

Abstract
Recent developments in chromatography, such as ultra-HPLC and superficially porous particles, offer significantly improved peptide separation. The narrow peak widths, often only several seconds, can permit a 15-min liquid chromatography run to have a similar peak capacity as a 60-min run using traditional HPLC approaches. In theory, these larger peak capacities should provide higher protein coverage and/or more protein identifications when incorporated into a proteomic workflow. We initially observed a decrease in protein coverage when implementing these faster chromatographic approaches, due to data-dependent acquisition (DDA) settings that were not properly set to match the narrow peak widths resulting from newly implemented, fast separation techniques. Oversampling of high-intensity peptides lead to low protein-sequence coverage, and tandem mass spectra (MS/MS) from lower-intensity peptides were of poor quality, as automated MS/MS events were occurring late on chromatographic peaks. These observations led us to optimize DDA settings to use these fast separations. Optimized DDA settings were applied to the analysis of Trypanosome brucei peptides, yielding peptide identifications at a rate almost five times faster than previously used methodologies. The described approach significantly improves protein identification workflows that use typical available instrumentation.
Keywords: liquid chromatography, mass spectrometry, proteomics, tandem mass spectrometry
INTRODUCTION
Mass spectrometry (MS) is at the center of many proteomic workflows, often times used with on-line liquid chromatography (LC) separation techniques.1,2 An issue with this approach is the large amount of instrument time required to run traditional LC tandem MS (LC-MS/MS) experiments. Recent developments in chromatography, such as sub-2 μm particles and superficially porous particles, can offer significantly improved peptide separation.3,4 High-efficiency columns allow increased mobile-phase velocities that shorten experimental analysis time and can provide narrow peak widths, typically a few seconds or less.5 Combined, these two factors can, permit a 15-min LC run to have similar peak capacity as a 60-min run using traditional HPLC approaches.6–8 A drawback sub-2 μm particles is the high back pressure created by these particles, which exceeds the pressure limit of most available standard HPLC instruments.9 Superficially porous particles offer similar efficiencies in terms of number of theoretical plates and improved plate height but operate at about one-half of the back pressures of sub-2 μm particles, which allow for use on a standard capillary LC system.10–13 These column packings exhibit rapid solute mass transfer with minimal loss of chromatographic resolution when operated with high flow rates to reduce experimental analysis time.14 Based on these characteristics and current instrumentation, superficially porous columns were integrated into our lab's proteomic workflow.
Fast separation methodologies were implemented with an expected outcome of increased peptide identifications in a shorter amount of time. However, we initially observed decreased protein-sequence coverage when implementing these separation methods. Analysis of peak capacities showed the fast-gradient conditions improved separation metrics, such as reduced peak widths, which should improve peptide identifications, as has been displayed previously in other proteomic applications.15 Detailed inspection of raw LC-MS data revealed that most MS/MS spectra were obtained on peptides of high abundance. This led to the acquisition of few MS/MS events being performed on peptide ions with low signal intensity, which in turn, led to poor protein-sequence coverage. It became apparent that improved chromatographic separations would not improve proteome coverage without enhanced MS/MS acquisition strategies.
The selection of peptide ions for MS/MS analysis is controlled by data-dependent acquisition (DDA) settings, which include repeat count, repeat duration, minimum MS signal, and dynamic exclusion. Previous work has shown that proteome coverage can be increased when DDA settings are optimized. Andrews et al.16 found that the ionization settings and the number of MS/MS events/cycle were the most important settings associated with improving proteome coverage on a linear quadrupole ion trap (LTQ) Orbitrap XL. They also stated that parameters, such as dynamic exclusion and minimum signal counts, had minimal influence on proteome coverage. Zhang et al.17 stated that increased dynamic exclusion can improve proteome coverage; however, this hinders spectral count quantitation. Whereas these reports provided good information for the basis of our experiments, they did not specifically apply to matching DDA settings with fast separation techniques and materials.
In this current work, we present a process of optimizing DDA settings on a LTQ to match fast LC separations with improved peak capacities and demonstrate that this combination can provide a large number of peptide identifications in significantly less time.
