compared with normal appearing white matter and control white matter (J), which is partly due to an increase in total neurofilaments (K). When corrected for β-actin and total neurofilament-H there is a 1.7-fold increase (P = 0.025, n = 6) in phosphorylated neurofilament-H in chronic lesions compared with control white matter. CON WM; control white matter. L = lesion. MS = multiple sclerosis; NAWM or N = normal appearing white matter; NF = neurofilaments.
Numerous mitochondrial elements, which are abundant in chronic lesions, were distributed parallel to the longitudinal axis of axons (Fig. 5D and G). The syntaphilin positive mitochondrial elements were prominent in chronic lesions compared with normal appearing white matter (Fig. 5E and H). As expected syntaphilin positive mitochondrial elements were not present in MHC class II positive macrophages or microglia. The triple labelling of porin, syntaphilin and SMI31 and confocal microscopy showed that the demyelinated axons are abundant in mitochondria relative to myelinated axons (Fig. 5D–I). When the area of axonal porin reactive elements as a percentage of axonal area was determined using confocal microscopy in large axons (>2.5 μm) with at least 50 μm continuous segments, SMI31 positive demyelinated axons in chronic lesions contained a significantly greater mitochondrial mass compared with the myelinated axons (Table 3). Based on the 95% confidence intervals of porin immunoreactivity within myelinated axons in control white matter, 34.8% and 37.1% (32 out of 92 and 49 out of 132) of chronically demyelinated axons in multiple sclerosis brain and spinal cord, respectively, showed the increase in mitochondrial mass.
Mitochondria interact with high molecular weight neurofilament-H, in particular to the phosphorylated side-arms of neurofilament-H (Wagner et al., 2003). The phosphorylation status of neurofilament-H side-arms influences the interaction between neurofilament and mitochondria in a manner that is dependent on the metabolic activity of mitochondria (Wagner et al., 2003). We found a significant increase in phosphorylated neurofilament-H compared with normal appearing white matter (1.83-fold in six cases, P = 0.033) and control (1.86-fold in six cases, P = 0.02) white matter when corrected for β-actin (Fig. 5J). When the same tissue homogenates were used to detect total neurofilament-H, the change in total neurofilament-H in multiple sclerosis lesions (1.35-fold increase in six cases, P = 0.58) compared with normal appearing white matter was not significant (Fig. 5K). The ratio between phosphorylated and total neurofilament-H, both corrected for β-actin, identified a significant increase in the phosphorylation status of neurofilament-H in chronic lesions compared with control white matter (1.70-fold increase in six cases, P = 0.025) and normal appearing white matter (1.34-fold increase in six cases, P = 0.048).
Complex IV activity correlates inversely with microglial and macrophage density in chronic multiple sclerosis lesions
Based on the facts that axonal injury correlates with inflammation, microglia/macrophages are a source of reactive oxygen species (ROS) in multiple sclerosis lesions and complex IV is susceptible to ROS-mediated damage, we determined the density of microglia and macrophages in active (n = 8) and inactive (n = 22) areas of chronic lesions and correlated with complex IV activity in serial sections. There was a significant inverse correlation between the density of microglia/macrophages and global (axonal and glial) complex IV activity in demyelinated areas relative to normal appearing white matter when densitometrically assessed at 20 × magnification (Fig. 6). The active rims of chronic lesions contained significantly less complex IV activity (densitometric value of −1.55 ± 2.54,n = 8) relative to the inactive areas of chronic lesions (13.15 ± 8.83, n = 22 and P < 0.001). The complex IV activity in chronic lesions did not correlate with post-mortem interval, which varied from 6–12 h (Table 1).
Figure 6.

Inverse correlation between global complex IV activity within multiple sclerosis lesions and the density of microglia/macrophages. When the global complex IV activity was densitometrically determined in 20× images, there is a significant inverse correlation (P = 0.001) between complex IV activity and the density of HLA reactive microglia and phagocytic macrophages. Furthermore, complex IV activity in the active rims or expanding edge of chronic active lesions (unfilled circles) is significantly lower (P < 0.001) compared with the inactive areas of chronic lesions (filled circles). The cell density is per 20× field. The densitometric values are corrected for background variation using normal appearing white matter.
