

Abstract
Patients with Barth syndrome (BTHS), a rare X-linked disease, suffer from skeletal and cardiomyopathy and bouts of cyclic neutropenia. The causative gene encodes tafazzin, a transacylase, which is the major determinant of the final acyl chain composition of the mitochondrial-specific phospholipid, CL. In addition to numerous frame shift and splice-site mutations, 36 missense mutations have been associated with BTHS. Previously, we established a BTHS-mutant panel in the yeast Saccharomyces cerevisiae that successfully models 18/21 conserved pathogenic missense mutations and defined the loss-of-function mechanism associated with a subset of the mutant tafazzins. Here, we report the biochemical and cell biological characterization of the rest of the yeast BTHS-mutant panel and in so doing identify three additional modes of tafazzin dysfunction. The largest group of mutant tafazzins is catalytically null, two mutants encode hypomorphic alleles, and another two mutants are temperature sensitive. Additionally, we have expanded the defects associated with previously characterized matrix-mislocalized-mutant tafazzins to include the rapid degradation of aggregation-prone polypeptides that correctly localize to the mitochondrial IMS. In sum, our in-depth characterization of the yeast BTHS-mutant panel has identified seven functional classes of BTHS mutation.
INTRODUCTION
Cardiolipin (CL) is made and retained exclusively in the mitochondrion (1–3). A diverse collection of mitochondrial-specific activities are influenced by CL including bioenergetic efficiency, mitochondrial biogenesis, division and fusion of mitochondria, mitochondrial morphology and processes that integrate mitochondrial activities into a broader cellular context (reviewed in 1,2).
CL is a dimeric phospholipid consisting of two phosphatidic acids bridged by a central glycerol; thus, CL has four total acyl chains (3). The biosynthesis of CL is conserved in eukaryotes and is executed by three enzymes that localize to the mitochondrial inner membrane (IM) (4–6). CL synthase catalyzes the final step in this cascade producing immature CL. Immature CL is not the same as mature CL. Whereas mature CL (e.g. the steady-state form) is enriched in unsaturated acyl chains that are symmetrically distributed across its chiral center, newly synthesized immature CL has a higher proportion of saturated acyl chains that are randomly attached to the molecule (7,8). The process of CL acyl chain remodeling involves up to three distinct pathways, one of which is evolutionarily conserved from yeast to humans and specifically affected in patients suffering from the X-linked disease Barth syndrome (BTHS) (1,8). BTHS, a rare childhood cardiomyopathy with cyclic neutropenia (8,9), is a potentially fatal disease if not diagnosed, with heart failure and opportunistic infections as major modes of patient mortality. TAFAZZIN, the BTHS gene (10), encodes a monolysocardiolipin (MLCL) transacylase that is the major contributor to the final acyl chain composition of mature CL (11,12). In the absence of tafazzin activity, the steady-state abundance of CL is reduced, the remaining CL has an abnormal acyl chain composition, and MLCL (CL with only three acyl chains) accumulates (13–18). Tafazzin-mediated CL remodeling is initiated by a lipase, in yeast identified as CL-specific deacylase 1 (19), which cleaves an acyl chain of CL, producing MLCL. Thus, the accumulation of MLCL in the absence of tafazzin activity results from the unimpeded initiation of CL remodeling and the subsequent inability to reacylate the remodeling intermediate, MLCL. Dysfunctional tafazzin is associated with defects in the assembly and/or function of the oxidative phosphorylation machinery (9,20–24), an increased production of reactive oxygen species (25) and the presence of mitochondria with altered IM cristae morphology (26–31).
To date, genotype–phenotype correlations are not evident for the BTHS patient population. In addition to numerous frameshift and splice-site mutations, 36 missense mutations that occur throughout the tafazzin polypeptide have been associated with BTHS (1). Further complicating matters is the absence of a known tafazzin structure and most of the molecular details of tafazzin function. In an effort to better understand tafazzin at a molecular level, we previously established a BTHS-mutant panel in the yeast Saccharomyces cerevisiae that successfully models 18/21 conserved pathogenic missense mutations (14). Moreover, our detailed characterization of a subset of these BTHS-mutant tafazzins identified three distinct loss-of-function mechanisms: submitochondrial mislocalization, altered macromolecular complex assembly and complex lability (13,14). Here, we report the biochemical and cell biological characterization of the remainder of the yeast BTHS-mutant panel and in the process identify three additional modes of tafazzin dysfunction: no catalytic activity, decreased catalytic activity and thermosensitivity. In addition, we expand the defects associated with the matrix-mislocalized-mutant tafazzins to include the rapid degradation of aggregation-prone polypeptides that correctly localize to membranes that line the mitochondrial intermembrane space (IMS).
RESULTS
Catalytic mutant tafazzins
Yeast tafazzin (Taz1p) normally associates with both the IM and the outer membrane (OM) always facing the IMS (13,21,32). While the knowledge of many of the molecular details of Taz1p biogenesis is lacking, it is known that the import of Taz1p into mitochondria does not require a membrane potential across the IM (21). Further, the proper targeting of Taz1p, which lacks a prototypical NH2-terminal sorting signal, to mitochondria is encoded by an unidentified internal sorting signal(s). As such, BTHS missense mutations could ablate an internal mitochondrial targeting signal in Taz1p resulting in an abnormal subcellular distribution. To test this hypothesis, yeast strains harboring uncharacterized BTHS-mutant tafazzins were subjected to subcellular fractionation (Fig. 1). Based on the co-fractionation of each mutant tafazzin with mitochondria (Cor2p), subcellular mislocalization is not the pathogenic mechanism for any of the tested BTHS missense mutations.
Figure 1.
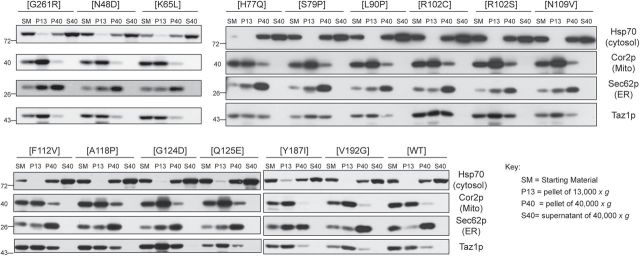
Every BTHS mutant still co-fractionates with mitochondria. Fractions were prepared from Δtaz1 yeast transformed with the indicated BTHS-mutant tafazzin through a series of differential centrifugations. Twenty-five micrograms of each fraction were separated by SDS-PAGE and analyzed by immunoblot using antisera specific for the indicated subcellular organelle.