MATERIALS AND METHODS
Data were acquired using an 1100 Capillary LC system (Agilent Technologies, Palo Alto, CA, USA), with on-line MS detection using a LTQ ion trap (Thermo-Fisher, San Jose, CA, USA), fitted with a Michrom Bioresources (Auburn, CA, USA) captive spray interface. Gradient delay volume was reduced by removing the mixing column in the Agilent LC system. These changes allowed for faster separations and re-equilibration of the chromatograph. Measured gradient delay volume determined by analytical column bypass was observed to be 56 μL. Analytical columns were either a 0.2 × 50-mm HALO peptide ES-C18 capillary column packed with 2.7 μm-diameter superficially porous particles (Advanced Materials Technology, Wilmington, DE, USA) or a 0.2 × 50-mm Magic C18AQ column packed with 3 μm-diameter porous particles (Michrom Bioresources). Proteomic sample analysis used the LTQ divert valve fitted with an EXP stem trap 2.6 μL cartridge packed with 2.7 μm-diameter superficially porous particles (Optimize Technologies, Oregon City, OR, USA) or with a 0.5 × 2-mm CapTrap packed with 3 μm-diameter porous particles (Michrom Bioresources). Experiments using trap columns were matched for stationary-phase materials of the analytical and guard columns. Slow-gradient conditions operated at a flow rate of 4 μL/min, increasing mobile-phase B concentration from 5% to 50% B over 30 min, with a total experiment time of 90 min. Fast-gradient conditions operated at a flow rate of 9 μL/min, increasing mobile-phase B concentration from 5% to 50% B over 12.5 min with a total experiment time of 21 min. Mobile phases used formic acid and acetonitrile from Sigma-Aldrich (St. Louis, MO, USA).
Tryptic peptides from BSA (Michrom Bioresources) were used to evaluate MS and separation metrics during the evaluation and optimization phase of this work. Peak-capacity data were analyzed using Xcalibur software. Peak widths at 50% height were measured by manual inspection with the use of extracted ion chromatograms. Calculations were made from duplicate experiments of 1 pmol injections, measuring both peak widths and retention times. Experiments were conducted without the collection of MS/MS spectra to obtain more MS1 data points to better define each chromatographic peak. Peak capacities were calculated using two methods, differing in the expression of gradient time (Tg).
Measured peak capacity:
| (1) |
Theoretical peak capacity
| (2) |
Equation 1, referred to as actual peak capacity, was calculated using the retention times of the first and last eluting peptides from Table 1. Equation 2 is referred to as theoretical peak capacity, using the actual length of gradient generation (Tg), which is independent of actual peptide-retention times. For fast-gradient conditions, this Tg value was 750 s, whereas for slow-gradient conditions, 1800 s was used for Tg. As a result of the use of multiple stationary phases and gradient conditions in this analysis, peak-capacity and proteomic experiments were conducted for each type of stationary-phase material under fast and slow-gradient conditions.
Table 1.
BSA Tryptic Peptides Used for Chromatographic Analysis and Peak-Capacity Calculations
| Sequence | Peptide mass (Da) | Retention time: fast gradient | Retention time: slow gradient |
|---|---|---|---|
| CASIQK | 705.8237 | 6.62 | 22.37 |
| LVTDLTK | 788.9290 | 7.80 | 25.87 |
| QTALVELLK | 1014.2164 | 8.92 | 27.29 |
| EACFAVEGPK | 1107.2371 | 6.59 | 21.58 |
| CCTESLVNR | 1139.2607 | 8.16 | 25.45 |
| SLHTLFGDELCK | 1419.6016 | 8.77 | 27.12 |
| LGEYGFQNALIVR | 1479.6783 | 9.39 | 28.70 |
| MPCTEDYLSLILNR | 1724.9953 | 11.12 | 33.25 |
| LFTFHADICTLPDTEK | 1908.1352 | 9.52 | 29.10 |
| DAIPENLPPLTADFAEDKDVCK | 2459.6801 | 9.63 | 29.75 |
| EYEATLEECCAKDDPHACYSTVFDK | 3039.2431 | 8.60 | 27.47 |
| TVMENFVAFVDKCCAADDKEACFAVEGPK | 3310.7085 | 11.20 | 32.95 |
Fast-gradient retention time provides the average retention time under the fast-gradient conditions with the use of the superficially porous particle (S.P.P.) column. Slow-gradient retention time displays the average retention time from slow-gradient conditions using the porous particle column.
Preferred operation conditions were tested for application to proteomic sample analysis using a whole cell lysate from the procyclic form Trypanosome brucei. Soluble proteins from T. brucei underwent electrophoresis through a NuPAGE 12% Bis(2-hydroxyethyl)-Tris gel (Invitrogen, Carlsbad, CA, USA). This step was aimed to remove undesirable, nonprotein containments with negligible protein separation; hence, the entire lane was treated as a single sample. Each gel lane was cut into 1 × 1-mm squares for digestion. Water was then added to the gel pieces and discarded to waste. Gel pieces were then washed with a mixture of 50% acetonitrile and 50% water, with solution removed as waste after 15 min. Ammonium bicarbonate (100 mM) was then added, and after 15 min, an equal volume of acetonitrile was added to make a 1:1 (v/v) solution. After incubation at room temperature for 15 min, the ammonium bicarbonate/acetonitrile solution was removed to waste. Acetonitrile was again added to the gel slices and incubated at room temperature for 15 min. This solution was then removed as waste. A solution of 10 mM DTT in 100 mM ammonium bicarbonate was then added and incubated in a 70°C water bath for 1 h. The reducing solution was then removed, and a 55-mM iodoacetamide in 100 mM ammonium bicarbonate-alkylating solution was added. Samples were incubated at room temperature for 1 h in the dark. The alkylating solution was then removed, and 100 mM ammonium bicarbonate was added. After 5 min, an equal volume of acetonitrile was added to make a 1:1 (v/v) solution. After 15 min of incubation, this solution was removed to waste. A solution of 0.1% sequencing-grade trypsin (Promega, San Luis Obispo, CA, USA) was made in 50 mM ammonium bicarbonate and added to the gel pieces at 1:50 (w/w) enzyme to protein and incubated overnight at 37°C. The following day peptides were extracted by pooling and saving the solution from gel pieces. A solution of 50% acetonitrile and 0.1% formic acid was then added to the gel pieces. After 15 min, this solution was extracted and added to the solution previously pulled from the digested gel slices. T. brucei sample analysis was conducted using 2-μg injections preformed in duplicate for each of the four gradient/stationary-phase conditions.
