

The structure of a SycH–YopH chaperone–effector complex from Yersinia reveals the bacterial state of a protein that adopts different folds in the host and pathogen environments.
Keywords: Yersinia pestis, type 3 secretion, YopH, SycH, bacterial pathogenesis, chaperones, virulence peptides
Abstract
Yersinia pestis injects numerous bacterial proteins into host cells through an organic nanomachine called the type 3 secretion system. One such substrate is the tyrosine phosphatase YopH, which requires an interaction with a cognate chaperone in order to be effectively injected. Here, the first crystal structure of a SycH–YopH complex is reported, determined to 1.9 Å resolution. The structure reveals the presence of (i) a nonglobular polypeptide in YopH, (ii) a so-called β-motif in YopH and (iii) a conserved hydrophobic patch in SycH that recognizes the β-motif. Biochemical studies establish that the β-motif is critical to the stability of this complex. Finally, since previous work has shown that the N-terminal portion of YopH adopts a globular fold that is functional in the host cell, aspects of how this polypeptide adopts radically different folds in the host and in the bacterial environments are analysed.
1. Introduction
Numerous Gram-negative pathogens of both animals and plants employ a protein-delivery system termed the type 3 secretion system (T3SS) to translocate bacterial proteins into the host cell (Galán & Wolf-Watz, 2006 ▶; Cornelis, 2006 ▶; Horn et al., 2004 ▶). T3SSs have been termed ‘organic nano-syringes’ owing to the macromolecular resemblance of the machinery to a syringe as well as the functional analogy to injection. The T3SSs allow the delivery of substrates, often called virulence factors or effectors, across three biological lipid membranes: the inner and outer membranes of the bacterium (and the periplasmic space between them) and the host-cell plasma membrane. The substrates possess a wide range of biochemical activities that modulate the eukaryotic cell in manners beneficial to the survival and replication of the pathogen (Stebbins & Galán, 2001a ▶; Barbieri et al., 2002 ▶; Stebbins, 2004 ▶; Galán, 2009 ▶).
The first 15–20 amino acids of T3SS substrates are required for secretion and are followed by a small 50–100-amino-acid ‘chaperone-binding domain’ (CBD) that is responsible for binding small acidic ‘secretion chaperones’ in the bacterium and targeting the virulence factors to the pathogenic secretion apparatus (Wattiau et al., 1996 ▶; Page & Parsot, 2002 ▶; Parsot et al., 2003 ▶; Stebbins & Galán, 2003 ▶; Ghosh, 2004 ▶; Lee & Galán, 2004 ▶).
Significant structural work over the last ten years has revealed that these chaperone–effector complexes possess unusual features: (i) the effector (T3SS substrate) adopts a nonglobular fold that wraps around the surface of the chaperone homodimer, (ii) a structural motif (the so-called β-motif) is present in all complexes characterized to date and is critical to a stable interaction and (iii) there is a hydrophobic pocket in the chaperone that is highly conserved that serves as a binding site for the β-motif (Lilic et al., 2006 ▶; Schubot et al., 2005 ▶; Singer et al., 2004 ▶; Phan et al., 2004 ▶; Evdokimov et al., 2002 ▶; Stebbins & Galán, 2001b ▶; Vogelaar et al., 2010 ▶).
YopH is a 468-amino-acid protein that is injected into host cells by the T3SS of several species of the Yersinia genus, including Y. pestis, the causative agent of bubonic and pneumonic plague (Guan & Dixon, 1990 ▶; Bliska et al., 1991 ▶). The C-terminal portion of the effector (spanning residues ∼160–468) functions as a protein tyrosine phosphatase (PTP) in the host cell, altering signal transduction pathways (Rosqvist et al., 1988 ▶; Andersson et al., 1996 ▶, 1999 ▶; Bartra et al., 2001 ▶; Black & Bliska, 1997 ▶; Black et al., 2000 ▶; Deleuil et al., 2003 ▶; Green et al., 1995 ▶; Hamid et al., 1999 ▶). The N-terminal region of the polypeptide has two functions: in the bacterium residues 1–130 bind to the SycH chaperone and are required for translocation into the host cell through the T3SS, whereas in the host cell this domain aids in targeting the PTP domain to its proper substrates (Woestyn et al., 1996 ▶; Black et al., 1998 ▶; Evdokimov et al., 2001 ▶; Montagna et al., 2001 ▶; Smith et al., 2001 ▶; Khandelwal et al., 2002 ▶).
An important question both from a biochemical/structural aspect as well as in general considerations of the host–pathogen interaction and evolutionary biology is whether this ‘reuse’ of the polypeptide in two different physiological environments is associated with dual structure–function states. In other words, does the polypeptide 1–130 of YopH adopt different folds in the bacterial versus the host environment that are associated with these different functions?
