

Abstract
Gibbons (Hylobatidae) shared a common ancestor with the other hominoids only 15–18 million years ago. Nevertheless, gibbons show very distinctive features that include heavily rearranged chromosomes. Previous observations indicate that this phenomenon may be linked to the attenuated epigenetic repression of transposable elements (TEs) in gibbon species. Here we describe the massive expansion of a repeat in almost all the centromeres of the eastern hoolock gibbon (Hoolock leuconedys). We discovered that this repeat is a new composite TE originating from the combination of portions of three other elements (L1ME5, AluSz6, and SVA_A) and thus named it LAVA. We determined that this repeat is found in all the gibbons but does not occur in other hominoids. Detailed investigation of 46 different LAVA elements revealed that the majority of them have target site duplications (TSDs) and a poly-A tail, suggesting that they have been retrotransposing in the gibbon genome. Although we did not find a direct correlation between the emergence of LAVA elements and human–gibbon synteny breakpoints, this new composite transposable element is another mark of the great plasticity of the gibbon genome. Moreover, the centromeric expansion of LAVA insertions in the hoolock closely resembles the massive centromeric expansion of the KERV-1 retroelement reported for wallaby (marsupial) interspecific hybrids. The similarity between the two phenomena is consistent with the hypothesis that evolution of the gibbons is characterized by defects in epigenetic repression of TEs, perhaps triggered by interspecific hybridization.
Keywords: gibbon, centromere, transposable element, SVA, hybrid
Introduction
Gibbon species shared a relatively recent common ancestor with other hominoids (15–18 million years ago). Despite this near relationship, gibbons show some distinctive traits, including a larger taxonomic diversity (17 species known to date), smaller body size than other hominoids, and monogamous behavior (Cunningham and Mootnick 2009; Thinh et al. 2010b). The most striking difference is their karyotype evolution, which has been marked by an exceptionally high frequency of chromosomal rearrangements (Van Tuinen and Ledbetter 1983; Koehler et al. 1995; Mrasek et al. 2003; Muller et al. 2003). Each of the four gibbon genera (Nomascus, Hylobates, Hoolock, and Symphalangus) has a distinct karyotype with different numbers of chromosomes ranging from 38 to 52. This characteristic is in contrast with the karyotype stability of the other apes, and generally of mammalian species (Wienberg 2004). Population genetics studies have shown that the evolutionary history of the gibbons has been complex and involved frequent migration events as indicated by the presence of gene flow between closely related species and incomplete lineage sorting (Thinh et al. 2010a; Kim et al. 2011). Furthermore, data on mitochondrial and nuclear DNA, as well as karyotype studies, indicate that the gibbons underwent particularly fast radiation events, possibly facilitated by a changing environment (Thinh et al. 2010a). The rainforests of Southeast Asia, the habitat of most gibbon species, have often been subjected to contractions and expansions, which favored repeated isolation and unification of different populations (Morley and Flenley 1987). These events explain the evidence of lineage sorting obtained in both genomic (Kim et al. 2011) and karyotype evolution studies (Capozzi O, Carbone L, Stanyon R, Marra AM, Yang F, Whelan C, de Jong PJ, Rocchi M, Archidiacono N, unpublished data). Difficulty in reconciling phylogenetic relationships based on different traits or datasets is a hallmark of “mosaic genomes,” which could have been generated by frequent hybridization events (Arnold and Meyer 2006). Examples of gibbon hybrids generating viable offspring (Myers and Shafer 1979) support this hypothesis.
Although successful interspecies hybridization has often been considered a rare event (Arnold and Meyer 2006), it is now understood to have played a substantial role in the evolution of many plant and animal species. Interestingly, studies with model organisms have shown that genome reorganization can occur rapidly after hybridization, and new chromosome forms can be fixed in a few generations, allowing quick recovery of fertility. Hybrids can therefore be regarded as sources of “evolutionary novelties” (Fontdevila 2005). Some understanding of the source of genome reshuffling after interspecific hybridization comes from a phenomenon observed by O’Neill et al. (1998) in the hybrid offspring of two kangaroo species (Macropus eugenii × Wallabia bicolor). The hybrid individual displayed grossly rearranged chromosomes, characterized by extended centromeres, as a consequence of the massive expansion of a lineage-specific retrotransposon (kangaroo endogenous retrovirus-1, KERV-1). Because the genome of the hybrid was found to be heavily hypomethylated, this observation also reinforced the notion that one of the roles of DNA methylation in mammals is to repress endogenous transposable elements (TEs) (Yoder et al. 1997; Szpakowski et al. 2009; van der Heijden and Bortvin 2009). Moreover, it suggested that one consequence of interspecific hybridization is the disruption of epigenetic repression of TEs, which can then be responsible for driving genomic reshuffling (Fontdevila 2005).
A large fraction of mammalian genomes is made of TEs, which represent a constant risk to genome stability (Cordaux and Batzer 2009; de Koning et al. 2011). In plants and vertebrates, DNA methylation preserves genome integrity by suppressing the transcription, and therefore, the transposition of TEs. When this repression is disrupted or absent, it can result in the unrestrained transposition and proliferation of TEs (Yoder et al. 1997; Aravin et al. 2008; Molaro et al. 2011). We recently uncovered further evidence of a link between genome hypomethylation, loss of TE repression, and chromosome remodeling by studying chromosomal rearrangements in the northern white-cheeked gibbon. In particular, Alu sequences located near chromosomal breakpoints show lower levels of CpG methylation than their orthologous counterparts in human (Carbone et al. 2009). This evidence suggests that the epigenetic mechanisms for TE repression were disrupted during the evolution of the gibbon lineage, compromising genome stability. The observed gene flow between, and migrations of, gibbon species support a scenario in which a peculiar evolutionary history characterized by frequent interspecific hybridization events is responsible for disrupting the epigenetic repression of TEs, leading to the genome reshuffling observed in gibbon species.