Raw tandem mass spectra were converted to mzXML files and then into Mascot generic files (MGFs) via the Trans-Proteomic Pipeline (Seattle Proteome Center, Seattle, WA, USA). MGFs were searched using Mascot (Matrix Scientific, Boston, MA, USA) against separate target and decoy databases obtained from the National Center for Biotechnology Information (Bethesda, MD, USA). The target database contained all T. brucei protein sequences, and the decoy database contained the reversed sequences from the target database. Mascot settings were as follows: tryptic enzymatic cleavages allowing for up to two missed cleavages, peptide tolerance of 1000 parts/million, fragment ion tolerance of 0.6 Da, fixed modification as a result of carboxyamidomethylation of cysteine (+57 Da), and variable modifications of oxidation of methionine (+16 Da) and deamidation of asparagine or glutamine (+0.98 Da). Mascot files were loaded into ProteoIQ (NuSep, Bogart, GA), where a 5% false discovery rate was applied for confirmation of protein or peptide identifications.
RESULTS
Separation Condition Analysis
The initial experiments were performed to evaluate the impact of fast-gradient separations when applied to LC-MS/MS-based proteomic experiments. Figure 1A shows a base peak chromatogram for slow-gradient conditions along with the timescale of events occurring during the experiment. The narrow window of peptide elution displayed under these conditions highlights a poor efficiency in the use of instrument time. This inefficiency can, in part, be attributed to the fixed LC system dwell volume, which when considered with a low flow rate, accounts for almost 15 min of experiment time. A high-efficiency gradient was developed, reducing experiment time from 90 min to 21 min using a superficially porous-particle stationary phase, allowing efficient separations at elevated flow rates. This reduction in experiment time was achieved by doubling the flow rate, optimizing gradient conditions, and using a highly efficient, reverse-phase column, packed with superficially porous particles. The increased flow rates affected the time for all aspects of the experiment; in particular, this halved the times required for loading, washing, and re-equilibrating of the chromatograph. This gradient included a 10× column-volume, high organic mobile-phase wash and a 10× column-volume re-equilibration of the chromatograph. Figure 1B displays a base peak chromatogram, along with a timescale of experimental events demonstrating the improvements in instrument time utilization. Although a further reduction in experiment time would be beneficial, the instrumentally fixed gradient-delay volume cannot be altered.
FIGURE 1.
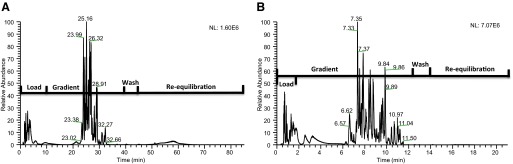
Base peak chromatograms with a superimposed timescale of experimental events from the LC-MS analysis of tryptic-digested BSA. (A) Chromatogram obtained using the 90-min experimental protocol, which included a 0.2 × 50-mm porous-particle column operated at 4 μL/min. (B) Chromatogram obtained using the 21-min experimental protocol, which included a 0.2 × 50-mm superficially porous particle operated at 9 μL/min.
The LC-resolving power was evaluated by calculating the peak capacity for both gradient conditions using both stationary-phase materials. To conduct this assessment, BSA tryptic digest was used as a result of its high availability and relative simplicity compared with a proteomic sample. Measured peak capacity allowed for an evaluation of separation conditions dependent on sample-retention times, whereas theoretical peak capacity allowed for evaluation independent of peptide-retention times. Theoretical peak-capacity calculations demonstrated the maximum peak capacity obtainable for each gradient/column condition.
Peak capacities showed significant improvement with the use of superficially porous packing material under both gradient conditions examined. This was achieved by substantial reduction in peak widths, as superficially porous particles produced peak widths of approximately one-half that of porous particles, as shown in Table 2. The use of superficially porous particles operated at elevated flow rates maintains high peak capacity, as shown by comparison of the results for fast and slow conditions. Figure 2 displays an extracted ion chromatogram for peptide QTALVELLK for all four experimental conditions, showing the reduction in peak width and improvement in peak shape using the superficially porous-particle, stationary-phase materials during operation with increased flow rates.