The complex IV dysfunction involves Na+/K+ ATPase -positive demyelinated axons
We determined the implications of a complex IV defect for nerve impulse transmission by identifying the population of Na+/K+ ATPase α-1 positive axons in chronic lesions, as Na+/K+ ATPase is energy dependent (Young et al., 2008). When demyelinated axons were identified using total neurofilament reactivity and captured in random 100× fields from the chronic inactive lesions (n = 6), the range of percentage complex IV active elements within Na+/K+ ATPaseα-1 positive demyelinated axons was 0–8.54 with an average of 2.56 (Fig. 7). Furthermore, there was a number (19 out of 59) of Na+/K+ ATPaseα-1 positive demyelinated axons without any complex IV active elements (Fig. 7).
Figure 7.
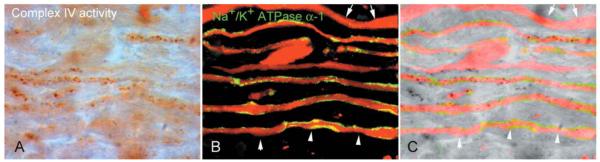
The complex IV dysfunction involves chronically demyelinated axons with Na+/K+ ATPase α-1 reactivity. (A–C) As a proportion of chronically demyelinated axons lacks Na+/K+ ATPase (Young et al., 2008), necessary for sodium extrusion from the axon, complex IV activity was determined within Na+/K+ ATPase α-1 positive chronically demyelinated axons. There are Na+/K+ ATPase α-1 positive demyelinated axons, identified with total neurofilaments, in inactive areas of chronic lesions showing a lack of complex IV active elements (arrowheads). Furthermore, there are axons with bulbous expansions lacking complex IV activity as well as axons lacking both Na+/K+ ATPase α-1 and complex IV activity (arrows).
Inhibition of complex IV augments glutamate-mediated axonal injury, in vitro
As the next step, we investigated the implication of complex IV inhibition for axonal integrity and susceptibility to excitotoxic damage in vitro, given the association between the lack of complex IV activity and axonal injury. We exposed neuronal cultures to sublethal concentrations of sodium azide, a specific inhibitor of complex IV, which reduced the activity by 36%, or glutamate as well as sodium azide followed by glutamate (Fig. 8). Interestingly, the inhibition of complex IV alone led to a slight but significant decrease in amyloid precursor protein immunoreactivity compared with controls, which may reflect reduced fast axonal transport in a state of partial energy deficiency (Fig. 8G). However, there was a striking synergistic effect on excitotoxic axonal injury (amyloid precursor protein and SMI32 immunoreactivity) when complex IV was inhibited preceding exposure to glutamate (Fig. 8F and G).
Figure 8.

Complex IV inhibition augments glutamate-mediated axonal injury, in vitro. (A–E) The axons in control neuron cultures, differentiated from mouse embryonic stem cells, in vitro, express both β-tubulin (A, green) as well as phosphorylated neurofilaments (SMI31 in a, red). The neuronal cultures were exposed to sublethal concentrations and durations of sodium azide (10 μM sodium azide for 120 min), a specific complex IV inhibitor, or glutamate (100 μM glutamate for 60 min) alone as well as sodium azide and glutamate sequentially (addition of 100 μM glutamate to the second 1 h of the 2 h 10 μM sodium azide exposure). Within β-tubulin positive unmyelinated axons not exposed to sodium azide or glutamate (B and D, red) there is minimal SMI32 (non-phosphorylated neurofilaments) and APP (amyloid precursor protein) reactivity (B and D, green). In contrast, neuron cultures exposed to sublethal concentrations of sodium azide and glutamate show abundant SMI32 (C, green) and APP (E, green) reactivity within β-tubulin positive (C and E, red) unmyelinated axons. (F, G) When acute injury of unmyelinated axons, in vitro, was densitometrically analysed the 10 μM sodium azide, which reduced complex IV activity by 36%, led to a significant decrease in APP immunoreactivity in β-tubulin positive axons (F). When the neuronal cultures were sequentially exposed to sodium azide there is a striking synergistic increase in axonal injury, judged by both SMI32 (F) and APP (G) immunoreactivity. *P < 0.001. Two hundred axons were analysed in four separate experiments for each group. CON = control; NaN3 = sodium azide.