We describe here another phenomenon that is consistent with this scenario. We observed the massive expansion of a repeat in almost all centromeres of the eastern hoolock (HLE, Hoolock leuconedys). This repeat is a novel, gibbon-specific, composite transposable element. Although the presence of satellite DNA is an almost universal characteristic of centromeres, accumulation of TEs is not. This feature has been reported in plants (Ma et al. 2007) and Drosophila (Garcia Guerreiro and Fontdevila 2007), but more rarely in mammals. Recently, centromeric satellites from genomes of multiple mammals (four Eutheria, one Methateria, and one Monotremata) have been analyzed and ERVs (endogenous retroviruses) have been found in only one species (armadillo) (Alkan et al. 2011). Similar observations were made in the tammar wallaby (Carone et al. 2009) and the opossum chromosomes (marsupials) (Gentles et al. 2007), indicating that interspersed repeats may have been the main source of centromeric DNA in the ancestral mammalian state (Alkan et al. 2011). The hoolock centromeres therefore represent a significant exception in placental mammals. Of note, the phenomenon we observe in the hoolock seems to mirror the centromeric TE expansion described in a wallaby hybrid (O'Neill et al. 1998). This similarity, together with the independent observations on the evolutionary history of gibbon species, suggests that interspecific hybridization might have been the driving force for the genomic reorganization experienced by gibbon species.
Materials and Methods
Fluorescent in situ Hybridization and Chromosome Painting
Chromosome preparations were obtained from peripheral blood following standard procedures. Briefly, blood was incubated with cell culture media and phytohemagglutinin (GIBCO) for 72 h (37°C, 5% CO2). Colcemid was then added (final concentration 0.05 µg/ml) and cells were harvested after a 1-h incubation. Cells were spun down by centrifugation, the media was discarded, and the pellet was resuspended in 8 ml of hypotonic solution (KCl 0.56%). After incubating for 20 min, the standard fixative solution (one part acetic acid, three parts methanol) was added and cells were centrifuged at 2,500 rpm for 5 min. The pellet was washed with fixative solution and cells were kept at 4°C overnight.
DNA from bacterial artificial chromosomes (BACs) was extracted using PureLink Miniprep kit (Invitrogen, Cat# K2100-10). Fluorescent in situ hybridization (FISH) experiments were performed essentially as described by Lichter et al. (1990). BACs and polymerase chain reaction (PCR) reactions were labeled either with Cy3-dUTP or FITC-dUTP by standard nick-translation assay. Images were acquired using a Nikon 80i microscope, equipped with a CCD camera Cool Snap HQ2 (Photometrics) and the Nis Elements Br (NIKON) software. Elaboration of the images was done using Photoshop.
Chromosome painting was performed using HLE-sorted chromosomes kindly provided by Dr Fengtang Yang (Sanger Institute). The HLE chromosomes were obtained from sorting lymphoblastoid and somatic hybrid cell line chromosomes followed by degenerate oligonucleotide primed (DOP) PCR. Each chromosome paint was amplified using a second DOP PCR reaction (Telenius et al. 1992) and labeled using the standard nick-translation reaction. BACs were hybridized together with chromosome-specific painting probes.
Fiber FISH
Fibers were prepared as described by the Current Protocols in Human Genetics (Supplement 44). Briefly, cells were harvested and resuspended in 1X PBS to a concentration of 5 × 104 − 2.5 × 106 cells/ml. A small volume (2 µl) of the cell suspension was placed on one end of the poly-l-lysine-treated glass slide and allowed to air dry. Subsequently, 7 µl of lysis buffer (2.5 ml 20% [w/v] SDS [0.5% final], 10 ml 0.5 M ethylenediaminetetraacetic acid [EDTA] [50 mM final], 20 ml 1 M TrisCl, pH 7.4 [200 mM final], 67.5 ml H2O) were applied to the cells on the slide and incubated for 5 min in a moist chamber. At the end of the incubation, the slide was tilted to vertical position, keeping the DNA at the upper end and allowing it to stream toward the end of the slide. The slide was air-dried almost completely and then covered with 400 µl of fixative (3:1 [v/v] methanol/glacial acetic acid). After 1 min, the excess fixative was drained off and the slide was air dried. FISH was performed as described above.
Quantitative PCR
Four intra-repeat element PCR assays were designed and primers were purchased from Sigma Aldrich. PCR evaluations of these assays, using a temperature gradient (annealing step) and agarose gel electrophoresis, revealed that two of the four assays were suitable for further analysis, in that they appeared to generate a strong and specific amplicon of the predicted size (Assay 1 is 153 bp; Assay 3 is 71 bp). The primer sequences are reported in supplementary table S1, Supplementary Material online. The sequence of the amplicon generated by these assays was subjected to a basic alignment search tool (Blast) against the nucleotide database as a preliminary test for specificity and seemed to detect the predicted targets. Conventional PCR was conducted on a DNA panel consisting of human, chimpanzee, gorilla, orangutan, 10 species of gibbons, as well as rhesus macaque and African green monkey as outgroups. Gel-based results indicated strong amplification in all gibbons, but weaker amplification in gorilla and orangutan, as compared with amplification in human. Optimal primer concentration for both assays was determined to be (200 nM) for Assay 1 and (500 nM) for Assay 3. Quantitative PCR (qPCR) reactions were carried out in 25 µl volumes (5 µl DNA template and 20 µl master mix) using 1X SYBR green buffer, 0.2 mM dNTPs, 2 mM MgCl2, optimized primer concentration, and 0.625 U AmpliTaq Gold DNA polymerase as recommended by the supplier. Each sample was subjected to an initial denaturation of 12 min at 95°C to activate the AmpliTaq Gold, followed by 40 amplification cycles of 95°C denaturation, 58°C (Assay 1) or 51°C (Assay 3) annealing, and 72°C extension, in steps of 30 s each. qPCR experiments were performed using an ABI Prism 7,000 sequence detection system with SDS software version 1.2.3. A preliminary experiment was performed to assess the feasibility of using whole-genome amplified DNA (GenomiPhi V2 DNA amplification kit, GE Healthcare) DNA in qPCR versus our relatively limited amount of stock DNA from cell culture. In this experiment, stock DNA from NLE and whole-genome amplified DNA from the same sample were assayed in duplicate for both Assay 1 and Assay 3, and within each assay the results were similar for stock versus GenomiPhied DNA, indicating that whole-genome amplified DNA could reliably be used for the qPCR experiments. GenomiPhied DNA from each species was quantified using a spectrophotometer and then adjusted to 100 ng/µl. The amount of input DNA was normalized for each species by performing a 10-fold serial dilution such that concentrations from 10 ng to 1 pg of each DNA template were assayed in duplicate in qPCR as described above. A no template control was also included for each dilution series and experimental condition to insure the validity of all data points used in our analyses.
