

Abstract
In the past 15 years, the quantitative trait locus (QTL) mapping approach has been applied to crosses between different inbred mouse strains to identify genetic loci associated with plasma HDL cholesterol levels. Although successful, a disadvantage of this method is low mapping resolution, as often several hundred candidate genes fall within the confidence interval for each locus. Methods have been developed to narrow these loci by combining the data from the different crosses, but they rely on the accurate mapping of the QTL and the treatment of the data in a consistent manner. We collected 23 raw datasets used for the mapping of previously published HDL QTL and reanalyzed the data from each cross using a consistent method and the latest mouse genetic map. By utilizing this approach, we identified novel QTL and QTL that were mapped to the wrong part of chromosomes. Our new HDL QTL map allows for reliable combining of QTL data and candidate gene analysis, which we demonstrate by identifying Grin3a and Etv6, as candidate genes for QTL on chromosomes 4 and 6, respectively. In addition, we were able to narrow a QTL on Chr 19 to five candidates.
Keywords: high-density lipoprotein, quantitative trait locus, meta-analysis
In the past decade, high-density lipoprotein (HDL) cholesterol has emerged as a new potential therapeutic target for the treatment of cardiovascular disease (CVD). The key role of HDL as a carrier of excess cellular cholesterol in the reverse cholesterol transport pathway is believed to provide protection against atherosclerosis (1). Several epidemiologic studies have demonstrated that HDL is a strong inverse predictor of CVD risk (2–5). Although much is known about HDL metabolism and reverse cholesterol transport, we still do not have a complete understanding of all the genetic factors involved in the regulation of HDL levels. Also, HDL is not a single entity but can be subdivided into different subpopulations. The relationships among these different subpopulations are not understood, and the genes responsible for the subpopulation differences among individuals are unknown.
Several approaches, including genetic mapping studies, have been used to identify novel genes involved in the regulation of HDL levels. In mice, the technique of quantitative trait locus (QTL) mapping, which uses crosses between different inbred strains of mice, has been heavily employed over the past 15 years. These results have been summarized in several reviews (6, 7). One limitation of traditional QTL analysis is low mapping resolution, which is a result of the limited genetic recombination possible in one-generation backcrosses [i.e., A×(A×B)] and two-generation intercrosses [i.e., (A×B)×(A×B)]. The 95% confidence interval (CI), the interval in which the causative gene is most likely to reside, is usually very broad. For example, one large study of bone mineral density QTL found that the average CI width for traditionally mapped QTL was 32 cM (8). As it can be assumed that there are, on average, 20 genes per cM (9), the number of candidate genes per QTL can be very large, making the identification of the causative gene very difficult. In the past few years, several methods have been developed that combine accumulated data from the different crosses, allowing for narrowing of the CI for QTL and reducing candidate gene lists (10). However, the success of these methods heavily depends on the accuracy of the QTL mapping.
The current standard genetic map for the mouse is curated and maintained by the Mouse Genome Informatics (MGI) Group at The Jackson Laboratory (www.informatics.jax.org) (11). Mapping QTL requires accurate genetic map information for both the relative order of markers and the distances between them (12). Recently, Shifman and colleagues published a new genetic map based on a large population of a heterogeneous stock (13). Cox and colleagues integrated a total of 7,080 standard, simple-sequence length polymorphism (SSLP) markers to this single-nucleotide polymorphism (SNP)-based map, generating a corrected mouse genetic map (14). This new map resolved inconsistencies between the physical and genetic maps and is now the standard MGI genetic map, providing highly accurate genetic distances. A recent mapping study, in which the new and traditional genetic maps were compared, suggests that up to 20% of published QTL may have been mislocalized due to marker order and positioning errors in the old genetic map (14).
In addition, many advances have been made over the years in the statistics and insights underlying QTL mapping. For example, in the past, males and females were analyzed as one population, disregarding possible sex-associated differences in the phenotype, or as two separate populations, resulting in a loss of power. We now analyze males and females as one population, using sex as a covariate. This method results in power compared with analyzing them separately, and it takes into account the obvious differences between males and females. Another issue is the direction of the cross. While, for most crosses, all F1 animals were produced by crossing a female from strain A with a male from strain B, in some crosses, the F1 animals originated from reciprocal crosses (A×B and B×A) and the F2 progeny were analyzed regardless of the parental grandmother. Now that we are aware of the epigenetic differences that can occur depending on the direction of the cross, we use the paternal grandmother as a covariate when analyzing the data (12).
The different HDL QTL datasets have been analyzed in various ways using different genetic maps and often without the above-mentioned covariates. This has resulted in misplaced QTL, missed QTL, or QTL that were only suggestive because of the loss of power. In this study, we made a concerted effort to identify and collect as many published, historical HDL QTL datasets as possible and reanalyze them. While most of the data used in this study were generated by our research group, several datasets were kindly provided by other investigators. All data underwent rigorous quality control and were then analyzed using current statistical methods. In all cases, the new corrected genetic mouse map was used. By analyzing the different datasets in the same way and by using the same genetic map, we created a more consistent and complete mouse HDL QTL map. As we demonstrate in our study, this improved map will aid in the search for novel HDL genes.
METHODS
Identification of mapping-cross datasets
First, a literature search to identify published reports of mouse HDL QTL was done using the following key words: “HDL,” “QTL,” and “mouse.” Second, the Mouse Genome Informatics Database (www.informatics.jax.org/) was searched for HDL QTL using the Genes and Markers Query form. Specifically, this database was searched using the keyword “HDL” in the Gene/Marker Symbol/Name field, “QTL” in the Type field, and “Any” in the ‘Chromosome field; the No Limit box was checked under the heading of Maximum Returned. The cross in which each HDL QTL was mapped was identified and compared with the list of crosses identified by conventional literature search. In short, the result was a list of mouse mapping crosses in which HDL QTL were mapped regardless of the method used to measure HDL.
Main-effects QTL mapping
Map positions for the markers for all datasets were updated to the new mouse genetic map using a mouse map converter tool (http://cgd.jax.org). All QTL analyses were done using the R/qtl software package (12) (R Version 2.6.2, qtl Library Version 1.09-43, www.rqtl.org/). Data were examined for phenotypic outliers and for genotyping errors, as previously described (8). A single locus main-scan for QTL was performed, and LOD scores were calculated at 2 cM intervals across the genome using the EM (or expectation-maximization) method in R/qtl for all datasets. The LOD thresholds for significant and suggestive QTL were determined in a cross-specific manner based on 1,000 permutations of the data. A QTL was considered to be suggestive if the LOD score exceeded the P < 0.63 threshold and significant if it exceeded the P < 0.05 threshold. These thresholds were chosen because they are the widely accepted cutoffs for suggestive and significant QTL.
For 5 of the 23 crosses, data were available for both male and female mice (Table 1). To account for the average differences between the males and females, we carried out scans using sex as an additive covariate. To identify sex-dependent QTL effects, we carried out additional scans using sex as an interactive covariate and computed the differences in LOD scores between these two scans (i.e., the ΔLOD) (12). The interactive scan model identified the most likely position of the sex-specific QTL. Calculating the ΔLOD score at the peak position is the secondary test for the QTL-by-sex interaction. This secondary test is carried out with no adjustment for multiple testing, and the threshold, based on the usual chi-square distribution of the likelihood ratio, is 2.0 on the LOD scale. However, sex specificity of QTL was not confirmed by further analysis, and thus all “sex-specificity” of QTL should be considered putative. For 6 of the 23 crosses, data were available for two different cross directions (i.e., B×A and A×B, Table 1). Putative cross-direction-specific QTL were identified using the strain of paternal grandmother (pgm) for each mouse as both an additive and interactive covariate, as was done for sex-specific QTL. In only one cross, both males and females were examined in crosses generated in a reciprocal fashion. In this last dataset, the interactive term of sex:pgm was examined to identify QTL that interacted with sex and were cross-direction specific; no such QTL, however, were identified in this study.
TABLE 1.
