

Abstract
Arsenic is a recognized human carcinogen, but the mechanism of carcinogenesis is not well understood. Oxidative stress and inhibition of DNA damage repair have been postulated as potential carcinogenic actions of arsenic. The present study tests the hypothesis that arsenite not only induces oxidative stress, but also inhibits the activity of the DNA base excision repair protein, poly(ADP-ribose) polymerase-1 (PARP-1), leading to exacerbation of the oxidative DNA damage induced by arsenic. HaCat cells were treated with arsenite for 24 hrs before measuring 8-hydroxyl-2′-deoxyguanosine (8-OHdG), PARP-1 activity, and reactive oxygen species (ROS). Zinc supplementation and PARP-1 siRNA were used to increase or decrease, respectively, the PARP-1 protein’s physiological function. At high concentrations (10μM or higher), arsenite greatly induced oxidative DNA damage, as indicated by 8-OHdG formation. At lower concentrations (1μM), arsenite did not produce detectable 8-OHdG, but was still able to effectively inhibit PARP-1 activity. Zinc supplementation reduced the formation of 8-OHdG, restored the PARP-1 activity inhibited by arsenite, but did not decrease ROS production. SiRNA knockdown of PARP-1 did not affect the 8-OHdG level induced by arsenic, while it greatly increased the 8-OHdG level produced by hydrogen peroxide indicating that PARP-1 is a molecular target of arsenite. Our findings demonstrate that in addition to inducing oxidative stress at higher concentrations, arsenite can also inhibit the function of a key DNA repair protein, PARP-1, even at very low concentrations, thus exacerbating the overall oxidative DNA damage produced by arsenite, and potentially, by other oxidants as well.
Keywords: arsenic, PARP-1, DNA damage repair, 8-OHdG, ROS, carcinogenesis
Introduction
Arsenic is a naturally occurring, widely distributed element, which is present in food, soil, and water (1–3). Environmental or occupational exposures to arsenic may result in both acute and chronic toxic effects in humans (4, 5). Arsenic and its methylated derivatives are distributed in different organs and tissues (6). It has been reported that human arsenic exposure causes many health problems, such as cardiovascular diseases, liver damage, dermatitis, and various nervous system disturbances (7–9). Moreover, arsenic has been identified as a risk factor for a variety of tumors, including those of the lung, bladder, and skin (10–15). The International Agency for Research on Cancer (IARC) has classified arsenic as a human carcinogen (16). However, the precise mechanism by which arsenic acts as a carcinogen is unclear.
Many studies have shown that arsenic contributes to carcinogenesis through multiple mechanisms (17–20). Among them, oxidative DNA damage is a widely-recognized mechanism underlying the carcinogenicity of arsenic (21–24). Our previous studies have demonstrated that arsenic caused DNA damage by generating superoxide anion, hydrogen peroxide (H2O2), and also hydroxyl radicals (25, 26). There are many markers of oxidative DNA damage, such as thymine glycol, 5-hydroxycytosine, 8-hydroxyadenine, and 8-hydroxyguanine. 8-hydroxyguanine is also commonly named 8-hydroxyl-2′-deoxyguanosine (8-OHdG) and it is one of the major products of reactive oxygen species (ROS)-induced DNA damage (27, 28). It is widely used as an important biomarker of oxidative DNA damage (29–31). 8-OHdG has been demonstrated to be a potential mutagen, due to misreading of 8-OHdG during DNA replication, which results in GC to TA transversion, a common kind of mutation frequently found in human tumors (31). Our previous study has shown that 10 μM arsenite can significantly induce the production of 8-OHdG in HaCat cells (26).
The cell has an array of elaborate DNA repair mechanisms, which play a crucial role in maintaining genomic stability. Poly(ADP-ribose) polymerase (PARP) family proteins play an important role in the regulation of DNA damage repair. Whereas 17 different PARP members have been identified, PARP-1 activity accounts for about 90% of the total cellular poly(ADP-ribose) formation (32–34). Using NAD+ as a substrate, PARP-1 builds up homopolymers of ADP ribose units on various nuclear proteins including PARP-1 itself (35). Activation of PARP-1 occurs as an immediate cellular response to DNA strand break, induced by ionizing radiation, alkylating agents, and oxidative stress (33). There is substantial evidence that PARP-1 directly participates in the repair of DNA strand breaks (36, 37). Separate studies also indicate that low concentrations of arsenite suppressed PARP-1 activity (38–40). Based on these reports, we speculate that since arsenite is able to induce oxidative DNA damage (such as generation of 8-OHdG) and suppress PARP-1 activity, it is possible that arsenite could inhibit the repair of oxidative DNA damage produced by arsenic itself. Therefore, the observed oxidative DNA damage upon arsenite exposure would be due to the combined result of DNA damage induction and inhibition of its repair. The present study is designed to test this hypothesis. 8-OHdG and PARP-1 activity were measured after cells were treated with arsenite. Zinc supplementation and PARP-1 siRNA were used to increase or decrease the PARP-1 protein’s physiological function. Our results demonstrate that PARP-1 is a molecular target of arsenite and its inhibition by arsenite significantly contributes to the overall level of oxidative DNA damage.
Materials and Methods
Cell culture and treatments
The human keratinocyte cell line (HaCat) was generously provided by Dr. Mitch Denning (Loyola University Medical Center, Maywood, IL). HaCat cells were maintained as described in our earlier report (26). The cells were cultured at 37 °C in 95% air/5% CO2 humidified incubators. Sodium arsenite (As) and zinc chloride (Zn) solutions were sterilized by passing through a 0.22 μm syringe filter. The working concentrations were diluted with serum-free DMEM/F12 medium. For all experiments involving incubation with arsenite and/or zinc, HaCat cells were rinsed with PBS and placed into serum-free DMEM/F12 medium containing concentrations of arsenite and/or zinc as indicated in the figure legends.
Immunocytochemistry detection of 8-OHdG
Immunocytochemistry detection of 8-OHdG was performed according to the procedure described by Yarborough (41) and Kessel (21). Briefly, cells grown on coverslips were fixed with methanol for 20 min at −20 °C and treated with RNase (100 μg/ml) (Sigma, St. Louis, MO) for 1 hr at 37 °C and Proteinase K (10 μg/ml) (Sigma) for 10 min at room temperature. DNA was denatured by treatment with 4 N HCl for 10 min followed by pH adjustment with 50 mM Tris (pH 10) for 5 min at room temperature. Endogenous peroxidase was blocked by treating the cells with 3% H2O2 in methanol for 30 min at room temperature. After washing with PBS, the cells were treated with 10% normal horse serum at 37 °C for 1 hr and then incubated with primary antibody (anti-8-OHdG) (Trevigen, Gaithersburg, MD) (1:200 dilution) at 4 °C overnight. Then cells were treated with goat anti-mouse IgG conjugated with biotin (Vector Labs, Burlingame, CA) at 37 °C for 30 min. ABC reagent, avidin conjugated to horseradish peroxidase (Vector Labs) was added and the coverslips were incubated at 37 °C for 30 min followed by extensive washing. To localize peroxidase, cells were treated with diaminobenzidine (Sigma) for 10 min. Finally, coverslips were washed with PBS, dehydrated by a series of 95% and 100% ethanol and xylene washes, then mounted to microscope slides using Permount. Images were obtained using an Olympus BH2-RFCA fluorescence microscope (Melville, NY) and Omegafire digital camera with MagnaFire 2.1 software (Optronix, Goleta, CA). The 8-OHdG level was represented by relative intensity of nucleus staining. It was quantified by measuring mean density of the nucleus using the Image-Pro Plus software (Media Cybernetics, Inc., Silver Spring, MD). 30–50 randomly selected nuclei on each slide were measured and a minimum of 3 independent slides in each group were analyzed.