The initial evidence would point to the dual structure–function possibility as the most likely hypothesis. YopH(1–130) is known to form a globular stable fold on its own, and this fold has been probed through structure-based mutagenesis to test the function of surface-exposed residues in the host cell, indicating that this domain as folded in the crystal structures is likely to be the functional form of the molecule in the host (Evdokimov et al., 2001 ▶; Smith et al., 2001 ▶; Khandelwal et al., 2002 ▶). However, all previous chaperone–effector studies from multiple T3SSs, including the related structure of the YopH chaperone SycH with the regulatory molecule YscM2 (which contains significant homology to YopH in its N-terminal domain), have shown that the interaction consists of a nonglobular polypeptide in the effector (Lilic et al., 2006 ▶; Schubot et al., 2005 ▶; Singer et al., 2004 ▶; Phan et al., 2004 ▶; Evdokimov et al., 2002 ▶; Stebbins & Galán, 2001b ▶; Vogelaar et al., 2010). This would suggest that YopH would also adopt the nonglobular fold and thus be utterly different from the fold that it adopts on its own in the host. However, the negative regulator of secretion YscM2, which interacts with SycH, is not itself translocated by the T3SS (Stainier et al., 1997 ▶; Cambronne & Schneewind, 2002 ▶; Phan et al., 2004 ▶). One could hypothesize that this different function, and in particular the unique engagement with the T3SS, could allow its interaction with SycH to be substantially different from the translocated YopH.
Therefore, to obtain data on the SycH–YopH interaction in order to distinguish between these hypotheses, we have determined the structure of a portion of YopH with the SycH chaperone to 1.9 Å resolution.
2. Materials and methods
2.1. Protein expression, purification and complex formation
YopH and SycH were PCR-amplified from the pIB102 virulence plasmid of Y. pseudotuberculosis (a generous gift from J. Bliska) using Pfu Turbo DNA polymerase (Stratagene). PCR products were cloned into a modified pCDF-1 Duet vector (Novagen) with an N-terminal hexahistidine tag and an engineered 3C protease-cleavage site using the SalI and NotI or NdeI and XhoI restriction sites. All constructs were verified by sequencing.
The proteins were expressed in Escherichia coli BL21 (DE3) cells (Stratagene). The cells were grown at 310 K in LB medium containing 50 µg ml−1 streptomycin with shaking at 200 rev min−1. When the OD600 reached 0.8, IPTG was added to a final concentration of 500 µM, the temperature was decreased to 293 K and the cells were shaken for 15 h. The cells were harvested by centrifugation, resuspended in lysis buffer (50 mM Tris pH 8.0, 200 mM NaCl) and lysed using an Emulsiflex C-5 cell homogenizer (Avestin Inc., Ottawa, Ontario, Canada). The insoluble fraction was pelleted by centrifugation at 30 000g and the soluble fraction was passed over Ni–NTA agarose resin (Qiagen). The resin was washed with lysis buffer containing 30 mM imidazole, followed by elution with lysis buffer containing 200 mM imidazole. The histidine tag was removed by overnight cleavage with the site-specific rhinovirus 3C protease using a 1:200(w:w) protease:protein ratio at 277 K in the presence of 1 mM EDTA. The eluted protein was concentrated and further purified on a 16/60 Superdex 75 size-exclusion column (GE Healthcare) pre-equilibrated in 25 mM Tris pH 8.0, 200 mM NaCl, 2 mM DTT using an ÄKTA purification system (GE Healthcare).
Protein-complex formation was assayed by a pull-down on Ni–NTA agarose resin using N-terminally histidine-tagged YopH and nontagged SycH1–141 co-expressed from the same plasmid (in the case of truncated YopH fragments) or by gel-filtration chromatography when YopH1–129 (wild type and triple mutant) and SycH1–141 were used. In the latter case, YopH and SycH were expressed as N-terminal histidine-tag fusions, purified separately and mixed overnight at 277 K.
2.2. Crystallization and structural determination of the YopH21–63–SycH1–141 complex
The YopH21–63–SycH1–141 complex was purified as described above. After final purification on a 16/60 Superdex 75 column, peak fractions were collected and concentrated to 38 mg ml−1 using Centricon protein concentrators (20 kDa cutoff). Crystals were obtained using the hanging-drop vapor-diffusion technique, mixing 1 µl protein solution with 1 µl well solution consisting of 0.2 M diammonium acetate, 20% PEG 3350. For cryo-preservation, the crystals were transferred into a drop consisting of 0.2 M diammonium acetate, 34% PEG 3350 and were allowed to equilibrate for approximately 2 min. For data collection, the crystals were captured in a loop and flash-cooled in a stream of nitrogen gas cooled to 93 K.
Data were collected on beamline X6A of the National Synchrotron Light Source at Brookhaven National Laboratory and were processed using HKL-2000 (Otwinowski & Minor, 1997 ▶). The crystals belonged to space group P41212, with unit-cell parameters a = b = 70.525, c = 71.517 Å. There was a single heterodimer of SycH–YopH in the asymmetric unit; the canonical secretion chaperone dimer was reproduced along a crystallographic twofold axis of symmetry. Phases were determined by molecular replacement using SycH from the SycH–YscM2 structure as a model (Phan et al., 2004 ▶) and the Phaser software (McCoy et al., 2007 ▶), and 90% of the final model was built by ARP/wARP (Langer et al., 2008 ▶). Cycles of manual building with O (Jones et al., 1991 ▶) and refinement with REFMAC5 (Murshudov et al., 2011 ▶) resulted in a model with an R and R free of 18.0% and 24.5%, respectively, to 1.9 Å resolution. The crystallographic statistics are summarized in Table 1 ▶.