Previously, we defined the relative expression of the BTHS mutants in yeast extracts (14). To extend these observations, we determined the expression of each BTHS mutant relative to plasmid-borne wild-type (WT) Taz1p in isolated mitochondria by quantitative immunoblotting (Fig. 2A and B). The expression level of the majority of the BTHS mutants was indistinguishable from Δtaz1 yeast transformed with WT Taz1p (Fig. 2B, Δtaz1[WT] expression indicated with dashed green line). As observed in yeast extracts, several BTHS mutants are expressed at drastically reduced levels including mutants that are mislocalized to the mitochondrial matrix (V223D, V224R and I226P; (13)) and mutants that localize and assemble normally but have accelerated rates of complex disassembly and are degraded by the mitochondrial protease, Yme1p (A88R/E, S140R, and L148H; (14)).
Figure 2.
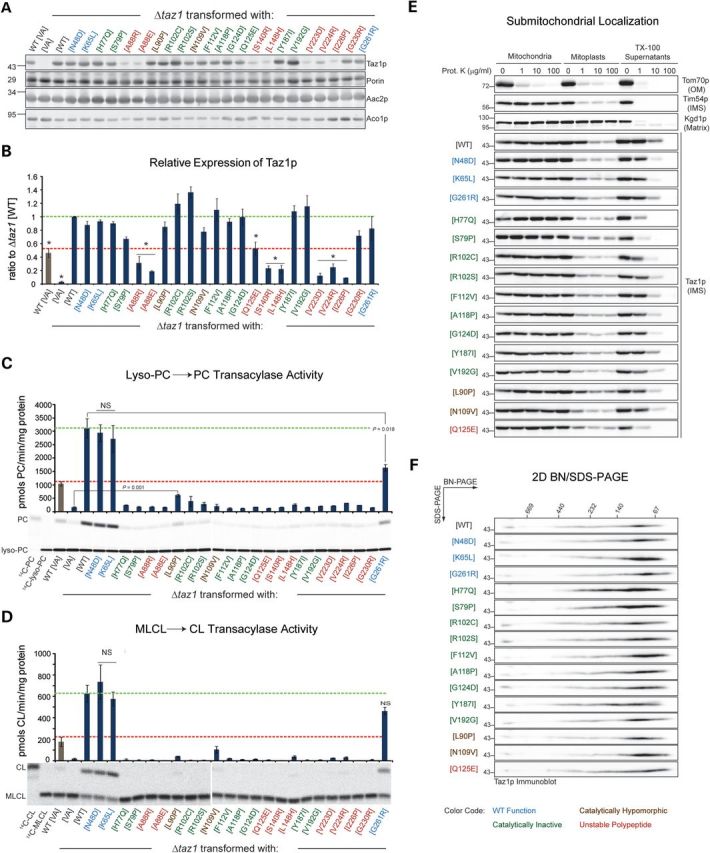
The majority of BTHS missense mutations disrupt tafazzin transacylase activity. (A) The relative expression of each of the BTHS mutants was determined in isolated mitochondria (20 μg) by fluorescent immunoblotting for Taz1p (top panel); Porin, Aac2p and Aco1p served as loading controls (lower panels). (B) The quantified band intensities are expressed as the ratio of the Taz1p signal in Δtaz1 [WT] (mean ± SEM, n = 3). Asterisks indicate significantly lower expression relative to Δtaz1 [WT] (P ≤ 0.001). Lyso-PC → PC (C) and MLCL → CL (D) transacylase activities were determined for each of the BTHS mutants (mean ± SEM, n = 3). Unless indicated as not significant (NS), all differences are significant relative to Δtaz1 [WT] (P ≤ 0.001). In B–D, the dashed green line indicates the value observed in Δtaz1 [WT] and the dashed red line indicate that measured in WT [VA] (WT yeast transformed with vector alone). (E) Submitochondrial localization of WT and BTHS-mutant tafazzins. Intact mitochondria, mitochondria subjected to osmotic shock (mitoplasts) or mitochondria solubilized with 0.1% Triton X–100 were incubated in the presence of the indicated concentration of proteinase K, resolved by SDS-PAGE and immunoblotted as indicated. n = 3. The controls for every source of mitochondria are provided in Supplementary Material, Fig. S1. (F) 1.5% (wt/vol) digitonin extracts from mitochondria derived from the indicated strains were resolved by 2D blue native/SDS-PAGE and Taz1p-complexes detected by immunoblot. n = 3.
In the initial characterization of the yeast BTHS-mutant panel, the functionality of a mutant polypeptide was inferred by its ability to rescue the CL and MLCL levels in a strain lacking endogenous tafazzin (Δtaz1). Based on this analysis, it was concluded that 18/21 BTHS-mutant tafazzins are dysfunctional (14). To directly determine the transacylase activity of each BTHS-mutant tafazzin, two transacylation reactions were performed employing two distinct substrates: 14C-lyso-phosphatidylcholine (Fig. 2C) and 14C-MLCL (Fig. 2D). Tafazzin is a transacylase that normally utilizes phosphatidylcholine (PC) or phosphatidylethanolamine (PE) as the acyl donor to reacylate MLCL (12). However, as transacylases lack inherent directionality, tafazzin can also catalyze the reverse reaction using CL (or another phospholipid) as the acyl chain donor to reacylate lyso-PC/PE. Regardless of the lyso-lipid substrate, the overexpression of WT Taz1p relative to endogenous Taz1p (Fig. 2B, compare Δtaz1[WT] and WT[VA] marked by green and red lines, respectively), resulted in a proportionate increase in transacylase activity (Fig. 2C and D, compare green and red lines). In contrast, the vast majority of BTHS-mutant tafazzins completely lacked any detectable transacylase activity above background (Δtaz1[VA]; Δtaz1 transformed with vector alone). Most of these inactive mutants, including H77Q, S79P, R102C, R102S, F112V, A118P, G124D, Y187I and V192G (highlighted in green), were expressed at WT levels (Fig. 2A and B) and localized correctly to membranes lining the IMS (Fig. 2E). Finally, each mutant formed normal Taz1p complexes as determined by blue native-polyacrylamide gel electrophoresis indicating that the quaternary structure of each mutant polypeptide was similar to WT Taz1p (Fig. 2F). As such, these mutants specifically inactivate tafazzin transacylase activity and are classified as catalytically null. Notably, H77 in the HX4D motif, a conserved structural motif in acyltransferases, is one of these catalytically dead mutants.