Detection of cellular poly (ADP-ribosyl)ation level
The level of poly (ADP-ribosyl)ation (PAR) was detected in HaCat cells according to Hartwig (39) with some modifications. Coverslips with cells were rinsed with ice-cold PBS and fixed in ice-cold 10% trichloroacetic acid (TCA) for 20 min followed by successive 10 min washes in 75%, 90%, and 100% ethanol (−20 oC). Then the coverslips were air-dried, rehydrated in PBS, and incubated in blocking reagent (1% BSA in PBS) at 37 oC for 1 hr. The coverslips were then incubated with mouse monoclonal antibody raised against poly(ADP-ribose) (Axxora, LLC. San Diego, CA), diluted to 10 μg/ml in blocking reagent, then placed in a humid chamber at 37 oC for 1 hr. The secondary, FITC-conjugated anti-mouse antibody (Chemicon, Temecula, CA) (diluted 1:200 in blocking reagent) was applied and the samples were incubated in a dark humid chamber at 37 oC for 1 hr. Then the coverslips were mounted on microscope slides using Vectashield mounting medium containing 2 μg/ml DAPI (Vector Labs) to visualize nuclei. Images were obtained using an Olympus BH2-RFCA fluorescence microscope and Omegafire digital camera with MagnaFire 2.1 software.
PARP activity assay
HaCaT cells were treated with arsenite and/or H2O2, with or without the presence of zinc, as described in the figure legends, then collected and the protein was extracted to assess PARP activity using the HT Colorimetric PARP/Apoptosis Assay kit (Trevigen) according to the manufacturer’s instruction. Briefly, protein was collected in cell extraction buffer and the concentration was determined with the BCA protein detection kit (Sigma). Extracts containing 200 ng/25 μl protein were prepared and added to histone coated strip wells in duplicate. PARP enzyme standards were prepared and assayed under identical conditions as the test samples. PARP substrate cocktail was added and the strip wells incubated for 30 min at room temperature. PARP activity was detected using anti-PAR monoclonal antibody followed by goat anti-mouse-HRP antibody. Activity was visualized by the addition of the TACS-Sapphire colorimetric substrate and the absorbance was read with a SpectraMax 340 plate reader at 450 nm. Absorbance values were averaged and activity was calculated from the standard curve generated.
Measurement of reactive oxygen species
Intracellular ROS production in HaCat cells was assessed using dihydroethidium (DHE, Molecular Probes, CA) as described (42). Briefly, when the HaCat cells reached 70% confluence, they were incubated witharsenite (20 μM) and/or zinc (20 μM) for 12 hrs. Thirty minutes prior to cell fixation, DHE (5 μM) was added as a fluorescent indicator of ROS generated in response to the indicated treatments. Coverslips were washed three times with PBS, fixed with 4% paraformaldehyde, and mounted to glass slides with Vecta-Shield (Vector Labs). Images were collected with an Olympus IX70 fluorescence microscope fitted with an Olympus camera and MagnaFire2.1 software. Relative fluorescence intensity was quantified by measuring pixel intensity along a line drawn through the center of a cluster of cells using Metamorph software (version 6.3r6). A minimum of 3 independent samples were analyzed per treatment. The upper 95% of each measurement was pooled and averaged, and the resulting data were analyzed for significance.
PARP-1 knockdown by transfection of PARP-1 siRNA
Transfection of siRNA was performed according to our reported procedure (43). Briefly, SMARTpool siRNA specific for human PARP-1 sequence (GenBank accession number: NM_001618) was obtained from Dharmacon Research, Inc. (Lafayette, CO). SiGLO RISC-Free siRNA (Dharmacon) was used as a control siRNA. HaCaT cells were seeded at a density of 2 × 105 cells/well in a 6-well plate the day before transfection in DMEM/F12 containing 10% FBS without antibiotics. Transfection of siRNAs was carried out using DharmaFECT transfection reagent (Dharmacon). DharmaFECT reagent was diluted 1:50 in serum free DMEM/F12 and incubated at room temperature for 5 min. In parallel, 2 μM siRNA in 1×siRNA buffer (Dharmacon) was diluted 1:1 in serum-free DMEM/F12. The two mixtures were combined and incubated for 20 min at room temperature prior to addition to cells for a final siRNA concentration of 100 nM. After 24 hrs, the medium was replaced with complete growth medium. After another 48 hrs, cells were treated with arsenite or H2O2, as indicated in the figure legends. Specific silencing was confirmed by Western Blot analysis.
Western Blot analysis of PARP-1 knockdown
Cells in 6-well plates were washed twice with ice-cold PBS and harvested with 50 μl Chaps Cell Extract Buffer (Cell Signaling, Danvers, MA) supplemented with complete proteinase inhibitor cocktail. Samples were lysed on ice for 30 min, subjected to freeze thaw cycles three times, and the extracts were clarified by centrifugation at 14,000 g for 15 min at 4 °C. Protein concentrations were determined by the Coomassie Plus Protein assay (Pierce, Rockford, IL). Cell lysate (30 μg of protein) was resolved in an 8% SDS-polyacrylamide gel and transferred onto nitrocellulose membrane (Bio-Rad, Hercules, CA) and incubated for 1 hr in TBST (10 mM Tris, pH 8.0, 150 mM NaCl and 0.1% Tween 20) containing 5% non-fat milk at room temperature. The membrane was then incubated with the rabbit polyclonal anti-PARP-1 antibody (1:500, Santa Cruz, CA) overnight at 4 °C. After washing with TBST, the membrane was incubated for 1 hr with horseradish peroxidase conjugated secondary antibody (1:1000, Cell Signaling, Danvers, MA), and signal was detected using the SuperSignal West Pico chemiluminescent kit (Pierce) on a Kodak Image Station 4000MM following the manufacturer’s instructions. In order to control for sample loading and protein transfer, the membrane was stripped and re-probed to detect β-actin (1:1000, Santa Cruz). The intensities were quantified by KODAK Molecular Imaging Software version 4.0. The PARP-1 protein levels were normalized to β-actin as compared with the untreated control (set to 1).
Data analysis
Data were presented as means ± S.D. Statistical analysis was carried out by analysis of variance (ANOVA) followed by multiple comparison tests (LSD). If the variance among groups is not homogeneity, Kruscal-wallis H test was performed. A value of P < 0.05 was considered statistically significant and labeled by an asterisk (*).
Results
8-OHdG formation induced by arsenite
8-OHdG is a biomarker of oxidative DNA damage (44). Using an HPLC-EC assay, we have previously shown a dose-dependent increase in 8-OHdG level following arsenite exposure (26). In the present study, we used immunocytochemistry to detect 8-OHdG and comparable results were obtained. After incubation with arsenite for 24 hrs, the 8-OHdG levels increased significantly in HaCat cells treated with arsenite at concentrations of 10 μM or higher (Figure 1). It is worthy to note that the formation of 8-OHdG increased exponentially with the increase of arsenite concentration above 10 μM (Figure 1B). These results indicate that arsenite induces oxidative DNA damage, but a statistically significant increase of 8-OHdG was detected only at concentrations of arsenite exceeding 5 μM in HaCat cells. This non-linear relationship may suggest an additional mechanism contributing to the accumulated DNA damage.
Fig. 1.