Table 1. Crystallographic statistics.
Values in parentheses are for the highest resolution shell.
| Data collection | |
| Space group | P41212 |
| Unit-cell parameters | |
| a () | 70.525 |
| b () | 70.525 |
| c () | 71.517 |
| Wavelength () | 1.0809 |
| Resolution () | 31.541.90 |
| No. of unique reflections | 13866 |
| R merge † | 4.0 (42.1) |
| I/(I) | 37.3 (2.6) |
| Completeness (%) | 98.8 (93.4) |
| Multiplicity | 6.7 (3.5) |
| Refinement | |
| Resolution () | 31.541.90 |
| No. of reflections | 13866 |
| R work/R free ‡ (%) | 18.0/24.5 |
| Total No. of atoms | 1361 |
| No. of protein atoms | 1171 |
| Waters | 190 |
| B factors (2) | |
| Average | 29.37 |
| Protein | 27.65 |
| Solvent | 39.96 |
| R.m.s. deviations§ | |
| Bond lengths () | 0.018 |
| Bond angles () | 1.890 |
| Ramachandran plot | |
| Favored regions (%) | 91.0 |
| Allowed regions (%) | 9.0 |
| Outliers (%) | 0 |
R
merge = 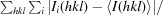
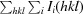 , for i observations of the intensity I(hkl) of reflection hkl.
, for i observations of the intensity I(hkl) of reflection hkl.
R = 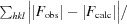
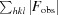 . R
free was calculated using 5% of the data, which were omitted from refinement.
. R
free was calculated using 5% of the data, which were omitted from refinement.
Bond and angle deviations from ideal values.
2.3. Mutagenesis
The triple mutant of YopH was made by in vitro site-directed mutagenesis using oligonucleotide primer pairs conferring Leu42Ala, Ile44Ala and Ser46Ala mutations. The amplification of the mutant plasmid was performed using Pfu Turbo DNA polymerase (Stratagene) in a thermal temperature cycler and the wild-type template plasmid was removed by overnight digestion with the DpnI enzyme before transformation. The introduced mutations were verified by DNA sequencing. The mutant was expressed in E. coli strain BL21 (DE3) and purification of the mutant protein was performed as described for the wild-type proteins.
3. Results and discussion
3.1. Domain determination
A minimal stable complex of YopH and SycH was identified by an iterative trial-and-error procedure combining protein-sequence analysis, structural comparisons with other chaperone–effector complexes and protease footprinting. While numerous constructs of YopH could be cloned and co-expressed with SycH to produce a stable complex, most did not crystallize or gave poorly diffracting crystals. In the end, a very small construct of YopH spanning residues 21–63 was co-purified with SycH (Fig. 1 ▶ a), well diffracting crystals of this complex were grown (Fig. 1 ▶ b) and the complex structure was solved to 1.9 Å resolution (Fig. 1 ▶ c, Table 1 ▶ and §2).
Figure 1.

Purification, crystallization and electron density of YopH–SycH. (a) Coomassie-stained SDS–PAGE analysis of the purified YopH–SycH complex used for crystallization. Lane St contains protein standard markers for assigning molecular weight (labeled in kDa). (b) Well diffracting crystals of the YopH21–63–SycH complex. (c) Domain schematic of YopH. (d) 2F o − F c model-phased electron-density maps contoured at 1σ shown in blue with the final refined model shown.
3.2. Overall structure of the complex
The crystal structure of the SycH–YopH21–63 complex reveals that the YopH–SycH interaction preserves the majority of the interactions between secretion chaperones and their cognate substrates observed in previous structures. Adopting a nonglobular conformation, the YopH chaperone-binding domain (CBD) wraps around SycH (Figs. 2 ▶ and 3 ▶). A three-amino-acid ‘plug’ (the so-called β-motif) on an intermolecular β-sheet inserts into a large hydrophobic patch in the chaperone (Figs. 4 ▶ and 5 ▶). Finally, an α-helix of the effector is clamped down on by a large hydrophobic patch produced by the concave surface of the SycH β-sheet (Figs. 2 ▶ and 4 ▶).
Figure 2.

Overall structure. (a) Overall fold shown as a cartoon diagram in two orientations related by a 90° rotation about a vertical axis. YopH is shown in salmon and SycH in purple. (b) Topology of the polypeptide chains. (c) Secondary-structure alignment with the polypeptide sequence. Key: blue H, helices; red β, strands; magenta β, β-turn; magenta γ, γ-turn; red loop, β-hairpin. (b) and (c) were generated by PDBSum (Laskowski, 2009 ▶).
Figure 3.