Two BTHS mutants are notable because they retained some residual catalytic activity (highlighted in brown). The L90P mutant had significant activity with lyso-PC as substrate (Fig. 2C) and some, albeit not significant, activity in the MLCL → CL transacylation reaction (Fig. 2D). Interestingly, whereas the N109V BTHS mutant had no detectable activity using lyso-PC as substrate, it had measurable MLCL → CL transacylase activity (P = 0.036 relative to Δtaz1[VA]). The mitochondrial expression (Fig. 2A and B), localization (Fig. 2E) and assembly (Fig. 2F) of both of the mutants were the same as WT Taz1p. Thus, these mutations appear to encode hypomorphic alleles. Finally, based on its low expression, absent activity with either lyso-lipid substrate, and normal submitochondrial localization and assembly, the Q125E mutation destabilizes and inactivates the mutant polypeptide.
BTHS mutants with combined biochemical defects
Based on submitochondrial localization studies, three BTHS mutations that occur in the middle of an unusual membrane anchor of Taz1p inactivate a stop-transfer signal resulting in their misincorporation into the mitochondrial matrix (13). Each of these mutants, V223D, V224R and I226P, is expressed at significantly lower levels than WT Taz1p. To test the possibility that these mutants are actively degraded, each of these mutants was expressed in the presence and absence of three IM proteases implicated in mitochondrial quality control: Oma1p, the m-AAA protease, and the i-AAA protease (Fig. 3A) (33,34). The m-AAA protease is a hetero-oligomer of Yta10p and Yta12p, and the i-AAA protease is a homomultimer of Yme1p. WT Taz1p expression was not changed by the presence or absence of any of these proteases. As previously shown (14), the expression of the L148H mutant, which is localized correctly to the IMS and assembles in complexes that are intrinsically unstable, is restored in the absence of Yme1p. Based on their mislocalization to the mitochondrial matrix (13), we hypothesized that deletion of the m-AAA protease, whose active site faces the matrix, would recover the expression of the V223D, V224R and I226P mutants. Surprisingly, while there was a modest increase in the expression of each of these mutants in the absence of Yta12p, there was a proportionately greater increase in the absence of the i-AAA protease (Fig. 3A).
Figure 3.

Aggregation-prone mutants with low-fidelity submitochondrial sorting. (A) Taz1p expression was determined in whole cell extracts derived from the indicated strains by immunoblotting for Taz1p, Yme1p, cytochrome c peroxidase (Ccp1p), and the loading control, Aac2p. n = 3 (B) Submitochondrial localization of WT and BTHS-mutant tafazzins in the presence and absence of Yme1p. n = 3. The controls for every source of mitochondria are provided in Supplementary Material, Fig. S2. (C and D) Solubility of Taz1p in detergents. Mitochondria isolated from the indicated strains were solubilized with digitonin and separated into a supernatant (S1) and pellet by centrifugation. Non-extracted material was solubilized with TX–100 and fractionated into a supernatant (S2) and pellet (P2) by centrifugation. (C) Fractions were resolved by SDS-PAGE and immunoblotted as indicated. (D) The percentage of Taz1p in TX–100 insoluble aggregates (light gray) and the percentage of Taz1p solubilized by digitonin (dark gray) (mean ± SEM, n = 3). Significant differences are indicated. (E) The mitochondrial MLCL:CL ratios following steady-state labeling with 32Pi (mean ± SEM, n = 6). Significant differences are indicated. (F and G) MLCL → CL transacylase assay. Lower panels in (F) show the expression of Taz1p in mitochondria (20 μg) by immunoblot. In (G), the calculated MLCL → CL activities were determined (mean ± SEM, n = 4) and significant differences are indicated.
As the active site of the i-AAA protease is on the IMS side of the IM, this result suggests that some fraction of the mutant polypeptides do, in fact, properly localize to the IMS side of the IM where they are actively degraded by Yme1p. To explore this possibility, the submitochondrial localization of WT, V223D, V224R and I226P was compared in the presence and absence of the i-AAA protease (Fig. 3B). WT Taz1p localized to IMS-facing leaflets (sensitive to added protease upon rupturing the OM; mitoplasts) regardless of the presence or absence of Yme1p. As shown previously, in the presence of Yme1p, the V223D, V224R and I226P mutants were only degraded by added proteinase K upon the inclusion of TX-100, consistent with their mislocalization to the mitochondrial matrix. In contrast, a significant proportion of each mutant polypeptide was degraded by proteinase K upon rupturing the OM when expressed in the absence of the i-AAA protease indicating that indeed, some fraction of each of these mutants is properly sorted to the IMS and degraded by Yme1p. Thus, Yme1p-mediated proteolysis masked the fact that some proportion of the V223D, V224R and I226P mutants was properly sorted to the IMS side of the IM.
To ascertain whether these mutants have a folding/assembly defect, a detergent-based aggregation protocol was performed (Fig. 3C and D). In this assay, protein aggregates are resistant to TX-100 solubilization (P2). WT Taz1p was almost completely extracted by digitonin in the presence or absence of Yme1p. In contrast, the known aggregation-prone mutant, L148H, showed some resistance to TX-100 solubilization in the presence of Yme1p; the fraction of L148H that was solubilized by digitonin was significantly less in the absence of Yme1p than in its presence and there was proportionately more L148H enriched in TX-100-resistant aggregates. This trend was recapitulated with the V223D, V224R and I226P mutants in both the presence and absence of the i-AAA protease. In the absence of Yme1p, each mutant was less well extracted by both digitonin and TX-100, indicating that in the absence of the i-AAA protease, the V223D, V224R and I226P-mutant polypeptides that reside on the IMS side of the IM have a tendency to form TX-100 insoluble aggregates.
