

Abstract
Despite their beneficial anti-inflammatory properties, eicosapentaenoic acid (EPA) and docosahexaenoic acid (DHA) may increase the infection risk at high doses, likely by generating an immune-depressed state. To assess the contribution of different immune cell populations to the immunomodulatory fatty acid effect, we comparatively investigated several aspects of inflammation in human T-helper (Th) cells and monocytes. Both fatty acids, but DHA to a lesser extent compared with EPA, selectively and dose-dependently reduced the percentage of cytokine-expressing Th cells in a peroxisome proliferator-activated receptor (PPAR)γ-dependent fashion, whereas the expression of the cell surface marker CD69 was unaltered on activated T cells. In monocytes, both EPA and DHA increased interleukin (IL)-10 without affecting tumor necrosis factor (TNF)-α and IL-6. Cellular incorporation of EPA and DHA occurred mainly at the expense of arachidonic acid. Concomitantly, thromboxane B (TXB)2 and leukotriene B (LTB)4 in supernatants decreased, while levels of TXB3 and LTB5 increased. This increase was independent of activation and in accordance with cyclooxygenase expression patterns in monocytes. Moreover, EPA and DHA gave rise to a variety of mono- and trihydroxy derivatives of highly anti-inflammatory potential, such as resolvins and their precursors. Our results suggest that EPA and DHA do not generally affect immune cell functions in an inhibitory manner but rather promote pro-resolving responses.
Keywords: cytokines, eicosapentaenoic acid, Th cells, docosahexaenoic acid
Long-chain polyunsaturated fatty acids of the omega-3 series (n-3 LC-PUFA), such as eicosapentaenoic acid (EPA, C20:5n-3) and docosahexaenoic acid (DHA, C22:6n-3), are unanimously considered to be health promoting due to their anti-inflammatory potential (reviewed in Ref. 1). Primarily, they are claimed to reduce the incidence and mortality of cardiovascular and angiopathy disorders (reviewed in Refs. 2, 3). In addition, a substantial number of studies have demonstrated their supportive efficacy to alleviate disease-specific symptoms in chronic immune-associated disorders, such as bronchial asthma (4, 5), psoriasis (6), rheumatoid arthritis (7), and inflammatory bowel disease (reviewed in Ref. 8). Moreover, EPA and DHA supplementation was shown to reduce episodes and duration of illnesses in children and might therefore be valuable for maintaining health (9).
On the other hand, a growing body of literature suggests that excessive dietary supplementation with EPA and DHA can be deleterious in certain immunocompromised disease states. Likely by generating a hyporesponsive host environment, high-dose fish oil consumption was shown to facilitate viral infections, to impair bacterial resistance, to delay pathogen clearance, to aggravate acute tissue injury (10–13), and even to be implicated in the development of pathogen-associated colitis and gastrointestinal cancer in rodents (14). Though evidence-based data for such an impairment of immunity in humans is limited, controversial, and still not conclusive (15, 16), the German Federal Research Institute for Risk Assessment recommended the setting of maximum levels for the fortification of foods with n-3 LC-PUFA to 1.5 g/day (17).
Investigating the mechanisms that underlie the fish oil dependent immunomodulation, most of the work has focused on the effects of orally administered EPA and DHA on immune markers reflected by ex vivo mitogen-induced lymphocyte proliferation, natural killer (NK) cell activity and cytokine production of T cells or monocytes. However, with respect to study population and design, methods of measurements and outcomes, available data are highly inconsistent and at times contradictory (thoroughly reviewed in Ref. 18). Moreover, studies on the effect of EPA and/or DHA on separate T-cell subsets, such as T-helper (Th) cells, are rare.
Therefore, the aim of the present study was to comparatively investigate several aspects of inflammation of pure EPA or DHA in primary human Th cells and monocytes in order to examine the contribution of these two immune cell populations, crucially involved in adaptive and innate immunity, to the immunomodulatory effect of n-3 LC-PUFA. For this purpose, we intended to match a largely realistic ex vivo representation on the one hand and highly standardized in vitro research on the other.
MATERIALS AND METHODS
Chemicals
EPA and DHA in free fatty acid form (both Larodan, Malmö, Sweden) were dissolved in sterile dimethylsulfoxide (DMSO) to produce a 100 mM stock solution and stored in aliquots at −20°C. Further, lyophilized 2-chloro-5-nitro-N-4-pyridinylbenzamide (T0070907), phorbol 12-myristate 13-acetate (PMA), ionomycin, brefeldin A (all Enzo, Lörrach, Germany), lipopolysaccharide (LPS from E.coli, Serotype 0111:B4, Sigma-Aldrich, Taufkirchen, Germany), and concanavalin A (ConA, Sigma-Aldrich) were solubilized in DMSO, aliquoted, and stored at −20°C.
Purification of PBMC
Peripheral blood mononuclear cells (PBMC) were isolated from buffy coats obtained from peripheral blood of healthy donors who gave their written consent. Buffy-coated blood was diluted with PBS (PAA, Cölbe, Germany) at a ratio of 1:1, layered onto lymphocyte separation medium (LSM) 1077 (1.077 g/ml; PAA; ratio 1:1) and centrifuged at 700 g for 20 min at 20°C. The PBMC interphase was collected, washed three times with PBS, and resuspended in Roswell Park Memorial Institute (RPMI) 1640 medium supplemented with 10% endotoxin-free heat-inactivated fetal bovine serum (PAA).
PBMC viability
To assess the impact of high-dose EPA and DHA, respectively, on cell viability, PBMC (1 × 106/ml) were incubated without or with 25, 50, 100, 150, or 200 µM of EPA or DHA for 24 h in a 5% CO2 humidified atmosphere at 37°C. Control cultures contained 0.2% DMSO vehicle, according to the maximal volume in the treatments. Cell viability was analyzed by annexin-V (Immunotech, Marseille, France) and propidium iodide (PI; Sigma-Aldrich) exclusion double staining. In brief, cells were washed with PBS, incubated in binding buffer and annexin-V or PI for 15 min at room temperature in the dark, and analyzed flow-cytometrically using an EPICS XL flow cytometer (Beckman Coulter, Krefeld, Germany).
Cell culture
For Th-cell assays, PBMC (1 × 106/ml) were incubated without or with increasing concentrations of EPA or DHA (25, 50, 100 µM) for 19 h. Subsequently, cells were alloreactively stimulated with PMA (2.5 ng/ml; induces cytokine production by activation of protein kinase C) and ionomycin (0.5 µg/ml; potentiates activation as calcium ionophore) in the presence of brefeldin A (5 µg/ml; increases sensitivity of intracellular cytokine detection by interfering with the function of the Golgi apparatus) for another 5 h. Supernatants were frozen at −80°C until lipid mediator analysis. For some experiments, cells were preincubated for 30 min with different concentrations of the selective peroxisome proliferator-activated receptor (PPAR)γ inhibitor T0070907 (0.4 or 2 µM) before 100 µM EPA or DHA and the stimulation cocktail were added as described above. For measurement of T-cell activation (expression of the cell surface marker CD69), PBMC were pretreated without or with increasing concentrations of EPA or DHA (25, 50, 100 µM) for 19 h and subsequently incubated with either 2.5 ng/ml PMA and 0.5 µg/ml ionomycin or 10 µg/ml ConA for further 5 h. For monocyte assays, PBMC were treated with fatty acids as indicated for 20 h before addition of 1 µg/ml LPS and 5 µg/ml brefeldin A for further 4 h. Control cultures contained maximum 0.2% DMSO. All experiments were performed under standard cell culture conditions.