SycH crystallographic dimer. Ribbon diagrams of the SicP and InvB effector chaperones of Salmonella (from PDB entries 1jyo and 2fm8, respectively; Stebbins & Galán, 2001b ▶; Lilic et al., 2006 ▶) alongside the reconstructed crystallographic SycH dimer from the complexes with YopH (this study) and YscM2. Each monomer in the dimers is colored either blue or purple.
Figure 4.

YopH–SycH interactions. (a) Hydrophobic patches in SycH (yellow) that bind the YopH polypeptide (shown as a ribbon cartoon in salmon). (b) Residues making contact between the S2 strand (β-motif) of YopH (salmon and orange) and SycH (purple and blue). (c) Contacts between the YopH H2 helix and the SycH helix-binding groove.
Figure 5.

YopH possesses a canonical β-motif. (a) Comparison of T3SS effector nonglobular polypeptides from several complexes with chaperones (the alignment was generated by aligning the complexes based on the conserved folds of the chaperones). Effector polypeptides include Salmonella SipA (PDB entry 2fm8, red; Lilic et al., 2006 ▶) and SptP (PDB entry 1jyo, purple; Stebbins & Galán, 2001b ▶), Yersinia YopN (PDB entry 1xkp, ellow; Schubot et al., 2005 ▶) and YopE (PDB entry 1l2w, cyan; Birtalan et al., 2002 ▶) and Pseudomonas syringae HopA1 (PDB entry 4g6t, green; C. E. Stebbins, R. Janjusevic & C. M. Quezada, unpublished work). Placed roughly for orienting the image is a surface rendering of SycH with hydrophobic patches shown in yellow. (b) Alignment of several β-motifs from animal and plant effectors. (c) Alignment of the β-motif binding pocket in chaperones from animal and plant pathogens. On the left are three key elements of structure in all chaperones characterized to date, consisting of six highly conserved hydrophobic amino acids which closely superimpose and which frequently make contacts with β-motif residues. On the right is a space-filling representation of SycH with the six conserved hydrophobic residues of the chaperone pocket shown in yellow and indicated by arrows. The main-chain trace of YopH is drawn as a tube in salmon; the β-motif is indicated.
In the crystals, SycH and YopH21–63 interact with a 1:1 stoichiometry. This is in contrast to the 2:1 chaperone:effector molar ratio which occurs when there are two intact β-motifs in the CBD, as found in SicP–SptP from Salmonella and YopE–SycE from Yersinia (Stebbins, 2005 ▶). This 1:1 arrangement has previously been observed in the structure of SycH with a truncated peptide of YscM2 (Phan et al., 2004 ▶), as well as in that of HopA1 bound to ShcA (PDB entry 4g6t; C. E. Stebbins, R. Janjusevic & C. M. Quezada, unpublished work). As biochemical studies show (see §3.5), YopH is likely to have two independent chaperone-binding sequences in the CBD sequence 1–130. One is a β-motif-containing region as identified in our crystal structure (YopH36–63). The second binding peptide (72–121) lacks a sequence identical to the first β-motif but contains several sequences that could serve as a second such motif by analogy to SptP and YopE. Therefore, the truncations in YopH are likely to have removed the second binding site. Moreover, the standard dimer interface observed in numerous secretion chaperones of this family is present in the crystal along a crystallographic twofold axis of symmetry (Fig. 3 ▶). This latter phenomenon was also observed in the crystals of SycH–YscM2, as well as in the structure of HopA1–ShcA, suggesting that the chaperone dimer is not a product of crystal packing. In all of these cases the portion of the effector that the authors crystallized was short and was equivalent to approximately half of the polypeptide known to bind the chaperone. In particular, the second β-motif was removed, freeing the conserved hydrophobic patch of the chaperone to bind to a second molecule of the truncated CBD.
3.3. Details of the SycH–YopH interface
The nonglobular interaction makes the interface of SycH and YopH extensive and significantly hydrophobic, burying approximately 1000 Å2 of surface area, a very high figure considering that fewer than 30 amino acids of YopH are present. In comparison, the interface in the chaperone homodimer buries approximately 1100 Å2 of surface area. Two primary contacts dominate the interaction between the chaperone and the effector.
The first is an intermolecular β-sheet between the proteins. The end of the five-stranded β-sheet of the chaperone is extended by two strands donated by YopH. The S2 strand of YopH extends the chaperone β-sheet, interacting with S1 of SycH. Both strands of YopH insert amino acids into an extended predominantly hydrophobic patch of SycH (Fig. 4 ▶ a). Glu36 and Phe39 of YopH strand S1 make several important contacts with SycH, primarily hydrophobic and van der Waals interactions with (primarily) hydrophobic residues in the effector-binding patch of the chaperone, but also hydrogen bonds such as that from Glu36 to Lys34 of SycH (Fig. 4 ▶ b).