In spite of its intrinsic lability, the expression of the L148H mutant in the absence of Yme1p was shown to partially restore the steady-state levels of CL and MLCL as well as the acyl chain composition of CL (14). A more sensitive measure of Taz1p activity is the MLCL:CL ratio (higher in the absence of tafazzin function) (16). The MLCL:CL ratio of mitochondria harboring WT Taz1p was worsened by the absence of Yme1p, whereas this ratio was improved for mitochondria expressing the L148H BTHS mutant specifically in the absence of the i-AAA protease. For the V223D, V224R and I226P mutants, the MLCL:CL ratio was not changed or only modestly altered upon deletion of the i-AAA protease, suggesting that they are nonfunctional independent of their expression levels and/or submitochondrial distribution. To formally test this, the MLCL → CL assay was performed (Fig. 3F and G). Consistent with the recovered acyl chain composition of CL and the steady-state abundance of CL and MLCL, the L148H mutant had significantly more transacylase activity when expressed in the absence of Yme1p. Thus, for this mutant, the increased expression that occurs in the absence of Yme1p is directly correlated with more tafazzin activity. For the V223D and I226P mutants, there was no change in their low transacylase activity when their expression levels were partially restored in the absence of the i-AAA protease. These mutants are, therefore, nonfunctional independent of their expression levels and/or submitochondrial localization. In contrast, the V224R mutant displayed a trend towards increased transacylase activity in the absence of Yme1p, indicating that this particular mutant may retain some catalytic potential. Interestingly, the transacylase activity of WT Taz1p was significantly less in the absence of the i-AAA protease than in its presence. Moreover, a slight and yet significant increase in WT Taz1p was detected in TX-100-resistant aggregates in the absence of the i-AAA protease (Fig. 3D). When combined with our previous determination that the import of WT Taz1p is more efficient in the absence of Yme1p than in its presence (14), these results suggest that the i-AAA protease is a major player in monitoring and enforcing tafazzin quality control.
Thermosensitive BTHS mutants
Three mutants, N48D, K65L and G261R, are functionally and biochemically indistinguishable from WT Taz1p when analyzed at 30°C (Fig. 2). To determine whether any or all of these mutants are temperature sensitive, their expression was compared in yeast grown at 30 or 37°C (Fig. 4A). Indeed, the expression of the N48D, K65L and G261R mutants was decreased by 30, 30, and 80%, respectively, at 37°C relative to 30°C. Further, a combined K65L/G261R mutant was even more thermolabile with <5% detected at 37°C compared with that at 30°C.
Figure 4.

Thermolabile BTHS alleles. Δtaz1 yeast transformed as indicated were grown at 30 or 37°C for 24 h. (A) Taz1p expression was determined in whole cell extracts by immunoblot; Pic1p and Tom70p are loading controls. The percentage of Taz1p at 37°C relative to 30°C is shown at the bottom (mean ± SEM, n = 3). (B) Mitochondrial phospholipids separated by thin-layer chromatography and revealed by phosphorimaging. (C) The derived MLCL:CL ratios (mean ± SEM n = 6). Significant differences are indicated. (D and E) MLCL → CL transacylase assay. Before initiating the reaction, mitochondria were pre-incubated at 4, 30 or 37°C for the indicated time. In (D), the calculated transacylase activities are expressed as a percentage of the maximal activity (mitochondria kept at 4°C) for a given source of mitochondria. (mean ± SEM, n = 3–4) The maximal activities were (pmols CL/min/mg protein): Δtaz1[WT] = 381.9 ± 45.7; Δtaz1[K65L] = 404.3 ± 25.8; Δtaz1[G261R] = 263.4 ± 10.7; and Δtaz1[K65L/G261R] = 119.3 ± 25.1. The significant differences relative to WT Taz1p treated the same are indicated. (E) The expression of Taz1p in the same samples (20 μg) by immunoblot; Porin, Aac2p, and Aco1p served as loading controls.
Next, the relative abundance of CL and MLCL, and the derived MLCL:CL ratio were determined at 30°C and 37°C (Fig. 4B and C). Importantly, for Δtaz1 yeast rescued with K65L, G261R or K65L/G261R, the drop in expression at 37°C correlated with the development of an abnormal MLCL:CL ratio (Fig. 4C). Moreover, for both the G261R and K65L/G261R mutants, the MLCL:CL ratio was significantly greater at both temperatures compared with WT. As virtually nothing is known about the half-life of CL or MLCL, the relatively modest change in the MLCL:CL ratio at 37°C compared with the drastic effect on mutant Taz1p abundance (Fig. 4A) suggests that the turnover of CL is slower than that of the mutant polypeptides.
The kinetics of tafazzin heat inactivation were determined by pre-incubating mitochondria at 30°C for 10 min or 37°C for 2.5, 5, or 10 min prior to initiating the transacylase reaction, performed at 30°C (Fig. 4D). The pre-incubation of WT Taz1p at 37°C resulted in a time-dependent drop in transacylase activity with only ∼45% of the initial activity (4°C samples) remaining after 10 min. For the K65L mutant, the rate of transacylase decay upon 37°C pre-incubation was faster and greater than for WT Taz1p. Pre-incubation at 37°C for just 2.5 min caused an 80–90% drop in the maximal activity of the G261R and K65L/G261R mutants. Moreover, a 10 min pre-incubation at 30°C resulted in a 60–70% reduction in their associated activity. Importantly, for WT and mutants alike, the decay in activity was not paralleled by any reduction in the levels of Taz1p polypeptide (Fig. 4E). Altogether, these results indicate that for the K65L, G261R and K65L/G261R mutants, the loss of transacylase activity precedes the decrease in steady-state polypeptide levels which occurs prior to major changes in the MLCL:CL ratio. Thus, the K65L and G261R mutants are temperature sensitive.