Discussion
We determined the mitochondrial respiratory chain complex IV activity and mitochondrial mass within axons in multiple sclerosis and control brain and spinal cord tissue, based on the hypothesis that mitochondrial defects lead to axonal degeneration and energy demand is greater in demyelinated axons. We then investigated the implications of a complex IV defect for axonal degeneration in vitro.
The exceptionally high rate of axonal injury in actively demyelinating multiple sclerosis lesions is well established (Ferguson et al., 1997; Trapp et al., 1998; Kornek et al., 2000). In chronic lesions, axonal degeneration correlates with the extent of inflammation and leads to axonal loss through a slow burning process (Dutta and Trapp, 2007). The pathological signs of axonal injury include intra-axonal accumulation of proteins such as amyloid precursor protein, a marker of fast axonal transport block, and de-phosphorylation of neurofilaments. The amyloid precursor protein positive segments of demyelinated axons located in active rims and relatively inactive centre of chronic lesions harbour mitochondria with a complex IV defect. Furthermore, the morphologically intact chronically demyelinated axons with strong non-phosphorylated neurofilament reactivity showed a significant decrease in complex IV activity, mostly due to mitochondrial depletion, compared with SMI31 positive chronically demyelinated axons. Mitochondrial depletion as a result of axonal transport disturbance is reported in microglia induced neuritic beading in vitro, motor neuron death in an in vivo mutant SOD-1 model of motor neuron disease, which targets mitochondria, as well as in syntaphilin knockout axons, affecting calcium signalling and leading to synaptic short-term facilitation (Takeuchi et al., 2005; De Vos et al., 2007; Kang et al., 2008).
For the first time, we report a significant increase in complex IV activity as well as mitochondrial mass within approximately half of large chronically demyelinated SMI31 positive axons located in relatively inactive areas of chronic (active and inactive) multiple sclerosis lesions in brain and spinal cord, respectively, compared with large myelinated axons. The increase in complex IV activity in mitochondria isolated from chronic lesions compared with normal appearing white matter provides support for the histochemical findings of this study (Lu et al., 2000). The lack of structural changes, in terms of accumulation of amyloid precursor protein and de-phosphorylation of neurofilament, in the chronically demyelinated axons with increased complex IV activity as well as previous reports of similar findings in unmyelinated axons with substantial densities of sodium channels and demyelinated axons suggest the above mitochondrial changes related to demyelination as a potentially adaptive rather than a pathogenic process (Hildebrand and Waxman, 1983; Bristow et al., 2002; Barron et al., 2004; Andrews et al., 2006). The mitochondrial adaptations to metabolic demand include changes in mitochondrial mass and activity (Waxman, 1982; Bristow et al., 2002; Hollenbeck and Saxton, 2005). Indeed, an adaptive change in axonal mitochondria in multiple sclerosis lesions offers an explanation for the predominant loss of small diameter demyelinated axons, which have relatively less volume to surface area or ‘energy to ions’ ratio compared with large diameter axons (DeLuca et al., 2004; Waxman, 2006b). Whilst local lesion environmental factors are likely to be the major drivers of such a mitochondrial change a role for neurons, which have been shown to contain increased mitochondrial DNA, is suggested by the significant increase in complex IV activity within myelinated axons in multiple sclerosis brain (Blokhin et al., 2008).