Strand S2 is particularly significant. Like its counterparts in previously determined structures of T3SS chaperone–effector complexes, it harbors the three-amino-acid β-motif. This structural motif is situated in a very similar fashion to the interactions observed in related complexes. The YopH β-motif interactions involve the main-chain to main-chain intermolecular contacts bridging the strands, as well as the motif residues Leu42, Ile44 and Ser46, which insert into the SycH hydrophobic patch contacting residues Leu15, Leu17, Ile20, Val30, Ile32, Asp33, Ala106, Leu109, Thr110 and Val113 (Fig. 4 ▶ b).
The second important interaction between YopH and its chaperone is on a proximal face of SycH. YopH inserts an α-helix into what has been termed the helix-binding groove of the chaperone (Stebbins & Galán, 2001b ▶). Created by the β-sheet of SycH, this concave groove is characterized by a large hydrophobic surface which is sequestered from solvent by the helix of YopH. Four key hydrophobic residues from the H2 helix of YopH (Phe55, Ala56, Val59 and Leu60) insert into the hydrophobic patch. In addition, the residue Glu52 makes significant contacts (Fig. 4 ▶ c).
3.4. The β-motif is conserved in the YopH–SycH complex
An intermolecular β-sheet analogous to that observed in the YopH–SycH complex is formed in all of the chaperone–effector interactions characterized by structural biology to date: a β-strand of the virulence factor in a position equivalent to the YopH S2 β-strand augments the chaperone β-sheet and inserts three amino acids (mostly hydrophobic) into the hydrophobic patch of the chaperone (Fig. 5 ▶ a). This β-motif was first recognized in the SipA–InvB complex from Salmonella (Lilic et al., 2006 ▶). The YopH β-motif (potentially one of two owing to the second chaperone-binding site between residues 64 and 129), consisting of Leu42, Ile44 and Ser46 and inserting into a SycH hydrophobic pocket, also aligns well with the motif residues in other pathogen effectors (Fig. 5 ▶ b).
Analogies to other effector–chaperone complexes can be made by noting the conserved nature of the hydrophobic pocket in SycH that interacts with the β-motif (Fig. 5 ▶ c). The hydrophobic residues at six positions that contribute significantly to the pocket also possess counterparts in the broad family of secretion chaperones (Fig. 5 ▶ c). This may be viewed as a complementary receptor ligand for the β-motif that is highly conserved throughout this system in many different pathogenic bacteria of plants and animals.
3.5. Structure-based mutagenesis
We created a series of truncations as well as loss-of-contact structure-based mutations of YopH to probe its interaction with SycH. To begin with, a series of constructs that were truncations of the larger 1–130 CBD of YopH were made to examine whether the 1:1 stoichiometry observed in the crystallographic asymmetric unit was indicative of a single binding site for YopH in SycH or whether the situation was similar to that observed in the SycH–YscM2 complex (Phan et al., 2004 ▶). In both these chaperone–effector structures the CBD was severely truncated in order to facilitate crystal growth. In the majority of chaperone–effector complexes the chaperone homodimer binds a single CBD at two different locations: the binding site on the identical chaperone and a β-motif present in both interactions. However, with the truncation of the second binding site it is possible that the first binding site could occupy both chaperones in the homodimer. This would allow a crystallographic twofold axis of symmetry to exist between each molecule of the chaperone homodimer (a symmetry that is broken when a 2:1 complex is formed in the other structures with more extensive CBDs containing the second binding site in the effector). This was in fact hypothesized for the SycH–YscM2 cocrystal structure (Phan et al., 2004 ▶).
Our results strongly argue that something similar is happening in the case of YopH. N-terminal CBD constructs (21–63 and 35–82) are able to bind to SycH, whereas a β-motif mutant of the N-terminal peptide (YopH21–63 L42A/I44A/S46A) as well as a construct that truncates the β-motif (YopH45–82) do not form stable complexes (Fig. 6 ▶). Similarly, YopH64–121 and YopH72–121, but not YopH77–121, were able to form a stable complex with the chaperone (Fig. 6 ▶). However, despite this evidence supporting the idea of an independent C-terminal second SycH-binding site, a mutant in the β-motif observed in the crystal structure (L42A/I44A/S46A) which abrogated complex formation of the crystallized construct (YopH21–63) also impaired complex formation of the larger YopH1–129 (Fig. 6 ▶). This indicates that while the second binding site is sufficient for binding the smaller C-terminal construct, without the intact N-terminal binding site the large polypeptide complex is destabilized in the assays that we have used.
Figure 6.

Structure-based mutagenesis of the YopH–SycH complex. Constructs of YopH tested for binding to SycH. Truncations and mutations are indicated, and binding (as assayed by wild-type levels of complex formation in recombinant co-expression and affinity pull-down; see §2) is indicated by + (colored green, complex formation) or − (colored red, no complex formation).
Together, these results suggest that not only is the β-motif critical for interaction with the chaperone (as has been observed in previous work), but that there are likely to be two binding sites in YopH spanning residues 36–63 and 72–121, respectively. We also hypothesize that the second binding site will also contain a canonical β-motif and that the true stoichiometry of the complex is a 2:1 ratio of the chaperone to the effector.