qPCR results were exported from the ABI Prism 7000 SDS software and the mean and standard deviation (SD) of each set of duplicates were calculated. The mean values for each pair of threshold PCR cycle numbers (Ct), were plotted as a scatter-plot line graph to form a standard curve for each species being evaluated. The difference between the Ct values for human and the other species (Δ Ct) were calculated for a minimum of three data points along each dilution series, where the plots were most parallel. Because PCR amplification occurs exponentially, the x-fold difference between samples can be calculated as 2ΔCt (i.e. if a difference is a Ct value of 6 cycles, then this indicates 26 or a 64-fold difference). For each point along a series in comparison with human, the corresponding x-fold value was multiplied by 6 (the known copy number in human) to estimate the copy number of composite element insertions within other species under investigation. The mean and SD were calculated and plotted as the estimated copy number for each species and assay condition (supplementary table S2 and supplementary fig. S1, Supplementary Material online).
Radioactive Screening of High Density Filters
To identify hoolock BACs containing the centromeric repeat, overgo probes of 40 bp (Thomas et al. 2002) were designed within the SINE-VNTR-Alu-like (SVA_A) element identified in CH271-340F4. All probes were pooled together and hybridized to high-density filters of CHORI-278 library (http://bacpac.chori.org/library.php?id=393) following procedures described on the BACPAC resources website (http://bacpac.chori.org/overgohyb.htm). The images were analyzed with the software ArrayVision Ver 6.0 (Imaging Research Inc). Subsequently, 12 of the clones obtained from this screening (CH278-317G23, CH278-321H13, CH278-322E2, CH278-324J8, CH278-319K15, CH278-295L2, CH278-336C20, CH278-305C4, CH278-317L19, CH278-325M8, CH278-311H11, and CH278-311E17) were selected for sequencing.
Illumina Sequencing of BACs and RepeatMasker Analysis of Reads
The BACs identified in the radioactive screening were used to generate Illumina libraries using the multiplexing strategy and standard protocols from the manufacturer. The 12 libraries were pooled together and sequenced in one lane of the Illumina GAIIX. A total of 66,763,300 100-bp sequencing reads were generated. Only 3,445,686 (5%) reads could be deconvoluted to a specific BAC based on barcodes giving an average of 287,140.5 reads per BAC and an average coverage of 191.43X per BAC assuming 170 kb inserts. All reads, including those that could not be deconvoluted, were used for analysis giving a hypothetical average of 5,563,608.3 reads per BAC and an average coverage of 3709.07X/BAC. All Illumina reads were run through RepeatMasker (http://www.repeatmasker.org/) using default parameters and RepeatMasker Database RELEASE 20090604. The sequences from unmasked portions of reads identified as containing SVA_A (28,007 reads or 0.042% of total sequenced reads) were filtered for Illumina adapters and primers. K-mer frequencies were then calculated from the unmasked portions of SVA_A containing reads and the top four most abundant k-mers (CTACCACAGAGGCCAGAAGCAA—2,588; GTCCAGCCCCCACATTGCTTCTGGCCTCTGTGGTAG—302; TTTCTATATTTAAATTCAACAATAATTACTAAACACCTGC—220; TGGTGTTTAGTAATTATTGTTGAATTTAAATATAGAAA—208) were used in a Blast search against Nomascus leucogenys (NLE) whole genome shotgun (WGS) sequences deposited in the NCBI Trace Archives (http://www.ncbi.nlm.nih.gov/Traces/home/). NLE WGS reads with the 100 top scoring alignments to each of the four k-mers were retrieved from the Trace Archives and ran through RepeatMasker using the same settings as for the Illumina reads. The counts of specific repeats adjacent to SVA_A repeats were calculated revealing 132 cases in which AluSz6 was adjacent to an SVA_A. Further examination of cases where SVA_A co-occurred with AluSz6 identified 57 cases where L1ME5 was adjacent to AluSz6. The order and strand orientation of the three elements were inferred from these cases and this pattern was used to identify the composite element in 458 fully sequenced NLE BACs downloaded from the National Center for Biotechnology Information (NCBI) nucleotide database (http://www.ncbi.nlm.nih.gov/nuccore).
Results
Centromeric Repeat Expansion in Hoolock Leuconedys
To study synteny relationships among chromosomes of different gibbon species, we performed numerous cross-species FISH experiments (see Materials and Methods) in which BACs from the NLE library were hybridized on metaphases from the other gibbon species. With this method we found that a number of BACs produce very bright centromeric signals on most chromosomes of Hoolock leuconedys (HLE) (fig. 1A). This phenomenon is exclusive to HLE. When hybridized to chromosomes of gibbon species from the other three genera, the same BACs do not produce the centromeric pattern (fig. 1B), although few weak and diffuse centromeric and pericentromeric signals are apparent in Nomascus and Hylobates (fig. 1B and supplementary fig. S1, Supplementary Material online). Furthermore, FISH with human BACs from regions orthologous to the NLE BACs generates single signals on HLE (data not shown), indicating that the sequence generating the centromeric signals is absent in the corresponding human regions. Taking this into account, we aligned the sequence of one of the BACs producing the centromeric signals (CH271-340F4, AC198183) to the human genome and identified a 12 kb region present exclusively in the gibbon BAC. The sequence of this entire region was recognized by RepeatMasker as an SVA_A element, a hominoid-specific TE composed of Short INterspersed Element (SINE), variable number tandem repeat (VNTR) and Alu-like elements. The VNTR portion has been found to be quite variable in length between different SVA elements (Wang et al. 2005) and in the sequence we identified, the VNTR extended for about 10 kb.