Intracellular cytokine and cyclooxygenase detection
Both unstimulated and stimulated cells were stained either with anti-human CD3 monoclonal antibody (mAb; PE-Dy647, clone MEM-57, Immunotools, Friesoythe, Germany) and anti-human CD4 mAb (FITC, clone MEM-241, Immunotools) for detection of Th cells or with anti-human CD14 mAb (PE-Dy647, clone MEM-15, Immunotools) for identification of monocytes before cells were fixed with 2% formaldehyde (Histofix, Roth, Karlsruhe, Germany). For intracellular cytokine quantification, cells were permeabilized by washing with PBS/0.1% BSA/0.1% saponine, stained with anti-human tumor necrosis factor (TNF)-α mAb (PE, clone MAb11, eBioscience, Frankfurt/Main, Germany), anti-human interleukin (IL)-2 mAb (PE, clone MQ1-17H12, eBioscience), anti-human IL-4 mAb (PE, clone 8D4-8, eBioscience), anti-human interferon (IFN)-γ mAb (PE, clone 4S.B3, eBioscience), anti-human IL-6 mAb (PE, clone MQ2-13A5, eBioscience), or anti-human IL-10 mAb (PE, clone JES3-9D7) and analyzed by means of flow cytometry. Intracellular levels of cyclooxygenase (COX)-1 and COX-2 in CD14+ cells were determined with multicolor anti-human COX-1-FITC/anti-human COX-2-PE mAb (clones AS70/AS57, Becton Dickinson, Heidelberg, Germany). To assess T-cell activation, cells were stained with anti-human CD3 mAb and anti-human CD69 mAb (PE, clone FN50, Biolegend/Biozol, Eching, Germany). When necessary, probes were analyzed in reference to FMO (fluorescence minus one)-PE controls. Nonspecific fluorescence was controlled by incubation with isotype-matched antibodies. Data were assessed and illustrated by WinMDI version 2.8 software (J. Trotter, Scripps Research Institute), except for COX-2 in stimulated monocytes that was measured in a FACSCalibur flow cytometer (Becton Dickinson, Heidelberg Germany) and analyzed using CELLQUEST software (BD).
Fatty acid profiles
PBMC (1 × 106/ml) were cultured in the presence of increasing concentrations (25, 50, 100 µM) of EPA and DHA, respectively, or DMSO vehicle for 24 h, as indicated above. Cells were harvested and washed twice with PBS, and then total lipids were extracted using a methanol/chloroform mixture according to Ref. 19. For fatty acid analysis, a base-catalyzed transesterification method was performed by incubating samples with 0.5 N sodium methylate at 100°C for 10 min followed by methanolic boron trifluoride (10% w/w, Supelco, Bellefonte, PA) treatment at 100°C for 5 min. Afterwards, fatty acid methyl esters (FAME) were extracted with n-hexan, separated in a gas chromatograph (GC 17a V3, Shimadzu, Kyoto, Japan) using a fused-silica capillary column with medium polarity (DB 225 MS, 60 m × 0.25 mm id, 0.25 µm film thickness, Agilent Technologies, Santa Clara, CA) and downstream detected by flame ionization (FID). GC conditions were as previously described (20). Peak area integration was accomplished using GC Solution software version 2.3 in comparison to previously measured reference standards (BR2, BR4, and Menhaden from Larodan/CPS-Chemie, Aachen, Germany; 463 and 674 from Nu-Chek-Prep, Elysian, MN).
Lipid mediator profiles
PBMC (1 × 106/ml) were cultured in the presence of 100 µM EPA or DHA for 19 h and subsequently activated for another 5 h with the stimulation mixture as described above. Corresponding control cultures contained the same volume of DMSO. Afterwards, 1 ml supernatant was added to 1 ml methanol containing BHT (0.1%) and spiked with an internal standard consisting of 15-HETE-d8, PGE2-d4, and leukotriene B (LTB)4-d4 (each 1 µg/ml). After addition of 2 ml 0.1 mM sodium acetate buffer to adjust the pH at 6 and centrifugation, the upper phase was subjected to a solid-phase extraction using anion exchange columns (Varian Bond Elute Certify II, Agilent), which were preconditioned with 3 ml methanol, followed by 3 ml 0.01 M sodium acetate buffer containing 5% methanol (v/v, pH 6). The columns were then washed twice with 3 ml methanol/water (1/1, v/v) each. For elution, 2 ml of an ethyl acetate:n-hexane extraction mixture (1/1, v/v) was added. The eluate was evaporated in N2 at 40°C, and the solid residues were dissolved in 50 µl acetonitrile. For HPLC analysis, an Agilent 1200SL system equipped with a Phenomenex Kinentex column and coupled with an Agilent 6460 Triplequad mass spectrometer with electrospray ionization source was used. The solvent system consisted of acetonitrile and aqueous formic acid (0.1%). The elution gradient was started with 5% acetonitrile for 30 s, which was increased within 10 min to 90% and held for 8 min. The injection volume was 7.5 µl, and the flow rate was set at 0.4 ml/min. Lipid mediator analysis was performed with dynamic multiple reaction monitoring (MRM) in negative mode. For the preparation of standards and peak calculation, including detailed MRM characterization of all measured compounds, see Ref. 21. Peak integration and signal-to-noise calculation was accomplished using Agilent MassHunter Workstation software.
Statistics
For the statistical analysis of the data concerning concentration-dependent fatty acid effects, including cellular fatty acid profiles, Th-cell cytokines in the absence or presence of T0070907, and lipid mediator profiles, linear mixed models were used with the concentration of the corresponding fatty acid/cytokine being the dependent variable, while the treatment (DHA versus EPA) and the concentration (of DHA/EPA or T0070907) were entered as independent variables (fixed effects). For the lipid mediator profiles, treatment and stimulation condition were used as independent variables. For all models, a random intercept per donor was integrated to control for interindividual differences. Visual inspection of QQ-plots of the residuals and plots of the residuals versus fitted response values allowed us to justify the assumptions of the linear mixed models, namely homoscedasticity and normal distribution of the residuals. In cases where the assumption of variance homogeneity could not be met, we extended the variance structure of the models by including a treatment-specific variance structure. We tested better model fit of the extended model by means of a likelihood ratio test (LRT). If the LRT was significant on the 0.05 level, the model with adapted variance structure was preferred. Statistical analysis was carried out using SAS software 9.2 and R 2.15.0 (R Foundation for Statistical Computing, Vienna, Austria). All other data were compared by means of 2-tailed Student t-test using SPSS software version 19.0 (SPSS Inc., Chicago, IL). All data are reported as means ± SEM (or SD as indicated) from at least five independent experiments (unless indicated otherwise). Significance of difference was set at P < 0.05.
RESULTS
Neither EPA nor DHA affect viability of human leukocytes
First we tested whether incubation with EPA or DHA at high concentration has an impact on the viability of primary lymphocytes and monocytes over a period of 24 h. Compared with the DMSO control, neither EPA nor DHA showed any cytotoxic effect at 100 µM (Fig. 1). A drop in viability was observed only at concentrations higher than 150 µM (data not shown). Based on these data, a maximum fatty acid concentration of 100 µM was used in all subsequent experiments.
Fig. 1.

EPA and DHA do not affect cell viability. Cell viability was flow-cytometrically assessed by annexin-V and propidium iodide exclusion double staining. (A) Forward scatter (FS) against side scatter (SS) dot plot of human PBMC after alloreactive stimulation. Data acquisition was set to lymphocytes (L) and monocytes (M) within the scatter gate R1 of 50,000 total counts. (B) Representative histograms of fluorescence intensities of PBMC treated without or with 100 µM fatty acid for 24 h. Relative to the control (ctrl), annexin-V-positive and PI-negative cells were defined as early apoptotic cells; annexin-V-positive and PI-positive cells were defined as late apoptotic and necrotic cells.