Description of the cross datasets
| Cross | Year (Reference) | Number (Sex) | Directa | Age (Weeks) | Notes on Diet |
| (NZB×SM)NZB | 2002 (40) | 90 (F) | Uni | 8, 12, 16, 26 | Chow diet first 8 weeks, followed by atherogenic diet for 18 weeks |
| D2×CAST | 2003 (41) | 278 (M) | Bi | 16 | Chow diet first 8 weeks, followed by atherogenic diet for 8 weeks |
| B6×CASA | 2003 (15) | 184 (F) 185 (M) | Uni | 11 | Chow diet |
| B6×D2 | 2003 (16) | 111 (F) | Uni | 52, 68 | Chow diet first 52 weeks, followed by atherogenic diet for 16 weeks |
| (B6×NZB)B6 | 2003 (42) | 100 (F) | Uni | 8, 12, 23 | Chow diet first 8 weeks, followed by atherogenic diet for 15 weeks |
| SM×NZB | 2004 (43) | 259 (F) 254 (M) | Uni | 8, 14, 20, 24 | Chow diet first 8 weeks, followed by atherogenic diet for 16 weeks |
| B6×129S1 | 2004 (44) | 294 (F) | Uni | 20 | Chow diet first 6 weeks, followed by atherogenic diet for 14 weeks |
| 129S1×CAST | 2004 (45) | 277 (M) | Bi | 16 | Chow diet first 8 weeks, followed by atherogenic diet for 8 weeks |
| RIII×129S1 | 2004 (46) | 150 (M) 180 (M) | Bi | 16 20 | Chow diet first 8 weeks, followed by atherogenic diet for 8 or 12 weeks |
| PERA×I | 2006 (47) | 164 (F) 141 (M) | Bi | 16 | Chow diet first 8 weeks, followed by atherogenic diet for 8 weeks |
| D2×PERA | 2006 (47) | 166 (M) 158 (M) | Bi | 16 | Chow diet first 8 weeks, followed by atherogenic diet for 8 weeks |
| B6×A | 2006 (48) | 343 (M) | Uni | 10 | Chow diet |
| B6×A | 2006 (48) | 271 (F) | Uni | 8 | Chow diet |
| NZB×RF | 2007 (49) | 542 (F) | Uni | 10 | Chow diet |
| D2×DU6i | 2007 (50) | 228 (M) 174 (F) | Uni | 6 | Chow diet |
| B6.Apoe−/−×C3H.Apoe−/− | 2008 (51) | 241(F) | Uni | 18 | Chow diet first 6 weeks, followed by western diet for 12 weeks |
| B6×C3H | 2009 (52) | 277 (F) | Uni | 14 | Chow diet first 8 weeks, followed by atherogenic diet for 6 weeks |
| (NZO×NON)NON | 2009 (52) | 204 (M) | Bi | 24 | Chow diet |
| B6×D2 | 2009 (52) | 340 (M) | Uni | 8 | Chow diet |
| B6×129S1 | 2009 (19) | 242 (F) 249 (M) | Uni | 11, 18 | Chow diet first 11 weeks, followed by atherogenic diet for 7 weeks |
| NOD(NOD×129.H2g7) | 2009 (53) | 159 (F) | Uni | 40 | Chow diet |
| B6×NOD | Unpublished | 139 (M) | Uni | 10 | Chow diet |
| B6×NZW | Unpublished | 143 (M) | Uni | 10 | Chow diet |
Uni = all F1 animals were produced using the direction as indicated in the first column, with the first strain being the female; bi = F1 animals were produced using both directions.
Resequencing of SNPs
High-quality genomic DNA from the different inbred mouse strains was obtained from the Jackson Laboratory's DNA Resource (www.jax.org/dnares/). Primers were designed spanning the exon that contained the SNP and ordered from Integrated DNA Technologies. PCR and Sanger sequencing were performed using standard protocols. Sequence data were analyzed using Sequencher 4.9.
RESULTS
Datasets used in this study
From our literature search, we determined that 28 datasets would be of interest to us for this project. However, 5 of these datasets were no longer available due to loss of records or the inability to contact the lead authors. In sum, we obtained a total of 23 independent datasets (summarized in Table 1). Of these datasets, 19 were from our own laboratory. Of the remaining 4 datasets, 2 were kindly provided by Drs. Breslow and Lusis, respectively (15, 16), and the other two were obtained from the QTL archive (www.qtlarchive.org), a free public access repository for QTL mapping datasets.
A large number of main-effects QTL were mapped
The analyses for single main effect QTL resulted in 143 QTL (Table 2, Fig. 1). In many instances, our analyses confirmed QTL that had been previously reported in the literature, but in other instances, our analyses yielded disparate results when compared with the published findings (Figs. 2, 3). For example, we observed that some QTL moved significantly on the same chromosome, the confidence interval width changed for some QTL, and peak LOD scores were different for many QTL. These dissimilarities were caused by the discrepancies between the genetic map we used and those used in the original studies (14) and because of methodological differences in the way we conducted our analyses compared with the original (8).
TABLE 2.
Main scan QTL peaks
| Chr | Peak (cM) | Interval (cM) | LOD | Sex | Age (Weeks) | Diet | High | Cross | Remark |
| 1 | 44 | 18–66 | 1.9 | F (M?) | 18 | Ath | SM | (NZB×SM)NZB | |
| 45 | 29–76 | 2.2 | M+F | 11 | Chow | CASA | B6×CASA | Add | |
| 63 | 16–74 | 3.2 | F | 10 | Chow | RF | NZB×RF | Add | |
| 68 | 54–88 | 4.5 | M (F?) | 8 | Chow | CAST | CAST×129S1 | ||
| 70 | 52–80 | 7.4 | F (M?) | 8 | Chow | A | B×A | ||
| 70 | 46–82 | 2.8 | M+F | 10 | Chow | NOD | B6×NOD | ||
| 70 | 63–88 | 3.9 | M (F?) | 8 | Chow | RIII | 129×RIII | ||
| 72 | 62–76 | 5.8 | M+F | 16 | Ath | PERA | D2×PERA | ||
| 78 | 64–90 | 2.7 | M (F?) | 8 | Chow | DBA | B6×D2 | ||
| 78 | 74–80 | 27.9 | M+F | 16 | Ath | 129 | B6×129S1 | ||
| 80 | 78–80 | 51.2 | M+F | 8 | Chow | 129 | B6×129S1 | ||
| 80 | 76–84 | 15.0 | M+F | 10 | Chow | NZW | B6×NZW | ||