Table 2.
Peak Widths and Peak Capacities Obtained from LC-MS Analysis of Tryptic BSA Peptides Using Each Chromatographic Condition
| Phase/particle size | Flow rate (μL/min) | Gradient length (min) | Average peak width: full width at one-half maximum(s) | Peak width sd | Measured peak capacity | Theoretical peak capacity |
|---|---|---|---|---|---|---|
| Porous 3 μm | 4 | 30 | 25.35 | 22.28 | 16.40 | 41.77 |
| Porous 3 μm | 9 | 12.5 | 8.68 | 4.12 | 18.71 | 50.85 |
| S.P.P. 2.7 μm | 4 | 30 | 11.35 | 5.89 | 32.62 | 93.29 |
| S.P.P. 2.7 μm | 9 | 12.5 | 5.08 | 0.84 | 32.16 | 86.93 |
FIGURE 2.
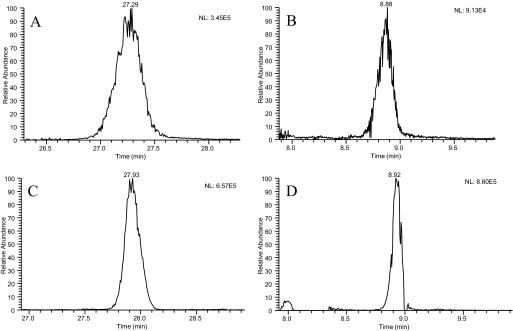
Extracted ion chromatograms for LC-MS analysis of BSA tryptic peptide QTALVELLK using each of the four chromatographic conditions analyzed. (A) Slow-gradient conditions using a 0.2 × 50-mm porous-particle column operated at 4 μL/min. (B) Fast-gradient conditions using a 0.2 × 50-mm porous-particle column operated at 9 μL/min. (C) Slow-gradient conditions using a 0.2 × 50-mm superficially porous-particle column operated at 4 μL/min. Fast-gradient conditions using a 0.2 × 50-mm superficially porous column operated at 9 μL/min.
Variation of the chromatographic peak widths was evaluated to assess the reliability of the separation system, as well as to inform the range of peak widths that should be considered for MS and MS/MS parameter optimization. The porous-particle column produced sds in the mean peak width approximately five times greater than the superficially porous particle-packed columns under identical gradient conditions. The small variance displayed by the superficially porous-particle column confirms conditions effective for the broad range of tryptic-digest peptides present in a complex proteomic sample.
Mass Spectral Condition Analysis
The overarching purpose of the present work was to evaluate the fitness of fast-gradient separations for high-throughput proteomic analysis. Whereas peak capacity is a useful measure of a separation, the use of fast-gradient separations was strongly dependent on appropriate MS settings. To acquire appropriate MS settings, mass spectral data generated from slow-gradient conditions with porous particles were compared with data collected using fast-gradient conditions with superficially porous particles. MS settings were evaluated by comparing Mascot scores and protein-sequence coverage using 1 pmol injections of BSA tryptic digests. Typical “lab favorite” MS instrument settings, used with slow-gradient conditions and porous-particle columns, produced a protein score of 10,109 with 82% sequence coverage. Application of fast-gradient conditions using superficially porous particles combined with the same MS settings produced a protein identification score of 2664 with 61% sequence coverage. The poor Mascot score and protein-sequence coverage from conditions displaying superior separation metrics demonstrated that MS/MS acquisition events were not properly matched to the narrow chromatographic peak widths.
The significant difference in Mascot scores and sequence coverage can be attributed to multiple factors associated with MS/MS acquisition. First, reducing Tg decreased the number of MS/MS spectra collected during an experiment. Experimentally measured duty cycles produced an average cycle time of ∼1.5 s. The duty cycle includes one MS1 acquisition of mass/charge ratio (m/z) 300–2000, with the top five most-intense precursor ions subjected to MS/MS analysis. From this data, slow-gradient conditions would produce ∼6000 MS/MS spectra during the 30-min gradient, whereas fast-gradient conditions would produce ∼2500 MS/MS spectra during the 12.5-min gradient. Based on these calculations, the number of MS/MS spectra collected for the fast-gradient conditions would be ∼2.5 times lower than obtained with slow-gradient conditions, indicating that more-efficient MS/MS spectra collection would be necessary to produce similar data.