3.6. Comparison of the YopH NTD monomer and SycH-bound states
We have analyzed the structural role of the residues in our chaperone–effector complex and compared them with their roles in the previously published globular host-cell fold of YopH. A caveat is that we only possess a short region of the YopH CBD (residues 36–63) and can only extend by analogy what we find for this region to the remainder of the polypeptide.
It is immediately obvious that the CBD peptide 36–63, either folded up on itself in the monomer or bound as a nonglobular peptide to the chaperone, adopts similar structures in these dramatically different contexts. To begin, the overall conformation of the polypeptide is similar (Fig. 7 ▶ a), although the strands that form by extending the chaperone β-sheet in the chaperone–effector complex adopt an ‘ordered-loop’ fold present as an extended loop in the monomeric structure of the YopH NTD. Because they do not form a similar fold in the two structures, the side chains of residues 36–49 do not pack in an analogous fashion and adopt divergent structural roles.
Figure 7.

Comparison of YopH peptides in the chaperone-bound and free states. (a) Ribbon diagram showing the YopH peptide 36–63 (yellow) in the context of the folded monomeric globular domain of YopH alone (left; YopH shown in purple outside of the 36–63 peptide) and in the context of the chaperone SycH (right; shown in blue). The sequence of the YopH peptide spanning residues 36–63 is shown below with secondary structure for each of the two structural contexts shown above them. Key: H, helices; S, strands; β, β-turn. (b) Helical wheel showing the interacting residues of YopH that bind in the SycH hydrophobic groove that are also used to form the hydrophobic core of the YopH globular fold. (c) The second SycH-binding peptide (shown in red) in YopH, with the hypothesized β-motif and amphipathic helix indicated.
However, the structure of the polypeptide that forms the helix that is bound in the hydrophobic groove of the chaperone (YopH residues 50–63) is far more conserved. In both structures it forms a similar amphipathic helix, with the predominantly hydrophobic face that binds the hydrophobic groove of SycH packing into the hydrophobic core of the globular fold in monomeric YopH (Figs. 7 ▶ a and 7 ▶ b). Therefore, the key YopH residues that contact SycH become central components of the internal packing and structure of the isolated YopH monomer.
4. Conclusions
Proteins are the core set of nanomachines in the cell and thereby underlie all life on earth. Composed of an extended polymer of amino-acid units, proteins possess a three-dimensional conformation in which this chain of residues adopts a complicated ‘fold’ stabilized by a number of different interatomic contacts. For most proteins, obtaining the proper, or functional, folded conformation is exquisitely dependent on the solvent conditions, temperature and the presence of other proteins that aid in the process. Understanding the processes by which proteins obtain their folded conformation is an important and actively pursued field. Here, we present an analysis of a virulence polypeptide from several species of the Yersinia genus that must fold in two highly divergent contexts in the bacterial or the host cell-like environments.
The structure of the YopH–SycH complex that we report shows that the nonglobular state of the effector polypeptide reported in all previous chaperone–effector complexes is maintained, despite the fact that the NTD of YopH by itself adopts a globular fold. How this could happen is readily explained by a comparison of the free and chaperone-bound states of YopH, in which the polypeptide is seen to nonetheless adopt a very similar conformation, secondary structure and hydrophobic amino-acid sequestration from solvent. It is almost as if one were to remove the CBD of YopH from the monomer, preserve most of its salient three-dimensional fold and paste it directly onto the chaperone.
While more speculative, this conceptualization is likely to be applicable to the more C-terminal portion of the polypeptide (YopH72–121) that we show binds to SycH independently of the N-terminal region (YopH21–63). To reiterate, the best model for these observations is likely to be analogous to previously determined chaperone–effector structures such as SicP–SptP and SycE–YopE in which there are two independent β-motifs at opposite ‘ends’ of the CBD, each of which binds to one of the two chaperone molecules in the chaperone homodimer. Our mutagenesis data support this in that we can make two independent peptides in YopH that bind to SycH, and when the β-motif that we identified structurally in this work (Leu42/Ile44/Ser46) is mutated the mutations abrogate complex formation in an N-terminal YopH construct (21–63) that lacks the second binding site (72–121).
Therefore, the remainder of the CBD that constitutes the second site would also be expected to bind in a similar fashion to the 36–63 peptide. In this case, it is reasonable to ask whether the observation that the 36–63 peptide adopts a fold in the chaperone complex similar to that in the globularly folded YopH1–130 monomeric fold might also apply to the second, more C-terminal peptide 72–121. Fig. 7 ▶(c) illustrates that this indeed may be the case. While not adopting a fold that is quite as easy to ‘drop onto the chaperone’ as the peptide 36–63, 72–121 possesses (i) the appropriate secondary structure in the globular fold (in fact it is more appropriate than that of 36–63), being composed of two adjacent β-strands followed by a helix, and (ii) the appropriate topological arrangement of the secondary-structural elements. While the N-terminal SycH-binding peptide requires an extended loop in YopH to rearrange into β-strands to bind SycH, the C-terminal polypetide would instead only require that the final helix flip 180° to bind analogously. Since this helix is connected to the β-strands via a very extended loop, this is a plausible scenario.