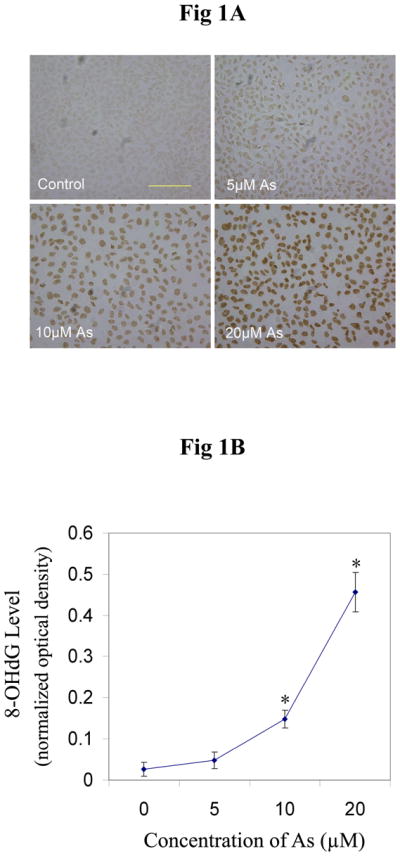
Production of 8-OHdG induced by different concentrations of arsenite. (A) HaCat cells were incubated with different concentrations of arsenite (0, 5, 10, 20, 40 μM) for 24 hrs before 8-OHdG was measured by immunocytochemistry. Scale bar = 200 μm. (B) The results (n ≥ 3) were quantified by measuring the optical density of the staining using Image-Pro Plus software. Data were presented as means ± S.D. * p<0.05 vs Control (0 μM).
Inhibition of PARP-1 activity by arsenite
Activated PARP-1 consumes NAD+ and produces PAR. Therefore measuring the level of PAR was used as one approach to evaluate PARP-1 activity. In the present study, H2O2 was used as a positive control for induction of oxidative DNA damage. As expected, the untreated cells did not show any visible PARP-1 activity (Figure 2A-Control). After the cells were treated with 0.1 mM H2O2, PARP-1 was activated and strong fluorescence intensity was observed (Figure 2A-H2O2). Pre-incubating the cells with 20 μM arsenite effectively abolished H2O2 -stimulated PARP-1 activation (Figure 2A-As+ H2O2). One plausible explanation for this observation is that arsenite inhibited the activity of PARP-1. To test this hypothesis, cells were treated with 20 μM arsenite alone. Again, there was no visible PARP-1 activity (Figure 2A-As). Since both 20 μM arsenite and 0.1 mM H2O2 generate oxidative DNA damage (Figure 1 and 2A-H2O2), which should lead to the activation of PARP-1, the absence of PARP-1 activity in both Fig 2A-As+ H2O2 and Fig 2A-As further indicates that arsenite can inhibit PARP-1 activity. In both cases, even though oxidative DNA damage has been caused by either H2O2 or arsenite, there is no detectable formation of PAR due to PARP-1 inhibition by arsenite. To confirm this explanation, the cells were incubated with 20 μM arsenite and 0.1 mM H2O2, in the presence of 20 μM zinc chloride. The rationale for zinc supplementation is that PARP-1 is a protein with two zinc finger domains that are essential for DNA binding (45). Literature reports suggest that arsenite could replace the zinc in the zinc finger (46–48), so we reasoned that zinc supplementation might be able to reverse the process, resulting in the revival of PARP-1 activity. As shown in Fig 2A-As+ H2O2+Zn, zinc treatment partially restored PARP-1 activity. Similarly, zinc treatment also partially restored the PARP-1 activity in cells treated with 20 μM arsenite alone (Fig 2A-As+Zn). These results suggest that 20 μM arsenite induces oxidative DNA damage, and concurrently inhibits the activity of PARP-1.
Fig. 2.
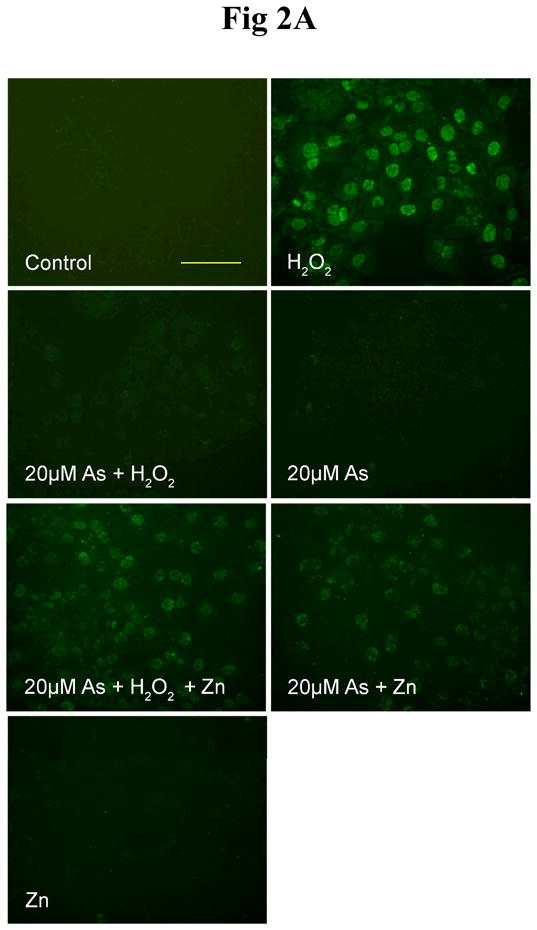
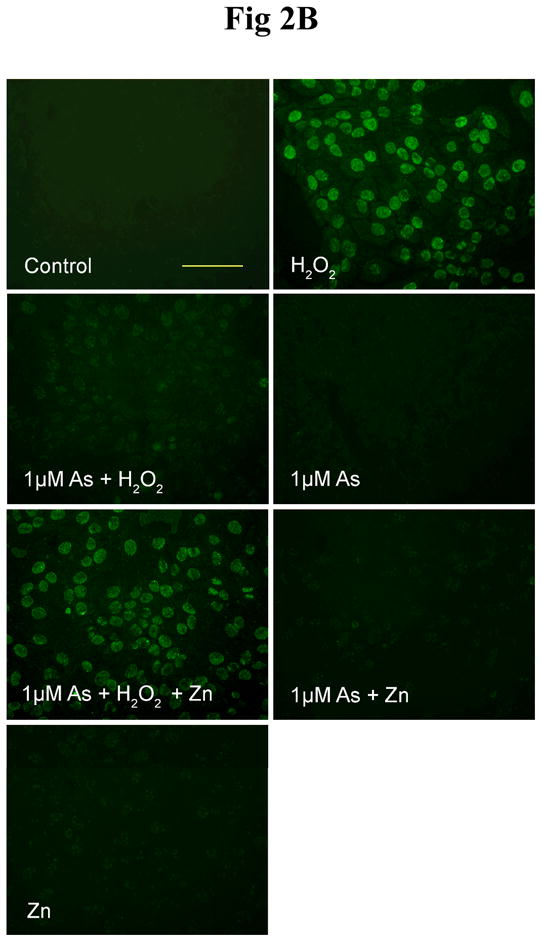
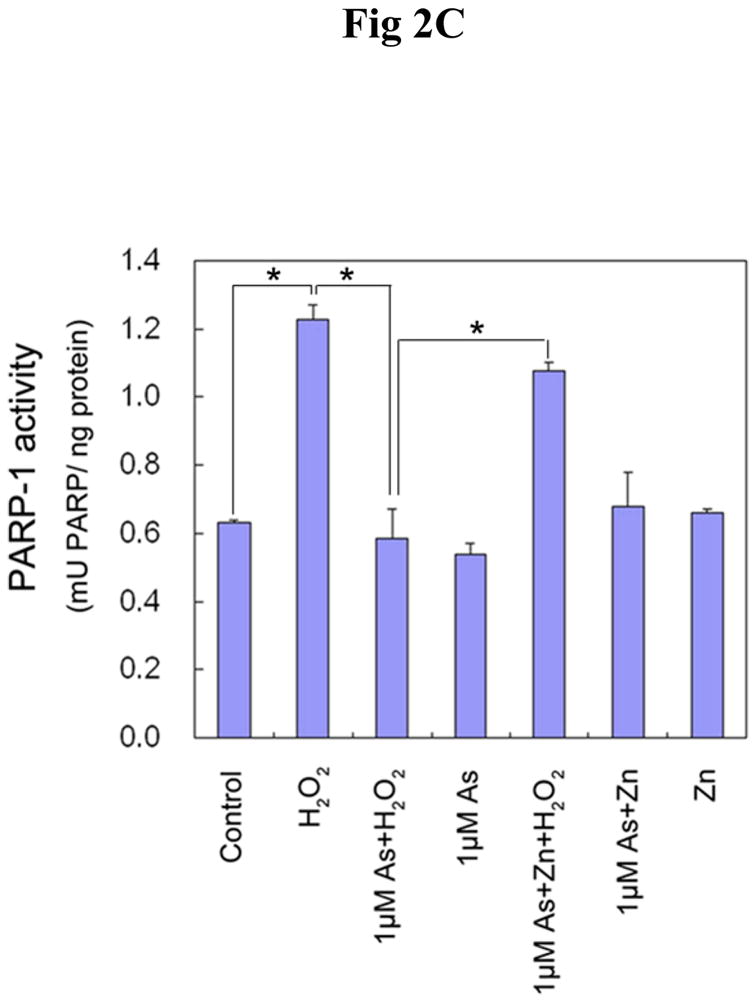
Effect of arsenite and/or zinc on PARP-1 activity. HaCat cells were incubated with 20 μM arsenite (A) or 1 μM arsenite (B) with or without 20 μM zinc for 24 hrs. Then 0.1 mM H2O2 was incubated in the indicated groups for 5 min. The PARP-1 activity was detected by measuring the production of PAR using immunofluorescence. Scale bar = 200 μm. (C). HaCat cells were incubated with 1 μM arsenite with or without 20 μM zinc for 24 hrs. Then 0.1 mM H2O2 was incubated in the indicated groups for 5 min. The PARP-1 activities were examined functionally by HT Colorimetric PARP Assay kit. Data (n ≥ 3) were presented as means ± S.D. * p<0.05 vs indicated group in the figure.