| 80 | 76–82 | 8.8 | F (M?) | 40 | Chow | Het | NOD×129S1 | ||
| 81 | 73–87 | 7.3 | F (M?) | 8 | Chow | Het | (B6×NZB)B6 | ||
| 81 | 73–85 | 8.3 | M+F | 8 | Chow | NZB | NZB×SM | ||
| 81 | 71–91 | 4.5 | M (F?) | 16 | Ath | RIII | 129×RIII | ||
| 82 | 74–88 | 9.8 | F (M?) | 14 | Western | C3H | B6×C3H | ||
| 82 | 66–86 | 6.8 | F (M?) | 18 | Ath | C3H | B6×C3H-Apoe KO | ||
| 84 | 84–90 | 10.6 | F (M?) | 20 | Ath | 129 | B6×129S1 | ||
| 85 | 63–89 | 2.2 | F (M?) | 23 | Ath | Het | (B6×NZB)B6 | ||
| 85 | 75–89 | 4.6 | M+F | 20 | Ath | NZB | NZB×SM | ||
| 85 | 77–91 | 3.0 | M+F | 24 | Ath | NZB | NZB×SM | ||
| 85 | 2–85 | 2.2 | M (F?) | 24 | Chow | NON | (NON×NZO)NON | ||
| 87 | 67–91 | 3.1 | M+F | 14 | Ath | NZB | NZB×SM | ||
| 89 | 75–89 | 5.6 | F (M?) | 23 | Ath | Het | B6×NZB | ||
| 2 | 44 | 28–84 | 2.3 | M+F | 14 | Ath | NZB | NZB×SM | |
| 46 | 37–95 | 1.8 | F (M?) | 40 | Chow | NOD | NOD×129S1 | ||
| 46 | 42–60 | 6.4 | M (F?) | 8 | Chow | CAST | D2×CAST | ||
| 60 | 32–100 | 2.5 | M+F | 10 | Chow | NZW | B6×NZW | ||
| 83 | 65–90 | 2.3 | M (F?) | 8 | Chow | D2 | B6×D2 | ||
| 83 | 78–88 | 4.1 | F (M?) | 18 | Western | Het | B6×C3H-Apoe KO | ||
| 83 | 74–103 | 2.8 | M+F | 11 | Chow | B6 | B6×CASA | ||
| 84 | 2–102 | 2.2 | F (M?) | 68 | Ath | D2 | B6×D2 | ||
| 96 | 8–96 | 2.3 | F (M?) | 14 | Ath | C3H | B6×C3H | ||
| 100 | 22–100 | 2.4 | F | 10 | Chow | RF | NZB×RF | ||
| 3 | 5 | 2–54 | 2.8 | M+F | 8 | Chow | NZB | NZB×SM | |
| 5 | 2–26 | 3.0 | M+F | 24 | Ath | NZB | NZB×SM | Add | |
| 17 | 13–79 | 2.1 | M+F | 16 | Ath | PERA | PERA×I | Sex-specific | |
| 33 | 23–79 | 2.6 | F (M?) | 52 | Chow | B6 | B6×D2 | Add | |
| 34 | 20–44 | 2.2 | M (F?) | 16 | Ath | Het | 129×RIII | ||
| 42 | 2–52 | 3.0 | M+F | 14 | Ath | NZB | NZB×SM | ||
| 55 | 53–59 | 5.4 | M | 8 | Chow | B6 | B6×129 | ||
| 56 | 44–68 | 2.7 | M (F?) | 8 | Chow | B6 | B6×D2 | ||
| 64 | 64–74 | 2.8 | M+F | 11 | Chow | B6 | B6×CASA | ||
| 4 | 17 | 7–29 | 3.9 | M | 10 | Chow | B6 | B6×NOD | Sex-specific |
| 21 | 11–31 | 4.3 | M+F | 16 | Ath | PERA | D2×PERA | Ad | |
| 26 | 12–38 | 3.9 | M+F | 16 | Ath | PERA | PERA×I | Ad | |
| 27 | 11–55 | 3.1 | M (F?) | 10 | Chow | B6 | B6×A | ||
| 27 | 15–31 | 7.4 | M (F?) | 8 | Chow | D2 | D2×CAST | ||
| 29 | 20–40 | 3.5 | M (F?) | 8 | Chow | 129 | CAST×129S1 | ||
| 48 | 16–76 | 2.7 | F | 8 | Chow | B6 | B6×A | ||
| 61 | 49–81 | 2.2 | M (F?) | 8 | Chow | D2 | B6×D2 | ||
| 62 | 54–68 | 3.7 | F (M?) | 68 | Ath | B6 | B6×D2 | ||
| 5 | 6 | 3–11 | 6.3 | M+F | 16 | Ath | PERA | PERA×I | |
| 10 | 3–49 | 4.3 | M+F | 16 | Ath | 129 | B6×129 | ||
| 35 | 11–43 | 4.6 | F (M?) | 23 | Ath | Het | (B6×NZB)B6 | ||
| 41 | 31–47 | 3.4 | M (F?) | 24 | Chow | Het | (NON×NZO)NON | ||
| 45 | 9–61 | 3.3 | M+F | 8 | Chow | B6 | B6×129 | ||
| 47 | 42–49 | 10.3 | M+F | 20 | Ath | NZB | NZB×SM | ||
| 47 | 21–49 | 8.7 | M+F | 24 | Ath | NZB | NZB×SM | ||
| 49 | 23–58 | 2.3 | F (M?) | 23 | Ath | Het | B6×NZB | ||
| 51 | 23–65 | 2.8 | F (M?) | 8 | Chow | Het | B6×NZB | ||
| 52 | 45–67 | 3.5 | F (M?) | 18 | Ath | NZB | (NZB×SM)NZB | ||
| 52 | 49–67 | 4.6 | F (M?) | 8 | Chow | NZB | (NZB×SM)NZB | ||
| 53 | 47–57 | 20 | F | 10 | Chow | NZB | NZB×RF | ||
| 59 | 41–61 | 10.6 | M+F | 14 | Ath | NZB | NZB×SM | ||
| 61 | 59–71 | 9.1 | M+F | 8 | Chow | NZB | NZB×SM | ||
| 65 | 51–80 | 2.2 | F (M?) | 18 | Ath | NZB | (NZB×SM)NZB | ||
| 79 | 55–80 | 2.8 | F (M?) | 8 | Chow | NZB | (NZB×SM)NZB | ||
| 6 | 29 | 16–67 | 2.3 | M+F | 10 | Chow | NOD | B6×NOD | Add |
| 33 | 20–46 | 2.7 | M (F?) | 8 | Chow | D2 | B6×D2 | Add | |
| 46 | 34–78 | 2.1 | M+F | 16 | Ath | PERA | PERA×I | Add | |
| 60 | 40–72 | 2.7 | F (M?) | 14 | Ath | B6 | B6×C3H | Add | |
| 70 | 58–76 | 4.0 | M+F | 8 | Chow | NZB | NZB×SM | ||
| 70 | 38–78 | 2.1 | M+F | 14 | Ath | NZB | NZB×SM | ||
| 72 | 48–78 | 4.4 | M (F?) | 8 | Chow | CAST | D2×CAST | ||
| 74 | 64–78 | 2.9 | F (M?) | 52 | Chow | Het | B6×D2 | ||
| 78 | 2–78 | 2.4 | F (M?) | 20 | Ath | B6 | B6×129S1 | ||
| 7 | 9 | 9–76 | 2.2 | M (F?) | 8 | Chow | Het | B6×A | |
| 74 | 62–80 | 4.0 | M | 10 | Chow | Het | B6×NOD | Sex-specific | |
| 87 | 75–89 | 3.0 | M+F | 24 | Ath | SM | NZB×SM | Add | |
| 88 | 50–88 | 3.9 | M+F | 16 | Ath | PERA | PERA×I | ||
| 8 | 2 | 2–34 | 2.7 | F | 10 | Chow | NZB | NZB×RF | |
| 3 | 2–52 | 3.3 | M+F | 24 | Ath | NZB | NZB×SM | Add | |
| 3 | 2–52 | 3.6 | M+F | 14 | Ath | NZB | NZB×SM | ||
| 36 | 26–44 | 6.1 | M (F?) | 10 | Chow | B6 | B6×A | ||
| 38 | 24–46 | 4.6 | F (M?) | 8 | Chow | B6 | B6×A | ||
| 46 | 34–66 | 2.3 | F (M?) | 20 | Ath | 129 | B6×129S1 | ||
| 50 | 34–66 | 3.8 | M+F | 10 | Chow | B6 | B6×NOD | ||
| 53 | 39–65 | 3.9 | M+F | 8 | Chow | B6 | B6×129S1 | ||
| 56 | 46–68 | 5.0 | M (F?) | 8 | Chow | B6 | B6×D2 | ||
| 73 | 3–76 | 4.3 | M+F | 11 | Chow | B6 | B6×CASA | ||
| 9 | 2 | 2–34 | 3.8 | M | 24 | Ath | NZB | NZB×SM | Sex-specific |
| 14 | 2–26 | 3.1 | M (F?) | 16 | Ath | RIII | 129×RIII | Add | |
| 18 | 18–25 | 8.1 | M+F | 11 | Chow | CASA | B6×CASA | ||
| 21 | 13–27 | 6.2 | M+F | 8 | Chow | 129 | B6×129S1 | ||
| 22 | 6–34 | 3.0 | F (M?) | 20 | Ath | 129 | B6×129 | ||
| 23 | 21–59 | 1.6 | F (M?) | 18 | Ath | Het | NZB×SM | ||