Fig. 1.—

FISH with NEL BACs generates bright centromeric signals in hoolock but not in the other gibbon genera. (A) FISH with NLE BAC clones produces bright signals on almost all the centromeres of HLE chromosomes. Five chromosome pairs (indicated by the arrows) show reduced or significantly less intense FISH signals. (B) The same BACs produced the main hybridization signal only on one pair of chromosomes in gibbon species from the other three genera. One of the BACs (CH271-457L13) is represented here (see also supplementary fig. S1, Supplementary Material online). Weaker repeated signals were observed in Nomascus and Hylobates.
To verify that the repetitive sequence in the HLE centromere corresponded to the SVA_A element identified by the human–gibbon alignment, we designed two sets of PCR primers (SVA_L1+R1 and SVA_L2 + SVA_R2) based on the sequence found in CH271-340F4 (supplementary table S1, Supplementary Material online). Both PCR products were fluorescently labeled and used as FISH probes on HLE metaphases, where they reproduced the pattern we originally observed with CH271-340F4 (fig. 2). This evidence suggests that a sequence similar to the SVA element has expanded in HLE centromeres.
Fig. 2.—
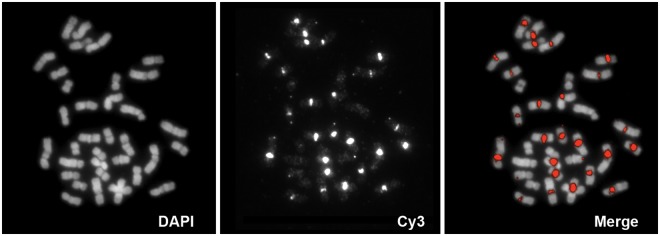
FISH with PCR-derived SVA probe generates centromeric signals. PCR primers were designed to amplify a portion of the SVA element. The PCR product was fluorescently labeled and hybridized by FISH on hoolock chromosomes. The hybridization pattern recapitulates the one obtained with the BACs.
Investigation of the Centromeric Repeat Reveals a New Transposable Element
In order to further characterize the HLE centromeric repeat and compare it with the traditional SVA_A element, we made use of the HLE BAC library (CHORI-278, http://bacpac.chori.org/library.php?id=393). Specifically, we designed overgo probes (Thomas et al. 2002) within the SVA_A element from CH271-340F4 and used them to screen high-density filters from CHORI-278 by radioactive hybridization (see Materials and Methods). This hybridization produced about 2,000 signals on each filter, some of which appeared weaker and were considered to be background. We selected 12 clones, based on their stronger signal intensity, and sequenced them with the Illumina GAIIX (see Materials and Methods). We did not attempt to assemble the HLE BACs. Instead, we analyzed all Illumina reads with RepeatMasker in order to identify the ones containing sequences recognized as SVA_A elements. The vast majority of these reads contained only a portion of an SVA_A element, mostly matching the region from base 425 to base 715 of the consensus sequence, which corresponds to the VNTR region. A portion of the reads also included the Alu-like portion (fig. 3A). We therefore hypothesized that the repeat that expanded in the HLE centromere was different from the standard full-length SVA element, although it included a portion of it.
Fig. 3.—

Structure of the LAVA element. (A) Illumina reads obtained from hoolock BACs and known for containing SVA sequences were mapped against the SVA consensus sequence. Most of the reads map in correspondence of the VNTR region of the SVA element. (B) Structure of the novel composite TE based on the sequence data obtained from NLE BACs. The arrows inside the different components indicate their orientation. (C) The PCR product spanning the junction between the AluSz6 and L1ME5 regions was labeled and used as probe for FISH on hoolock metaphases. The bright centromeric signals confirm that the novel TE is expanded in the hoolock centromeres.
Defining the full structure of repeats with short Illumina reads is challenging, and we thus used an alternative approach (see Materials and Methods). First, we isolated all reads from the HLE BACs containing sequences recognized as “SVA” by RepeatMasker. We then identified highly represented k-mers within the unmasked portions of these reads. Subsequently, we used Blast to query these highly represented k-mers against the NLE whole-genome shotgun sequences deposited in the NCBI Trace Archives (http://www.ncbi.nlm.nih.gov/Traces/home/). Finally, we ran RepeatMasker on the reads identified by Blast as containing the highly represented k-mer. We noticed that many of the results included portions of AluSz6 and L1ME5. In particular, we identified 132 instances in which the SVA_A portion was followed by a sequence recognized as part of an AluSz or AluSz6 element, and 57 instances in which a L1ME5 element was also present. Additionally, the junctions between the three different repeats were similar in size. These are all evidence of a composite TE that we named LAVA (L1ME5 - AluSz6 - VNTR- Alu-like).
Characterization of the LAVA Element
In order to better characterize the LAVA element and obtain evidence that it has been retrotransposing in the gibbon genome, we investigated whether the copies found in the gibbon genome were flanked by Target Site Duplications (TSDs), duplicated genomic sequences that would have been introduced through the integration process (Cordaux and Batzer 2009). We searched for the combination SVA_A-AluSz6-L1ME5 within 458 NLE BACs that have been fully sequenced by us and other groups (Birney et al. 2007; Carbone et al. 2009; Girirajan et al. 2009). We identified 46 instances of LAVA, 33 of which are flanked by TSDs. In the remaining 13, the identification of TSDs was not possible due to the presence of A-rich regions and the insertion of another repeat (most often Alu elements) or the presence of a sequence gap at the 5′ end of the element (supplementary table S3, Supplementary Material online). Furthermore, all the LAVA elements are flanked by a poly-A tail. These features indicate that LAVA elements mobilize by retrotransposition. Analysis of the LAVA copies found in the BACs enabled a more detailed description of this element (fig. 3B) and the generation of a consensus sequence (supplementary information, Supplementary Material online). The 5′ sequence of the full-length LAVA element closely resembles the 5′ sequence of the traditional SVA_A element, starting with a CT-rich sequence, similar to the hexamer region characteristic of SVA_A elements, although it appears to be more variable and overall more enriched in Ts than Cs. Since the first 30 base pairs of the elements found in the BACs are poorly conserved and present in only 28 of the 46 elements, we did not include them in the LAVA consensus sequence. The CT-rich sequence is followed by the Alu-like sequence and VNTR region, whereas the SINE-R region typical of the SVA_A element, is missing. Instead, the VNTR region is followed by a short sequence (U1) and a 3′ truncated AluSz6 sequence in positive strand orientation, followed by another stretch of sequence (U2) and a portion of the L1ME5 element in the opposite strand orientation (fig. 3B). The intervening sequences, U1 and U2, are mostly of a constant length (24 nt and 156 nt, respectively) (fig. 3B and supplementary table S3, Supplementary Material online). The element ends in a polyA-tail that also contains a poly-adenylation signal. In 20 elements, RepeatMasker also identifies simple repeats between the AluSz6 and L1ME5 regions, which are most likely the relics of the original poly-A tail of the L1ME5 elements that accumulated mutations with time and are no longer recognizable. The length of the full element is variable as it depends on the extension of the VNTR portion but it is ∼1000–1300 bp long. We amplified one full-length element from BAC CH271-261H3 (CT954299) and NLE genomic DNA confirming its size (supplementary fig. S2, Supplementary Material online).