EPA reduces intracellular IL-2, TNF-α, and IL-4, but not IFN-γ, more strongly than does DHA in Th cells
The expression kinetics of IL-2 and TNF-α and their stable presence in relatively high proportions within Th cells following short-term stimulation enable comparable and reliable measurements in vitro (22). In activated cell cultures, 18.7 ± 1.7% of the T cells (CD3+) were identified as Th cells (CD3+CD4+) positive for IL-2 and 25.6 ± 2.2% were positive for TNF-α Fig. 2A, B). After 24 h incubation with EPA, the percentage of IL-2-positive and TNF-α-positive Th cells dose-dependently decreased by up to 81% and 73% to 2.7 ± 0.5% and 4.2 ± 0.9%, respectively (P < 0.001). Simultaneously, the mean fluorescence intensity (MFI) reflecting the cytokine levels on a per-cell basis dose-dependently decreased. DHA also significantly reduced the intracellular content of IL-2 and TNF-α in activated Th cells by 61% and 71% to 7.3 ± 1.2% and 7.5 ± 1.2%, respectively (P < 0.001). The reduction in cytokine levels caused by DHA treatment was significantly weaker than that ascertained for EPA (P = 0.005 for IL-2 and P = 0.032 for TNF-α) at all concentrations tested. Correlation analysis revealed a stronger negative association between the cellular uptake of EPA in PBMC and the reduction in the IL-2- and TNF-α-positive Th-cell population, respectively, than of DHA (Fig. 2C). Regarding the Th-cell population positive for IL-4, a signature cytokine produced by Th2-type T cells (23), also a significant fatty acid-mediated and concentration-dependent decrease was found (P = 0.002). However, the reduction observed after incubation with EPA tended to be more pronounced than in the presence of DHA (P = 0.087; Fig. 2A). The fatty acid-mediated inhibition of cytokine production in Th cells was not general, as neither EPA nor DHA affected the level of IFN-γ, classically characterizing the Th1 lineage (23), at any tested concentration (Fig. 2A).
Fig. 2.



Effect of EPA and DHA on cytokine production of Th cells (CD3+CD4+). (A) PBMC from buffy coats were incubated without or with increasing concentrations of EPA or DHA for 19 h and activated by PMA and ionomycin for subsequent 5 h. Th cells were identified by staining for CD3 and CD4. Expression of IL-2, TNF-α, IL-4, and IFN-γ was analyzed by means of flow cytometry. Right scales denote mean fluorescence intensity (MFI) depicted as dots. (B) Representative dot plots of lymphocytes stained for CD3, CD4, and TNF-α. (C) Levels of IL-2 (left) and TNF-α (right) are negatively associated with cellular incorporation of EPA and DHA as determined by gas chromatographic analysis. Nonlinear curve-fitting was performed, and P values were calculated with regression analysis.
Both EPA- and DHA-mediated cytokine effects involve PPARγ
To investigate whether the fatty acids exert cytokine expression reducing effects via a PPARγ ligand-like action, cells were pretreated with the selective PPARγ antagonist T0070907 in different concentrations before 100 µM EPA or DHA and the alloreactive stimulation mixture were added. In the example of IL-2 and TNF-α, in both EPA- and DHA-treated cultures, the reduced cytokine-positive Th-cell population dose-dependently reexpanded in the presence of T0070907 (P < 0.001 for both IL-2 and TNF-α Fig. 3). These results clearly imply that PPARγ was involved as a mediator of the anti-inflammatory fatty acid effect.
Fig. 3.

Pre-treatment with T0070907 reverses the inhibitory effect of EPA and DHA on cytokine production of Th cells (CD3+CD4+). PBMC were pretreated for 30 min with 0.4 or 2 µM T0070907 before 100 µM fatty acids were added. After 19 h, cells were activated for subsequent 5 h. Cytokine expression of CD4+ T cells was flow-cytometrically analyzed. ***P < 0.001: main effect of T0070907 with no significant difference determined between EPA and DHA; n = 4.
Neither EPA nor DHA affect the expression of the T-cell activation marker CD69
Mitogen activation of PBMC resulted in a significant increase in the expression of the activation marker CD69 on T cells. In the presence of PMA/ionomycin, the percentage of CD69+ rapidly increased from less than 1% to greater than 95% of CD3+ lymphocytes (data not shown). Neither EPA nor DHA altered this percentage at any tested concentration. Likewise, the corresponding MFI remained unaltered (data not shown). To evaluate whether any fatty acid effect might have been overlaid by the strong stimulation effect, we gradually reduced the PMA concentration to one tenth of the initially used 2.5 ng/ml. However, the percentage of CD3+CD69+ constantly remained greater than 95% (data not shown). Thus, to ensure that the lacking effect of EPA and DHA on CD69 expression on T cells was true and not due to nonoptimal stimulation conditions for displaying any fatty acid effect, we repeated this experimental part with ConA as the mitogen stimulant. Upon ConA stimulation, the percentage of CD69+ increased to 35.4 ± 10.2% of CD3+ lymphocytes and remained unaltered in the presence of either EPA or DHA at any tested concentration (Fig. 4). Likewise, no changes in the corresponding MFI were observed (Fig. 4).
Fig. 4.
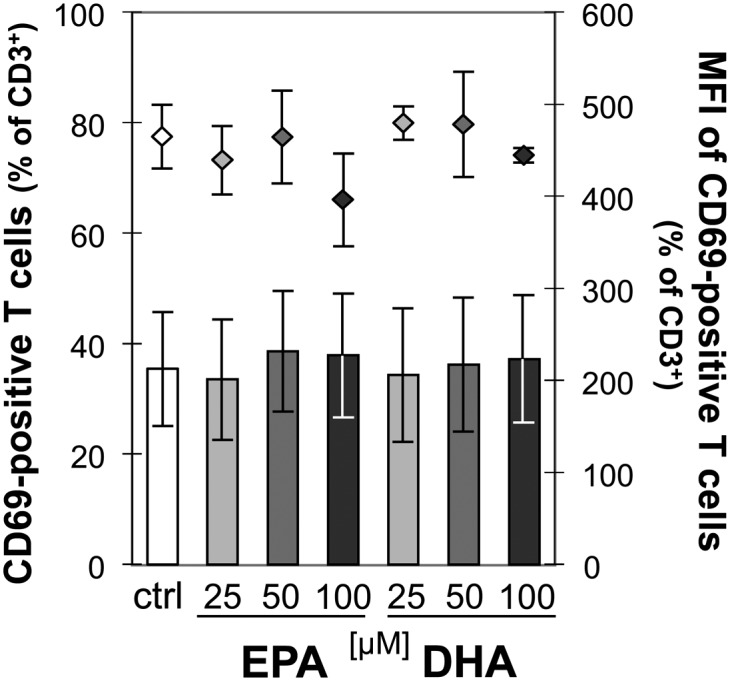
EPA and DHA do not affect CD69 expression on activated T cells. PBMC were incubated for 19 h without or with increasing concentrations of EPA or DHA before stimulation with ConA for subsequent 5 h. Cells were then stained for CD3 and CD69. Expression of cell surface markers was flow-cytometrically analyzed. Data are expressed as means ± SD of n = 3. Right scales denote mean fluorescence intensity (MFI) depicted as dots.
Both EPA and DHA increase anti-inflammatory IL-10 in monocytes
IL-10 is known to have immunoregulatory and suppressive function on T cells and to be important for the generation of regulatory T cells (Treg) (24). The spontaneous expression of IL-10 in monocytes was low but detectable: 6.2 ± 0.4% of the CD14+ cell population was determined to be IL-10 positive, and LPS activation of the cells did not alter this percentage (5.8 ± 0.1%; Fig. 5). However, the population of IL-10-positive monocytes significantly increased in the presence of EPA (9.3 ± 0.5%, P < 0.05 compared with the activated control) as well as DHA (10.5 ± 0.6%, P < 0.01).
Fig. 5.