The significant differences in Mascot scores are also attributed to the high number of redundant MS/MS spectra collected under slow-gradient conditions. Poor separation performance conditions, resulting in wide peaks and significant peak tailing, which allowed peptide precursor ions to go on and off dynamic exclusion lists multiple times, provided multiple opportunities to collect redundant MS/MS spectra. Slow-gradient conditions matched 741 MS/MS spectra to 97 peptides. This produced an average of 7.7 MS/MS spectra/peptide identification. Fast-gradient conditions matched 152 MS/MS spectra to 70 peptides, averaging 2.2 spectra/identification. Figure 3A shows the chromatographic peak for peptide LFTFHADICTLPDTEK using slow-gradient conditions with an X indicating the location where MS/MS acquisition occurred. The majority of MS/MS spectra for this peptide was collected on the tail of the peak, as seen in Fig. 3B, which displays the occurrence of MS/MS acquisition and dynamic exclusion on the timescale of this chromatographic peak. The collection of nine MS/MS spectra on the peak tail provided redundant spectra that increased the protein score. Alternately, fast-gradient conditions showed significantly less peak tailing of this peptide, as shown in Fig. 4. The result is fewer MS/MS events performed on this peptide, leading to a reduction in the protein score. The wide chromatographic peaks associated with the slow-gradient conditions led to the collection of redundant spectra, accounting for more matching MS/MS spectra and higher protein scores.
FIGURE 3.

(A) An extracted ion chromatogram and (B) a magnification of this chromatogram obtained from the LC-MS/MS analysis of the BSA tryptic peptide LFTFHADICTLPDTEK. Data were collected using slow-gradient conditions, which included a 0.2 × 50-mm porous-particle column operated at 4 μL/min. An “X” on the chromatogram indicates each location that a MS/MS spectrum was collected for this peptide. A superimposed timescale for the application of dynamic exclusion is also shown.
FIGURE 4.

An extracted ion chromatogram for the BSA peptide LFTFHADICTLPDTEK using the fast-gradient conditions, which included a 0.2 × 50-mm superficially porous-particle column operated at 9 μL/min. An X on the chromatogram indicates each location that a MS/MS spectrum was collected for this peptide. A superimposed timescale for the application of dynamic exclusion is also shown.
The poor sequence coverage obtained using fast-gradient conditions can be attributed to the reduced experiment time, which decreased the total number of MS/MS events during a LC-MS/MS experiment. The reduced experiment time will require more-efficient precursor ion selection for MS/MS acquisition so that low-abundance ions are less likely to be missed. For example, the slow-gradient conditions produced an average peak width at a one-half height of 25.4 s, during which time 17 duty cycles acquiring 85 MS/MS spectra occurred. Fast-gradient conditions were not afforded the same luxury. In this case, the average peak widths were 5.1 s, allowing approximately three duty cycles to obtain 15 MS/MS spectra. Fast-gradient conditions have smaller windows of opportunity to collect MS/MS spectra on individual peptides during their chromatographic peak elution, which necessitates collection of MS/MS spectra on a wider variety of precursor ions. This can be seen in the fast LC-MS/MS run: 152 MS/MS spectra were matched to 70 peptides, and 22 of these peptides were identified with three or more spectra. Spectra matching these 22 peptides accounted for 126 of the 152 (83%)-matched MS/MS spectra, demonstrating the oversampling of high-abundance peptides, leading to poor sequence coverage.
Peptides identified with the slow-gradient conditions but not identified using the fast-gradient conditions were examined via extracted ion chromatograms. These missed peptides typically had the following characteristics: low signal intensity, coelution with high-signal intensity peptides, and very narrow peak widths. These issues are demonstrated by an extracted ion chromatogram for peptide NYQEAK (Fig. 5A), which elutes in a peak that is 2.7-s wide. For this peptide to be identified, this ion would need to be selected in one of the ∼10 MS/MS spectra collected during this time. The MS1 spectrum (Fig. 5B) acquired during this peak displays an ion at m/z 752.3, which corresponds to the M + H+ of this peptide, and at least six ions with higher abundance. These other ions would have been subjected to MS/MS analysis prior to the ion at m/z 752.3, because of their higher abundance. Furthermore, the repeat count setting allowed up to three MS/MS spectra to be obtained on these higher-abundance ions; consequently, the ion at m/z 752.3 was not subjected to MS/MS analysis during the two cycles that it eluted from the LC. For other low-abundance peptides, the presence of coeluting peptides caused the MS/MS event to occur past the apex of the chromatographic peak, as shown in the extracted ion chromatogram for peptide YICDNQDTISSK and the corresponding MS spectrum for this peak (Fig. 6A and B, respectively). This peptide has a wider chromatographic peak than the peptide NYQEAK, discussed above, permitting more MS/MS acquisitions, which in turn, allowed the higher-abundance peptides to be placed on the dynamic-exclusion list. Although this peptide was subjected to MS/MS analysis, the spectrum was of low quality as a result of its acquisition late on the chromatographic peak. Although signal intensity is not directly factored into a peptide score via database searching, poor signal intensity will produce less-distinguishable fragment ions, leading to lower scores, and decrease the chance of correct identification.
FIGURE 5.

(A) The extracted ion chromatogram for m/z 752.30, which corresponds to the BSA peptide NYQEAK, obtained with the fast-gradient conditions. The extracted ion chromatogram contains a narrow chromatographic peak for this peptide. These data also show that no MS/MS spectra were collected on this peptide while it was eluting. (B) A mass spectrum collected during the elution of peptide NYQEAK, showing that other ions, several of which had higher abundance, were present, along with the ion of interest at m/z 752.30.