The increase in complex IV activity in relation to demyelination is associated with an increase in mitochondrial mass within axons. In addition, the majority of mitochondrial elements within axons contain syntaphilin, which was recently identified as an axon specific mitochondrial-docking protein (Kang et al., 2008). Syntaphilin immobilizes axonal mitochondria through targeting of outer mitochondrial membrane by the carboxyl terminal tail and binding with microtubules. The immunohistochemical detection of syntaphilin is a reliable tool to identify immobile mitochondria within axons and provides further support for the increased mitochondrial mass within demyelinated axons in multiple sclerosis. The detection of syntaphilin immunoreactivity without porin may be due to the amplification of syntaphilin and not porin and the presence of syntaphilin unbound to mitochondria within demyelinated axons. Mitochondria also interact with neurofilaments dependent on their metabolic activity and the phosphorylation status of neurofilaments, with energized mitochondria binding to more phosphorylated neurofilaments (Wagner et al., 2003). There is an increase in hyperphosphorylated neurofilaments in acute lesions and phosphorylated neurofilaments immunoreactivity in chronic lesions (Shintaku et al., 1988; Petzold et al., 2008). In this study we have identified a significant increase in phosphorylated relative to total neurofilament-H in chronic multiple sclerosis lesions compared with adjacent myelinated areas in multiple sclerosis and corresponding areas in control brains using western blotting. Whether the interaction between mitochondria and phosphorylated neurofilaments affects mitochondrial movement and activity within axons is not known. However, the increase in syntaphilin in chronic lesions suggests that mitochondria within large chronically demyelinated axons are relatively immobile compared with myelinated axons and decreased mitochondrial movement may partly determine their accumulation within demyelinated axons (Kang et al., 2008). In addition, mitochondrial fission, fusion as well as biogenesis within axons may contribute to the increase in axonal mitochondrial mass following demyelination (Amiri and Hollenbeck, 2008).
Interestingly, not all chronic multiple sclerosis lesions showed an increase in complex IV activity, with the active rims of chronic active lesions showing significantly less complex IV activity compared with relatively inactive lesion centre. Inflammation is a potent inducer of mitochondrial dysfunction and activated microglia and macrophages are sources of nitric oxide and reactive oxygen species in lesions (Qi et al., 2006). Nitric oxide and reactive oxygen species may irreversibly inhibit complex IV activity through nitration of catalytic subunits, post-translational modification as well as oxidative damage to mitochondrial DNA (Lu et al., 2000; Smith and Lassmann, 2002; Wei et al., 2002). In EAE, nitration of mitochondrial respiratory chain complex I and complex IV subunits and reduced ATP synthesis was mediated intrinsically within the central nervous system preceding the arrival of inflammatory infiltrate (Qi et al., 2006). Furthermore, the inhibition of complex IV is recognized in microglia induced neuritic beading, in vitro (Takeuchi et al., 2005). We recently identified a complex IV defect within acutely demyelinated axons in cases with fulminant multiple sclerosis and Balo’s type lesions, where microglial activation is an early feature and innate immunity is implicated early in the pathogenesis (Marik et al., 2007; Mahad et al., 2008). In this study, the inverse correlation between the global complex IV activity and density of microglia/macrophages (HLA-positive cells) identifies innate immunity (microglia) as a probable determinant of complex IV dysfunction in chronic lesions from brain and spinal cord. On a technical aspect, the above inverse correlation suggests the inclusion of microglial complex IV active elements overlapping axons in bright field images as an unlikely contributor to the apparent increase in axonal complex IV activity in chronic lesions.
In the 1960s, Hodgkin and colleagues identified complex IV as an essential component for efflux of sodium by Na+/K+ ATPase and maintenance of resting membrane potential (Hodgkin, 1964). Nitric oxide, an inhibitor of complex IV, has been elegantly demonstrated to cause conduction block (Redford et al., 1997). Hence, the mitochondrial respiratory chain complex IV is necessary for nerve impulse transmission. The lack of sodium channels and Na+/K+ ATPase on a subset of chronically demyelinated axons with the implications for nerve impulse transmission has been suggested as a contributor to disease progression in multiple sclerosis (Black et al., 2007; Young et al., 2008). We identified a complete lack of complex IV activity within a number of Na+/K+ ATPase positive chronically demyelinated axons, which will not be able to maintain their resting membrane potential. We propose mitochondrial complex IV dysfunction as a cause of conduction failure during the progressive stage of multiple sclerosis, possibly accounting for part of the persistent neurological impairment, and identify mitochondria as a potential therapeutic target.
Besides an energy defect, a disturbance in calcium handling capacity is an important consequence of mitochondrial respiratory chain defects (Rizzuto and Pozzan, 2006; von Kleist-Retzow et al., 2007). In multiple sclerosis, the loss of inhibitory interneurons and their processes increases excitatory neurotransmission and firing of demyelinated axons (Dutta et al., 2006; Clements et al., 2008). Furthermore, the increase in mitochondrial mass infers a greater role for mitochondria as an intracellular source of calcium buffering in demyelinated compared with myelinated axons in multiple sclerosis (Nikolaeva et al., 2005). Using sublethal doses of sodium azide, a specific inhibitor of complex IV, and glutamate in neuron cultures we show an augmentation of glutamate induced axonal injury judged by amyloid precursor protein accumulation and non-phosphorylated neurofilaments. The in vitro findings in this study, which is unlikely to be an indirect affect on axons by glial cells due to the paucity of glia in cultures, together with the degeneration of axons conducting impulses at physiological frequencies when exposed to nitric oxide, suggest that a complex IV defect within demyelinated axons in multiple sclerosis may increase the susceptibility to excitotoxicity (Smith et al., 2001).