DISCUSSION
While the biological function of tafazzin as a transacylase with a central role in the process of CL acyl chain remodeling is firmly established, the molecular details of how it executes this function are lacking. Moreover, there is currently no published structure of tafazzin from any species. To begin to fill this void, we established a BTHS-mutant tafazzin panel in which every pathogenic mutation occurring at conserved residues between the human and yeast orthologs was individually modeled in yeast Taz1p (14). Yeast was chosen as the model because CL biosynthesis and tafazzin-mediated acyl chain remodeling are conserved from yeast to humans and the cell biology of endogenous WT tafazzin has been defined (13). In contrast, nothing is currently known about the basic cell biology and biochemistry of human tafazzin as expressed endogenously and a cell culture model that recapitulates the basic biochemical parameters of tafazzin deficiency (e.g. decreased CL and increased MLCL) has not been described. Thus, at present, yeast is the best model to define the pathogenic mechanism of mutant alleles. Through our in-depth characterization of the BTHS panel, we have defined the loss-of-function mechanism of each pathogenic mutation, information that has provided potential therapeutic strategies, may aid in the recognition of genotype–phenotype correlations for BTHS patients, and forms the basis for future structure–function studies based on a homology model of tafazzin.
In all, we have defined seven functional classes of BTHS mutation (Fig. 5A). Class 1 mutations, numerically the largest class, include the numerous frameshift and splice-site mutations that result in the production of little or no functional tafazzin. Class 2 mutations were originally thought to be only mistargeted to the mitochondrial matrix (13); however, we demonstrated here that these mutants have pleiotropic biochemical defects. Specifically, the V223D, V224R and I226P mutants display low-fidelity sorting to the mitochondrial IMS with some mutant polypeptides mistargeted to the mitochondrial matrix, as originally annotated. A significant fraction of each mutant polypeptide is localized correctly to the IMS but has a tendency to form detergent-resistant aggregates that are degraded by the i-AAA protease. Class 3 mutations specifically impact the macromolecular assembly of Taz1p; G230R is the only documented BTHS mutation in this class (13). Class 4 mutations result in a catalytically inactive protein that is otherwise normal (expression, localization and assembly); the majority of missense BTHS mutations are class 4 mutants. BTHS mutations that are biochemically normal and retain residual transacylase activity are class 5 mutants. So far, only two mutations, L90P and N109V, encode such hypomorphic alleles. Class 6 mutants localize to and within mitochondria as expected, but they are degraded by the i-AAA component of the mitochondrial quality control apparatus due to folding and assembly defects that shift the equilibrium of the mutant polypeptides towards disassembly (14). Interestingly, two class 6 mutants, A88R and L148H, retain significant transacylase activity when their expression is restored in the absence of the i-AAA protease, suggesting that small molecules that stabilize these mutant polypeptides and prevent their degradation may be a viable therapeutic strategy. Finally, class 7 mutants are temperature sensitive. For class 7 mutants, activity loss precedes the degradation of the mutant polypeptide; changes in the steady-state phospholipid profile occur with delayed kinetics presumably reflecting, at least in part, the half-life of mature CL.
Figure 5.
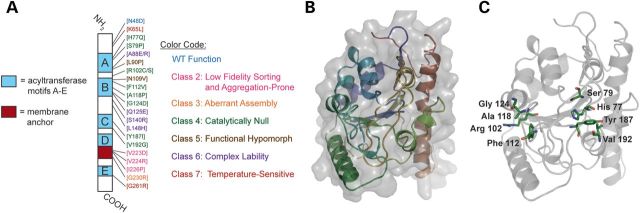
Model of yeast Taz1p. (A) Schematic representation of Taz1p with the position and functional class of each BTHS missense mutation indicated. Light blue boxes, predicted acyl transferase motifs A–E; blood red box, interfacial membrane anchor. (B and C) A homology model of yeast Taz1p was generated using the Phyre server, with the squash glycerol-3-phosphate acyltransferase as a template. (B) Taz1p in cartoon format colored from the N-terminus (blue) to the C-terminus (red). Surface representation is shown in grey. (C) The location of the class 4 mutant residues (green).
In total, 20 of 21 BTHS mutations are dysfunctional when modeled in yeast Taz1p. Still, it is reasonable to ask whether the defined loss-of-function mechanisms in the yeast panel are conserved with equivalent mutations in human tafazzin in the mammalian setting. While the answer to this question requires direct testing in an appropriate mammalian model, perhaps the best evidence for the accuracy of the yeast BTHS-mutant panel is provided by the one mutation, N48D, which according to every tested parameter behaves exactly like WT Taz1p. Interestingly, two brothers harboring the corresponding mutation, N40D, suffered from cardiomyopathy in infancy (A. Bowron and C. Steward, personal communication). They had 3-methylglutaconic aciduria, which lead to their formal BTHS diagnosis. However, their clinical course has been comparatively mild, and neutropenic episodes have never been documented. Moreover, these two boys have CL levels within the normal range. Still, their MLCL levels were comparable with those of other BTHS patients which resulted in a modestly altered MLCL:CL ratio. Thus, the absence of any discernible defect in the yeast N48D mutant is consistent with human patients harboring the equivalent mutation. As tafazzin can modify more than just CL (12), the observations with the N48D/N40D mutant tafazzins may potentially uncover novel conserved activities of tafazzin independent of CL metabolism.
Tafazzin belongs to a protein superfamily that includes acyltransferases which utilize glycerolphosphate derivatives as substrates, although tafazzins form a distinct subgroup within this superfamily (8,35). Thus, the Phyre server (36) was used to generate a homology model of Taz1p using the squash glycerol-3-phosphate acyltransferase (PDBID:1iuq) as a template (Fig. 5B). When the class 4 (catalytically inactive) mutants were mapped on the homology model (Fig. 5C), four residues (H77, S79, Y187 and V192) faced the predicted catalytic pocket. Thus, mutating these residues is expected to either directly inhibit catalysis or prevent substrate binding. The remaining class 4 mutants cluster relatively close to each other, but are not immediately adjacent to the predicted catalytic pocket; therefore, mutations in these residues may affect substrate binding, but they are unlikely to directly affect catalysis. Future structure–function studies based on this tentative structural model are expected to provide significant molecular insights into tafazzin function.