In order to investigate whether lower concentration of arsenite could still inhibit PARP-1 activity, identical experiments were conducted using 1 μM arsenite. As shown in Fig 2B, very similar results to Fig 2A were obtained, indicating that even at a low concentration of 1 μM, which did not produce detectable increase in 8-OHdG formation (Fig 1), arsenite can significantly inhibit PARP-1 activity. Comparing the results obtained with 20 and 1 μM arsenite (Fig 2A and 2B), the following observation is worth pointing out: addition of 20 μM zinc to cells treated with 20 μM arsenite revealed the PARP-1 activity (Fig 2A-As+Zn), whereas no PARP-1 activity was visible in cells treated with 1 μM arsenite (Fig 2B-As+Zn). The results are consistent with the findings in Figure 1 that 20 μM, but not 1 μM, arsenite caused oxidative DNA damage.
In order to further confirm the inhibition of PARP-1 activity by arsenite at low concentration, a colorimetric assay kit was used to functionally detect PARP-1 activity. H2O2 was used to induce oxidative DNA damage under identical conditions to Fig 2B. Figure 2C shows that 0.1 mM H2O2 treatment greatly activated PARP-1 activity. Pre-incubation with 1 μM arsenite abolished this increase, while 1 μM arsenite alone did not affect PARP-1 activity. If the cells were pre-treated with zinc along with the arsenite, the PARP-1 activity was mostly restored. These data are highly consistent with results obtained from the immunostaining measurement of PARP-1 activity in Fig 2B. Taken together, these results provide direct evidence that low concentrations of arsenite effectively inhibit PARP-1 activity.
Zinc-dependent reversal of 8-OHdG production induced by arsenite
PARP-1 has been reported to be necessary for base excision repair of oxidative DNA damage (49). Since our results in Figure 2 show that arsenite inhibition of PARP-1 activity could be reversed by zinc, we next examined whether the restoration of PARP-1 activity by zinc is functional in terms of oxidative DNA damage repair. The cells were co-treated with 20 μM arsenite and/or 20 μM zinc. After 24 hrs incubation, the 8-OHdG levels in cells treated with arsenite alone increased greatly (Figure 3). Zinc supplementation significantly decreased the 8-OHdG production induced by arsenite, while zinc treatment alone did not affect the 8-OHdG level in cells. These results suggest that restoration of PARP-1 activity by zinc leads to significantly reduced 8-OHdG levels in cells treated with arsenite, most likely because more 8-OHdG damage was repaired due to increased PARP-1 activity.
Fig. 3.

Effect of zinc on the production of 8-OHdG induced by arsenite. (A). HaCat cells were incubated with 20 μM arsenite and/or 20 μM zinc for 24 hrs before 8-OHdG was measured by immunocytochemistry. Scale bar = 200 μm. (B). The results were quantified by measuring the optical density of the staining using Image-Pro Plus software. Data (n ≥ 3) were presented as means ± S.D. * p<0.05 vs arsenite alone group.
Effect of zinc on ROS generation induced by arsenite
Zinc is a common element in human and natural environments and plays an important part in many biological processes (50). Its antioxidant properties have been reported in some papers (51, 52). In order to rule out the possibility that zinc decreased the 8-OHdG level by decreasing ROS production through antioxidant activities, the effect of zinc on ROS production was examined. After 12 hrs incubation, arsenite treatment greatly induced ROS level in cells, on which zinc supplement did not have significant effect (Figure 4A). Fluorescence was measured using Metamorph software, and graphical representation of the measured fluorescence intensity was shown in Figure 4B. These results demonstrate that zinc did not affect the production of ROS by arsenite, suggesting that zinc reduced 8-OHdG level most likely through restoration of PARP-1 activity (Figure 2), resulting in increased repair of 8-OHdG lesions.
Fig. 4.
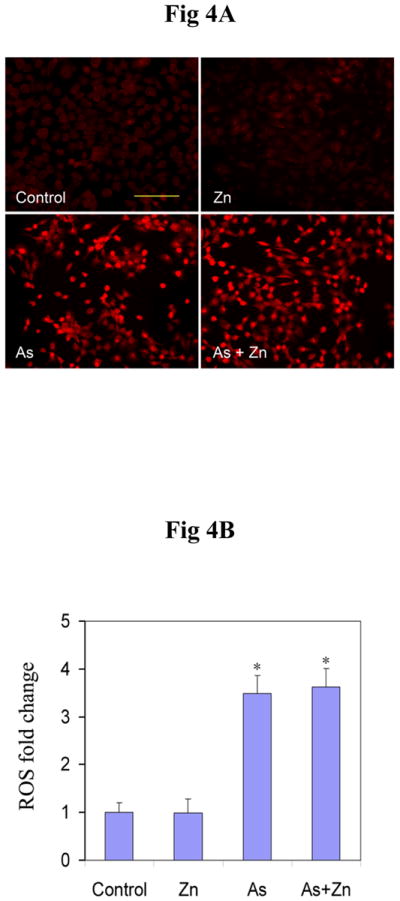
Effect of zinc on the production of ROS induced by arsenite. (A). HaCat cells were incubated with 20 μM arsenite and/or 20 μM zinc for 12 hrs. DHE (5 μM) was added as a fluorescent indicator of ROS for 30 min. Scale bar = 200 μm. (B). Relative fluorescence intensity was quantified using Metamorph software. The changes of ROS (n ≥ 3) were presented as means ± S.D. * p<0.05 vs Control group.