| 25 | 18–25 | 9.0 | M+F | 11 | Chow | CASA | B6×CASA | ||
| 25 | 22–50 | 6.4 | F (M?) | 40 | Chow | Het | NOD×129 | ||
| 25 | 12–32 | 6.6 | M (F?) | 16 | Ath | RIII | 129×RIII | ||
| 33 | 12–50 | 2.6 | M+F | 24 | Ath | NZB | NZB×SM | ||
| 37 | 17–49 | 5.4 | M+F | 16 | Ath | 129 | B6×129 | ||
| 10 | 2 | 2–66 | 2.4 | M+F | 10 | Chow | Het | B6×NZW | |
| 63 | 53–72 | 3.2 | F/M | 8 | Chow | NZB/SM | NZB×SM | Sex-specific | |
| 68 | 39–68 | 2.2 | M (F?) | 10 | Chow | B6 | B6×A | Sex-specific | |
| 72 | 61–72 | 3.9 | F | 14 | Ath | NZB | NZB×SM | ||
| 11 | 18 | 14–66 | 2.7 | M+F | 14 | Ath | Het | NZB×SM | |
| 27 | 17–43 | 2.7 | M (F?) | 10 | Chow | B6 | B6×A | Add | |
| 30 | 18–54 | 3.4 | M+F | 16 | Ath | PERA | PERA×I | Add | |
| 36 | 14–50 | 3.4 | M+F | 16 | Ath | PERA | D2×PERA | Add | |
| 41 | 35–55 | 4.0 | M+F | 6 | Chow | DU6i | D2×DU6i | ||
| 56 | 43–72 | 2.9 | F (M?) | 14 | Ath | C3H | B6×C3H | ||
| 76 | 58–83 | 2.5 | F (M?) | 68 | Ath | D2 | B6×D2 | ||
| 81 | 73–83 | 3.9 | M+F | 16 | Ath | 129 | B6×129 | ||
| 81 | 3–81 | 2.2 | M+F | 10 | Chow | Het | B6×NZW | ||
| 12 | 10 | 6–44 | 3.4 | F | 10 | Chow | NZB | NZB×RF | Sex-specific |
| 20 | 12–28 | 5.9 | F (M?) | 20 | Ath | 129 | B6×129S1 | ||
| 28 | 10–40 | 4.8 | M (F?) | 16 | Ath | RIII | 129×RIII | ||
| 30 | 17–45 | 7.4 | M | 8 | Chow | I | PERA×I | ||
| 38 | 12–54 | 2.1 | M (F?) | 8 | Chow | B6 | B6×D2 | ||
| 54 | 26–68 | 2.2 | M+F | 8 | Chow | NZB | NZB×SM | ||
| 56 | 10–74 | 2.8 | M+F | 20 | Ath | NZB | NZB×SM | ||
| 13 | 10 | 2–28 | 3.0 | M+F | 8 | Chow | 129 | B6×129 | Add |
| 14 | 19 | 15–29 | 3.1 | M (F?) | 8 | Chow | B6 | B6×D2 | Add |
| 38 | 18–56 | 2.6 | M+F | 10 | Chow | Het | B6×NOD | ||
| 15 | 16 | 4–34 | 2.6 | F (M?) | 14 | Ath | B6 | B6×C3H | Add |
| 18 | 8–28 | 3.0 | M (F?) | 8 | Chow | CAST | CAST×129 | ||
| 23 | 15–35 | 3.5 | F (M?) | 8 | Chow | B6 | B6×A | ||
| 38 | 37–54 | 2.6 | M+F | 8 | Chow | NZB | NZB×SM | ||
| 54 | 24–59 | 2.1 | M+F | 16 | Ath | PERA | PERA×I | ||
| 16 | 11 | 7–21 | 2.7 | F (M?) | 52 | Chow | B6 | B6×D2 | |
| 22 | 20–28 | 2.6 | F (M?) | 18 | Ath | Het | (NZB×SM)NZB | ||
| 28 | 2–44 | 3.6 | M+F | 8 | Chow | NZB | NZB×SM | ||
| 40 | 24–50 | 2.4 | M+F | 24 | Ath | NZB | NZB×SM | ||
| 40 | 6–54 | 2.5 | M+F | 14 | Ath | NZB | NZB×SM | ||
| 49 | 32–49 | 1.6 | F (M?) | 18 | Ath | NZB | (NZB×SM)NZB | ||
| 58 | 3–58 | 2.1 | M+F | 16 | Ath | PERA | PERA×I | ||
| 17 | 9 | 5–31 | 2.2 | F (M?) | 20 | Ath | B6 | B6×129S1 | |
| 12 | 8–61 | 2.4 | M+F | 6 | Chow | D2 | D2×DU6i | Sex-specific | |
| 18 | 14–61 | 2.2 | F | 10 | Chow | RF | NZB×RF | Add | |
| 21 | 5–29 | 2.2 | F (M?) | 8 | Chow | Het | B6×A | Add | |
| 22 | 14–40 | 4.4 | M | 14 | Ath | NZB | NZB×SM | ||
| 22 | 22–50 | 1.7 | F (M?) | 18 | Ath | NZB | (NZB×SM)NZB | ||
| 28 | 8–42 | 2.4 | M+F | 8 | Chow | NZB | NZB×SM | ||
| 30 | 14–40 | 2.2 | M+F | 20 | Ath | NZB | NZB×SM | ||
| 30 | 14–40 | 2.8 | M+F | 24 | Ath | NZB | NZB×SM | ||
| 32 | 16–44 | 2.4 | M+F | 14 | Ath | NZB | NZB×SM | ||
| 39 | 31–51 | 2.6 | M+F | 16 | Ath | D2 | D2×PERA | ||
| 53 | 39–55 | 2.3 | M+F | 16 | Ath | I | PERA×I | ||
| 58 | 40–60 | 2.8 | F (M?) | 18 | wester | C3H | B6×C3H-Apoe KO | ||
| 18 | 17 | 13–39 | 2.6 | F (M?) | 18 | Wester | C3H | B6×C3H-Apoe KO | |
| 19 | 13–39 | 3.7 | F (M?) | 8 | Chow | NZB | (NZB×SM)NZB | ||
| 26 | 10–42 | 2.1 | M (F?) | 8 | Chow | CAST | D2×CAST | ||
| 28 | 15–45 | 2.7 | M+F | 8 | Chow | 129 | B6×129 | ||
| 30 | 16–48 | 2.5 | M | 10 | Chow | B6 | B6×A | ||
| 39 | 21–43 | 2.0 | F (M?) | 18 | Ath | NZB | (NZB×SM)NZB | ||
| 39 | 11–58 | 3.0 | M+F | 8 | Chow | NZB | NZB×SM | ||
| 53 | 41–58 | 3.0 | F (M?) | 14 | Ath | C3H | B6×C3H | ||
| 55 | 39–58 | 2.8 | F (M?) | 52 | Chow | D2 | B6×D2 | ||
| 58 | 41–58 | 2.8 | M+F | 14 | Ath | NZB | NZB×SM | ||
| 58 | 7–58 | 4.0 | M+F | 20 | Ath | NZB | NZB×SM | ||
| 19 | 3 | 3–57 | 1.9 | F (M?) | 40 | Chow | Het | NOD×129 | Sex-specific |
| 14 | 6–52 | 2.4 | F (M?) | 8 | Chow | B6 | B6×A | ||
| 34 | 26–56 | 3.2 | F | 20 | Ath | NZB | NZB×SM | ||
| 34 | 24–54 | 2.6 | M+F | 10 | Chow | NZW | B6×NZW | ||
| 36 | 20–56 | 2.9 | M+F | 14 | Ath | NZB | NZB×SM | ||
| 49 | 35–50 | 5.6 | F | 10 | Chow | NZB | NZB×RF | ||
| 56 | 18–56 | 2.2 | M+F | 8 | Chow | NZB | NZB×SM | ||
| 56 | 38–56 | 3.0 | M+F | 20 | Ath | NZB | NZB×SM | ||
| 56 | 28–56 | 3.0 | M+F | 24 | Ath | NZB | NZB×SM |
Ath, atherogenic; KO, knockout.
Fig. 1.

Mouse chromosome map with the recalculated HDL QTL. Each bar represents the 95% confidence interval, with the black dot indicating the peak position of the QTL.
Fig. 2.

Chromosome 1 with the old map positions on the right and the new QTL map positions on the left. Each bar represents the confidence interval of a QTL within a cross, with the black dot indicating the peak. Bars with the same number on the left and right are the QTL in the same cross. Careful examination of the QTL mapped to distal Chr 1 in this study shows three discrete clusters of comapping QTL: one cluster of five QTL centered at 70 cM, one cluster of nine QTL centered at 80 cM, and one cluster of four QTL centered at 85 cM.
Fig. 3.