FIGURE 6.

(A) The extracted ion chromatogram for BSA peptide YICDNQDTISSK, obtained with the fast-gradient conditions. An X on the chromatogram indicates each location that a MS/MS spectrum was collected for this peptide. (B) A mass spectrum collected during the elution of peptide YICDNQDTISSK, showing that other ions, several of which had higher abundance, were present along with the ion of interest at m/z 1444.65.
These issues made it apparent that DDA settings needed to be optimized for the sharper chromatographic peaks observed using fast-gradient conditions. Specifically, minimum MS signal, repeat count, repeat duration, and dynamic exclusion were examined to improve MS/MS acquisition and quality. Minimum MS signal controls the minimum signal intensity required for a precursor ion to be selected for MS/MS acquisition. After a m/z is subjected to MS/MS acquisition, repeat count is the number of times that specific m/z is eligible for additional MS/MS acquisition. Repeat duration is the length of time the m/z is eligible for MS/MS acquisition before it is no longer subjected to further fragmentation spectra. Dynamic exclusion is the amount of time a m/z is put on an exclusion list preventing it from MS/MS fragmentation to allow other m/z to be subjected to MS/MS fragmentation.
These experiments were performed using a DDA setting to select the “top five” most-abundant ions for MS/MS analysis. Changes to this setting were not explored in this work. Increasing this DDA setting would allow for the selection of lower-abundance ions but at the expense of increased cycle time. The increase in cycle time was not conducive for fast-gradient conditions producing very narrow peak widths. For example, the cycle time increases to over 3 s with the selection of the top nine most-abundant ions, which would not allow for multiple cycles to occur over the average 5-s-wide chromatographic peaks obtained using these conditions. The reduction of this DDA setting was also not explored as a result of the reduction in the number of MS/MS spectra that would be collected during each experiment.
Systematic optimization of DDA settings to match peak widths with decreased numbers of duty cycles acquired will lead to a desired condition of operation. Table 3 presents the DDA setting procedure used for fast-gradient separation conditions and the impact this systematic change had on the number of matched MS/MS spectra, Mascot score, and protein-sequence coverage. Reduction of dynamic exclusion time to 10 s was selected to match the average peak width at base, while maintaining the dynamic exclusion time to peak-width ratio used with the slow gradient. This change improved sequence coverage and increased the number of matched MS/MS spectra; however, this also led to an increased number of redundant MS/MS spectra collected. The next variable investigated was the minimum MS signal, which was decreased from 1500 to 1000. This change further increased the number of matched MS/MS spectra, peptide identifications, and improved sequence coverage. Further reductions in the value were not considered useful, with the expectation that MS/MS spectra quality would suffer significantly. The number of redundant MS/MS spectra was decreased by lowering the repeat count parameter. A value of one for this setting was found to produce similar sequence coverage to slow-gradient conditions, although the collection of redundant spectra was still prevalent. The repeat duration parameter was also examined, but changes to this value appeared to have an insignificant effect on evaluation metrics. A value of 10 s for repeat duration was selected to correspond with observed average peak width at base.
Table 3.
Mascot Search Results from LC-MS/MS Analysis of Tryptic-Digested BSA Using Different DDA Parameters
| Column/gradient length | Condition change | Mascot sc | Sequence coverage | Matched MS/MS Spectra | Peptides identified | Spectra/peptide |
|---|---|---|---|---|---|---|
| Porous 50 mm/90 min | RC = 3 | 10,109 | 82% | 741 | 97 | 7.7 |
| RD = 10 s | ||||||
| DX = 60 s | ||||||
| Min MS = 1500 | ||||||
| S.P.P. 50 mm/20 min | RC = 3 | 2664 | 61% | 152 | 60 | 2.5 |
| RD = 10 s | ||||||
| DX = 60 s | ||||||
| Min MS = 1500 | ||||||
| S.P.P. 50 mm/20 min | RC = 3 | 7322 | 72% | 406 | 68 | 6.0 |
| RD = 10 s | ||||||
| *DX = 10 s | ||||||
| Min MS = 1500 | ||||||
| S.P.P. 50 mm/20 min | RC = 3 | 7372 | 78% | 469 | 75 | 6.3 |
| RD = 10 s | ||||||
| DX = 60 s | ||||||
| *Min MS = 1000 | ||||||
| S.P.P. 50 mm/20 min | *RC = 2 | 6083 | 70% | 381 | 72 | 5.3 |
| RD = 10 s | ||||||
| DX = 60 s | ||||||
| Min MS = 1500 | ||||||
| S.P.P. 50 mm/20 min | *RC = 1 | 4651 | 82% | 365 | 85 | 4.3 |
| RD = 10 s | ||||||
| DX = 60 s | ||||||
| Min MS = 1500 | ||||||
| S.P.P. 50 mm/20 min | RC = 3 | 7065 | 71% | 325 | 63 | 5.2 |
| *RD = 5 s | ||||||
| DX = 60 s | ||||||
| Min MS = 1500 | ||||||
| S.P.P. 50 mm/20 min | RC = 3 | 6657 | 73% | 347 | 67 | 5.2 |
| *RD = 15 s | ||||||
| DX = 60 s | ||||||
| Min MS = 1500 |
Experimental conditions used for analysis and metrics from Mascot database searching to assess the impact of each setting.