To confirm that LAVA is the repeat that expanded in the hoolock centromeres, we designed a forward primer in the AluSz6 portion (AluSF) and a reverse primer in the L1ME5 portion (L1MR) (fig. 3B and supplementary table S1, Supplementary Material online) using as a template, one of the full-length elements identified in NLE. The PCR on the HLE genomic DNA generated a 400 bp single band which was then used as probe for FISH on HLE chromosomes and generated bright centromeric signals (fig. 3C).
We also looked for a possible correlation between the presence of LAVA elements and the high rate of chromosome rearrangements in NLE. Human–NLE synteny breakpoints have been extensively characterized (Carbone et al. 2009; Girirajan et al. 2009). Within the 458 fully sequenced NLE BACs that we used to identify the LAVA elements, 42 BACs span at least one chromosomal breakpoint. Only five of these BACs (∼12%) contain one LAVA insertion. Moreover, the LAVA elements are always very distant (>1 Mb) from the breakpoint location. This evidence seems to exclude a direct association between the chromosomal breakpoints and LAVA insertions.
Measuring the Copy Number of LAVA Elements
An in silico search for LAVA elements in the human, chimpanzee, orangutan, and rhesus macaque assemblies does not find any sequence matching the entire element. However, in the human genome (hg19), we find six copies of the U1-AluS-U2-L1ME5 combination. We constructed a sequence alignment of 18 gibbon-specific insertions from the NLE BAC clones and the six human-specific sequences. Within the LAVA element, the U1, U2, and L1ME5 regions appeared to be the most conserved among the aligned gibbon elements, as well as between the gibbon and human elements. Our intra-repeat element PCR assays were designed to facilitate effective amplification of both gibbon and human elements, while also preventing cross-amplification with any other type of mobile element or genomic sequence. We selected two assays as they generated a strong and specific amplicon of the predicted size (Assay 1 = 153 bp; Assay 3 = 71 bp). Primer sequences for these assays are reported in supplementary table S1, Supplementary Material online. After optimizing the conditions for qPCR, we assayed 10 gibbon species (see Materials and Methods, supplementary table S3 and supplementary fig. S3, Supplementary Material online). These results yielded an estimate of 600–1,200 copies of LAVA elements in gibbon genomes. Nevertheless, our copy number estimates are based on a very short segment of the composite element (∼150 bp) and these numbers may or may not extrapolate to “full-length” copies in the genomes of gibbon species. The analysis of the NLE genome, currently being performed by the Gibbon Genome Sequencing and Analysis Consortium, will enable a more precise estimate of LAVA copy number.
Relationship Between the LAVA Element and Other Centromeric Repeats
FISH indicated that not all HLE centromeres display the same levels of amplification of LAVA sequence: some chromosomes show very weak or no hybridization signals. These chromosomes were identified as HLE4, HLE11, HLE15, HLE17, and HLE18 by chromosome painting (see Materials and Methods and supplementary fig. S4, Supplementary Material online). We attempted to explain this observation by investigating the evolutionary history of these chromosomes (Capozzi et al., unpublished data). First, all five chromosomes derive from hoolock-specific chromosomal rearrangements. In particular, HLE4 is the result of a Robertsonian fusion between a chromosome homologous to human 2q and a small chromosome from the gibbon ancestor which carried the centromere corresponding to human chromosome 12. HLE15 and HLE18 derive from a reciprocal translocation between the ancestral gibbon chromosomes 9 and 19, and consequently originated at the same time. However, it is not clear why these chromosomes display a reduced repeat content. Finally, HLE17 and HLE 11 are characterized by hoolock-specific, evolutionarily new centromeres (Montefalcone et al. 1999; Rocchi et al. 2009) that may have emerged after the LAVA centromeric invasion.
The α-satellite is the main centromeric satellite in primates (Manuelidis 1978). To investigate the relationship between LAVA elements and the α-satellite in HLE centromeres, we generated a PCR-derived probe for the α-satellite using primers designed in the most conserved region of the human α-satellite consensus, and HLE genomic DNA as template (supplementary table S1, Supplementary Material online). We then performed a dual color FISH experiment with the α-satellite probe and CH271-340F4 on HLE chromosomes. We observed a strong centromeric signal from the α-satellite probe on HLE 4, and weaker signals on the centromeres of all other chromosomes (fig. 4A). Since HLE 4 is depleted of LAVA sequences, this pattern seem complementary to the one produced by CH271-340F4.
Fig. 4.—

Co-hybridization experiments to investigate the relationship between LAVA elements and centromeric satellites. (A) Hoolock chromosomes were co-hybridized with α-satellite and LAVA probe. All centromeres, except that of chromosome 4, show depletion of α-satellite. (B) The SVA probe co-hybridizes with a probe designed on the SATR-1 satellite. (C) The relationship between LAVA elements and SATR-1 sequences is shown at higher resolution by the fiber-FISH experiment.