Effects of PARP-1 knockdown on 8-OHdG induced by arsenite or H2O2
Findings from the above experiments suggest that arsenite can inhibit the activity of PARP-1, which could lead to decreased oxidative DNA damage repair, ultimately resulting in an increase in the observed 8-OHdG level. In order to further verify the role of PARP-1 in 8-OHdG repair, and to investigate the interaction of arsenite with PARP-1, PARP-1 expression was knocked down in HaCat cells with siRNA. After 72 hrs transfection of PARP-1 siRNA, the PARP-1 protein level was decreased by 80% (Figure 5A), demonstrating the efficiency of PARP-1 siRNA. Then the cells were incubated with arsenite for another 24 hrs to determine whether PARP-1 knockdown would affect the 8-OHdG level in HaCat cells. As shown in Figure 5B, 20 μM arsenite treatment considerably increased the 8-OHdG level in untreated and control siRNA-treated cells. Importantly, PARP-1 knockdown did not cause any further increase of 8-OHdG. These results demonstrate that PARP-1 knockdown did not affect 8-OHdG level induced by arsenite. There are two potential explanations for the observation: one is that PARP-1 plays a key role in the oxidative DNA damage repair, as reported both in the literature (49) and in our own results (Fig 2 and Fig 3), but since arsenite treatment had already inhibited PARP-1 function, PARP-1 knockdown did not cause any additional change in 8-OHdG level. The second explanation is that under our experimental conditions, PARP-1 did not participate in 8-OHdG repair process. In order to clarify the situation, H2O2 was again used as the positive control to induce oxidative DNA damage. After 72 hrs transfection with PARP-1 siRNA, the cells were incubated with H2O2 for 24 hrs before measurement of 8-OHdG formation. As expected, treatment with 0.1 mM H2O2 markedly increased the level of 8-OHdG in untreated or control siRNA-treated cells (Fig 5C). However, knockdown of PARP-1 further increased the level of 8-OHdG, demonstrating that PARP-1 played an important role in the repair of 8-OHdG under our experimental conditions. These results rule out the possibility of the second explanation for Figure 5B, thus validating our hypothesis that the inhibition of PARP-1 activity by arsenite contributes to the overall level of 8-OHdG formation.
Fig. 5.
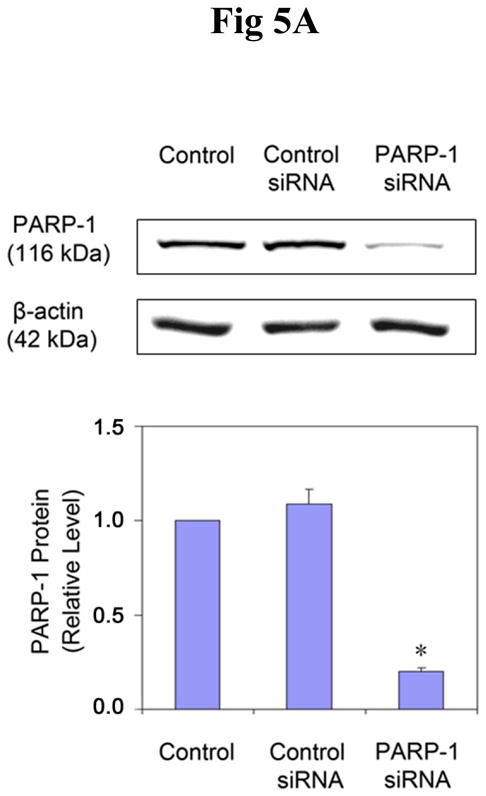


Effects of PARP-1 knockdown on the production of 8-OHdG induced by arsenite or H2O2. (A). Effect of PARP-1 siRNA on PARP-1 protein level. HaCat cells were incubated with PARP-1 siRNA or control siRNA for 24 hrs. Then the medium was replaced with a complete growth medium. After another 48 hrs, the cells were collected to measure the PARP-1 protein by Western-Blot. Data (n ≥ 3) were presented as means ± S.D. * p<0.05 vs control group. (B) and (C). Effect of PARP-1 knockdown on the production of 8-OHdG induced by arsenite or H2O2, respectively. After the PARP-1 was knocked down by siRNA described as (A), the cells were incubated with 20 μM arsenite or 0.1 mM H2O2 for 24 hrs before 8-OHdG was measured by immunocytochemistry. Scale bar = 200 μm. The results were quantified by measuring the optical density of the staining using Image-Pro Plus software. Data (n ≥ 3) were presented as means ± S.D. * p<0.05 vs control group, # p<0.05 vs H2O2 alone group.
Discussion
The results presented in this study demonstrate that arsenite-mediated oxidative DNA damage results from two distinct mechanisms: induction of oxidative DNA damage, and inhibition of PARP-1 activity, thus exacerbating the overall oxidative DNA damage. Furthermore, zinc supplementation was found to restore PARP-1 activity that was inhibited by arsenite, leading to increased repair of 8-OHdG lesions.
It is well established that oxidative DNA damage is an important contributing factor in cancer etiology. The oxidative DNA damage induced by arsenic has been documented by several reports, in which ROS generation has been demonstrated to be an important mechanism (17, 24, 53). Our previous studies demonstrated that arsenite caused DNA damage in keratinocyte cells via generation of hydroxyl radicals (25), and that arsenite-mediated ROS generation resulted in oxidative damage to DNA and protein (26). It was also reported that arsenite activated the growth arrest and DNA damage protein 45 alpha (GADD45a) gene via the formation of H2O2 (54). The results presented in the current study confirmed arsenite’s ability to induce oxidative DNA damage, as measured by the formation of 8-OHdG.
The overall steady-state level of oxidative DNA damage is determined by two factors: one is the DNA damage by toxic agents, mainly via generating ROS; the other is the activity of the DNA damage repair system. In oxidative DNA repair systems, especially in BER, PARP-1 is the most important nuclear zinc finger protein (48, 49, 55). This enzyme possesses two zinc fingers within its DNA-binding domain through which it binds tightly to the damaged DNA. There is a growing body of evidence that zinc finger proteins are a potential target and a novel molecular mechanism in arsenite-induced carcinogenesis (46, 48, 56, 57). The proposed modes of zinc finger damage by toxic metals include isostructural substitution, substitution with altered geometry, mixed complex formation, and catalysis of thiol oxidation (56). Recent reports showed that arsenite could inhibit PARP-1 activity stimulated by H2O2 (39, 58). In the present study, H2O2 was used as a probe to induce oxidative DNA damage so that we could investigate the inhibitory effect of arsenite on PARP-1. Our results demonstrate that PARP-1 was activated by oxidative DNA damage and this activation was inhibited in the presence of arsenite, even at concentrations as low as 1 μM (Figure 2). Treatment with 20 μM arsenite alone induced oxidative DNA damage (Figure 1), but the immunofluorescence detection of PARP-1 activity did not show any positive staining (Figure 2A), due to the inhibition of PARP-1 activity by arsenite. The restoration of PARP-1 activity by zinc treatment provided additional evidence that PARP-1 was inhibited by arsenite. The functionality assay of PARP-1 activity also shows that low levels (1 μM) of arsenite could effectively inhibit PARP-1 activity (Figure 2C). However, 1 μM arsenite treatment did not show any PARP-1 activity even when combined with zinc, indicating that there was no significant DNA damage. The dose-response result (Figure 1) also shows low concentrations of arsenite did not induce detectable oxidative DNA damage. Collectively, these results demonstrate that at high concentrations (≥ 10 μM), arsenite could induce DNA damage, and also inhibit its repair. However, at low concentrations (≤ 1 μM), arsenite did not generate detectable oxidative DNA damage, and yet, it still was able to effectively inhibit PARP-1 activity. The net outcome is that the generation of 8-OHdG increased dramatically with the increase of arsenite concentrations, because at higher concentrations two distinct mechanisms, induction of oxidative DNA damage and inhibition of its repair, are both in operation. It is noteworthy that the restoration of PARP-1 activity by zinc supplement (Fig 2) provides an important insight into the potential molecular mechanism of interaction of arsenite and zinc finger protein.