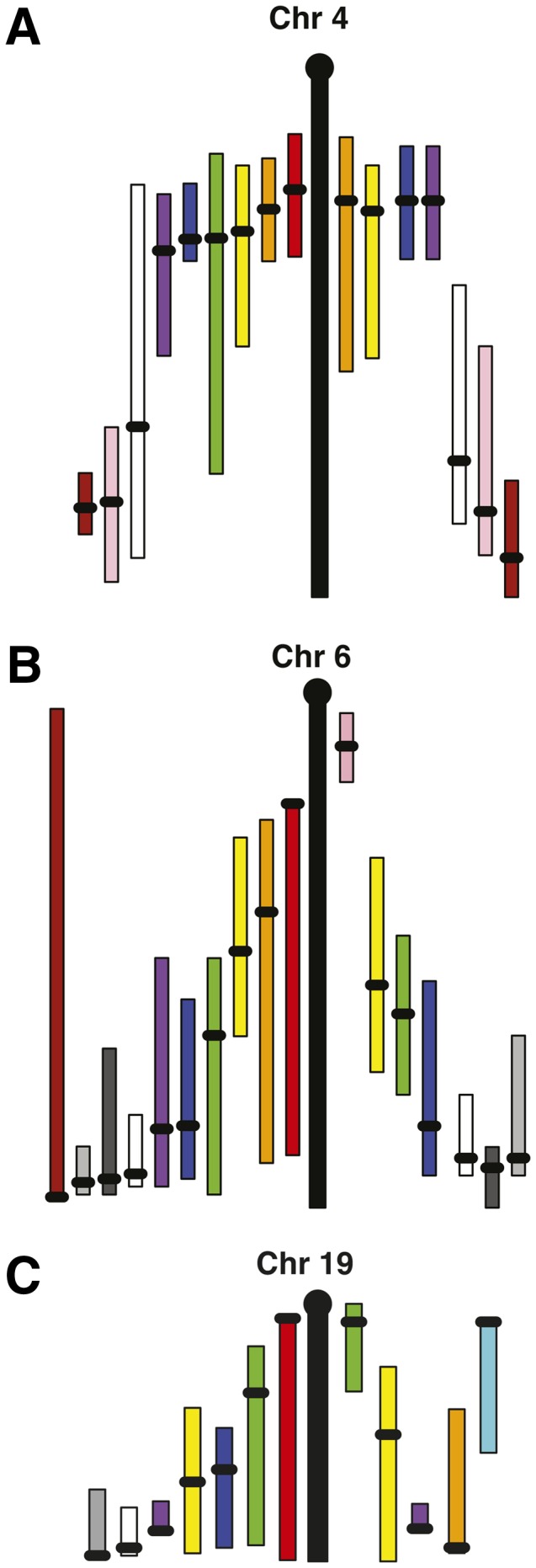
Chromosomes 4 (A), 6 (B), and 19 (C) with the old map positions on the right and the new QTL map positions on the left. Each bar represents the confidence interval of a QTL within a cross, with the black dot indicating the peak. Bars with the same color on the left and right are QTL in the same cross.
New HDL QTL map is required to determine the number of independent HDL QTL
As previously reported, many of the QTL comap to the same loci, suggesting a common underlying causative gene (7). The most striking example of HDL QTL clustering is observed on the distal end of Chromosome (Chr) 1 (Fig. 2). By using bioinformatics techniques, such as haplotyping, which leverages these comapping QTL clusters, we previously showed that a single nucleotide polymorphism (SNP) in Apoa2 is the most likely causative SNP underlying the QTL mapped to distal Chr 1 in all crosses where the two strains were polymorphic for this SNP (17). However, Apoa2 is not the only distal Chr 1 gene to influence HDL (18). Careful examination of the QTL mapped to distal Chr 1 in this study showed three clusters of comapping QTL: one cluster of five QTL centered at 70 cM, one cluster of nine QTL centered at 80 cM, and one cluster of four QTL centered at 85 cM. The Apoa2 gene (80 cM) is the most likely candidate for this middle cluster, whereas Soat1 (67.7 cM) is the most likely candidate for the upper cluster (19). The gene underlying the most distal cluster remains unknown.
Grin3a is a candidate gene for an HDL QTL on mouse Chr 4
We mapped nine QTL to mouse Chr 4, including a cluster of four QTL with peak locations between 26 and 29 cM (Table 2, Fig. 3A). This QTL cluster consisted of QTL mapped in the following crosses (underlined strains indicate the high allele for each QTL): PERA×I (peak at 26 cM), B6×A (peak at 27 cM), D2×129 (peak at 27 cM), and 129×CAST (peak at 29 cM). The minimum shared interval for these QTL extended from 20 to 31 cM (38.6–56.2 Mb), a region containing 343 genes with assigned MGI IDs (http://www.informatics.jax.org), as determined using the Biomart tool available from the Ensembl genome database [http://www.ensembl.org/index.html, Ensembl Genes 69 dataset:Mus musculus genes (GRCm38)]. The raw centimorgan positions for the confidence interval were converted to genome coordinates (NCBI, Bld37) using the Mouse Map Converter tool (http://cgd.jax.org). We then examined this interval to identify SNPs that met the following allele conditions:
| (Eq. 1) |
The SNP dataset used for this analysis was the CGD-MDA1 dataset, which is publically available (http://phenome.jax.org); all SNP in the above QTL interval in this database were examined. While this SNP dataset does not contain all known mouse SNP, it is the most complete, publically available SNP dataset containing data for all of the strains listed in Equation 1. Another limitation is that we excluded all SNPs that did not have complete genotype information for our strains of interest. Therefore, until complete genome sequences for all involved strains is available, we cannot exclude the possibility that candidate genes are missed. Of the 3,483 SNP for which data was available for the minimal QTL interval, only 8 SNPs met the allele distribution pattern defined by Equation 1. These SNP were all located in a narrow region between 49.86 and 50.58 Mb. This narrow region does not contain the coding region for any gene. However, the most proximal of the SNPs is located 937 bp distal to the start codon for the Grin3a gene. The Grin3a gene runs on the negative strand, and our SNPs overlap with its predicted regulatory/promoter region. There are no polymorphisms in the coding region of Grin3a that match the strain pattern shown in Equation 1.
Identification of Etv6 as a candidate gene for an HDL QTL on distal mouse Chr 6
On the distal end of Chr 6, we identified a cluster of three comapping QTL with peak locations between 70 and 74 cM (Table 2, Fig. 3B). These QTL were mapped in the following crosses (underlined strains have the high allele): SM×NZB (peak at 70 cM), D2×CAST (peak at 72 cM), and B6×D2 (peak at 74 cM). The B6×D2 QTL was only a suggestive QTL, and the allele effects for this QTL were not conclusive. Very few mice were phenotyped in the B6×D2 cross, severely underpowering the mapping in this study. However, the D2 strain was used in another cross with a QTL mapping to this location, suggesting that this B6×D2 QTL is valid. Using methods similar to that which was used for the Chr 4 locus, we then attempted to narrow this QTL to a single candidate gene. The minimum shared interval for this cluster on distal Chr 6 extended from 64 to 76 cM (131.92–143.65 Mb). This relatively gene-dense region contains 124 known or predicted genes. Using SNP comparison such that the conditions in Equation 2 were met, we were able to narrow this QTL to a single region spanning from 134.00 to 134.09 Mb.
| (Eq. 2) |
This region contains a single gene, Etv6. No polymorphisms are found in the coding region of Etv6 that match the strain pattern shown in Equation 2. This gene appears to be moderately expressed in the liver (20). Therefore, we examined the expression pattern for this gene in our previously published liver expression strain survey (21) using the reported Q values (http://cgd.jax.org/datasets/expression/10strain.shtml), but it did not show differential expression between strains in the three crosses defining this QTL. Thus, it appears that this gene either does not function directly in the liver to modulate HDL or that the causative mutation(s) in this gene do not affect transcription levels but may still influence protein levels (22).
Cluster of QTL on distal Chr 19 can be narrowed to a small number of candidate genes
The new QTL map for Chr 19 differs significantly from the previous map (Fig. 3C). In addition to shifts in the peaks of QTL identified in several crosses, we identified several new peaks in other crosses but could not find some of the peaks reported in the literature. We did identify an interesting cluster of three QTL at the distal end of Chr 19 in the following crosses (underlined strains have the high allele): NZB×RF (peak at 49 cM), NZW×B6 (peak at 54 cM), and NZB×SM (peak at 56 cM). The last marker genotyped in the NZB×RF cross is located at 50 cM. The LOD score at this marker is nearly identical to the LOD score at the location of peak association. Thus, while the proximal boundary of the confidence interval for this QTL can be determined, the distal boundary cannot. By using the distal limit of the confidence interval for the B6×NZW cross, we determined that the narrowest common region for this QTL extends from 42 to 56 cM (48.5–59.5 Mb). As for the other QTL clusters described above, we used SNP comparison to narrow the list of candidate genes. The conditions for the comparison were:
| (Eq. 3) |
Unlike the two previous QTL clusters, we were unable to narrow this QTL to a single candidate gene. Fifteen known genes (Sorcs3, Sorcs1, Add3, Mxi1, 5830416P10Rik, Rbm20, Pdcd4, Shoc2, Acsl5, Vti1a, Tcf7l2, B230217O12Rik, Fam160b1, Trub1, and Atrnl1) and two predicted genes (1700001K23Rik and Gm16745) were identified as containing at least one SNP for which the alleles met the conditions defined in Equation 3.