Conditional change. RC, Repeat count; RD, repeat duration; DX, dynamic exclusion; Min MS, minimum MS signal. All experiments included Microscans = 1; Max inject time = 50 ms.
The ultimate DDA setting to be optimized was dynamic exclusion. Preferred conditions for repeat count, repeat duration, and minimum MS signal were applied for dynamic exclusion data collection to determine a best fit for fast-gradient conditions. Whereas longer dynamic exclusion times produced less-redundant spectra, fewer peptide identifications were observed, shown in Table 4. Most tryptic peptides carrying a +2 charge state will have a m/z in the range of 400–700. Long dynamic exclusion times, when combined with the limited resolution of the LTQ MS will significantly hamper peptide identifications in a proteomic application, with many peptides displaying a similar m/z for a complex tryptic digest. The preferred dynamic exclusion time was 30 s. This was selected as approximately three times the average peak width at base, thereby providing sufficient time for exclusion of peaks displaying some degree of tailing, whereas not being too long, and thereby, covered a significant portion of the short gradient. Although additional dynamic exclusion times could be examined in more detail, the objective was to improve MS/MS spectra collection, not to optimize DDA settings specifically to BSA. This strategy improved MS/MS acquisition for application to fast-gradient conditions, permitting reduction of experiment time. Based on preferred MS parameter settings, fast-gradient conditions produced similar peptide identifications and protein-sequence coverage, as did the previous “typical” conditions, when used for standard tryptic-digest peptides.
Table 4.
Mascot Search Results from LC-MS/MS Analysis of Tryptic-Digested BSA Using Different Dynamic Exclusion Times
| Application of preferred settings | Condition change | Mascot score | Sequence coverage | Matched spectra | Peptides identified | Spectra/peptide identification |
|---|---|---|---|---|---|---|
| S.P.P. 50 mm/20 min | DX = 15 s | 2749 | 83% | 248 | 83 | 3.0 |
| S.P.P. 50 mm/20 min | DX = 30 s | 2845 | 83% | 221 | 89 | 2.5 |
| S.P.P. 50 mm/20 min | DX = 60 s | 1699 | 82% | 152 | 74 | 2.1 |
| S.P.P. 50 mm/20 min | DX = 120 s | 1385 | 72% | 114 | 64 | 1.8 |
Data were obtained using the optimum DDA settings determined in Table 3. Specifically, minimum MS signal 1000, repeat count one, and repeat duration 10 s were applied.
Proteomic Sample Analysis
The preferred DDA settings and separation conditions were evaluated with an authentic proteomic sample mixture, using peptides from a tryptic digest of a whole cell lysate of procyclic T. brucei, applying all four experimental conditions described above. The superficially porous particles displayed increased peptide and protein identifications, as shown in Table 5. With the use of experimentally optimized DDA settings, fast-gradient conditions with superficially porous particles produced the highest number of peptide and protein identifications. This condition also produced the fewest number of redundant spectra, as shown by the lowest spectra/peptide identification average. An analysis of peptide identifications/min also confirmed that the most efficient use of instrument time was with the use of fast-gradient conditions with superficially porous particles, with an improvement of nearly sixfold relative to previously used separation conditions and instrument settings.
Table 5.
Proteomic Results from T. brucei Analysis under Various Chromatographic Conditions with the Application of Optimized DDA Settings
| Column | Flow rate | Exp. time | Protein identifications | Matched spectra | Peptide identifications | Spectra/peptide | Peptide/min |
|---|---|---|---|---|---|---|---|
| Porous 50 mm | 4 μL/min | 90 min | 22 | 370 | 121 | 3.1 | 2.1 |
| Porous 50 mm | 9 μL/min | 21 min | 15 | 376 | 86 | 4.4 | 9.4 |
| S.P.P. 50 mm | 4 μL/min | 90 min | 43 | 439 | 139 | 3.2 | 2.4 |
| S.P.P. 50 mm | 9 μL/min | 21 min | 45 | 477 | 185 | 2.6 | 11.9 |
Results from the analysis of soluble proteins from T. brucei using all four experimental conditions examined. Each condition displays data from duplicate analysis with protein and peptide identifications validated using a 5% false discovery rate. Peptide/min reflects the total number of peptides identified for each condition divided by total experiment time for duplicate sample analysis.