In addition to the similarities in nonglobular interaction, the complex also shows that the β-motif (the structural unit of an intermolecular β-sheet formation centered on a strand that inserts three predominantly hydrophobic residues into a conserved chaperone-binding pocket) is central to the interaction. Mutagenesis reveals that the β-motif residues play a critical role in stabilizing the chaperone–effector interaction.
Overall, these results confirm the initial discovery and characterization of the β-motif in chaperone–effector interactions and also provide insight into how YopH is able to adopt both a globular and a nonglobular fold in the host and bacterial environments, respectively.
One could thus view the YopH globular versus nonglobular conundrum as a false one when seen from the right perspective: the YopH polypeptide only appears to be ‘nonglobular’ in the context of the chaperone–effector complex because it is viewed as ‘distinct’ from the chaperone molecule. A more illuminating perspective is that, like the remainder of the YopH NTD monomeric structure onto which the peptide folds, the chaperone acts as a proteinaceous framework on which the peptide can also fold, in both cases primarily burying clustered hydrophobic amino acids (such as those in the H1 helix). The hydrophobic patches in the chaperone are therefore analogous to the hydrophobic ‘patches’ in the YopH NTD that are present when the peptide 36–63 is removed. In this context, whether or not ‘folding units’ of a protein are covalently linked (e.g. the YopH NTD by itself) or not (YopH–SycH) is less important than how the separate units interact.
Supplementary Material
PDB reference: SycH–YopH, 4gf3
Acknowledgments
We thank D. Oren at Rockefeller University and Vivian Stojanoff of Brookhaven beamline X6A for access to and assistance with crystallographic equipment. This work was funded in part by NIH Grant R01AI052182 to CES.
References
- Andersson, K., Carballeira, N., Magnusson, K. E., Persson, C., Stendahl, O., Wolf-Watz, H. & Fällman, M. (1996). Mol. Microbiol. 20, 1057–1069. [DOI] [PubMed]
- Andersson, K., Magnusson, K. E., Majeed, M., Stendahl, O. & Fällman, M. (1999). Infect. Immun. 67, 2567–2574. [DOI] [PMC free article] [PubMed]
- Barbieri, J. T., Riese, M. J. & Aktories, K. (2002). Annu. Rev. Cell Dev. Biol. 18, 315–344. [DOI] [PubMed]
- Bartra, S., Cherepanov, P., Forsberg, A. & Schesser, K. (2001). BMC Microbiol. 1, 22. [DOI] [PMC free article] [PubMed]
- Birtalan, S. C., Phillips, R. M. & Ghosh, P. (2002). Mol. Cell, 9, 971–980. [DOI] [PubMed]
- Black, D. S. & Bliska, J. B. (1997). EMBO J. 16, 2730–2744. [DOI] [PMC free article] [PubMed]
- Black, D. S., Marie-Cardine, A., Schraven, B. & Bliska, J. B. (2000). Cell. Microbiol. 2, 401–414. [DOI] [PubMed]
- Black, D. S., Montagna, L. G., Zitsmann, S. & Bliska, J. B. (1998). Mol. Microbiol. 29, 1263–1274. [DOI] [PubMed]
- Bliska, J. B., Guan, K. L., Dixon, J. E. & Falkow, S. (1991). Proc. Natl Acad. Sci. USA, 88, 1187–1191. [DOI] [PMC free article] [PubMed]
- Cambronne, E. D. & Schneewind, O. (2002). J. Bacteriol. 184, 5880–5893. [DOI] [PMC free article] [PubMed]
- Cornelis, G. R. (2006). Nature Rev. Microbiol. 4, 811–825. [DOI] [PubMed]
- Deleuil, F., Mogemark, L., Francis, M. S., Wolf-Watz, H. & Fällman, M. (2003). Cell. Microbiol. 5, 53–64. [DOI] [PubMed]
- Evdokimov, A. G., Tropea, J. E., Routzahn, K. M., Copeland, T. D. & Waugh, D. S. (2001). Acta Cryst. D57, 793–799. [DOI] [PubMed]
- Evdokimov, A. G., Tropea, J. E., Routzahn, K. M. & Waugh, D. S. (2002). Acta Cryst. D58, 398–406. [DOI] [PubMed]
- Galán, J. E. (2009). Cell Host Microbe, 5, 571–579. [DOI] [PMC free article] [PubMed]