EPA and DHA increase IL-10 production in monocytes. Intracellular levels of IL-10 were flow-cytometrically analyzed in LPS-stimulated compared with unstimulated monocytes. (A) Data are expressed as means ± SEM, a versus b: P < 0.05, 2-tailed Student's t-test. (B) Representative histograms of monocytes stained for CD14 and IL-10. Marker was set in reference to the isotype control.
Neither EPA nor DHA affects TNF-α and IL-6 production in monocytes
We next measured the levels of TNF-α and IL-6, as these cytokines are the most important produced by monocytes and macrophages. Their overexpression is implicated in several inflammatory conditions, e.g., in the context of the acute phase response (25). Upon activation by LPS, 71.8 ± 7.5% of the CD14+ cell population were found to be TNF-α positive and 19.5 ± 6.0% expressed IL-6 (Fig. 6). Since both EPA and DHA exerted a strong inhibition on the expression of pro-inflammatory cytokines, such as TNF-α, in Th cells, it was surprising that the percentages of TNF-α− and IL-6-positive monocytes remained completely unaffected in the presence of EPA and DHA at 100 µmol/l (Fig. 6) or less (data not shown), respectively.
Fig. 6.

EPA and DHA have no effects on TNF-α and IL-6 production in monocytes. Intracellular levels of TNF-α and IL-6 were flow-cytometrically analyzed in LPS-stimulated monocytes. (A) Data are expressed as means ± SEM. (B) Representative histograms of monocytes stained for CD14 and TNF-α. w/o stim, without stimulant. Marker was set in reference to the isotype control.
Cellular incorporation of both EPA and DHA is negatively associated with arachidonic acid
Since the strong reduction in TNF-α was clearly associated with the cellular incorporation of EPA and DHA (Fig. 2C), but only present in the T-cell population, we took a closer look at effects other than cytokine. We determined the impact of EPA and DHA on the metabolism of arachidonic acid whose derivatives are crucially involved in inflammation and known to frequently counteract those derived from EPA and DHA (26).
The base levels of EPA and DHA in native primary PBMC were comparatively low (0.3 ± 0.1% and 2.1 ± 0.2% of total FAME, respectively). Stearic acid (C18:0) was identified as main fatty acid (29.5 ± 0.7% of total FAME), followed by palmitic acid (C16:0; 15.3 ± 0.8%) and arachidonic acid (C20:4n-6; 10.4 ± 1.2%). The increase in the proportion of EPA within the cellular lipid fraction, which almost exclusively consists of phospholipids (supplementary Fig. I), entailed a dose-dependent reduction in the percentage of C20:4n-6 (P < 0.001), whereas the main fatty acid C18:0 (31.3 ± 1.6% at 100 µM EPA) did not quantitatively change (Fig. 7). Interestingly, the reduction of n-6 PUFA in sum (from 24.2 ± 1.6% to 18.9 ± 0.6% at 100 µM EPA, P < 0.05, 2-tailed Student t-test) was solely due to the decrease in C20:4n-6, as the percentages of C18:2n-6, C18:3n-6, C22:2n-6, and C22:4n-6 remained unaltered (data not shown). DHA likewise dose-dependently caused a drop in the proportion of C20:4n-6 (Fig. 7). This DHA-caused effect tended to be less pronounced than that seen for EPA (P = 0.087). However, the cellular enrichment of DHA in response to the supplementation was disproportionately higher compared with EPA (17.5 ± 2.3% versus 7.4 ± 1.7% at 100 µM; Fig. 7), mainly at the expense of saturated fatty acid (in sum, 46.3 ± 2.2% at 100 µM DHA, P < 0.01 versus both ctrl: 55.5 ± 2.6% and 100 µM EPA: 56.8 ± 3.7%, 2-tailed Student t-test), in particular of C18:0 (P = 0.003 versus EPA; Fig. 7A). For C18:0, an interaction effect between fatty acid treatment and concentration was found (P = 0.008). However, as DHA usually is in the sn-2 position, whereas C18:0 is positioned at sn-1, it is likely that DHA replaced C18:0 in neutral lipids rather than the phospholipids.
Fig. 7.

Cellular incorporation of EPA and DHA dose-dependently decreases arachidonic acid (C20:4n-6). Total lipids were extracted from PBMC after 24 h incubation with increasing concentrations of EPA or DHA, and fatty acid profiles were determined by gas chromatographic analysis. (A) Data are expressed as means ± SEM relating to 100% detectable fatty acids (as FAME). (B) Partial GC chromatograms of PBMC treated either with EPA (upper chart) or DHA (lower chart) in increasing concentrations. n.d., not detected.
EPA inhibits COX-2 expression in monocytes and formation of arachidonic acid oxygenation products more strongly than does DHA
As the drop in C20:4n-6 was likely not due to its increased catabolism (percentages of shorter chain n-6 fatty acids remained unaltered), we next examined whether an increased metabolism (e.g., via cyclo- or lipoxygenation) took place.
Upon stimulation, the formation of bioactive lipid mediators deriving from C20:4n-6, such as PGE2, thromboxane B (TXB)2 (both metabolized by COX-2), LTB4 [generated via 5-lipoxygenase (LO) pathway], and 12-HETE (generated via 12-LO pathway), which are often causally involved in pathophysiological processes (27), increased (Fig. 8A). At 100 µM, EPA, more clearly than DHA, prevented this increase (significant for TXB2: P = 0.021 versus ctrl, and LTB4: P = 0.017 versus ctrl; Fig. 8A). In accordance, COX-2 abundance in stimulated monocytes decreased in the presence of EPA (Fig. 8B, C). However, this reduction was only by trend (P = 0.1, 2-tailed Student t-test).
Fig. 8.

EPA impairs arachidonic acid metabolism stronger than DHA. (A) Enzyme-dependent oxygenation products of arachidonic acid converted by COX-2 (PGE2 and TXB2), 12-LO [12(S)-HETE] and 5-LO (LTB4) determined in PBMC supernatants. Gray bars, PBMC were alloreactively activated; open bars, unstimulated PBMC. Means ± SEM of n = 4. (B) COX-2 expression in stimulated monocytes. PBMC from buffy coats were incubated without or with 100 µM EPA or DHA for 20 h and subsequently activated for another 4 h with LPS. Monocytes were identified by staining for CD14. Expression of COX-2 was flow-cytometrically analyzed by intracellular staining. Upper graph: Data are expressed as means ± SEM of n = 5, t: P = 0.102 versus ctrl. Right scales denote mean fluorescence intensity (MFI) depicted as dots. Lower graph: Representative histograms of LPS-activated monocytes stained for CD14 and COX-2. Marker was set in reference to the unstimulated control.
Increase in both EPA- and DHA-derived lipid metabolite formations is independent of stimulation
In contrast to the arachidonic acid derived cyclo- and lipoxygenation products, the corresponding EPA products, namely TXB3 (COX-2) and LTB5 (5-LO), augmented significantly (Fig. 9A). Interestingly, the increase in EPA metabolite formation was independent of stimulation. Thus, we supposed that the presence of EPA sufficed to activate the oxygenation pathway. Indeed, here we show that COX-2 expression was induced by 100 µM EPA, but not DHA, in monocytes, even in the absence of any stimulant (P = 0.012; Fig. 9B).
Fig. 9.