Our study has identified a mitochondrial respiratory chain complex IV defect, possibly mediated by microglia and macrophages, within injured axons and an increase in mitochondrial mass and complex IV activity within chronically demyelinated axons in inactive areas of chronic lesions. The complex IV defect is likely to impair Na+/K+ ATPase activity, resting membrane potential and nerve impulse transmission, possibly accounting for part of the persistent neurological disability in patients with progressive multiple sclerosis. Furthermore, a complex IV defect increases the susceptibility to glutamate-mediated axonal injury. Thus, preservation of mitochondrial respiratory chain activity within demyelinated axons may have a therapeutic benefit in terms of improving the function and prevention of axonal degeneration in patients with multiple sclerosis.
Acknowledgements
We thank Professor Richard Reynolds, UK MS Tissue Bank, and Newcastle Brain Bank for providing tissue, Dr T Booth and Dr A Trevelyan for assistance with confocal microscopy and Professor RN Lightowlers for guidance on mitochondrial subunit detection.
Funding Welcome Trust and Multiple Sclerosis Society of Great Britain and Northern Ireland.
References
- Ames A., 3rd CNS energy metabolism as related to function. Brain Res Brain Res Rev. 2000;34:42–68. doi: 10.1016/s0165-0173(00)00038-2. [DOI] [PubMed] [Google Scholar]
- Amiri M, Hollenbeck PJ. Mitochondrial biogenesis in the axons of vertebrate peripheral neurons. Dev Neurobiol. 2008;68:1348–61. doi: 10.1002/dneu.20668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andrews H, White K, Thomson C, Edgar J, Bates D, Griffiths I, et al. Increased axonal mitochondrial activity as an adaptation to myelin deficiency in the Shiverer mouse. J Neurosci Res. 2006;83:1533–9. doi: 10.1002/jnr.20842. [DOI] [PubMed] [Google Scholar]
- Andreyev AY, Kushnareva YE, Starkov AA. Mitochondrial metabolism of reactive oxygen species. Biochemistry. 2005;70:200–14. doi: 10.1007/s10541-005-0102-7. [DOI] [PubMed] [Google Scholar]
- Barron MJ, Griffiths P, Turnbull DM, Bates D, Nichols P. The distributions of mitochondria and sodium channels reflect the specific energy requirements and conduction properties of the human optic nerve head. Br J Ophthalmol. 2004;88:286–90. doi: 10.1136/bjo.2003.027664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Black JA, Newcombe J, Trapp BD, Waxman SG. Sodium channel expression within chronic multiple sclerosis plaques. J Neuropathol Exp Neurol. 2007;66:828–37. doi: 10.1097/nen.0b013e3181462841. [DOI] [PubMed] [Google Scholar]
- Blokhin A, Vyshkina T, Komoly S, Kalman B. Variations in Mitochondrial DNA Copy Numbers in MS Brains. J Mol Neurosci. 2008;35:283–7. doi: 10.1007/s12031-008-9115-1. [DOI] [PubMed] [Google Scholar]
- Bristow EA, Griffiths PG, Andrews RM, Johnson MA, Turnbull DM. The distribution of mitochondrial activity in relation to optic nerve structure. Arch Ophthalmol. 2002;120:791–6. doi: 10.1001/archopht.120.6.791. [DOI] [PubMed] [Google Scholar]
- Caldwell PC, Hodgkin AL, Keynes RD, Shaw TL. The effects of injecting ‘energy-rich’ phosphate compounds on the active transport of ions in the giant axons of Loligo. J Physiol. 1960;152:561–90. doi: 10.1113/jphysiol.1960.sp006509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark KM, Bindoff LA, Lightowlers RN, Andrews RM, Griffiths PG, Johnson MA, et al. Reversal of a mitochondrial DNA defect in human skeletal muscle. Nat Genet. 1997;16:222–4. doi: 10.1038/ng0797-222. [DOI] [PubMed] [Google Scholar]