- Galán, J. E. & Wolf-Watz, H. (2006). Nature (London), 444, 567–573. [DOI] [PubMed]
- Ghosh, P. (2004). Microbiol. Mol. Biol. Rev. 68, 771–795. [DOI] [PMC free article] [PubMed]
- Green, S. P., Hartland, E. L., Robins-Browne, R. M. & Phillips, W. A. (1995). J. Leukoc. Biol. 57, 972–977. [DOI] [PubMed]
- Guan, K. L. & Dixon, J. E. (1990). Science, 249, 553–556. [DOI] [PubMed]
- Hamid, N., Gustavsson, A., Andersson, K., McGee, K., Persson, C., Rudd, C. E. & Fällman, M. (1999). Microb. Pathog. 27, 231–242. [DOI] [PubMed]
- Horn, M., Collingro, A., Schmitz-Esser, S., Beier, C. L., Purkhold, U., Fartmann, B., Brandt, P., Nyakatura, G. J., Droege, M., Frishman, D., Rattei, T., Mewes, H. W. & Wagner, M. (2004). Science, 304, 728–730. [DOI] [PubMed]
- Jones, T. A., Zou, J.-Y., Cowan, S. W. & Kjeldgaard, M. (1991). Acta Cryst. A47, 110–119. [DOI] [PubMed]
- Khandelwal, P., Keliikuli, K., Smith, C. L., Saper, M. A. & Zuiderweg, E. R. (2002). Biochemistry, 41, 11425–11437. [DOI] [PubMed]
- Langer, G., Cohen, S. X., Lamzin, V. S. & Perrakis, A. (2008). Nature Protoc. 3, 1171–1179. [DOI] [PMC free article] [PubMed]
- Laskowski, R. A. (2009). Nucleic Acids Res. 37, D355–D359. [DOI] [PMC free article] [PubMed]
- Lee, S. H. & Galán, J. E. (2004). Mol. Microbiol. 51, 483–495. [DOI] [PubMed]
- Lilic, M., Vujanac, M. & Stebbins, C. E. (2006). Mol. Cell, 21, 653–664. [DOI] [PubMed]
- McCoy, A. J., Grosse-Kunstleve, R. W., Adams, P. D., Winn, M. D., Storoni, L. C. & Read, R. J. (2007). J. Appl. Cryst. 40, 658–674. [DOI] [PMC free article] [PubMed]
- Montagna, L. G., Ivanov, M. I. & Bliska, J. B. (2001). J. Biol. Chem. 276, 5005–5011. [DOI] [PubMed]
- Murshudov, G. N., Skubák, P., Lebedev, A. A., Pannu, N. S., Steiner, R. A., Nicholls, R. A., Winn, M. D., Long, F. & Vagin, A. A. (2011). Acta Cryst. D67, 355–367. [DOI] [PMC free article] [PubMed]
- Otwinowski, Z. & Minor, W. (1997). Methods Enzymol. 276, 307–326. [DOI] [PubMed]
- Page, A. L. & Parsot, C. (2002). Mol. Microbiol. 46, 1–11. [DOI] [PubMed]
- Parsot, C., Hamiaux, C. & Page, A. L. (2003). Curr. Opin. Microbiol. 6, 7–14. [DOI] [PubMed]
- Phan, J., Tropea, J. E. & Waugh, D. S. (2004). Acta Cryst. D60, 1591–1599. [DOI] [PubMed]
- Rosqvist, R., Bölin, I. & Wolf-Watz, H. (1988). Infect. Immun. 56, 2139–2143. [DOI] [PMC free article] [PubMed]
- Schubot, F. D., Jackson, M. W., Penrose, K. J., Cherry, S., Tropea, J. E., Plano, G. V. & Waugh, D. S. (2005). J. Mol. Biol. 346, 1147–1161. [DOI] [PubMed]
- Singer, A. U., Desveaux, D., Betts, L., Chang, J. H., Nimchuk, Z., Grant, S. R., Dangl, J. L. & Sondek, J. (2004). Structure, 12, 1669–1681. [DOI] [PubMed]
- Smith, C. L., Khandelwal, P., Keliikuli, K., Zuiderweg, E. R. & Saper, M. A. (2001). Mol. Microbiol. 42, 967–979. [DOI] [PubMed]
- Stainier, I., Iriarte, M. & Cornelis, G. R. (1997). Mol. Microbiol. 26, 833–843. [DOI] [PubMed]
- Stebbins, C. E. (2004). Curr. Opin. Struct. Biol. 14, 731–740. [DOI] [PubMed]
- Stebbins, C. E. (2005). Cell. Microbiol. 7, 1227–1236. [DOI] [PubMed]
- Stebbins, C. E. & Galán, J. E. (2001a). Nature (London), 412, 701–705. [DOI] [PubMed]
- Stebbins, C. E. & Galán, J. E. (2001b). Nature (London), 414, 77–81. [DOI] [PubMed]
- Stebbins, C. E. & Galán, J. E. (2003). Nature Rev. Mol. Cell Biol. 4, 738–743. [DOI] [PubMed]
- Vogelaar, N. J., Jing, X., Robinson, H. H. & Schubot, F. D. (2010). Biochemistry 49, 5870–5879. [DOI] [PubMed]
- Wattiau, P., Woestyn, S. & Cornelis, G. R. (1996). Mol. Microbiol. 20, 255–262. [DOI] [PubMed]
- Woestyn, S., Sory, M. P., Boland, A., Lequenne, O. & Cornelis, G. R. (1996). Mol. Microbiol. 20, 1261–1271. [DOI] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
PDB reference: SycH–YopH, 4gf3