We also carried out restriction site-associated DNA sequencing (RAD-seq) using Illumina (Wall JD, Kim SK, Luca F, Carbone L, Mootnick AR, de Jong PJ, Di Rienzo A, unpublished data) and generated 1.5 Mb of orthologous sequence from one individual of each of the four gibbon genera. RepeatMasker on this dataset revealed that SATR1 (Jurka 2000; Costa et al. 2006) is the most abundant satellite sequence in HLE. SATR1 is a satellite that has been found to localize in human centromeres (Ventura et al. 2003; Wong et al. 2004). Because this feature might be related to the unique structure of HLE centromeres, and possibly the expansion of LAVA elements, we investigated it further. We used PCR primers based on the SATR1 consensus sequence to generate a probe (supplementary table S1, Supplementary Material online) and performed dual color FISH with the LAVA probe and CH271-340F4. The two repeats displayed overlapping patterns and they are depleted from the same loci (fig. 4B). As expected, centromeres enriched with SATR1 were also depleted of α-satellite and vice versa (data not shown). To better understand the spatial relationship between SATR1 and LAVA sequences, we performed dual color FISH on chromatin fibers obtained from HLE lymphoblastoid cells (see Materials and Methods). As shown in figure 4C, the signals from the two repeats are always in close proximity to each other and are interspersed throughout the chromatin fibers. This scenario is remarkably similar to that observed in wallaby hybrids for the repeat KERV 1 and the satellite sat23 (Carone et al. 2009).
Discussion
Gibbons have strikingly rearranged karyotypes (Muller et al. 2003). Here we report additional observations supporting the notion that the genomes of these species underwent a peculiar evolutionary history. First, we describe the gross accumulation of a transposable element in most of the centromeres of the Eastern hoolock gibbon. This phenomenon seems to have occurred exclusively in HLE, although previous findings point to the presence of other types of repeats in the centromeres of the other gibbon genera (Chen et al. 2007; Cellamare et al. 2009). Second, we found that the centromeric repeat is a new composite transposable element generated by combination of the 3′ portion of the traditional SVA element with portions of other two repetitive elements commonly found in primate genomes (AluS and L1ME5). We observed that the new element is present only in the gibbon lineage. The genomes of gibbons also carry traditional SVA_A elements, but their copy number is considerably lower than in the other hominoids (Wang et al. 2005). SVA_A originated around 13.56 million years ago (Wang et al. 2005), which places its emergence just prior to the divergence of gibbons from the other hominoids. We estimate that the copy number of LAVA insertions in gibbon genomes is 600–1,200. This is a rough assessment that will be refined when analysis of the NLE genome assembly is complete by the Gibbon Genome Sequencing and Analysis Consortium. The likely active retrotransposition of SVA_A during the divergence of gibbons from other hominoids together with the presence of LAVA in all the gibbons we assayed suggests LAVA originated in the gibbons’ common ancestor. When we isolated all sequences recognized as SVA elements from the NLE trace archives, the majority of them contain ∼30% of the full-length element, which roughly corresponds to the portion included in the LAVA element (data not shown). Thus, in the gibbon lineage, LAVA element has been more successful than the traditional SVA_A element. In vitro trans-mobilization assays show that human SVA elements that acquired an AluS element had a higher mobilization rate than the SVA element alone (Raiz et al. 2011). In this case, the distance between the AluSp and the SVA human elements is 31 bp, therefore comparable with the U1 of the LAVA element (fig. 3B). Moreover, the two elements are in the same orientation like we found for the Alu-like-VNTR and the AluSz6 in the LAVA element. We speculate that the presence of the AluSz6 region in the LAVA element makes it a better substrate for the L1 retrotransposition apparatus normally used by SVA insertions. Additional assays, however, will be needed to demonstrate that the current LAVA element is still able to retrotranspose, and at what rate.
The in silico search for the LAVA element in the human, chimpanzee, orangutan, rhesus macaque, and common marmoset genomes do not retrieve any sequence matching the entire element. However, one copy of the U1-AluSz6-U2-L1M5 combination can be found on human chromosome 9 (chr9:99,026,502-99,026,980) and orthologous positions in all other primate genomes investigated. This locus is not flanked by TSDs, indicating that it may not have arisen through the normal reverse transcription mechanism. The same locus also exists in the gibbon (NLE). We may speculate that in the gibbon ancestor, a copy of the U1-AluSz5-U2-L1M5 combination existed and, at this locus, a portion of an SVA element inserted just 5′ to U1, creating a new combination, which was able to retrotranspose efficiently. More investigations, however, are needed to reconstruct in detail the origin of the LAVA element. As well as being present in the genomes of all gibbon species for which DNA samples were available, LAVA elements expanded in most of the centromeres of HLE. Despite this, qPCR did not detect an increased copy number of LAVA insertions in HLE as compared with the other species in which the centromeric expansion did not occur. This could be the result of gene conversion within the centromeric copies, causing their sequences to diverge more rapidly from copies dispersed in the genome. Capturing and analyzing centromeric sequences is challenging, as they are underrepresented in large insert clones and cannot easily be sequenced due to their repetitive nature.
We can only speculate on the events responsible for the centromeric accumulation of LAVA elements in HLE. Given its similarity to the centromeric expansion of the K-ERV retrotransposon observed in the hybrid wallaby (O'Neill et al. 1998), which may have been triggered by global hypomethylation, we hypothesize that a similar loss of epigenetic repression occurred in HLE, possibly as consequence of interspecific hybridization. This scenario is supported by population genetic data that indicate frequent migration and gene flow between closely related gibbon species (Kim et al. 2011). We have observed these centromeric LAVA elements in three wild-born unrelated HLE individuals, suggesting that this trait has been fixed in the species, and possibly the whole genus (the lack of chromosome specimens from the western hoolock prevented us from determining if this is a genus- or species-specific phenomenon). It is therefore likely that this phenomenon did not interfere with centromeric function. Centromeres are almost always heavily enriched in repeated sequences, mostly represented by highly repeated satellites. Exceptions to this association are neocentromeres in human (Choo 1997), the “point” centromeres of budding yeast (Henikoff et al. 2001), the polymorphic centromere in orangutan (Locke et al. 2011), and the evolutionarily new centromere in horses (Wade et al. 2009). Repetitive DNA may not be a “precursor” of centromere function and identity, but rather a consequence of the evolution of a locus with centromeric function (Eichler 1999). The accumulation of repeats in the centromere is unlikely to have a detrimental effect, unless it interferes with the ability of the centromere to guarantee correct movement and segregation of the sister chromatids.