- Clements RJ, McDonough J, Freeman EJ. Distribution of parvalbumin and calretinin immunoreactive interneurons in motor cortex from multiple sclerosis post-mortem tissue. Exp Brain Res. 2008;187:459–65. doi: 10.1007/s00221-008-1317-9. [DOI] [PubMed] [Google Scholar]
- Coleman M. Axon degeneration mechanisms: commonality amid diversity. Nat Rev Neurosci. 2005;6:889–98. doi: 10.1038/nrn1788. [DOI] [PubMed] [Google Scholar]
- Compston A. McAlpine’s Multiple Sclerosis. Vol. 1. Churchill Livingstone; London: 2005. [Google Scholar]
- Craner MJ, Newcombe J, Black JA, Hartle C, Cuzner ML, Waxman SG. Molecular changes in neurons in multiple sclerosis: altered axonal expression of Nav1.2 and Nav1.6 sodium channels and Na+/Ca2+ exchanger. Proc Natl Acad Sci U S A. 2004;101:8168–73. doi: 10.1073/pnas.0402765101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Vos KJ, Chapman AL, Tennant ME, Manser C, Tudor EL, Lau KF, et al. Familial amyotrophic lateral sclerosis-linked SOD1 mutants perturb fast axonal transport to reduce axonal mitochondria content. Hum Mol Genet. 2007;16:2720–8. doi: 10.1093/hmg/ddm226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeLuca GC, Ebers GC, Esiri MM. Axonal loss in multiple sclerosis: a pathological survey of the corticospinal and sensory tracts. Brain. 2004;127:1009–18. doi: 10.1093/brain/awh118. [DOI] [PubMed] [Google Scholar]
- DiMauro S, Schon EA. Mitochondrial respiratory-chain diseases. N Engl J Med. 2003;348:2656–68. doi: 10.1056/NEJMra022567. [DOI] [PubMed] [Google Scholar]
- Dutta R, McDonough J, Yin X, Peterson J, Chang A, Torres T, et al. Mitochondrial dysfunction as a cause of axonal degeneration in multiple sclerosis patients. Ann Neurol. 2006;59:478–89. doi: 10.1002/ana.20736. [DOI] [PubMed] [Google Scholar]
- Dutta R, Trapp BD. Pathogenesis of axonal and neuronal damage in multiple sclerosis. Neurology. 2007;68:S22–31. doi: 10.1212/01.wnl.0000275229.13012.32. discussion S43–54. [DOI] [PubMed] [Google Scholar]
- Ebers GC, Heigenhauser L, Daumer M, Lederer C, Noseworthy JH. Disability as an outcome in MS clinical trials. Neurology. 2008;71:624–31. doi: 10.1212/01.wnl.0000313034.46883.16. [DOI] [PubMed] [Google Scholar]
- Ferguson B, Matyszak MK, Esiri MM, Perry VH. Axonal damage in acute multiple sclerosis lesions. Brain. 1997;120(Pt 3):393–9. doi: 10.1093/brain/120.3.393. [DOI] [PubMed] [Google Scholar]
- Hevner RF, Liu S, Wong-Riley MT. A metabolic map of cytochrome oxidase in the rat brain: histochemical, densitometric and biochemical studies. Neuroscience. 1995;65:313–42. doi: 10.1016/0306-4522(94)00514-6. [DOI] [PubMed] [Google Scholar]
- Hildebrand C, Waxman SG. Regional node-like membrane specializations in non-myelinated. Brain Research. 1983;258:23–32. doi: 10.1016/0006-8993(83)91222-2. [DOI] [PubMed] [Google Scholar]
- Hodgkin AL. The Ionic Basis of Nervous Conduction. Science. 1964;145:1148–54. doi: 10.1126/science.145.3637.1148. [DOI] [PubMed] [Google Scholar]