Enzyme-dependent formation of oxygenated metabolites of EPA and DHA is independent of stimulation. (A) Enzyme-dependent oxygenation products of EPA converted by COX-2 (TXB3) and 5-LO (LTB5) determined in PBMC supernatants. Gray bars, PBMC were alloreactively activated; open bars, unstimulated PBMC. Means ± SEM of n = 4. (B) COX-2 expression in unstimulated monocytes. PBMC from buffy coats were incubated without or with 100 µM EPA or DHA for 24 h. Monocytes were identified by staining for CD14. Expression of COX-2 was flow-cytometrically analyzed by intracellular staining. Left: Data are expressed as means ± SEM of n = 5, a versus b: P < 0.05. Right: Representative histograms of monocytes stained for CD14 and COX-2. Marker was set in reference to the isotype control. (C) Monohydroxy products of EPA: (1) 5-HEPE, (2) 8-HEPE, (3) 9-HEPE, (4) 12-HEPE, (5) 15-HEPE, and (6) 18-HEPE. (D) Mono- (HDHA) and Trihydroxy products (Resolvin D1, RvD1) of DHA: (7) 4-HDHA, (8) 7-HDHA, (9) 8-HDHA, (10) 11-HDHA, (11) 14-HDHA, (12) 17-HDHA, and (13) 20-HDHA. Means ± SEM of n = 4.
COX-1 expression in monocytes was low (0.2–0.8% of CD14+) and unaffected by either fatty acid treatment or stimulation (data not shown).
Moreover, we detected a variety of monohydroxy derivatives in the supernatants of unstimulated and stimulated cultures after 24 h incubation with the fatty acids. Of the numerous HEPE generated from EPA, 18-HEPE, which can be further metabolized to the inflammation-dampening resolvins of the E-series (28), was predominant with 171.5 ± 7.5 ng/ml (Fig. 9C). Monohydroxy products from DHA were likewise abundantly detectable. Beside 4-, 20-, 17-, 8-, 7-, and 11-HDHA (quantitatively in descending order; Fig. 9D), 10-, 13-, and 16-HDHA were found in relevant quantities (data not shown), accounting for a total HDHA amount of 936.2 ± 326.6 ng/ml. Moreover, DHA gave rise to resolvin D1 (RvD1; Fig. 9D), a lipoxygenated trihydroxy position isomer with high anti-inflammatory potential (29). Likewise, alloreactive stimulation of PBMC did not further increase hydroxy product formation (Fig. 9C, D). Analysis of the medium supplemented with EPA or DHA in increasing concentrations but without PBMC revealed that HEPE as well as HDHA and subsequent RvD1 formation occurs nonenzymatically (supplementary Fig. II).
DISCUSSION
Th cells and monocytes are intrinsically involved in adaptive and innate immune responses. The present study aimed at distinctively elucidating the contribution of these two key mediators of cellular immunity to the immunomodulatory effect of EPA and DHA. For this purpose, we focused on the early production of cytokines and lipid mediators, since these strategies belong to the first being pursued in the acute state of defense.
Herein, we show that both EPA and DHA, but DHA to a lesser extent than EPA, dose-dependently decreased the intracellular amounts of immunostimulatory cytokines in Th cells. The extent of cytokine reduction was cytokine specific, affecting some (IL-2, TNF-α, IL-4), but not others (IFNγ) (Fig. 2). This finding corroborates previous reports on an EPA-mediated strong suppression of IL-2, whereas other cytokines, such as IFNγ, remained unaltered (30). Induction of IL-2 production in Th cells by alloreactive stimulation mimics the sophisticated response to T-cell receptor (TCR) engagement (31) and reflects the convergence of multiple signal transduction pathways on at least four unrelated families of transcription factors, such as nuclear factor of activated T cells (NFAT) and nuclear factor κB (NF-κB) (32). IL-2 itself is stimulus for further cytokines. Its signals are transduced by heterodimerized IL-2 receptors (IL-2R) via activation of phosphatidylinositol 3-kinase (PI 3-K)/AKT, Ras/mitogen-activated protein kinase (MAPK), and Janus kinase (JAK)/signal transducer and activator of transcription (STAT) pathways (33), and finally result in a specific cellular response (e.g., augmented cytokine production, including TNF-α but likely not IFNγ) (34). Therefore, the reduction in TNF-α herein likely depends on the reduction in IL-2.
PPARγ (NR1C3) is predominately expressed in Th cells (35), and the expression of its splice variant γ1 is therein inducible by agonist ligation (36). Since PPARγ activation by ligand binding antagonizes the pro-inflammatory capability of several transcription factors, such as NF-κB, STATs (37, 38), and NFAT, to control the expression of, e.g., IL-2 (39) and IL-4 (40) on the one hand, and pretreatment of the cultures with the selective PPARγ antagonist T0070907 (41) largely reverted the effect EPA and DHA on the other (Fig. 3), it is plausible that the fatty acids acted at least in part through PPARγ. Moreover, our results are in line with previous data on anti-inflammatory effects exerted by EPA and DHA via activation of PPARγ (42). However, it has also been shown that n-3 LC-PUFA PPARγ independently decrease activation of NF-κB in immune-competent cells (43).
A second mechanism underlying an altered T-cell function is that n-3 LC-PUFA induce biophysical and biochemical changes within the cell membrane, affecting both detergent-insoluble and nonraft membrane domains (44). For instance, the reorganization of these domains results in displacement of signal proteins, such as IL-2R, and/or impairment of relocalization of integrins, such as lymphocyte function-associated antigen-1 (LFA-1). Interference with the formation of the immunological synapse has substantial consequences for Ca2+-dependent cytoskeletal rearrangement, transcriptional regulation, and cytokine secretion (45–49). Moreover, it has been shown that EPA and DHA inhibit PMA-induced membrane recruitment of protein kinases C (PKC)-α and PKC-ϵ, which downstream leads to a decrease in nuclear translocation of NF-κB, resulting in an inhibition of IL-2 gene expression and cell proliferation (50). Although EPA and DHA treatment herein caused significant changes regarding the cellular fatty acid profiles (Fig. 7), we did not observe any fatty acid-mediated effect on the surface expression of CD25, the ligand-specific α-chain of the IL-2R, on activated T cells (unpublished findings). However, others showed a reduction in CD25 expression following DHA but not EPA treatment at 25 µM (51). In accordance with our findings, Zeyda et al. (30) and Kew et al. (52) observed no significant effect of EPA or DHA on the mitogen-induced expression of the early T-cell activation marker CD69, when expressed as percentage of CD69-positive cells (Fig. 4). In the latter study, however, confined to the MFI, which is related to the number of CD69 molecules expressed per T cell, a DHA-dependent inhibitory effect was shown, which we did not observe. Nevertheless, although the experimental design (mode of application, composition of supplemented fatty acids, concentration of EPA and DHA) substantially differed between that study and ours, the data concerning the percentage of CD69+ T cells obtained following ConA stimulation are largely comparable to one another.
Interestingly, with regard to cytokine production, a cell-type-specific fatty acid effect also existed, since neither EPA nor DHA reduced TNF-α or IL-6 (Fig. 6) and they even stimulated the expression of the immunoregulatory IL-10 (Fig. 5) in monocytes. The latter outcome might be of substantial significance as it indicates that EPA and DHA do not primarily act as immunosuppressives; rather, they promote resolution by selectively stimulating the production of pro-resolving cytokines. For instance, IL-10 induces the expression of the suppressor of cytokine signaling-3 (SOCS3), which inhibits JAK by acting as a pseudo-substrate and appears, therefore, to be responsible for the termination of IL-10 effects (reviewed in Ref. 24). Moreover, IL-10 has been suggested to directly and indirectly interfere with the NF-κB pathway primarily mediated by the activation of STAT3 (reviewed in Ref. 31). However, why IL-10 induces anti-inflammatory effects, whereas other cytokines, such as IL-6, which also signal through STAT3 do not, remains elusive (31). It is conceivable that EPA and DHA herein indirectly affect the monocyte response by maintaining IFNγ in Th cells (Fig. 2): IFNγ-polarized monocytes have been shown to preferably produce TNF-α and IL-6 in the presence of LPS (53). One group reported a negative association between the LPS-stimulated production of TNF-α and IL-6 by PBMC and n-3 PUFA intake, which, however, appeared to be characterized by a “U-shaped” dose-response relationship with maximum inhibitory effects noted at comparably low doses (54). This could be another explanation for the lacking effect of EPA and DHA herein on TNF-α and IL-6 in primary monocytes at high doses, as are generally used in vitro. However, since EPA and DHA in the aforementioned study were orally given as capsuled formula and no distinction has been made regarding different immune cell populations or subsets, the limitation for data comparison resides again in the differences between the approaches. In general, available data on the effects of EPA or DHA exerted on monocyte cytokine production are highly inconsistent. The majority of the studies have employed heterogeneous blends of EPA and DHA, which preclude examination of the individual effects.