MATERIALS AND METHODS
Generating 14C-labeled phospholipid
∼600 OD600's Δtaz1 yeast grown in rich lactate medium (1% yeast extract, 2% tryptone, 0.05% dextrose, 2% lactic acid, 3.4 mm CaCl2 · 2H2O, 8.5 mm NaCl, 2.95 mm MgCl2 · 6H2O, 7.35 mm KH2PO4, 18.7 mm NH4Cl, pH 5.5) was sedimented, the supernatant sterilely aspirated and the yeast pellet resuspended in SInC medium (0.17% yeast nitrogen base minus amino acids and ammonium sulfate, 0.5% ammonium sulfate, 0.2% dropout mix synthetic) at 5 OD600's/ml. 5 μCi/ml 14C-acetic acid sodium salt (PerkinElmer Life Sciences) was added and the yeast was incubated at 30°C for 6 h shaking at 300RPM. Isolation of mitochondria and phospholipid extraction was as described (13). Phospholipids were resuspended in 93 μl chloroform and 10 aliquots spotted onto a Soft Layer Silica Gel GF TLC plate (Alltech) pretreated with 1.8% boric acid in ethanol. One hundred micrograms of bovine CL and PE (Sigma) were loaded as migration standards. Following resolution in chloroform:ethanol:water:triethylamine (30:35:7:35), phospholipids were imaged using the ethidium bromide setting of a Pharos Imager (Bio-Rad Laboratories) and CL, MLCL, and PC were recovered post-scraping. Dried down lipids were resuspended in chloroform and stored at −20°C. Phosphate quantification was performed as described (37) and the amount of 14C-incorporation determined by liquid scintillation.
Transacylation assays
The transacylase assays were performed as previously described (12) with minor modifications. In brief, per reaction, 1 nmol of 1-[14C]palmitoyl-2-lyso-PC (0.05 μCi; PerkinElmer Life Sciences) or [14C]-MLCL (1.22 nCi) was evaporated in a glass tube under a stream of N2. Following addition of 0.2 ml reaction buffer (10 mm 2-mercaptoethanol, 0.5 mm EDTA, 50 mm Tris, pH 7.4) and incubation at 30°C for at least 1 h, the reactions were sonicated in a water bath sonicator for 1 min. CHAPs (8 mm) were also included in the MLCL → CL reaction buffer. Reactions were initiated by adding mitochondria (25–50 μg with the exact amount determined in parallel by the BCA assay) followed by a ∼1 second vortex on high. In some experiments, mitochondria (5–10 mg/ml) was incubated in pre-warmed SEH buffer (250 mm sucrose, 5 mm EDTA, 10 mm HEPES pH7.4) at the indicated temperature for the designated time prior to beginning the transacylase reactions. The reaction mixture was incubated at 30°C for 5 (lyso-PC → PC) to 30 (MLCL → CL) min shaking at 220 rpm and stopped by the addition of 1 ml methanol. Lipids were extracted with 2 ml of chloroform, phase separation initiated with 0.6 ml of 0.9% NaCl and lower organic phases washed with 0.5 ml of 1:1 methanol:ddH2O. The organic phases were collected and dried down under streams of N2. Phospholipids were separated on silica gel 60 plates with the solvent chloroform:methanol:water (65:25:4). Labeled phospholipids were revealed using a K-screen and Pharos Imager (Bio-Rad Laboratories), quantitation was executed using the affiliated Quantity One software and statistical analyses performed using SigmaPlot 11.
Antibodies
Most of the antibodies used in this work were generated in the Schatz or Koehler laboratories and have been described previously. Antibodies were raised in rabbits using His6Pic1 as antigen. In brief, the entire open reading frame of MIR1 was cloned into the pET28a vector (Novagen), downstream of the His6 tag, induced in BL21-CodonPlus(DE3)-RIL Escherichia coli and purified using Ni2+ agarose (Qiagen). Other antibodies used were mouse anti-Sec62p (kind gift of Dr David Meyers, University of California, Los Angeles, USA), mouse anti-yAAC2 (clone 6H8; (38)) and horseradish peroxidase-conjugated (Figs. 1, 2E and F, 3B, 4A; Supplementary Material, Figs. S1 and S2) or fluorescent-conjugated (Fig. 2A, 3A, C, F and 4E) secondary antibodies (Pierce).
Miscellaneous
The yeast BTHS-mutant tafazzin panel has been described (14). Isolation of mitochondria, subcellular fractionation, preparation of yeast cell extracts, submitochondrial localization, 2D BN/sodium dodecyl sulphate-polyacrylamide gel electrophoresis (SDS-PAGE), phospholipid analyses, detergent-based aggregation and immunoblotting were performed as previously described (13,14,22). The Taz1p model was generated using the Phyre server (36).
SUPPLEMENTARY MATERIAL
Supplementary Material is available at HMG online.
ACKNOWLEDGEMENTS
We would like to thank Dr Jeff Schatz and Carla Koehler (UCLA) for antibodies, and Drs Michael Schlame (NYU) and Wayne Riekhof (National Jewish) for help with the transacylase assays and isolating radiolabeled yeast phospholipids. This work was supported by National Institutes of Health (Grant number R00HL089185 to S.M.C.). M.G.B. is a predoctoral fellow of the American Heart Association.
Conflict of Interest statement. None declared.