DISCUSSION
The increase in instrument use efficiency using fast-separation conditions required optimization of MS instrument DDA settings to acquire good-quality MS/MS spectra. Relevant DDA settings included minimum MS signal counts, dynamic exclusion, repeat count, and repeat duration. An appropriate scheme to optimize DDA parameters to match the narrow chromatographic peaks, resulting from fast separation conditions, was uncovered. The change from columns packed with porous particles to columns of superficially porous particles allowed for operation at elevated flow rates while doubling the peak capacity. Analysis of T. brucei tryptic digests demonstrated the use of optimized DDA settings and fast separation techniques. In the evaluation of optimized conditions, the number of protein identifications doubled, the number of peptide identifications increased by >50%, and the experiment time was reduced by a factor of five.
ACKNOWLEDGMENTS
This work was supported by U.S. National Institutes of Health (GM077688 and GM093747) and the U.S. National Institutes of Health Integrated Technology Resource for Biomedical Glycomics grant P41RR018502.
REFERENCES
- 1. Aebersold R, Mann M. Mass spectrometry-based proteomics. Nature 2003;422:198–207 [DOI] [PubMed] [Google Scholar]
- 2. Chen CHW. Review of a current role of mass spectrometry for proteome research. Anal Chim Acta 2008;624:16–36 [DOI] [PubMed] [Google Scholar]
- 3. Salisbury JJ. Fused-core particles: a practical alternative to sub-2 micron particles. J Chromatogr Sci 2008;46:883–886 [DOI] [PubMed] [Google Scholar]
- 4. Fekete S, Fekete J, Ganzler K. Characterization of new types of stationary phases for fast liquid chromatographic applications. J Pharm Biomed Anal 2009;50:703–709 [DOI] [PubMed] [Google Scholar]
- 5. DeStefano J, Langlois T, Kirkland J. Characteristics of superficially-porous silica particles for fast HPLC: some performance comparisons with sub-2-m particles. J Chromatogr Sci 2008;46:254–260 [DOI] [PubMed] [Google Scholar]
- 6. Ali I, Gaitonde VD, Grahn A. Halo columns: new generation technology for high speed liquid chromatography. J Chromatogr Sci 2010;48:386–394 [DOI] [PubMed] [Google Scholar]
- 7. Kiss I, Bacskay I, Kilár F, Felinger A. Comparison of the mass transfer in totally porous and superficially porous stationary phases in liquid chromatography. Anal Bioanal Chem 2010;397:1307–1314 [DOI] [PubMed] [Google Scholar]
- 8. Marchetti N, Guiochon G. High peak capacity separations of peptides in reversed-phase gradient elution liquid chromatography on columns packed with porous shell particles. J Chromatogr A 2007;1176:206–216 [DOI] [PubMed] [Google Scholar]
- 9. Nguyen DTT, Guillarme D, Rudaz S, Veuthey JL. Fast analysis in liquid chromatography using small particle size and high pressure. J Sep Sci 2006;29:1836–1848 [DOI] [PubMed] [Google Scholar]
- 10. Hsieh Y, Duncan CJG, Brisson JM. Fused-core silica column high-performance liquid chromatography/tandem mass spectrometric determination of rimonabant in mouse plasma. Anal Chem 2007;79:5668–5673 [DOI] [PubMed] [Google Scholar]
- 11. Schuster SA, Boyes BE, Wagner BM, Kirkland JJ. Fast high performance liquid chromatography separations for proteomic applications using Fused-Core® silica particles. J Chromatogr A 2011;1228:232–241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Kirkland JJ, Langlois TJ, DeStefano JJ. Fused core particles for HPLC. American Laboratory. South San Francisco, CA, USA: American Laboratory, 2007 [Google Scholar]
- 13. Schuster S, Wagner B, Boye B, Kirkland J. Wider pore superficially porous particles for peptide separations by HPLC. J Chromatogr Sci 2010;48:566–571 [DOI] [PubMed] [Google Scholar]
- 14. Gritti F, Leonardis I, Abia J, Guiochon G. Physical properties and structure of fine core-shell particles used as packing materials for chromatography: relationships between particle characteristics and column performance. J Chromatogr A 2010;1217:3819–3843 [DOI] [PubMed] [Google Scholar]
- 15. Fairchild JN, Walworth MJ, Horváth K, Guiochon G. Correlation between peak capacity and protein sequence coverage in proteomics analysis by liquid chromatography-mass spectrometry/mass spectrometry. J Chromatogr A 2010;1217:4779–4783 [DOI] [PubMed] [Google Scholar]
- 16. Andrews GL, Dean RA, Hawkridge AM, Muddiman DC. Improving proteome coverage on a LTQ-Orbitrap using design of experiments. J Am Soc Mass Spectrom 2011;22:773–783 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Zhang Y, Wen Z, Washburn M, Florens L. Effect of dynamic exclusion duration on spectral count based quantitative proteomics. Anal Chem 2009;81:6317–6326 [DOI] [PubMed] [Google Scholar]