Monocytes not only contribute to the immune response via the secretion of cytokines but also are a main population of immune cells with respect to COX-2-catalyzed eicosanoid formation. A rapid increase in the expression of COX-2, classically inducible by LPS in monocytes, has also been shown in T cells following PMA/ionomycin stimulation (55). The fact that EPA and DHA were not very effective in reducing COX-2 expression in monocytes (Fig. 8B, C) may, in some respect, be considered positive, as they were allowed to maintain their function to suppress T-cell immune responses and to govern the conversion of resting Th cells into such with a suppressive phenotype (56). Interestingly, the presence of substrate, here EPA, sufficed to increase the percentage of COX-2-positive cells (Fig. 9B), albeit to a much lesser extent than did the stimulation procedure. It has previously been shown that EPA induces COX-2, even in the absence of any stimulant, and that this induction is PPARγ-mediated (57).
Moreover, we show that EPA and DHA gave rise to a bunch of monohydroxylated derivatives (Fig. 9C, D) that can be further metabolized to highly bioactive compounds. Polyhydroxylated and conjugated di-, tri,- or tetraene derivatives of EPA and DHA are subjects of a new concept of resolution-phase mediators that elicit their anti-inflammatory and protective properties at concentrations in the nano- and picomolar range. The mechanisms underlying the action of such specialized pro-resolving mediators, collectively referred to as resolvins (resolution-phase interaction products, Rv), protectins (PD), and, most recently, maresins (macrophage mediators in resolving inflammation, MaR), are being systematically elucidated (58).
By means of gene array analyses, the body of data that contributes to the understanding of the molecular mechanisms leading to the effects of EPA and DHA has grown rapidly. A substantial number of genes have been revealed to be altered in human PBMC following middle- to long-term ingestion of EPA+DHA, of which many are involved in inflammatory processes, including several NF-κB target genes, pro-inflammatory cytokines, and genes crucial for eicosanoid synthesis (59, 60). However, we can conclude from our data that it is mandatory to distinguish between subpopulations of PBMC, since EPA and DHA differently influenced Th cells and monocytes and did not generally affect immune responses in a suppressive fashion. Moreover, our findings indicate that differences exist in the extent of the fatty acid-mediated effects. The conclusion to be drawn from our data, however, cannot be generally transferred to courses of inflammation, since the model system used here does not allow discriminating between acute and chronic states. This appears to be crucial, as available data show that, with respect to infections, n-3 LC-PUFA even improve host response under chronic conditions (61).
Altogether, our data may contribute to understanding of the regulatory action of EPA and DHA in immune cells and, therefore, to increasing knowledge in terms of fatty acid assessment from an immunological point of view.
Supplementary Material
Acknowledgments
The authors thank Lars Uhlmann, Gisela Schultheiss, Susanne Merkel, and Marina Giorgi from the Department of Physiology and Biochemistry of Nutrition, Federal Research Institute of Nutrition and Food, Karlsruhe, Germany, for their excellent technical assistance.
Footnotes
Abbreviations:
- CD
- cluster of differentiation
- COX
- cyclooxygenase
- DHA
- docosahexaenoic acid
- EPA
- eicosapentaenoic acid
- IFN-γ
- interferon gamma
- IL
- interleukin
- mAb
- monoclonal antibody
- LO
- lipoxygenase
- LPS
- lipopolysaccharide
- LTB
- leukotriene B
- MFI
- mean fluorescence intensity
- NK
- natural killer
- PBMC
- peripheral blood mononuclear cell
- PI
- propidium iodide
- PMA
- phorbol 12-myristate 13-acetate
- PPARγ
- peroxisome proliferator-activated receptor gamma
- T0070907
- 2-Chloro-5-nitro-N-4-pyridinylbenzamide
- Th
- T-helper
- TNF-α
- tumor necrosis factor alpha
- TXB
- thromboxane B
This work was supported by a grant from the German Research Foundation (DFG Ja 893/5).
The online version of this article (available at http://www.jlr.org) contains supplementary data in the form of two tables and four figures.
REFERENCES
- 1.Calder P. C. 2006. N-3 polyunsaturated fatty acids, inflammation, and inflammatory diseases. Am. J. Clin. Nutr. 83: 1505S–1519S. [DOI] [PubMed] [Google Scholar]
- 2.Wang C., Harris W. S., Chung M., Lichtenstein A. H., Balk E. M., Kupelnick B., Jordan H. S., Lau J. 2006. N-3 fatty acids from fish or fish-oil supplements, but not alpha-linolenic acid, benefit cardiovascular disease outcomes in primary- and secondary-prevention studies: a systematic review. Am. J. Clin. Nutr. 84: 5–17. [DOI] [PubMed] [Google Scholar]
- 3.León H., Shibata M. C., Sivakumaran S., Dorgan M., Chatterley T., Tsuyuki R. T. 2008. Effect of fish oil on arrhythmias and mortality: systematic review. BMJ. 337: a2931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Nagakura T., Matsuda S., Shichijyo K., Sugimoto H., Hata K. 2000. Dietary supplementation with fish oil rich in omega-3 polyunsaturated fatty acids in children with bronchial asthma. Eur. Respir. J. 16: 861–865. [DOI] [PubMed] [Google Scholar]
- 5.Mickleborough T. D., Lindley M. R., Ionescu A. A., Fly A. D. 2006. Protective effect of fish oil supplementation on exercise-induced bronchoconstriction in asthma. Chest. 129: 39–49. [DOI] [PubMed] [Google Scholar]
- 6.Balbás G. M., Regaña M. S., Millet P. U. 2011. Study on the use of omega-3 fatty acids as a therapeutic supplement in treatment of psoriasis. Clin. Cosmet. Investig. Dermatol. 4: 73–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Caughey G. E., James M. J., Proudman S. M., Cleland L. G. 2010. Fish oil supplementation increases the cyclooxygenase inhibitory activity of paracetamol in rheumatoid arthritis patients. Complement. Ther. Med. 18: 171–174. [DOI] [PubMed] [Google Scholar]
- 8.Calder P. C. 2008. Polyunsaturated fatty acids, inflammatory processes and inflammatory bowel disease. Mol. Nutr. Food Res. 52: 885–897. [DOI] [PubMed] [Google Scholar]
- 9.Thienprasert A., Samuhaseneetoo S., Popplestone K., West A. L., Miles E. A., Calder P. C. 2009. Fish oil n-3 polyunsaturated fatty acids selectively affect plasma cytokines and decrease illness in Thai schoolchildren: a randomized, double-blind, placebo-controlled intervention trial. J. Pediatr. 154: 391–395. [DOI] [PubMed] [Google Scholar]
- 10.Byleveld P. M., Pang G. T., Clancy R. L., Roberts D. C. 1999. Fish oil feeding delays influenza virus clearance and impairs production of interferon-gamma and virus-specific immunoglobulin A in the lungs of mice. J. Nutr. 129: 328–335. [DOI] [PubMed] [Google Scholar]
- 11.Auvin S., Collet F., Gottrand F., Husson M. O., Leroy X., Beermann C., Guery B. P. 2005. Long-chain polyunsaturated fatty acids modulate lung inflammatory response induced by Pseudomonas aeruginosa in mice. Pediatr. Res. 58: 211–215. [DOI] [PubMed] [Google Scholar]
- 12.McFarland C. T., Fan Y. Y., Chapkin R. S., Weeks B. R., McMurray D. N. 2008. Dietary polyunsaturated fatty acids modulate resistance to Mycobacterium tuberculosis in guinea pigs. J. Nutr. 138: 2123–2128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Schwerbrock N. M., Karlsson E. A., Shi Q., Sheridan P. A., Beck M. A. 2009. Fish oil-fed mice have impaired resistance to influenza infection. J. Nutr. 139: 1588–1594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Woodworth H. L., McCaskey S. J., Duriancik D. M., Clinthome J. F., Langohr I. M., Gardner E. M., Fenton J. I. 2010. Dietary fish oil alters T lymphocyte cell populations and exacerbates disease in a mouse model of inflammatory colitis. Cancer Res. 70: 7960–7969. [DOI] [PubMed] [Google Scholar]
- 15.Anderson M., Fritsche K. L. 2002. (n-3) fatty acids and infectious disease resistance. J. Nutr. 132: 3566–3576. [DOI] [PubMed] [Google Scholar]
- 16.Hooper L., Thompson R. L., Harrison R. A., Summerbell C. D., Ness A. R., Moore H. J., Worthington H. V., Durrington P. N., Higgins J. P., Capps N. E., et al. 2006. Risks and benefits of omega 3 fats for mortality, cardiovascular disease, and cancer: systematic review. BMJ. 332: 752–760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.BfR Opinion No. 030/2009. 2009. BfR recommends the setting of maximum levels for the fortification of foods with omega-3 fatty acids. Accessed December 3, 2012 at http://www.bfr.bund.de/cm/349/bfr_recommends_the_setting_of_maximum_levels_for_the_fortification_of_foods_with_omega_3_fatty_acids.pdf.