In conclusion, we have discovered a new composite transposable element, the LAVA element, which formed and thrived exclusively in gibbon species. Together with a high frequency of chromosomal rearrangements, the LAVA element is a sign of the exceptional genomic plasticity of the gibbons. Nevertheless, at first glance, the evolution of the LAVA element and the high rate of chromosomal breakpoints in gibbons do not seem to be correlated. The centromeric expansion of LAVA elements in hoolock chromosomes is an indication that the epigenetic repression of transposable elements was attenuated during the evolution of gibbon lineages.
Supplementary Material
Supplementary tables S1–S3, figures S1–S4 and supplementary information are available at Genome Biology and Evolution online (http://www.gbe.oxfordjournals.org/).
Acknowledgments
This work is dedicated to our colleague and friend Alan R. Mootnick, who passed away during the preparation of the manuscript, and without whom this work would not have been possible. He will be missed.
The whole genome shotgun sequencing of the northern white-cheeked gibbon was performed at the Washington University Genome Sequencing Center and the Baylor College of Medicine Human Genome Sequencing Center. We also thank Dr Jeff Wall (UCSF) for sharing the gibbon RAD-seq datasets. We finally thank Thomas J. Meyer for his contribution to the analysis.
Part of this work was supported by National Institutes of Health (NIH) Grant RR000163 and NIH Grant RO1 GM59290 (M.A.B.).
Literature Cited
- Alkan C, et al. Genome-wide characterization of centromeric satellites from multiple mammalian genomes. Genome Res. 2011;21:137–145. doi: 10.1101/gr.111278.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aravin AA, et al. A piRNA pathway primed by individual transposons is linked to de novo DNA methylation in mice. Mol Cell. 2008;31:785–799. doi: 10.1016/j.molcel.2008.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arnold ML, Meyer A. Natural hybridization in primates: one evolutionary mechanism. Zoology. 2006;109:261–276. doi: 10.1016/j.zool.2006.03.006. [DOI] [PubMed] [Google Scholar]
- Birney E, et al. Identification and analysis of functional elements in 1% of the human genome by the ENCODE pilot project. Nature. 2007;447:799–816. doi: 10.1038/nature05874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carbone L, et al. Evolutionary breakpoints in the gibbon suggest association between cytosine methylation and karyotype evolution. PLoS Genet. 2009;5:e1000538. doi: 10.1371/journal.pgen.1000538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carone DM, et al. A new class of retroviral and satellite encoded small RNAs emanates from mammalian centromeres. Chromosoma. 2009;118:113–125. doi: 10.1007/s00412-008-0181-5. [DOI] [PubMed] [Google Scholar]
- Cellamare A, et al. New insights into centromere organization and evolution from the white-cheeked gibbon and marmoset. Mol Biol Evol. 2009;26:1889–1900. doi: 10.1093/molbev/msp101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen L, et al. Construction, characterization, and chromosomal mapping of a fosmid library of the white-cheeked gibbon (Nomascus leucogenys) Genomics Proteomics Bioinform. 2007;5:207–215. doi: 10.1016/S1672-0229(08)60008-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choo KH. Centromere DNA dynamics: latent centromeres and neocentromere formation. Am J Hum Genet. 1997;61:1225–1233. doi: 10.1086/301657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cordaux R, Batzer MA. The impact of retrotransposons on human genome evolution. Nat Rev Genet. 2009;10:691–703. doi: 10.1038/nrg2640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costa FF, et al. SATR-1 hypomethylation is a common and early event in breast cancer. Cancer Genet Cytogenet. 2006;165:135–143. doi: 10.1016/j.cancergencyto.2005.07.023. [DOI] [PubMed] [Google Scholar]
- Cunningham C, Mootnick A. Gibbons. Curr Biol. 2009;19:R543–R544. doi: 10.1016/j.cub.2009.05.013. [DOI] [PubMed] [Google Scholar]
- de Koning AP, Gu W, Castoe TA, Batzer MA, Pollock DD. Repetitive elements may comprise over two-thirds of the human genome. PLoS Genet. 2011;7:e1002384. doi: 10.1371/journal.pgen.1002384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eichler EE. Repetitive conundrums of centromere structure and function. Human Mol Genet. 1999;8:151–155. doi: 10.1093/hmg/8.2.151. [DOI] [PubMed] [Google Scholar]
- Fontdevila A. Hybrid genome evolution by transposition. Cytogenet Genome Res. 2005;110:49–55. doi: 10.1159/000084937. [DOI] [PubMed] [Google Scholar]
- Garcia Guerreiro MP, Fontdevila A. Molecular characterization and genomic distribution of Isis: a new retrotransposon of Drosophila buzzatii. Mol Genet Genomics. 2007;277:83–95. doi: 10.1007/s00438-006-0174-0. [DOI] [PubMed] [Google Scholar]
- Gentles AJ, et al. Evolutionary dynamics of transposable elements in the short-tailed opossum Monodelphis domestica. Genome Res. 2007;17:992–1004. doi: 10.1101/gr.6070707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Girirajan S, et al. Sequencing human-gibbon breakpoints of synteny reveals mosaic new insertions at rearrangement sites. Genome Res. 2009;19:178–190. doi: 10.1101/gr.086041.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henikoff S, Ahmad K, Malik HS. The centromere paradox: stable inheritance with rapidly evolving DNA. Science. 2001;293:1098–1102. doi: 10.1126/science.1062939. [DOI] [PubMed] [Google Scholar]
- Jurka J. Repbase update: a database and an electronic journal of repetitive elements. Trends Genet. 2000;16:418–420. doi: 10.1016/s0168-9525(00)02093-x. [DOI] [PubMed] [Google Scholar]