A recent report showed that the activity of PARP-1 isessential for 8-OHdG repair (49). Since zinc is found to restore the activity of PARP-1 inhibited by arsenite (Figure 2), it was reasoned that zinc supplementation could result in a reduction in 8-OHdG formation induced by arsenite. The positive effect of zinc on 8-OHdG formation (Figure 3) confirmed this hypothesis, as well as validated the key role of PARP-1 in the repair of 8-OHdG. One potential alternative explanation of these results is that since arsenite induces oxidative DNA damage via generation of ROS (21, 59), it is possible that zinc, which has been reported to possess certain antioxidant properties (51, 52), could reduce the ROS level, leading to reduction of 8-OHdG generation. Our result show that zinc supplement did not affect the ROS level induced by arsenite (Figure 4), ruling out the possibility that zinc reduced the level of 8-OHdG through reduction of ROS generation. However, it is not clear how zinc supplement restores the activity of PARP-1 inhibited by arsenite, which will require further study to investigate this fascinating observation.
In order to definitively demonstrate that PARP-1 inhibition by arsenite contributes to the overall level of arsenite-mediated 8-OHdG geneation, PARP-1 expression was knocked down in HaCat cells. PARP-1 knockdown increased the production of 8-OHdG induced by H2O2, demonstrating PARP-1’s involvement in the repair of 8-OHdG (Fig 5C). However, PARP-1 knockdown did not cause any significant change in the level of 8-OHdG formation in cells treated with arsenite alone (Fig 5B). The only reasonable explanation is that PARP-1 is a target of arsenite and PARP-1 activity has already been inhibited by arsenite treatment alone. Further treatment with PARP-1 siRNA does not produce any meaningful effects.
Signal transduction and gene induction by arsenic are also considered important mechanisms. Some key pathways activated by arsenic, such as PTP-MAPK-AP-1 pathway, IKKβ-NF-κB pathway, and Keap1-Nrf2 pathway, have been well demonstrated in many type of cells (reviewed by (60)). Another important action of low arsenic is altered DNA methylation of many genes, either in their promoter regions or within exons, which are important in carcinogenesis(61). Our finding on inhibition of PARP-1 activity by low concentrations of arsenite is consistent with the literature report that cell signaling can be affected by arsenite via low levels of oxidants that do not cause DNA damage (reviewed in (62)). Similarly, low arsenite concentrations increased oxidant signaling and oxidant-dependent activation of NF-κB without activating stress effector pathways in human endothelial cells (63). Together, these observations suggest that low concentrations of arsenite could interfere with a host of cellular and molecular responses, which may play a greater role in the mechanism of arsenic carcinogenesis than those events that only occur at high concentrations, because environmentally relevant concentration of arsenite is usually ≤ 1 μM. In the case of oxidative DNA damage associated with arsenite exposure, inhibiting DNA damage repair may be of greater significance in arsenite carcinogenesis than inducing DNA damage since a much lower concentration is sufficient. Further studies are needed to fully understand and appreciate the carcinogenic actions of arsenite at environmentally relevant concentrations.
In summary, the results presented here show that arsenite induces oxidative DNA damage through two mechanisms (Summarized as Figure 6): induction of DNA damage, and inhibition of DNA damage repair. At high concentrations (≥ 10 μM), both mechanisms are in operation. At concentrations lower than 1 μM, direct oxidative damage due to arsenite-mediated ROS generation is diminished. However, arsenite could still effectively inhibit the repair of oxidative damage induced by other oxidants through inhibiting the PARP-1 activity. Therefore, inhibiting DNA damage repair could be a highly significant mechanism in arsenic toxicity and carcinogenesis.
Fig. 6.

Scheme depicting dual roles of arsenic involved in carcinogenesis: induction of ROS, and inhibition of DNA damage repair. At high concentrations, both roles are in operation. At concentrations, direct oxidative damage due to arsenite-mediated ROS generation is diminished. However, arsenite could still effectively inhibit the repair of oxidative damage induced by other oxidants through inhibiting the PARP-1 activation.
Acknowledgments
This study was funded by a Department of Health and Human Services grant from the U.S. National Institutes of Health R01 ES012938 and R01 ES015826. Support was also provided by the UNM Cancer Research and Treatment Center P30 CA118100 and the UNM NIEHS Center P30 ES-012072.
References
- 1.Hewitt DJ, Millner GC, Nye AC, Simmons HF. Investigation of arsenic exposure from soil at a superfund site. Environ Res. 1995;68:73–81. doi: 10.1006/enrs.1995.1010. [DOI] [PubMed] [Google Scholar]
- 2.McKone TE, Daniels JI. Estimating human exposure through multiple pathways from air, water, and soil. Regul Toxicol Pharmacol. 1991;13:36–61. doi: 10.1016/0273-2300(91)90040-3. [DOI] [PubMed] [Google Scholar]
- 3.Applebaum KM, Karagas MR, Hunter DJ, Catalano PJ, Byler SH, Morris S, Nelson HH. Polymorphisms in nucleotide excision repair genes, arsenic exposure, and non-melanoma skin cancer in New Hampshire. Environ Health Perspect. 2007;115:1231–1236. doi: 10.1289/ehp.10096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Edelman P. Environmental and workplace contamination in the semiconductor industry: implications for future health of the workforce and community. Environ Health Perspect. 1990;86:291–295. doi: 10.1289/ehp.9086291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Akter KF, Owens G, Davey DE, Naidu R. Arsenic speciation and toxicity in biological systems. Rev Environ Contam Toxicol. 2005;184:97–149. doi: 10.1007/0-387-27565-7_3. [DOI] [PubMed] [Google Scholar]
- 6.Chattopadhyay S, Bhaumik S, Nag Chaudhury A, Das Gupta S. Arsenic induced changes in growth development and apoptosis in neonatal and adult brain cells in vivo and in tissue culture. Toxicol Lett. 2002;128:73–84. doi: 10.1016/s0378-4274(01)00535-5. [DOI] [PubMed] [Google Scholar]
- 7.Rodriguez VM, Jimenez-Capdeville ME, Giordano M. The effects of arsenic exposure on the nervous system. Toxicol Lett. 2003;145:1–18. doi: 10.1016/s0378-4274(03)00262-5. [DOI] [PubMed] [Google Scholar]