REFERENCES
- 1.Claypool S.M., Koehler C.M. The complexity of cardiolipin in health and disease. Trends Biochem. Sci. 2012;37:32–41. doi: 10.1016/j.tibs.2011.09.003. doi:10.1016/j.tibs.2011.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Osman C., Voelker D.R., Langer T. Making heads or tails of phospholipids in mitochondria. J. Cell Biol. 2011;192:7–16. doi: 10.1083/jcb.201006159. doi:10.1083/jcb.201006159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Schlame M., Rua D., Greenberg M.L. The biosynthesis and functional role of cardiolipin. Prog. Lipid Res. 2000;39:257–288. doi: 10.1016/s0163-7827(00)00005-9. doi:10.1016/S0163-7827(00)00005-9. [DOI] [PubMed] [Google Scholar]
- 4.Dzugasova V., Obernauerova M., Horvathova K., Vachova M., Zakova M., Subik J. Phosphatidylglycerolphosphate synthase encoded by the PEL1/PGS1 gene in Saccharomyces cerevisiae is localized in mitochondria and its expression is regulated by phospholipid precursors. Curr. Genet. 1998;34:297–302. doi: 10.1007/s002940050399. doi:10.1007/s002940050399. [DOI] [PubMed] [Google Scholar]
- 5.Osman C., Haag M., Wieland F.T., Brugger B., Langer T. A mitochondrial phosphatase required for cardiolipin biosynthesis: the PGP phosphatase Gep4. EMBO J. 2010;29:1976–1987. doi: 10.1038/emboj.2010.98. doi:10.1038/emboj.2010.98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Schlame M., Haldar D. Cardiolipin is synthesized on the matrix side of the inner membrane in rat liver mitochondria. J. Biol. Chem. 1993;268:74–79. [PubMed] [Google Scholar]
- 7.Schlame M., Ren M., Xu Y., Greenberg M.L., Haller I. Molecular symmetry in mitochondrial cardiolipins. Chem. Phys. Lipids. 2005;138:38–49. doi: 10.1016/j.chemphyslip.2005.08.002. doi:10.1016/j.chemphyslip.2005.08.002. [DOI] [PubMed] [Google Scholar]
- 8.Schlame M., Ren M. Barth syndrome, a human disorder of cardiolipin metabolism. FEBS Lett. 2006;580:5450–5455. doi: 10.1016/j.febslet.2006.07.022. doi:10.1016/j.febslet.2006.07.022. [DOI] [PubMed] [Google Scholar]
- 9.Barth P.G., Scholte H.R., Berden J.A., Van der Klei-Van Moorsel J.M., Luyt-Houwen I.E., Van 't Veer-Korthof E.T., Van der Harten J.J., Sobotka-Plojhar M.A. An X-linked mitochondrial disease affecting cardiac muscle, skeletal muscle and neutrophil leucocytes. J. Neurol. Sci. 1983;62:327–355. doi: 10.1016/0022-510x(83)90209-5. doi:10.1016/0022-510X(83)90209-5. [DOI] [PubMed] [Google Scholar]
- 10.Bione S., D'Adamo P., Maestrini E., Gedeon A.K., Bolhuis P.A., Toniolo D. A novel X-linked gene, G4.5, is responsible for Barth syndrome. Nat. Genet. 1996;12:385–389. doi: 10.1038/ng0496-385. doi:10.1038/ng0496-385. [DOI] [PubMed] [Google Scholar]
- 11.Schlame M., Kelley R.I., Feigenbaum A., Towbin J.A., Heerdt P.M., Schieble T., Wanders R.J., DiMauro S., Blanck T.J. Phospholipid abnormalities in children with Barth syndrome. J. Am. Coll. Cardiol. 2003;42:1994–1999. doi: 10.1016/j.jacc.2003.06.015. doi:10.1016/j.jacc.2003.06.015. [DOI] [PubMed] [Google Scholar]
- 12.Xu Y., Malhotra A., Ren M., Schlame M. The enzymatic function of tafazzin. J. Biol. Chem. 2006;281:39217–39224. doi: 10.1074/jbc.M606100200. doi:10.1074/jbc.M606100200. [DOI] [PubMed] [Google Scholar]
- 13.Claypool S.M., McCaffery J.M., Koehler C.M. Mitochondrial mislocalization and altered assembly of a cluster of Barth syndrome mutant tafazzins. J. Cell Biol. 2006;174:379–390. doi: 10.1083/jcb.200605043. doi:10.1083/jcb.200605043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Claypool S.M., Whited K., Srijumnong S., Han X., Koehler C.M. Barth syndrome mutations that cause tafazzin complex lability. J. Cell Biol. 2011;192:447–462. doi: 10.1083/jcb.201008177. doi:10.1083/jcb.201008177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gu Z., Valianpour F., Chen S., Vaz F.M., Hakkaart G.A., Wanders R.J., Greenberg M.L. Aberrant cardiolipin metabolism in the yeast taz1 mutant: a model for Barth syndrome. Mol. Microbiol. 2004;51:149–158. doi: 10.1046/j.1365-2958.2003.03802.x. doi:10.1046/j.1365-2958.2003.03802.x. [DOI] [PubMed] [Google Scholar]
- 16.Houtkooper R.H., Rodenburg R.J., Thiels C., van Lenthe H., Stet F., Poll-The B.T., Stone J.E., Steward C.G., Wanders R.J., Smeitink J., et al. Cardiolipin and monolysocardiolipin analysis in fibroblasts, lymphocytes, and tissues using high-performance liquid chromatography-mass spectrometry as a diagnostic test for Barth syndrome. Anal. Biochem. 2009;387:230–237. doi: 10.1016/j.ab.2009.01.032. doi:10.1016/j.ab.2009.01.032. [DOI] [PubMed] [Google Scholar]
- 17.Valianpour F., Mitsakos V., Schlemmer D., Towbin J.A., Taylor J.M., Ekert P.G., Thorburn D.R., Munnich A., Wanders R.J., Barth P.G., et al. Monolysocardiolipins accumulate in Barth syndrome but do not lead to enhanced apoptosis. J. Lipid Res. 2005;46:1182–1195. doi: 10.1194/jlr.M500056-JLR200. doi:10.1194/jlr.M500056-JLR200. [DOI] [PubMed] [Google Scholar]
- 18.Vaz F.M., Houtkooper R.H., Valianpour F., Barth P.G., Wanders R.J. Only one splice variant of the human TAZ gene encodes a functional protein with a role in cardiolipin metabolism. J. Biol. Chem. 2003;278:43089–43094. doi: 10.1074/jbc.M305956200. doi:10.1074/jbc.M305956200. [DOI] [PubMed] [Google Scholar]
- 19.Beranek A., Rechberger G., Knauer H., Wolinski H., Kohlwein S.D., Leber R. Identification of a cardiolipin-specific phospholipase encoded by the gene CLD1 (YGR110W) in yeast. J. Biol. Chem. 2009;284:11572–11578. doi: 10.1074/jbc.M805511200. doi:10.1074/jbc.M805511200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Barth P.G., Van den Bogert C., Bolhuis P.A., Scholte H.R., van Gennip A.H., Schutgens R.B., Ketel A.G. X-linked cardioskeletal myopathy and neutropenia (Barth syndrome): respiratory-chain abnormalities in cultured fibroblasts. J. Inherit. Metab. Dis. 1996;19:157–160. doi: 10.1007/BF01799418. doi:10.1007/BF01799418. [DOI] [PubMed] [Google Scholar]