- 18.Sijben J. W., Calder P. C. 2007. Differential immunomodulation with long-chain n-3 PUFA in health and chronic disease. Proc. Nutr. Soc. 66: 237–259. [DOI] [PubMed] [Google Scholar]
- 19.Bligh E. G., Dyer W. J. 1959. A rapid method of total lipid extraction and purification. Can. J. Biochem. Physiol. 37: 911–917. [DOI] [PubMed] [Google Scholar]
- 20.Jaudszus A., Möckel P., Hamelmann E., Jahreis G. 2010. Trans-10,cis-12-CLA-caused lipodystrophy is associated with profound changes of fatty acid profiles of liver, white adipose tissue and erythrocytes in mice: possible link to tissue-specific alterations of fatty acid desaturation. Ann. Nutr. Metab. 57: 103–111. [DOI] [PubMed] [Google Scholar]
- 21.Gomolka B., Siegert E., Blossey K., Schunck W. H., Rothe M., Weylandt K. H. 2011. Analysis of omega-3 and omega-6 fatty acid-derived lipid metabolite formation in human and mouse blood samples. Prostaglandins Other Lipid Mediat. 94: 81–87. [DOI] [PubMed] [Google Scholar]
- 22.Mascher B., Schlenke P., Seyfarth M. 1999. Expression and kinetics of cytokines determined by intracellular staining using flow cytometry. J. Immunol. Methods. 223: 115–121. [DOI] [PubMed] [Google Scholar]
- 23.Mosmann T. R., Coffman R. L. 1989. Th1 and Th2 cells: Different patterns of lymphokine secretion lead to different functional properties. Annu. Rev. Immunol. 7: 145–173. [DOI] [PubMed] [Google Scholar]
- 24.Sabat R., Grütz G., Warszawska K., Kirsch S., Witte E., Wolk K., Geginat J. 2010. Biology of interleukin-10. Cytokine Growth Factor Rev. 21: 331–344. [DOI] [PubMed] [Google Scholar]
- 25.Haimovich B., Calvano J., Haimovich A. D., Calvano S. E., Coyle S. M., Lowry S. F. 2010. In vivo endotoxin synchronizes and suppresses clock gene expression in human peripheral blood leukocytes. Crit. Care Med. 38: 751–758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Calder P. C. 2001. Polyunsaturated fatty acids, inflammation and immunity. Lipids. 36: 1007–1024. [DOI] [PubMed] [Google Scholar]
- 27.Samuelsson B. 1991. Arachidonic acid metabolism: role in inflammation. Z. Rheumatol. 50: 3–6. [PubMed] [Google Scholar]
- 28.Isobe Y., Arita M., Matsueda S., Iwamoto R., Fujihara T., Nakanishi H., Taguchi R., Masuda K., Sasaki K., Urabe D., et al. 2012. Identification and structure determination of novel anti-inflammatory mediator resolvin E3, 17,18-dihydroxyeicosapentaenoic acid. J. Biol. Chem. 287: 10525–10534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Serhan C. N., Petasis N. A. 2011. Resolvins and protectins in inflammation resolution. Chem. Rev. 111: 5922–5943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zeyda M., Szekeres A. B., Säemann M. D., Geyeregger R., Stockinger H., Zlabinger G. J., Waldhäusl W., Stulnig T. M. 2003. Suppression of T cell signaling by polyunsaturated fatty acids: selectivity in inhibition of mitogen-activated protein kinase and nuclear factor activation. J. Immunol. 170: 6033–6039. [DOI] [PubMed] [Google Scholar]
- 31.Akdis M., Burgler S., Crameri R., Eiwegger T., Fujita H., Gomez E., Klunker S., Meyer N., O'Mahony L., Palomares O., et al. 2011. Interleukins, from 1 to 37, and interferon-γ: receptors, functions, and roles in diseases. J. Allergy Clin. Immunol. 127: 701–721. [DOI] [PubMed] [Google Scholar]
- 32.Jain J., Loh C., Rao A. 1995. Transcriptional regulation of the IL-2 gene. Curr. Opin. Immunol. 7: 333–342. [DOI] [PubMed] [Google Scholar]
- 33.Liao W., Lin J. X., Leonard W. J. 2011. IL-2 family cytokines: new insights into the complex roles of IL-2 as a broad regulator of T helper cell differentiation. Curr. Opin. Immunol. 23: 598–604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Reddy J., Chastagner P., Fiette L., Liu X., Thèze J. 2001. IL-2-induced tumor necrosis factor (TNF)-beta expression: further analysis in the IL-2 knockout model, and comparison with TNF-alpha, lymphotoxin-beta, TNFR1 and TNFR2 modulation. Int. Immunol. 13: 135–147. [DOI] [PubMed] [Google Scholar]
- 35.Clark R. B., Bishop-Bailey D., Estrada-Hernandez T., Hla T., Puddington L., Padula S. J. 2000. The nuclear receptor PPAR gamma and immunoregulation: PPAR gamma mediates inhibition of helper T cell responses. J. Immunol. 164: 1364–1371. [DOI] [PubMed] [Google Scholar]
- 36.Norazmi M. N., Mohamed R., Nurul A. A., Yaacob N. S. 2012. The modulation of PPARγ1 and PPARγ2 mRNA expression by ciglitazone in CD3/CD28-activated naïve and memory CD4+ T cells. Clin. Dev. Immunol. 2012: 849195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Delerive P., Fruchart J. C., Staels B. 2001. Peroxisome proliferator-activated receptors in inflammation control. J. Endocrinol. 169: 453–459. [DOI] [PubMed] [Google Scholar]
- 38.Wang P., Anderson P. O., Chen S., Paulsson K. M., Sjogren H. O., Li S. 2001. Inhibition of the transcription factors AP-1 and NF-kappaB in CD4 T cells by peroxisome proliferator-activated receptor gamma ligands. Int. Immunopharmacol. 1: 803–812. [DOI] [PubMed] [Google Scholar]
- 39.Yang X. Y., Wang L. H., Chen T., Hodge D. R., Resau J. H., DaSilva L., Farrar W. L. 2000. Activation of human T lymphocytes is inhibited by peroxisome proliferator-activated receptor gamma (PPARgamma) agonists. PPARgamma co-association with transcription factor NFAT. J. Biol. Chem. 275: 4541–4544. [DOI] [PubMed] [Google Scholar]
- 40.Chung S. W., Kang B. Y., Kim T. S. 2003. Inhibition of interleukin-4 production in CD4+ T cells by peroxisome proliferator-activated receptor-gamma (PPAR-gamma) ligands: involvement of physical association of between PPAR-gamma and the nuclear factor of activated T cells transcription factor. Mol. Pharmacol. 64: 1169–1179. [DOI] [PubMed] [Google Scholar]