- 8.Navas-Acien A, Sharrett AR, Silbergeld EK, Schwartz BS, Nachman KE, Burke TA, Guallar E. Arsenic exposure and cardiovascular disease: a systematic review of the epidemiologic evidence. Am J Epidemiol. 2005;162:1037–1049. doi: 10.1093/aje/kwi330. [DOI] [PubMed] [Google Scholar]
- 9.Wang CH, Hsiao CK, Chen CL, Hsu LI, Chiou HY, Chen SY, Hsueh YM, Wu MM, Chen CJ. A review of the epidemiologic literature on the role of environmental arsenic exposure and cardiovascular diseases. Toxicol Appl Pharmacol. 2007;222:315–326. doi: 10.1016/j.taap.2006.12.022. [DOI] [PubMed] [Google Scholar]
- 10.Hayes RB. The carcinogenicity of metals in humans. Cancer Causes Control. 1997;8:371–385. doi: 10.1023/a:1018457305212. [DOI] [PubMed] [Google Scholar]
- 11.Chiu HF, Ho SC, Wang LY, Wu TN, Yang CY. Does arsenic exposure increase the risk for liver cancer? J Toxicol Environ Health A. 2004;67:1491–1500. doi: 10.1080/15287390490486806. [DOI] [PubMed] [Google Scholar]
- 12.Steinmaus C, Yuan Y, Bates MN, Smith AH. Case-control study of bladder cancer and drinking water arsenic in the western United States. Am J Epidemiol. 2003;158:1193–1201. doi: 10.1093/aje/kwg281. [DOI] [PubMed] [Google Scholar]
- 13.Yu HS, Liao WT, Chai CY. Arsenic carcinogenesis in the skin. J Biomed Sci. 2006;13:657–666. doi: 10.1007/s11373-006-9092-8. [DOI] [PubMed] [Google Scholar]
- 14.Guo HR. Cancer risks associated with arsenic in drinking water. Environ Health Perspect. 2007;115:A339–340. doi: 10.1289/ehp.9927. author reply A340–331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cohen SM, Arnold LL, Eldan M, Lewis AS, Beck BD. Methylated arsenicals: the implications of metabolism and carcinogenicity studies in rodents to human risk assessment. Crit Rev Toxicol. 2006;36:99–133. doi: 10.1080/10408440500534230. [DOI] [PubMed] [Google Scholar]
- 16.IARC, I. A. f. R. O. C. Arsenic in drinking-water. IARC Monogr Eval Carcinog Risks Hum. 2004;84:39–267. [PMC free article] [PubMed] [Google Scholar]
- 17.Huang C, Ke Q, Costa M, Shi X. Molecular mechanisms of arsenic carcinogenesis. Mol Cell Biochem. 2004;255:57–66. doi: 10.1023/b:mcbi.0000007261.04684.78. [DOI] [PubMed] [Google Scholar]
- 18.Kitchin KT. Recent advances in arsenic carcinogenesis: modes of action, animal model systems, and methylated arsenic metabolites. Toxicol Appl Pharmacol. 2001;172:249–261. doi: 10.1006/taap.2001.9157. [DOI] [PubMed] [Google Scholar]
- 19.Schoen A, Beck B, Sharma R, Dube E. Arsenic toxicity at low doses: epidemiological and mode of action considerations. Toxicol Appl Pharmacol. 2004;198:253–267. doi: 10.1016/j.taap.2003.10.011. [DOI] [PubMed] [Google Scholar]
- 20.Rossman TG. Mechanism of arsenic carcinogenesis: an integrated approach. Mutat Res. 2003;533:37–65. doi: 10.1016/j.mrfmmm.2003.07.009. [DOI] [PubMed] [Google Scholar]
- 21.Kessel M, Liu SX, Xu A, Santella R, Hei TK. Arsenic induces oxidative DNA damage in mammalian cells. Mol Cell Biochem. 2002;234–235:301–308. [PubMed] [Google Scholar]
- 22.Shi H, Shi X, Liu KJ. Oxidative mechanism of arsenic toxicity and carcinogenesis. Mol Cell Biochem. 2004;255:67–78. doi: 10.1023/b:mcbi.0000007262.26044.e8. [DOI] [PubMed] [Google Scholar]
- 23.Valko M, Rhodes CJ, Moncol J, Izakovic M, Mazur M. Free radicals, metals and antioxidants in oxidative stress-induced cancer. Chem Biol Interact. 2006;160:1–40. doi: 10.1016/j.cbi.2005.12.009. [DOI] [PubMed] [Google Scholar]
- 24.Hughes MFaKKT. Arsenic, oxidative stress and carcinogenesis. In: Singh KK, editor. Oxidative Stress, Disease and Cancer. Imperial College Press; London: 2006. pp. 825–850. [Google Scholar]
- 25.Shi H, Hudson LG, Ding W, Wang S, Cooper KL, Liu S, Chen Y, Shi X, Liu KJ. Arsenite causes DNA damage in keratinocytes via generation of hydroxyl radicals. Chem Res Toxicol. 2004;17:871–878. doi: 10.1021/tx049939e. [DOI] [PubMed] [Google Scholar]
- 26.Ding W, Hudson LG, Liu KJ. Inorganic arsenic compounds cause oxidative damage to DNA and protein by inducing ROS and RNS generation in human keratinocytes. Mol Cell Biochem. 2005;279:105–112. doi: 10.1007/s11010-005-8227-y. [DOI] [PubMed] [Google Scholar]
- 27.Kasai H, Nishimura S. Hydroxylation of deoxyguanosine at the C-8 position by ascorbic acid and other reducing agents. Nucleic Acids Res. 1984;12:2137–2145. doi: 10.1093/nar/12.4.2137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mei N, Kunugita N, Hirano T, Kasai H. Acute arsenite-induced 8-hydroxyguanine is associated with inhibition of repair activity in cultured human cells. Biochem Biophys Res Commun. 2002;297:924–930. doi: 10.1016/s0006-291x(02)02309-4. [DOI] [PubMed] [Google Scholar]
- 29.Kasai H. Analysis of a form of oxidative DNA damage, 8-hydroxy-2′-deoxyguanosine, as a marker of cellular oxidative stress during carcinogenesis. Mutat Res. 1997;387:147–163. doi: 10.1016/s1383-5742(97)00035-5. [DOI] [PubMed] [Google Scholar]
- 30.Helbock HJ, Beckman KB, Ames BN. 8-Hydroxydeoxyguanosine and 8-hydroxyguanine as biomarkers of oxidative DNA damage. Methods Enzymol. 1999;300:156–166. doi: 10.1016/s0076-6879(99)00123-8. [DOI] [PubMed] [Google Scholar]
- 31.Hwang ES, Kim GH. Biomarkers for oxidative stress status of DNA, lipids, and proteins in vitro and in vivo cancer research. Toxicology. 2007;229:1–10. doi: 10.1016/j.tox.2006.10.013. [DOI] [PubMed] [Google Scholar]
- 32.Christmann M, Tomicic MT, Roos WP, Kaina B. Mechanisms of human DNA repair: an update. Toxicology. 2003;193:3–34. doi: 10.1016/s0300-483x(03)00287-7. [DOI] [PubMed] [Google Scholar]
- 33.Burkle A, Diefenbach J, Brabeck C, Beneke S. Ageing and PARP. Pharmacol Res. 2005;52:93–99. doi: 10.1016/j.phrs.2005.02.008. [DOI] [PubMed] [Google Scholar]
- 34.Schreiber V, Dantzer F, Ame JC, de Murcia G. Poly(ADP-ribose): novel functions for an old molecule. Nat Rev Mol Cell Biol. 2006;7:517–528. doi: 10.1038/nrm1963. [DOI] [PubMed] [Google Scholar]
- 35.Andrabi SA, Kim NS, Yu SW, Wang H, Koh DW, Sasaki M, Klaus JA, Otsuka T, Zhang Z, Koehler RC, Hurn PD, Poirier GG, Dawson VL, Dawson TM. Poly(ADP-ribose) (PAR) polymer is a death signal. Proc Natl Acad Sci U S A. 2006;103:18308–18313. doi: 10.1073/pnas.0606526103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Fisher AE, Hochegger H, Takeda S, Caldecott KW. Poly(ADP-ribose) polymerase 1 accelerates single-strand break repair in concert with poly(ADP-ribose) glycohydrolase. Mol Cell Biol. 2007;27:5597–5605. doi: 10.1128/MCB.02248-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wang M, Wu W, Wu W, Rosidi B, Zhang L, Wang H, Iliakis G. PARP-1 and Ku compete for repair of DNA double strand breaks by distinct NHEJ pathways. Nucleic Acids Res. 2006;34:6170–6182. doi: 10.1093/nar/gkl840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hartwig A, Groblinghoff UD, Beyersmann D, Natarajan AT, Filon R, Mullenders LH. Interaction of arsenic(III) with nucleotide excision repair in UV-irradiated human fibroblasts. Carcinogenesis. 1997;18:399–405. doi: 10.1093/carcin/18.2.399. [DOI] [PubMed] [Google Scholar]