- Kim SK, et al. Patterns of genetic variation within and between Gibbon species. Mol Biol Evol. 2011;28:2211–2218. doi: 10.1093/molbev/msr033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koehler U, Bigoni F, Wienberg J, Stanyon R. Genomic reorganization in the concolor gibbon (Hylobates concolor) revealed by chromosome painting. Genomics. 1995;30:287–292. doi: 10.1006/geno.1995.9875. [DOI] [PubMed] [Google Scholar]
- Lichter P, Ledbetter SA, Ledbetter DH, Ward DC. Fluorescence in situ hybridization with Alu and L1 polymerase chain reaction probes for rapid characterization of human chromosomes in hybrid cell lines. Proc Natl Acad Sci U S A. 1990;87:6634–6638. doi: 10.1073/pnas.87.17.6634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Locke DP, et al. Comparative and demographic analysis of orang-utan genomes. Nature. 2011;469:529–533. doi: 10.1038/nature09687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma J, Wing RA, Bennetzen JL, Jackson SA. Plant centromere organization: a dynamic structure with conserved functions. Trends Genet. 2007;23:134–139. doi: 10.1016/j.tig.2007.01.004. [DOI] [PubMed] [Google Scholar]
- Manuelidis L. Chromosomal localization of complex and simple repeated human DNAs. Chromosoma. 1978;66:23–32. doi: 10.1007/BF00285813. [DOI] [PubMed] [Google Scholar]
- Molaro A, et al. Sperm methylation profiles reveal features of epigenetic inheritance and evolution in primates. Cell. 2011;146:1029–1041. doi: 10.1016/j.cell.2011.08.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montefalcone G, Tempesta S, Rocchi M, Archidiacono N. Centromere repositioning. Genome Res. 1999;9:1184–1188. doi: 10.1101/gr.9.12.1184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morley RJ, Flenley JR. Late Cainozoic vegetational and environmental changes in the Malay Archipelago. In: TC Whitmore., editor. Biogeographical evolution of the Malay Archipelago. Oxford: Clarendon Press; 1987. pp. 50–59. [Google Scholar]
- Mrasek K, et al. Detailed Hylobates lar karyotype defined by 25-color FISH and multicolor banding. Int J Mol Med. 2003;12:139–146. [PubMed] [Google Scholar]
- Muller S, Hollatz M, Wienberg J. Chromosomal phylogeny and evolution of gibbons (Hylobatidae) Hum Genet. 2003;113:493–501. doi: 10.1007/s00439-003-0997-2. [DOI] [PubMed] [Google Scholar]
- Myers RH, Shafer DA. Hybrid ape offspring of a mating of gibbon and siamang. Science. 1979;205:308–310. doi: 10.1126/science.451603. [DOI] [PubMed] [Google Scholar]
- O'Neill RJ, O'Neill MJ, Graves JA. Undermethylation associated with retroelement activation and chromosome remodelling in an interspecific mammalian hybrid. Nature. 1998;393:68–72. doi: 10.1038/29985. [DOI] [PubMed] [Google Scholar]
- Raiz J, et al. The non-autonomous retrotransposon SVA is trans-mobilized by the human LINE-1 protein machinery. Nucleic Acids Res. 2011;40:1666–1683. doi: 10.1093/nar/gkr863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rocchi M, Stanyon R, Archidiacono N. Evolutionary new centromeres in primates. Prog Mol Subcell Biol. 2009;48:103–152. doi: 10.1007/978-3-642-00182-6_5. [DOI] [PubMed] [Google Scholar]
- Szpakowski S, et al. Loss of epigenetic silencing in tumors preferentially affects primate-specific retroelements. Gene. 2009;448:151–167. doi: 10.1016/j.gene.2009.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Telenius H, et al. Cytogenetic analysis by chromosome painting using DOP-PCR amplified flow-sorted chromosomes. Genes Chromosome Canc. 1992;4:257–263. doi: 10.1002/gcc.2870040311. [DOI] [PubMed] [Google Scholar]
- Thinh VN, et al. Mitochondrial evidence for multiple radiations in the evolutionary history of small apes. BMC Evol Biol. 2010a;10:74. doi: 10.1186/1471-2148-10-74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thinh VN, et al. Mitochondrial evidence for multiple radiations in the evolutionary history of small apes. BMC Evol Biol. 2010b;10:74. doi: 10.1186/1471-2148-10-74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas JW, et al. Parallel construction of orthologous sequence-ready clone contig maps in multiple species. Genome Res. 2002;12:1277–1285. doi: 10.1101/gr.283202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Heijden GW, Bortvin A. Transient relaxation of transposon silencing at the onset of mammalian meiosis. Epigenetics. 2009;4:76–79. doi: 10.4161/epi.4.2.7783. [DOI] [PubMed] [Google Scholar]
- Van Tuinen P, Ledbetter H. Cytogenetic comparison and phylogeny of three species of Hylobatidae. Am J Phys Anthropol. 1983;61:453–466. doi: 10.1002/ajpa.1330610408. [DOI] [PubMed] [Google Scholar]
- Ventura M, et al. Neocentromeres in 15q24-26 map to duplicons which flanked an ancestral centromere in 15q25. Genome Res. 2003;13:2059–2068. doi: 10.1101/gr.1155103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wade CM, et al. Genome sequence, comparative analysis, and population genetics of the domestic horse. Science. 2009;326:865–867. doi: 10.1126/science.1178158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H, et al. SVA elements: a hominid-specific retroposon family. J Mol Biol. 2005;354:994–1007. doi: 10.1016/j.jmb.2005.09.085. [DOI] [PubMed] [Google Scholar]
- Wienberg J. The evolution of eutherian chromosomes. Curr Opin Genet Dev. 2004;14:657–666. doi: 10.1016/j.gde.2004.10.001. [DOI] [PubMed] [Google Scholar]
- Wong A, et al. Diverse fates of paralogs following segmental duplication of telomeric genes. Genomics. 2004;84:239–247. doi: 10.1016/j.ygeno.2004.03.001. [DOI] [PubMed] [Google Scholar]
- Yoder JA, Walsh CP, Bestor TH. Cytosine methylation and the ecology of intragenomic parasites. Trends Genet. 1997;13:335–340. doi: 10.1016/s0168-9525(97)01181-5. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}