To further narrow the list of genes for this QTL, we examined liver expression for strains included in our previously published gene expression strain survey (21). While liver expression data are not available for two of the strains used to map QTL identified in this cluster (NZW and RF), data are available for NZB, B6, and SM. Statistically significant differential expression was noted for Add3, Shoc2, and Fam160b1, and nearly statistically significant differential expression was noted for Rbm20. The differential expression profile for Fam160b1, Rbm20, and Shoc2 showed the expected profile such that the two low allele strains (B6 and SM) were expressed at levels different than that for the high allele NZB strain, but expression was not different when comparing B6 with SM. The expression profile for the Add3 gene did not follow this pattern, as expression was not different when comparing the high allele NZB strain to the low allele B6 strain.
As coding region polymorphisms can affect gene function in a way that is not necessarily reflected in expression profiles, we looked for coding region polymorphisms (nonsynonymous, nonsense) and splice site SNPs in which the allele pattern met the requirements defined in Equation 3; none were found, however, in the SNP databases available for these five strains. We then examined the Sanger resequencing databases to find any known nonsynonymous (Cn) SNPs in our 15 candidate genes in the classical inbred strains (i.e., not wild derived). Of the five strains of interest for this QTL cluster, only B6 has been resequenced to date. Nevertheless, given the number of strains that are resequenced, examination of this database should give a more complete listing of Cn SNP than other available databases. Nonsynonymous SNPs are listed in the Sanger database in Add3, Rbm20, Sorcs3, Sorcs1, Fam160b1, Atrnl1, and Trub1. The only Cn SNP listed in Add3 appeared to be private to the NON/ShiLtJ strain, and the only two in Rbm20 appeared to be private to the C57BL substrains (C57BL/6J and C57BL/6NJ). We then used the SIFT tool (http://sift.jcvi.org/) to determine whether the resulting amino acid changes for the remaining Cn SNPs were predicted to have a functional consequence(s). We found one such Cn SNP (rs37375751) in the Sorcs3 gene, but this SNP was not predicted to affect protein function. Four Cn SNPs are listed in the Sorcs1 gene in the classical inbred strains. One SNP (at 50255976 Mb, no rs number available, NCBI, Bld37) appeared to be a private SNP in the AKR/J strain. Two others, rs37453589 and a SNP at 50752804 Mb (no rs number available), were predicted to be tolerated. A fourth SNP in this gene, at 50752876 Mb (no rs number available), was predicted to result in an A-to-V substitution. This substitution was predicted not to be tolerated and thus had the potential to impact protein function. While this SNP appears quite polymorphic among inbred strains, the alanine amino acid at this location is quite conserved among other mammals. We resequenced this SNP in the five strains of interest for this QTL cluster and determined that the genotypes for the five strains fit Equation 3. Five Cn SNPs are listed in the Fam160b1 gene, for the classical inbred strains (rs36742147, rs31128381, rs38343270, rs37035338, and rs36675440). However, none of these SNPs is predicted to affect protein function. Likewise, a single Cn SNP is listed in the Atrnl1 gene (rs36943069), but again this SNP is expected to be tolerated with regard to protein function. A single Cn SNP is listed in the Trub1 gene. This SNP at 57532595 Mb (no rs number available), results in a change from S to F at amino acid position 40, which is predicted not to be tolerated with regard to protein function. While serine at this position is well conserved among mammals, a phenylalanine is found at this position in opossums. A number of strains are polymorphic for this SNP, including the NZO/HILtJ strain, a close relative of the NZB/B1NJ strain. We then resequenced this SNP in the five strains of interest for this QTL cluster and determined that the genotypes for the five strains fit Equation 3. In sum, the expression data provide support for Fam160b1, Shoc2, and Rbm20, and the presence of Cn SNPs provide support for Sorcs1 and Trub1 as candidates for this Chr 19 QTL. Further work will be required to further narrow this QTL.
DISCUSSION
QTL analysis is a reliable method to identify loci involved in the variation of a phenotype of interest, and since the key publication by Lander and Botstein (23), thousands of QTL studies have been performed for many different phenotypes and utilizing different species. A disadvantage of the method is the low resolution of mapping, as it is dependent on the number of recombinations in the population, which is inherently low in a two-generation cross between inbred strains. Because of this low resolution, a typical QTL spans 10–20 cM and contains hundreds of genes. We and others have developed statistical approaches that combine the data from multiple crosses to increase mapping resolution yielding narrower QTL intervals and bioinformatics methods, such as haplotyping, that allow to decrease the number of possible candidate genes within an interval (10). Using these methods, either in isolation or in combination, has allowed for the identification of causative genes for QTLs. However, these approaches strongly depend on accurate QTL mapping, and inclusion of an inaccurately mapped QTL or exclusion of an accurately mapped QTL can lead to false conclusions.
The literature reports a number of clusters of putatively comapping HDL QTL for which no candidate gene has been identified. For many of these clusters, our analysis suggests the existence of additional comapping QTL (i.e., an addition of QTL to a locus) or inappropriate conclusions regarding comapping (i.e., the eliminated QTL from a locus). In the first situation, the addition of a QTL can provide additional haplotypes, which might allow for further narrowing of the interval. In the second situation, candidate genes may have been inappropriately eliminated by using the “false” strains that were included in the haplotype comparison.
The advantages of our reanalysis are demonstrated in the three examples presented above. In the first example highlighting a QTL on Chr 4, we were able to narrow the interval to one single gene, Glutamate receptor ionotropic NMDA3A (Grin3a). Grin3a, which is also known as NR3A, codes for an inhibitory subunit of NMDA-type glutamate receptors. Traditional NMDA receptors are key players in glutamate-facilitated neurotransmission (24). Studies have suggested that expression of Grin3a is highest in selected regions of the brain in the postnatal period and in the adult retina, where the GRIN3A/NR3A subunit is thought to inhibit NMDA receptor Ca2+ permeability and has a neuroprotective function (25). Similarly, expression of Grin3a is high in the neonatal kidney, but expression does persist into adulthood. As in the brain, Grin3a appears to serve a protective role for the cells of the collecting duct of the kidney (26). While mechanism by which Grin3a influences HDL levels remains unknown, this gene was recently identified to be associated with HDL in a human genome-wide association study (27).
In the second example, for a Chr 6 QTL, we were also able to narrow the interval to a single gene (Etv6) using the QTL data. Etv6 is an ubiquitously expressed gene encoding a transcription repressor, and ETV6 is a member of the ETS family of transcription factors (28). Most studies on ETV6 have focused on the fusion proteins that can arise from chromosomal rearrangements between Etv6 and other genes encoding transcription factors, such as Runx1, and which have been implicated as being causative in some forms of leukemia (29). While the mechanism by which ETV6 regulates HDL levels is unclear, studies of gene networks associated with ETV6 action have suggested that this transcription repressor regulates genes associated with cholesterol biogenesis (30).
Finally, in our third example, Chr 19, combining the newly calculated QTL narrowed the region to a cluster of 15 genes, for which additional evidence exists supporting 5 as candidates for this QTL. This QTL is different from the other two examples, as it seems to be exclusive to NZ strains. Both the NZB/B1NJ and the NZW/LacJ strains can be traced back to a common set of mice brought to Australia in the 1930s, and recent analysis of the relatedness of inbred strains shows that the NZW/LacJ and the NZB/B1NJ strains are indeed genetically very closely related (31). In all three crosses contributing to the QTL cluster on distal Chr 19, one of the NZ strains was the high allele, suggesting that the alleles underlying this QTL are private to the NZ strains. As the modern NZO/H1LtJ is related to both the NZW/LacJ and the NZB/B1NJ strains, it is possible that this strain also carries the private “NZ” allele at this locus. The addition of the NZO/H1LtJ strain to the haplotyping for this locus does eliminate the Add3, 170001K23Rik, Mxi1I, and Tcf7l2 genes. Future work must be conducted, however, to determine whether this QTL is indeed caused by private NZ alleles. The 5 candidate genes for this QTL are Fam160b1, Shoc2, Rbm20, Sorcs1, and Trub1. Of these 5, Sorcs1 is the most likely candidate gene based on the existing evidence. This gene codes for a Vsp10p-D receptor family member transmembrane protein, which functions to sort and traffic proteins (32). Genetic variation in this gene has been associated with both the development of type II diabetes (33, 34) and risk for development of Alzheimer disease (35). These two diseases are correlated with serum lipid levels and genetic variation in another Vsp10p-D receptor gene, Sort1, which has been linked to serum LDL levels (36, 37). The remaining 4 genes have been poorly studied, and thus it is unclear how or whether these genes could function to modulate serum HDL levels. Mutations in SHOC2 can cause Noonan syndrome, an autosomal dominant disorder characterized by short stature, down-slanted eyes, cardiovascular defects, and increased tumor risks (38). The Rbm20 gene codes for RNA binding protein/RNA splicing protein, which shows particularly high expression in muscle, heart, and brain, and recent studies have shown that null mutations in this gene are associated with hereditary cardiomyopathies (39). Little is known about Trub1; it has been identified as a tRNA pseudouridinilation gene.