- 41.Lee G., Elwood F., McNally J., Weiszmann J., Lindstrom M., Amaral K., Nakamura M., Miao S., Cao P., Learned R. M., et al. 2002. T0070907, a selective ligand for peroxisome proliferator-activated receptor gamma, functions as an antagonist of biochemical and cellular activities. J. Biol. Chem. 277: 19649–19657. [DOI] [PubMed] [Google Scholar]
- 42.Li H., Ruan X. Z., Powis S. H., Fernando R., Mon W. Y., Wheeler D. C., Moorhead J. F., Varghese Z. 2005. EPA and DHA reduce LPS-induced inflammation responses in HK-2 cells: evidence for a PPAR-gamma-dependent mechanism. Kidney Int. 67: 867–874. [DOI] [PubMed] [Google Scholar]
- 43.Draper E., Reynolds C. M., Canavan M., Mills K. H., Loscher C. E., Roche H. M. 2011. Omega-3 fatty acids attenuate dendritic cell function via NF-κB independent of PPARγ. J. Nutr. Biochem. 22: 784–790. [DOI] [PubMed] [Google Scholar]
- 44.Raza Shaikh S. 2010. Diet-induced docosahexaenoic acid non-raft domains and lymphocyte function. Prostaglandins Leukot. Essent. Fatty Acids. 82: 159–164. [DOI] [PubMed] [Google Scholar]
- 45.Li Q., Wang M., Tan L., Wang C., Ma J., Li N., Li Y., Xu G., Li J. 2005. Docosahexaenoic acid changes lipid composition and interleukin-2 receptor signaling in membrane rafts. J. Lipid Res. 46: 1904–1913. [DOI] [PubMed] [Google Scholar]
- 46.Geyeregger R., Zeyda M., Zlabinger G. J., Waldhäusl W., Stulnig T. M. 2005. Polyunsaturated fatty acids interfere with formation of the immunological synapse. J. Leukoc. Biol. 77: 680–688. [DOI] [PubMed] [Google Scholar]
- 47.Li Q., Tan L., Wang C., Li N., Li Y., Xu G., Li J. 2006. Polyunsaturated eicosapentaenoic acid changes lipid composition in lipid rafts. Eur. J. Nutr. 45: 144–151. [DOI] [PubMed] [Google Scholar]
- 48.Yog R., Barhoumi R., McMurray D. N., Chapkin R. S. 2010. N-3 polyunsaturated fatty acids suppress mitochondrial translocation of the immunological synapse and modulate calcium signaling in T cells. J. Immunol. 184: 5864–5873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Shaik S. R., Jolly C. A., Chapkin R. S. 2012. N-3 polyunsaturated fatty acids exert immunomodulatory effects on lymphocytes by targeting plasma membrane molecular organization. Mol. Aspects Med. 33: 46–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Denys A., Hichami A., Khan N. A. 2005. N-3 PUFAs modulate T-cell activation via protein kinase C-alpha and -epsilon and the NF-kappaB signaling pathway. J. Lipid Res. 46: 752–758. [DOI] [PubMed] [Google Scholar]
- 51.Gorjão R., Hirabara S. M., de Lima T. M., Cury-Boaventura M. F., Curi R. 2007. Regulation of interleukin-2 signaling by fatty acids in human lymphocytes. J. Lipid Res. 48: 2009–2019. [DOI] [PubMed] [Google Scholar]
- 52.Kew S., Mesa M. D., Tricon S., Buckley R., Minihane A. M., Yaqoob P. 2004. Effects of oils rich in eicosapentaenoic and docosahexaenoic acids on immune cell composition and function in healthy humans. Am. J. Clin. Nutr. 79: 674–681. [DOI] [PubMed] [Google Scholar]
- 53.Ambarus C. A., Santegoets K. C., van Bon L., Wenink M. H., Tak P. P., Radstake T. R., Baeten T. L. 2012. Soluble immune complexes shift the TLR-induced cytokine production of distinct polarized human macrophage subsets towards IL-10. PLoS ONE. 7: e35994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Trebble T., Arden N. K., Strout M. A., Wootton S. A., Burdge G. C., Miles E. A., Ballinger A. B., Thompson R. L., Calder P. C. 2003. Inhibition of tumour necrosis factor-alpha and interleukin 8 production by mononuclear cells following dietary fish-oil supplementation in healthy men and response to antioxidant co-supplementation. Br. J. Nutr. 90: 405–412. [DOI] [PubMed] [Google Scholar]
- 55.Lee J. Y., Choi A. Y., Oh Y. T., Choe W., Yeo E. J., Ha J., Kang I. 2012. AMP-activated protein kinase mediates T cell activation-induced expression of FasL and COX-2 via protein kinase C theta-dependent pathway in human Jurkat T leukemia cells. Cell. Signal. 24: 1195–1207. [DOI] [PubMed] [Google Scholar]
- 56.Bryn T., Yaqub S., Mahic M., Henjum K., Aandahl E. M., Taskén E. 2008. LPS-activated monocytes suppress T-cell immune responses and induce FOXP3+ T cells through a COX-2–PGE2-dependent mechanism. Int. Immunol. 20: 235–245. [DOI] [PubMed] [Google Scholar]
- 57.Chêne G., Dubourdeau M., Balard P., Escoubet-Lozach L., Orphila C., Berry A., Bernad J., Aries M. F., Charveron M., Pipy B. 2007. n-3 and n-6 polyunsaturated fatty acids induce the expression of COX-2 via PPARgamma activation in human keratinocyte HaCaT cells. Biochim. Biophys. Acta. 1771: 576–589. [DOI] [PubMed] [Google Scholar]
- 58.Bannenberg G., Serhan C. N. 2010. Specialized pro-resolving lipid mediators in the inflammatory response: an update. Biochim. Biophys. Acta. 1801: 1260–1273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Bouwens M., van de Rest O., Dellschaft N., Bromhaar M. G., de Groot L. C., Geleijnse J. M., Müller M., Afman L. A. 2009. Fish oil supplementation induces antiinflammatory gene expression profiles in human blood mononuclear cells. Am. J. Clin. Nutr. 90: 415–424. [DOI] [PubMed] [Google Scholar]
- 60.Vedin I., Cederholm T., Freund-Levi Y., Basun H., Garlind A., Irving G. F., Eriksdotter-Jönhagen M., Wahlund L. O., Dahlmann I., Palmblad J. 2012. Effects of DHA-rich n-3 fatty acid supplementation on gene expression in blood mononuclear leukocytes: the OmegAD study. PLoS ONE. 7: e35425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Pierre M., Husson M. O., Le Berre R., Desseyn J. L., Galabert C., Béghin L., Beermann C., Dagenais A., Berthiaume Y., Cardinaud B., et al. 2007. Omega-3 polyunsaturated fatty acids improve host response in chronic Pseudomonas aeruginosa lung infection in mice. Am. J. Physiol. Lung Cell. Mol. Physiol. 292: L1422–L1431. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.