- Hollenbeck PJ, Saxton WM. The axonal transport of mitochondria. J Cell Sci. 2005;118:5411–9. doi: 10.1242/jcs.02745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang JS, Tian JH, Pan PY, Zald P, Li C, Deng C, et al. Docking of axonal mitochondria by syntaphilin controls their mobility and affects short-term facilitation. Cell. 2008;132:137–48. doi: 10.1016/j.cell.2007.11.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirkinezos IG, Bacman SR, Hernandez D, Oca-Cossio J, Arias LJ, Perez-Pinzon MA, et al. Cytochrome c association with the inner mitochondrial membrane is impaired in the CNS of G93A-SOD1 mice. J Neurosci. 2005;25:164–72. doi: 10.1523/JNEUROSCI.3829-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kornek B, Storch MK, Weissert R, Wallstroem E, Stefferl A, Olsson T, et al. Multiple sclerosis and chronic autoimmune encephalomyelitis: a comparative quantitative study of axonal injury in active, inactive, and remyelinated lesions. Am J Pathol. 2000;157:267–76. doi: 10.1016/S0002-9440(10)64537-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu F, Selak M, O’Connor J, Croul S, Lorenzana C, Butunoi C, et al. Oxidative damage to mitochondrial DNA and activity of mitochondrial enzymes in chronic active lesions of multiple sclerosis. J Neurol Sci. 2000;177:95–103. doi: 10.1016/s0022-510x(00)00343-9. [DOI] [PubMed] [Google Scholar]
- Mahad D, Ziabreva I, Lassmann H, Turnbull D. Mitochondrial defects in acute multiple sclerosis lesions. Brain. 2008;131:1722–35. doi: 10.1093/brain/awn105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marik C, Felts PA, Bauer J, Lassmann H, Smith KJ. Lesion genesis in a subset of patients with multiple sclerosis: a role for innate immunity? Brain. 2007;130:2800–15. doi: 10.1093/brain/awm236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nikolaeva MA, Mukherjee B, Stys PK. Na+-dependent sources of intra-axonal Ca2+ release in rat optic nerve during in vitro chemical ischemia. J Neurosci. 2005;25:9960–7. doi: 10.1523/JNEUROSCI.2003-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petzold A, Gveric D, Groves M, Schmierer K, Grant D, Chapman M, et al. Phosphorylation and compactness of neurofilaments in multiple sclerosis: indicators of axonal pathology. Exp Neurol. 2008;213:326–35. doi: 10.1016/j.expneurol.2008.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qi X, Lewin AS, Sun L, Hauswirth WW, Guy J. Mitochondrial protein nitration primes neurodegeneration in experimental autoimmune encephalomyelitis. J Biol Chem. 2006;281:31950–62. doi: 10.1074/jbc.M603717200. [DOI] [PubMed] [Google Scholar]
- Redford EJ, Kapoor R, Smith KJ. Nitric oxide donors reversibly block axonal conduction: demyelinated axons are especially susceptible. Brain. 1997;120(Pt 12):2149–57. doi: 10.1093/brain/120.12.2149. [DOI] [PubMed] [Google Scholar]
- Rizzuto R, Pozzan T. Microdomains of intracellular Ca2+: molecular determinants and functional consequences. Physiol Rev. 2006;86:369–408. doi: 10.1152/physrev.00004.2005. [DOI] [PubMed] [Google Scholar]
- Shintaku M, Hirano A, Llena JF. Increased diameter of demyelinated axons in chronic multiple sclerosis of the spinal cord. Neuropathol Appl Neurobiol. 1988;14:505–10. doi: 10.1111/j.1365-2990.1988.tb01341.x. [DOI] [PubMed] [Google Scholar]
- Smith KJ. Sodium channels and multiple sclerosis: roles in symptom production, damage and therapy. Brain Pathol. 2007;17:230–42. doi: 10.1111/j.1750-3639.2007.00066.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith KJ, Kapoor R, Hall SM, Davies M. Electrically active axons degenerate when exposed to nitric oxide. Ann Neurol. 2001;49:470–6. [PubMed] [Google Scholar]
- Smith KJ, Lassmann H. The role of nitric oxide in multiple sclerosis. Lancet Neurol. 2002;1:232–41. doi: 10.1016/s1474-4422(02)00102-3. [DOI] [PubMed] [Google Scholar]