In conclusion, our reanalysis of the QTL data for 23 crosses results in an improved map for HDL QTL and facilitates the identification of HDL genes in the mouse. We show that using this approach, we can identify previously missed QTL and locate QTL on different regions of a chromosome. The new HDL QTL map allows for reliable combining of QTL data and candidate gene analysis, which we demonstrate by identifying Grin3a, Etv6, and a cluster of five genes as likely candidate genes for QTL on Chr 4, 6, and 19, respectively.
Supplementary Material
Footnotes
Abbreviations:
- Chr
- chromosome
- LOD
- logarithm of the odds
- MGI
- Mouse Genome Informatics
- QTL
- quantitative trait locus
- SNP
- single-nucleotide polymorphism
This work was supported by the National Institutes of Health Grants R01 HL-077796, R01 HL-081162, R01 HL-095668, and R01 AR-060234.
REFERENCES
- 1.Brewer H. B. 2004. High-density lipoproteins: a new potential therapeutic target for the prevention of cardiovascular disease. Arterioscler. Thromb. Vasc. Biol. 24: 387–391. [DOI] [PubMed] [Google Scholar]
- 2.Gordon T., Castelli W. P., Hjortland M. C., Kannel W. B., Dawber T. R. 1977. High density lipoprotein as a protective factor against coronary heart disease. The Framingham Study. Am. J. Med. 62: 707–714. [DOI] [PubMed] [Google Scholar]
- 3.Castelli W. P., Garrison R. J., Wilson P. W., Abbott R. D., Kalousdian S., Kannel W. B. 1986. Incidence of coronary heart disease and lipoprotein cholesterol levels. The Framingham Study. JAMA. 256: 2835–2838. [PubMed] [Google Scholar]
- 4.Assmann G., Schulte H., von Eckardstein A., Huang Y. 1996. High-density lipoprotein cholesterol as a predictor of coronary heart disease risk. The PROCAM experience and pathophysiological implications for reverse cholesterol transport. Atherosclerosis. 124(Suppl.): S11–S20. [DOI] [PubMed] [Google Scholar]
- 5.Sharrett A. R., Ballantyne C. M., Coady S. A., Heiss G., Sorlie P. D., Catellier D., Patsch W., Atherosclerosis Risk in Communities Study Group. 2001. Coronary heart disease prediction from lipoprotein cholesterol levels, triglycerides, lipoprotein(a), apolipoproteins A-I and B, and HDL density subfractions: The Atherosclerosis Risk in Communities (ARIC) Study. Circulation. 104: 1108–1113. [DOI] [PubMed] [Google Scholar]
- 6.Wang X., Paigen B. 2002. Quantitative trait loci and candidate genes regulating HDL cholesterol: a murine chromosome map. Arterioscler. Thromb. Vasc. Biol. 22: 1390–1401. [DOI] [PubMed] [Google Scholar]
- 7.Wang X., Paigen B. 2005. Genetics of variation in HDL cholesterol in humans and mice. Circ. Res. 96: 27–42. [DOI] [PubMed] [Google Scholar]
- 8.Ackert-Bicknell C. L., Karasik D., Li Q., Smith R. V., Hsu Y-H., Churchill G. A., Paigen B. J., Tsaih S-W. 2010. Mouse BMD quantitative trait loci show improved concordance with human genome-wide association loci when recalculated on a new, common mouse genetic map. J. Bone Miner. Res. 25: 1808–1820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ridgway W. M., Healy B., Smink L. J., Rainbow D., Wicker L. S. 2007. New tools for defining the “genetic background” of inbred mouse strains. Nat. Immunol. 8: 669–673. [DOI] [PubMed] [Google Scholar]
- 10.DiPetrillo K., Wang X., Stylianou I. M., Paigen B. 2005. Bioinformatics toolbox for narrowing rodent quantitative trait loci. Trends Genet. 21: 683–692. [DOI] [PubMed] [Google Scholar]
- 11.Blake J. A., Richardson J. E., Davisson M. T., Eppig J. T. 1997. The Mouse Genome Database (MGD). A comprehensive public resource of genetic, phenotypic and genomic data. The Mouse Genome Informatics Group. Nucleic Acids Res. 25: 85–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Broman K. W., Sen S. 2009. A Guide to QTL Mapping with R/qtl. Springer, New York. [Google Scholar]
- 13.Shifman S., Bell J. T., Copley R. R., Taylor M. S., Williams R. W., Mott R., Flint J. 2006. A high-resolution single nucleotide polymorphism genetic map of the mouse genome. PLoS Biol. 4: e395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cox A., Ackert-Bicknell C. L., Dumont B. L., Ding Y., Bell J. T., Brockmann G. A., Wergedal J. E., Bult C. J., Paigen B., Flint J., et al. 2009. A new standard genetic map for the laboratory mouse. Genetics. 182: 1335–1344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sehayek E., Duncan E. M., Yu H. J., Petukhova L., Breslow J. L. 2003. Loci controlling plasma non-HDL and HDL cholesterol levels in a C57BL/6J x CASA/Rk intercross. J. Lipid Res. 44: 1744–1750. [DOI] [PubMed] [Google Scholar]
- 16.Colinayo V. V., Qiao J-H., Wang X., Krass K. L., Schadt E., Lusis A. J., Drake T. A. 2003. Genetic loci for diet-induced atherosclerotic lesions and plasma lipids in mice. Mamm. Genome. 14: 464–471. [DOI] [PubMed] [Google Scholar]
- 17.Wang X., Korstanje R., Higgins D., Paigen B. 2004. Haplotype analysis in multiple crosses to identify a QTL gene. Genome Res. 14: 1767–1772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Su Z., Cox A., Shen Y., Stylianou I. M., Paigen B. 2009. Farp2 and Stk25 are candidate genes for the HDL cholesterol locus on mouse chromosome 1. Arterioscler. Thromb. Vasc. Biol. 29: 107–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Su Z., Wang X., Tsaih S-W., Zhang A., Cox A., Sheehan S., Paigen B. 2009. Genetic basis of HDL variation in 129/SvImJ and C57BL/6J mice: importance of testing candidate genes in targeted mutant mice. J. Lipid Res. 50: 116–125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Visel A., Thaller C., Eichele G. 2004. GenePaint.org: an atlas of gene expression patterns in the mouse embryo. Nucleic Acids Res. 32: D552–D556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Shockley K. R., Witmer D., Burgess-Herbert S. L., Paigen B., Churchill G. A. 2009. Effects of atherogenic diet on hepatic gene expression across mouse strains. Physiol. Genomics. 39: 172–182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ghazalpour A., Bennett B., Petyuk V. A., Orozco L., Hagopian R., Mungrue I. N., Farber C. R., Sinsheimer J., Kang H. M., Furlotte N., et al. 2011. Comparative analysis of proteome and transcriptome variation in mouse. PLoS Genet. 7: e1001393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lander E. S., Botstein D. 1989. Mapping mendelian factors underlying quantitative traits using RFLP linkage maps. Genetics. 121: 185–199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Fitzgerald P. J. 2012. The NMDA receptor may participate in widespread suppression of circuit level neural activity, in addition to a similarly prominent role in circuit level activation. Behav. Brain Res. 230: 291–298. [DOI] [PubMed] [Google Scholar]