- 39.Hartwig A, Pelzer A, Asmuss M, Burkle A. Very low concentrations of arsenite suppress poly(ADP-ribosyl)ation in mammalian cells. Int J Cancer. 2003;104:1–6. doi: 10.1002/ijc.10911. [DOI] [PubMed] [Google Scholar]
- 40.Yager JW, Wiencke JK. Inhibition of poly(ADP-ribose) polymerase by arsenite. Mutat Res. 1997;386:345–351. doi: 10.1016/s1383-5742(97)00011-2. [DOI] [PubMed] [Google Scholar]
- 41.Yarborough A, Zhang YJ, Hsu TM, Santella RM. Immunoperoxidase detection of 8-hydroxydeoxyguanosine in aflatoxin B1-treated rat liver and human oral mucosal cells. Cancer Res. 1996;56:683–688. [PubMed] [Google Scholar]
- 42.Cooper KL, Liu KJ, Hudson LG. Contributions of reactive oxygen species and mitogen-activated protein kinase signaling in arsenite-stimulated hemeoxygenase-1 production. Toxicol Appl Pharmacol. 2007;218:119–127. doi: 10.1016/j.taap.2006.09.020. [DOI] [PubMed] [Google Scholar]
- 43.Liu W, Rosenberg GA, Liu KJ. AUF-1 mediates inhibition by nitric oxide of lipopolysaccharide-induced matrix metalloproteinase-9 expression in cultured astrocytes. J Neurosci Res. 2006;84:360–369. doi: 10.1002/jnr.20895. [DOI] [PubMed] [Google Scholar]
- 44.Dizdaroglu M. Oxidative damage to DNA in mammalian chromatin. Mutat Res. 1992;275:331–342. doi: 10.1016/0921-8734(92)90036-o. [DOI] [PubMed] [Google Scholar]
- 45.Ikejima M, Noguchi S, Yamashita R, Ogura T, Sugimura T, Gill DM, Miwa M. The zinc fingers of human poly(ADP-ribose) polymerase are differentially required for the recognition of DNA breaks and nicks and the consequent enzyme activation. Other structures recognize intact DNA. J Biol Chem. 1990;265:21907–21913. [PubMed] [Google Scholar]
- 46.Kitchin KT, Wallace K. Arsenite binding to synthetic peptides based on the Zn finger region and the estrogen binding region of the human estrogen receptor-alpha. Toxicol Appl Pharmacol. 2005;206:66–72. doi: 10.1016/j.taap.2004.12.010. [DOI] [PubMed] [Google Scholar]
- 47.Schwerdtle T, Walter I, Hartwig A. Arsenite and its biomethylated metabolites interfere with the formation and repair of stable BPDE-induced DNA adducts in human cells and impair XPAzf and Fpg. DNA Repair (Amst) 2003;2:1449–1463. doi: 10.1016/j.dnarep.2003.09.001. [DOI] [PubMed] [Google Scholar]
- 48.Kitchin KT, Wallace K. The role of protein binding of trivalent arsenicals in arsenic carcinogenesis and toxicity. J Inorg Biochem. 2008;102:532–539. doi: 10.1016/j.jinorgbio.2007.10.021. [DOI] [PubMed] [Google Scholar]
- 49.Le Page F, Schreiber V, Dherin C, De Murcia G, Boiteux S. Poly(ADP-ribose) polymerase-1 (PARP-1) is required in murine cell lines for base excision repair of oxidative DNA damage in the absence of DNA polymerase beta. J Biol Chem. 2003;278:18471–18477. doi: 10.1074/jbc.M212905200. [DOI] [PubMed] [Google Scholar]
- 50.Frassinetti S, Bronzetti G, Caltavuturo L, Cini M, Croce CD. The role of zinc in life: a review. J Environ Pathol Toxicol Oncol. 2006;25:597–610. doi: 10.1615/jenvironpatholtoxicoloncol.v25.i3.40. [DOI] [PubMed] [Google Scholar]
- 51.Powell SR. The antioxidant properties of zinc. J Nutr. 2000;130:1447S–1454S. doi: 10.1093/jn/130.5.1447S. [DOI] [PubMed] [Google Scholar]
- 52.Rostan EF, DeBuys HV, Madey DL, Pinnell SR. Evidence supporting zinc as an important antioxidant for skin. Int J Dermatol. 2002;41:606–611. doi: 10.1046/j.1365-4362.2002.01567.x. [DOI] [PubMed] [Google Scholar]
- 53.Kitchin KT, Ahmad S. Oxidative stress as a possible mode of action for arsenic carcinogenesis. Toxicol Lett. 2003;137:3–13. doi: 10.1016/s0378-4274(02)00376-4. [DOI] [PubMed] [Google Scholar]
- 54.Bower JJ, Leonard SS, Chen F, Shi X. As(III) transcriptionally activates the gadd45a gene via the formation of H2O2. Free Radic Biol Med. 2006;41:285–294. doi: 10.1016/j.freeradbiomed.2006.04.007. [DOI] [PubMed] [Google Scholar]
- 55.Virag L. Structure and function of poly(ADP-ribose) polymerase-1: role in oxidative stress-related pathologies. Curr Vasc Pharmacol. 2005;3:209–214. doi: 10.2174/1570161054368625. [DOI] [PubMed] [Google Scholar]
- 56.Hartwig A. Zinc finger proteins as potential targets for toxic metal ions: differential effects on structure and function. Antioxid Redox Signal. 2001;3:625–634. doi: 10.1089/15230860152542970. [DOI] [PubMed] [Google Scholar]
- 57.Witkiewicz-Kucharczyk A, Bal W. Damage of zinc fingers in DNA repair proteins, a novel molecular mechanism in carcinogenesis. Toxicol Lett. 2006;162:29–42. doi: 10.1016/j.toxlet.2005.10.018. [DOI] [PubMed] [Google Scholar]
- 58.Walter I, Schwerdtle T, Thuy C, Parsons JL, Dianov GL, Hartwig A. Impact of arsenite and its methylated metabolites on PARP-1 activity, PARP-1 gene expression and poly(ADP-ribosyl)ation in cultured human cells. DNA Repair (Amst) 2007;6:61–70. doi: 10.1016/j.dnarep.2006.08.008. [DOI] [PubMed] [Google Scholar]
- 59.Lantz RC, Hays AM. Role of oxidative stress in arsenic-induced toxicity. Drug Metab Rev. 2006;38:791–804. doi: 10.1080/03602530600980108. [DOI] [PubMed] [Google Scholar]
- 60.Kumagai Y, Sumi D. Arsenic: signal transduction, transcription factor, and biotransformation involved in cellular response and toxicity. Annu Rev Pharmacol Toxicol. 2007;47:243–262. doi: 10.1146/annurev.pharmtox.47.120505.105144. [DOI] [PubMed] [Google Scholar]
- 61.Klein CB, Leszczynska J, Hickey C, Rossman TG. Further evidence against a direct genotoxic mode of action for arsenic-induced cancer. Toxicol Appl Pharmacol. 2007;222:289–297. doi: 10.1016/j.taap.2006.12.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Simeonova PP, Luster MI. Arsenic and atherosclerosis. Toxicol Appl Pharmacol. 2004;198:444–449. doi: 10.1016/j.taap.2003.10.018. [DOI] [PubMed] [Google Scholar]
- 63.Barchowsky A, Roussel RR, Klei LR, James PE, Ganju N, Smith KR, Dudek EJ. Low levels of arsenic trioxide stimulate proliferative signals in primary vascular cells without activating stress effector pathways. Toxicol Appl Pharmacol. 1999;159:65–75. doi: 10.1006/taap.1999.8723. [DOI] [PubMed] [Google Scholar]