- Stys PK. General mechanisms of axonal damage and its prevention. J Neurol Sci. 2005;233:3–13. doi: 10.1016/j.jns.2005.03.031. [DOI] [PubMed] [Google Scholar]
- Takeuchi H, Mizuno T, Zhang G, Wang J, Kawanokuchi J, Kuno R, et al. Neuritic beading induced by activated microglia is an early feature of neuronal dysfunction toward neuronal death by inhibition of mitochondrial respiration and axonal transport. J Biol Chem. 2005;280:10444–54. doi: 10.1074/jbc.M413863200. [DOI] [PubMed] [Google Scholar]
- Taylor RW, Turnbull D. Laboratory diagnosis of mitochondrial disease. In: Applegarth DA, Dimmick J, Hall JG, editors. Organelle diseases. Chapman & Hall; London: 1997. pp. 341–50. [Google Scholar]
- Taylor RW, Turnbull DM. Mitochondrial DNA mutations in human disease. Nat Rev Genet. 2005;6:389–402. doi: 10.1038/nrg1606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trapp BD, Peterson J, Ransohoff RM, Rudick R, Mork S, Bo L. Axonal transection in the lesions of multiple sclerosis. N Engl J Med. 1998;338:278–85. doi: 10.1056/NEJM199801293380502. [DOI] [PubMed] [Google Scholar]
- van der Valk P, De Groot CJ. Staging of multiple sclerosis (MS) lesions: pathology of the time frame of MS. Neuropathol Appl Neurobiol. 2000;26:2–10. doi: 10.1046/j.1365-2990.2000.00217.x. [DOI] [PubMed] [Google Scholar]
- von Kleist-Retzow JC, Hornig-Do HT, Schauen M, Eckertz S, Dinh TA, Stassen F, et al. Impaired mitochondrial Ca2+ homeostasis in respiratory chain-deficient cells but efficient compensation of energetic disadvantage by enhanced anaerobic glycolysis due to low ATP steady state levels. Exp Cell Res. 2007;313:3076–89. doi: 10.1016/j.yexcr.2007.04.015. [DOI] [PubMed] [Google Scholar]
- Wagner OI, Lifshitz J, Janmey PA, Linden M, McIntosh TK, Leterrier JF. Mechanisms of mitochondria-neurofilament interactions. J Neurosci. 2003;23:9046–58. doi: 10.1523/JNEUROSCI.23-27-09046.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waxman SG. Membranes, myelin, and the pathophysiology of multiple sclerosis. N Engl J Med. 1982;306:1529–33. doi: 10.1056/NEJM198206243062505. [DOI] [PubMed] [Google Scholar]
- Waxman SG. Axonal conduction and injury in multiple sclerosis: the role of sodium channels. Nat Rev Neurosci. 2006a;7:932–41. doi: 10.1038/nrn2023. [DOI] [PubMed] [Google Scholar]
- Waxman SG. Ions, energy and axonal injury: towards a molecular neurology of multiple sclerosis. Trends Mol Med. 2006b;12:192–5. doi: 10.1016/j.molmed.2006.03.001. [DOI] [PubMed] [Google Scholar]
- Waxman SG. Axonal dysfunction in chronic multiple sclerosis: meltdown in the membrane. Ann Neurol. 2008;63:411–3. doi: 10.1002/ana.21361. [DOI] [PubMed] [Google Scholar]
- Waxman SG, Black JA, Stys PK, Ransom BR. Ultrastructural concomitants of anoxic injury and early post-anoxic recovery in rat optic nerve. Brain Res. 1992;574:105–19. doi: 10.1016/0006-8993(92)90806-k. [DOI] [PubMed] [Google Scholar]
- Wei J, Guo H, Kuo PC. Endotoxin-stimulated nitric oxide production inhibits expression of cytochrome c oxidase in ANA-1 murine macrophages. J Immunol. 2002;168:4721–7. doi: 10.4049/jimmunol.168.9.4721. [DOI] [PubMed] [Google Scholar]
- Young EA, Fowler CD, Kidd GJ, Chang A, Rudick R, Fisher E, et al. Imaging correlates of decreased axonal Na+/K+ ATPase in chronic multiple sclerosis lesions. Ann Neurol. 2008;63:428–35. doi: 10.1002/ana.21381. [DOI] [PubMed] [Google Scholar]