- 21.Brandner K., Mick D.U., Frazier A.E., Taylor R.D., Meisinger C., Rehling P. Taz1, an outer mitochondrial membrane protein, affects stability and assembly of inner membrane protein complexes: implications for Barth Syndrome. Mol. Biol. Cell. 2005;16:5202–5214. doi: 10.1091/mbc.E05-03-0256. doi:10.1091/mbc.E05-03-0256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Claypool S.M., Oktay Y., Boontheung P., Loo J.A., Koehler C.M. Cardiolipin defines the interactome of the major ADP/ATP carrier protein of the mitochondrial inner membrane. J. Cell Biol. 2008;182:937–950. doi: 10.1083/jcb.200801152. doi:10.1083/jcb.200801152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ma L., Vaz F.M., Gu Z., Wanders R.J., Greenberg M.L. The human TAZ gene complements mitochondrial dysfunction in the yeast taz1Delta mutant. Implications for Barth syndrome. J. Biol. Chem. 2004;279:44394–44399. doi: 10.1074/jbc.M405479200. doi:10.1074/jbc.M405479200. [DOI] [PubMed] [Google Scholar]
- 24.McKenzie M., Lazarou M., Thorburn D.R., Ryan M.T. Mitochondrial respiratory chain supercomplexes are destabilized in Barth Syndrome patients. J. Mol. Biol. 2006;361:462–469. doi: 10.1016/j.jmb.2006.06.057. doi:10.1016/j.jmb.2006.06.057. [DOI] [PubMed] [Google Scholar]
- 25.Chen S., He Q., Greenberg M.L. Loss of tafazzin in yeast leads to increased oxidative stress during respiratory growth. Mol. Microbiol. 2008;68:1061–1072. doi: 10.1111/j.1365-2958.2008.06216.x. doi:10.1111/j.1365-2958.2008.06216.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Claypool S.M., Boontheung P., McCaffery J.M., Loo J.A., Koehler C.M. The cardiolipin transacylase, tafazzin, associates with two distinct respiratory components providing insight into Barth syndrome. Mol. Biol. Cell. 2008;19:5143–5155. doi: 10.1091/mbc.E08-09-0896. doi:10.1091/mbc.E08-09-0896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Acehan D., Khuchua Z., Houtkooper R.H., Malhotra A., Kaufman J., Vaz F.M., Ren M., Rockman H.A., Stokes D.L., Schlame M. Distinct effects of tafazzin deletion in differentiated and undifferentiated mitochondria. Mitochondrion. 2009;9:86–95. doi: 10.1016/j.mito.2008.12.001. doi:10.1016/j.mito.2008.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Acehan D., Malhotra A., Xu Y., Ren M., Stokes D.L., Schlame M. Cardiolipin affects the supramolecular organization of ATP synthase in mitochondria. Biophys. J. 2011;100:2184–2192. doi: 10.1016/j.bpj.2011.03.031. doi:10.1016/j.bpj.2011.03.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Acehan D., Vaz F., Houtkooper R.H., James J., Moore V., Tokunaga C., Kulik W., Wansapura J., Toth M.J., Strauss A., et al. Cardiac and skeletal muscle defects in a mouse model of human Barth syndrome. J. Biol. Chem. 2011;286:899–908. doi: 10.1074/jbc.M110.171439. doi:10.1074/jbc.M110.171439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Acehan D., Xu Y., Stokes D.L., Schlame M. Comparison of lymphoblast mitochondria from normal subjects and patients with Barth syndrome using electron microscopic tomography. Lab. Invest. 2007;87: 40–48. doi: 10.1038/labinvest.3700480. doi:10.1038/labinvest.3700480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Soustek M.S., Falk D.J., Mah C.S., Toth M.J., Schlame M., Lewin A.S., Byrne B.J. Characterization of a transgenic short hairpin RNA-induced murine model of tafazzin deficiency. Hum. Gene Ther. 2011;22:865–871. doi: 10.1089/hum.2010.199. doi:10.1089/hum.2010.199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gebert N., Joshi A.S., Kutik S., Becker T., McKenzie M., Guan X.L., Mooga V.P., Stroud D.A., Kulkarni G., Wenk M.R., et al. Mitochondrial cardiolipin involved in outer-membrane protein biogenesis: implications for Barth syndrome. Curr. Biol. 2009;19:2133–2139. doi: 10.1016/j.cub.2009.10.074. doi:10.1016/j.cub.2009.10.074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Khalimonchuk O., Jeong M.Y., Watts T., Ferris E., Winge D.R. Selective Oma1 protease-mediated proteolysis of Cox1 subunit of cytochrome oxidase in assembly mutants. J. Biol. Chem. 2012;287: 7289–7300. doi: 10.1074/jbc.M111.313148. doi:10.1074/jbc.M111.313148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tatsuta T. Protein quality control in mitochondria. J. Biochem. 2009;146:455–461. doi: 10.1093/jb/mvp122. doi:10.1093/jb/mvp122. [DOI] [PubMed] [Google Scholar]
- 35.Neuwald A.F. Barth syndrome may be due to an acyltransferase deficiency. Curr. Biol. 1997;7:R465–466. doi: 10.1016/s0960-9822(06)00237-5. doi: [DOI] [PubMed] [Google Scholar]
- 36.Kelley L.A., Sternberg M.J. Protein structure prediction on the Web: a case study using the Phyre server. Nat. Protoc. 2009;4:363–371. doi: 10.1038/nprot.2009.2. doi:10.1038/nprot.2009.2. [DOI] [PubMed] [Google Scholar]
- 37.Rouser G., Fkeischer S., Yamamoto A. Two dimensional then layer chromatographic separation of polar lipids and determination of phospholipids by phosphorus analysis of spots. Lipids. 1970;5:494–496. doi: 10.1007/BF02531316. doi:10.1007/BF02531316. [DOI] [PubMed] [Google Scholar]
- 38.Panneels V., Schussler U., Costagliola S., Sinning I. Choline head groups stabilize the matrix loop regions of the ATP/ADP carrier ScAAC2. Biochem. Biophys. Res. Commun. 2003;300:65–74. doi: 10.1016/s0006-291x(02)02795-x. doi:10.1016/S0006-291X(02)02795-X. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.