- 25.Nakanishi N., Tu S., Shin Y., Cui J., Kurokawa T., Zhang D., Chen H-S. V., Tong G., Lipton S. A. 2009. Neuroprotection by the NR3A subunit of the NMDA receptor. J. Neurosci. 29: 5260–5265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sproul A., Steele S. L., Thai T. L., Yu S., Klein J. D., Sands J. M., Bell P. D. 2011. N-methyl-D-aspartate receptor subunit NR3a expression and function in principal cells of the collecting duct. Am. J. Physiol. Renal Physiol. 301: F44–F54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Willer C. J., Sanna S., Jackson A. U., Scuteri A., Bonnycastle L. L., Clarke R., Heath S. C., Timpson N. J., Najjar S. S., Stringham H. M., et al. 2008. Newly identified loci that influence lipid concentrations and risk of coronary artery disease. Nat. Genet. 40: 161–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Coyne H. J., De S., Okon M., Green S. M., Bhachech N., Graves B. J., McIntosh L. P. 2012. Autoinhibition of ETV6 (TEL) DNA binding: appended helices sterically block the ETS domain. J. Mol. Biol. 421: 67–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bohlander S. K. 2005. ETV6: a versatile player in leukemogenesis. Semin. Cancer Biol. 15: 162–174. [DOI] [PubMed] [Google Scholar]
- 30.Boily G., Beaulieu P., Healy J., Sinnett D. 2008. Connections between ETV6-modulated genes: identification of shared features. Cancer Inform. 6: 183–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Petkov P. M., Graber J. H., Churchill G. A., Dipetrillo K., King B. L., Paigen K. 2005. Evidence of a large-scale functional organization of mammalian chromosomes. PLoS Genet. 1: e33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Nielsen M. S., Keat S. J., Hamati J. W., Madsen P., Gutzmann J. J., Engelsberg A., Pedersen K. M., Gustafsen C., Nykjaer A., Gliemann J., et al. 2008. Different motifs regulate trafficking of SorCS1 isoforms. Traffic. 9: 980–994. [DOI] [PubMed] [Google Scholar]
- 33.Goodarzi M. O., Lehman D. M., Taylor K. D., Guo X., Cui J., Quiñones M. J., Clee S. M., Yandell B. S., Blangero J., Hsueh W. A., et al. 2007. SORCS1: a novel human type 2 diabetes susceptibility gene suggested by the mouse. Diabetes. 56: 1922–1929. [DOI] [PubMed] [Google Scholar]
- 34.Clee S. M., Yandell B. S., Schueler K. M., Rabaglia M. E., Richards O. C., Raines S. M., Kabara E. A., Klass D. M., Mui E. T-K., Stapleton D. S., et al. 2006. Positional cloning of Sorcs1, a type 2 diabetes quantitative trait locus. Nat. Genet. 38: 688–693. [DOI] [PubMed] [Google Scholar]
- 35.Reitz C., Tokuhiro S., Clark L. N., Conrad C., Vonsattel J-P., Hazrati L-N., Palotás A., Lantigua R., Medrano M., Jiménez-Velázquez I. Z., et al. 2011. SORCS1 alters amyloid precursor protein processing and variants may increase Alzheimer's disease risk. Ann. Neurol. 69: 47–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Shirts B. H., Hasstedt S. J., Hopkins P. N., Hunt S. C. 2011. Evaluation of the gene-age interactions in HDL cholesterol, LDL cholesterol, and triglyceride levels: the impact of the SORT1 polymorphism on LDL cholesterol levels is age dependent. Atherosclerosis. 217: 139–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Willer C. J., Mohlke K. L. 2012. Finding genes and variants for lipid levels after genome-wide association analysis. Curr. Opin. Lipidol. 23: 98–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Denayer E., Peeters H., Sevenants L., Derbent M., Fryns J. P., Legius E. 2012. NRAS Mutations in Noonan Syndrome. Mol Syndromol. 3: 34–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Guo W., Schafer S., Greaser M. L., Radke M. H., Liss M., Govindarajan T., Maatz H., Schulz H., Li S., Parrish A. M., et al. 2012. RBM20, a gene for hereditary cardiomyopathy, regulates titin splicing. Nat. Med. 18: 766–773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Pitman W. A., Korstanje R., Churchill G. A., Nicodeme E., Albers J. J., Cheung M. C., Staton M. A., Sampson S. S., Harris S., Paigen B. 2002. Quantitative trait locus mapping of genes that regulate HDL cholesterol in SM/J and NZB/B1NJ inbred mice. Physiol. Genomics. 9: 93–102. [DOI] [PubMed] [Google Scholar]
- 41.Lyons M. A., Wittenburg H., Li R., Walsh K. A., Churchill G. A., Carey M. C., Paigen B. 2003. Quantitative trait loci that determine lipoprotein cholesterol levels in DBA/2J and CAST/Ei inbred mice. J. Lipid Res. 44: 953–967. [DOI] [PubMed] [Google Scholar]
- 42.Wang X., Le Roy I., Nicodeme E., Li R., Wagner R., Petros C., Churchill G. A., Harris S., Darvasi A., Kirilovsky J., et al. 2003. Using advanced intercross lines for high-resolution mapping of HDL cholesterol quantitative trait loci. Genome Res. 13: 1654–1664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Korstanje R., Li R., Howard T., Kelmenson P., Marshall J., Paigen B., Churchill G. 2004. Influence of sex and diet on quantitative trait loci for HDL cholesterol levels in an SM/J by NZB/BlNJ intercross population. J. Lipid Res. 45: 881–888. [DOI] [PubMed] [Google Scholar]
- 44.Ishimori N., Li R., Kelmenson P. M., Korstanje R., Walsh K. A., Churchill G. A., Forsman-Semb K., Paigen B. 2004. Quantitative trait loci analysis for plasma HDL-cholesterol concentrations and atherosclerosis susceptibility between inbred mouse strains C57BL/6J and 129S1/SvImJ. Arterioscler. Thromb. Vasc. Biol. 24: 161–166. [DOI] [PubMed] [Google Scholar]
- 45.Lyons M. A. 2004. Quantitative trait loci that determine lipoprotein cholesterol levels in an intercross of 129S1/SvImJ and CAST/Ei inbred mice. Physiol. Genomics. 17: 60–68. [DOI] [PubMed] [Google Scholar]
- 46.Lyons M. A. 2004. Genetic contributors to lipoprotein cholesterol levels in an intercross of 129S1/SvImJ and RIIIS/J inbred mice. Physiol. Genomics. 17: 114–121. [DOI] [PubMed] [Google Scholar]
- 47.Wittenburg H., Lyons M. A., Li R., Kurtz U., Wang X., Mössner J., Churchill G. A., Carey M. C., Paigen B. 2006. QTL mapping for genetic determinants of lipoprotein cholesterol levels in combined crosses of inbred mouse strains. J. Lipid Res. 47: 1780–1790. [DOI] [PubMed] [Google Scholar]
- 48.Stylianou I. M., Tsaih S-W., Dipetrillo K., Ishimori N., Li R., Paigen B., Churchill G. A. 2006. Complex genetic architecture revealed by analysis of high-density lipoprotein cholesterol in chromosome substitution strains and F2 crosses. Genetics. 174: 999–1007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wergedal J. E., Ackert-Bicknell C. L., Beamer W. G., Mohan S., Baylink D. J., Srivastava A. K. 2007. Mapping genetic loci that regulate lipid levels in a NZB/B1NJxRF/J intercross and a combined intercross involving NZB/B1NJ, RF/J, MRL/MpJ, and SJL/J mouse strains. J. Lipid Res. 48: 1724–1734. [DOI] [PubMed] [Google Scholar]
- 50.Brockmann G. A., Karatayli E., Neuschl C., Stylianou I. M., Aksu S., Ludwig A., Renne U., Haley C. S., Knott S. 2007. Genetic control of lipids in the mouse cross DU6i x DBA/2. Mamm. Genome. 18: 757–766. [DOI] [PubMed] [Google Scholar]
- 51.Li Q., Li Y., Zhang Z., Gilbert T. R., Matsumoto A. H., Dobrin S. E., Shi W. 2008. Quantitative trait locus analysis of carotid atherosclerosis in an intercross between C57BL/6 and C3H apolipoprotein E-deficient mice. Stroke. 39: 166–173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Su Z., Ishimori N., Chen Y., Leiter E. H., Churchill G. A., Paigen B., Stylianou I. M. 2009. Four additional mouse crosses improve the lipid QTL landscape and identify Lipg as a QTL gene. J. Lipid Res. 50: 2083–2094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Leiter E. H., Reifsnyder P. C., Wallace R., Li R., King B., Churchill G. C. 2009. NOD x 129.H2(g7) backcross delineates 129S1/SvImJ-derived genomic regions modulating type 1 diabetes development in mice. Diabetes. 58: 1700–1703. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.