

An X-ray crystallographic structure is described for unliganded Vaccinia virus poly(A) polymerase monomer (VP55), showing the first domain-level structural isoforms among either VP55, it’s processivity factor VP39, or the VP55-VP39 heterodimer. The occurrence of domain-level motion specifically in monomeric VP55 is consistent with the finding that the monomeric protein undergoes saltatory translocation whereas the heterodimer does not.
Keywords: VP55, Vaccinia virus poly(A) polymerase, polymerases
Abstract
Vaccinia virus poly(A) polymerase (VP55) is the only known polymerase that can translocate independently with respect to single-stranded nucleic acid (ssNA). Previously, its structure has only been solved in the context of the VP39 processivity factor. Here, a crystal structure of unliganded monomeric VP55 has been solved to 2.86 Å resolution, showing the first backbone structural isoforms among either VP55 or its processivity factor (VP39). Backbone differences between the two molecules of VP55 in the asymmetric unit indicated that unliganded monomeric VP55 can undergo a ‘rocking’ motion of the N-terminal domain with respect to the other two domains, which may be ‘rigidified’ upon VP39 docking. This observation is consistent with previously demonstrated experimental molecular dynamics of the monomer during translocation with respect to nucleic acid and with different mechanisms of translocation in the presence and absence of processivity factor VP39. Side-chain conformational changes in the absence of ligand were observed at a key primer contact site and at the catalytic center of VP55. The current structure completes the trio of possible structural forms for VP55 and VP39, namely the VP39 monomer, the VP39–VP55 heterodimer and the VP55 monomer.
1. Introduction
Vaccinia virus is a large cytoplasmic DNA virus which encodes a poly(A) polymerase (VP55) and an associated processivity factor (VP39) among its ∼200 genes. Among the known polymerases (Edmonds, 1982 ▶, 1990 ▶, 2002 ▶), VP55 seems uniquely able to translocate on ssNA. Moreover, both VP55 and VP39 possess features that are not found in any other known enzyme. VP55 is able to extend poly(A) tails to a net length of just ∼30 nt before abruptly ceasing processive tail elongation, albeit primers with oligo(A) tails greater than ∼30 nt in length can be elongated in a very slow distributive manner (Gershon et al., 1991 ▶; Gershon & Moss, 1992 ▶). In contrast, the VP55–VP39 heterodimer is able to elongate tails to several hundred nucleotides in length in a ‘semi-processive’ fashion (i.e. in a handful of prolonged rounds of processive tail elongation; Gershon & Moss, 1993 ▶) with no discernable pausing.
VP55 requires a ribouridylate-containing nucleic acid primer that is at least ∼34 nt in length (Gershon & Moss, 1992 ▶). VP55 can bind 34 nt nucleic acids containing a minimum of three ribouridylates, optimally arranged as UU-N15-U (two regions of riboU spaced 15 nt apart; Deng et al., 1997 ▶). During the addition of ∼30 nt poly(A) tails, monomeric VP55 translocates in a discontinuous fashion with respect to the primer (Yoshizawa et al., 2007 ▶, and references therein) in a manner that requires additional uridylates (Johnson & Gershon, 1999 ▶). To accomplish this, the primer apparently flows around the surface of VP55 from the catalytic center in the central domain to the proximal primer U-binding site in the N-terminal domain and from there to the primer UU-binding site on the opposite face of the N-terminal domain (Li et al., 2009 ▶). Consistent with this, evidence from fluorescence experiments suggests a physical looping of the primer around the end of the N-terminal domain (Li et al., 2009 ▶). In order to bind the VP55–VP39 heterodimer, the minimal primer length increases from ∼34 to ∼44 nt and the two U-rich patches are optimally spaced 10 nt further apart (25 nt total spacing). A key property of the heterodimer is its ability to interact much more strongly than VP55 alone with non-uridylate-containing RNA such as poly(A) (Johnson & Gershon, 1999 ▶). In experiments comparable to those showing VP55 translocation to be discontinuous with respect to the primer and growing poly(A) tail (Yoshizawa et al., 2007 ▶), polyadenylation by the VP55–VP39 heterodimer was found to be continuous (unpublished data). The apparent ability of VP39 to change the mode of translocation of VP55 from discontinuous to continuous raised the question of whether VP39 might, as part of its mechanism for conferring processivity, ‘rigidify’ VP55 and/or re-route the primer around the polymerase to alternative binding sites. An extensive molecular-dynamics simulation and experimental analysis by hydrogen–deuterium exchange mass spectrometry detected widespread but only small-scale (not domain-level) changes in the conformation of VP55 upon VP39 binding (Li et al., 2010 ▶). The absence of evidence for major domain-level conformational remodeling therefore left the ‘rigidification’ hypothesis open.
Understanding the conformational characteristics of VP55 requires atomic-level structures. Although crystal structures have been solved of the VP55–VP39 heterodimer with bound 3′-dATP in the absence (Moure et al., 2006 ▶) and presence (Li et al., 2009 ▶) of RNA primer segments (the two structures were very similar), a missing piece of the puzzle (in determining whether VP39 ‘remodels’ or ‘rigidifies’ VP55) has been the atomic structure of the VP55 monomer owing to our difficulty in obtaining crystals of the latter. In this communication, issues in obtaining well diffracting crystals of VP55 monomer were solved, leading to X-ray crystal structures of monomeric VP55. These structures are described and interpreted.
2. Materials and methods
2.1. Protein purification, crystallization and data collection
6His-VP55 was expressed from plasmid pPG192a in Escherichia coli strain Rosetta(DE3) pLysS as described by Yoshizawa et al. (2007 ▶) and purified from the cell extract using cobalt-chelating chromatography followed by SP Sepharose chromatography (Yoshizawa et al., 2007 ▶). Samples of VP55 (3–5 mg ml−1) were mixed with reservoir solution [12–16%(w/v) polyethylene glycol 3350, 200 mM sodium citrate pH ∼9, 5% glycerol, 1 mM DTT] in a 1:1(v:v) ratio and crystals were allowed to grow via sitting-drop vapor diffusion at 277 K (in a cold room). Rod-like crystals were subjected to cryo-soaks in up to 25%(v/v) glycerol in the reservoir solution followed by flash-cooling in liquid nitrogen. X-ray diffraction data were collected at 100 K using the ADSC Q315 CCD detector on beamline 9-1 of the Stanford Synchrotron Radiation Lightsource (SSRL). Data were collected over a 100° scanning angle for the orthorhombic lattice system. Data frames were integrated and scaled using HKL-2000 (Otwinowski & Minor, 1997 ▶) in space group P2 1212.
2.2. Structure determination and refinement
The search model for molecular-replacement calculations using Phaser (McCoy et al., 2007 ▶) was the VP55 molecule from the previously determined 2.3 Å resolution structure of the heterodimer (PDB entry 2ga9; Moure et al., 2006 ▶) after stripping all nonprotein atoms. Good solutions were found with two molecules of VP55 in the asymmetric unit in space group P21212, with TFZ = 29.6 and LLG = 1571. Coordinates were imported to CNS (Brünger et al., 1998 ▶) for refinement using the simulated-annealing protocol with NCS restraints (weight = 100) imposed between the two copies of the VP55 molecule in the asymmetric unit and this was followed by group B-factor refinement. The resulting 2F o − F c and F o − F c maps were used for model inspection and rebuilding with the graphics programs O (Jones et al., 1991 ▶) or Coot (Emsley et al., 2010 ▶). Progress in model refinement was gauged by the decrease in the free R factor. Towards the later stages of refinement, the NCS restraints for some protein surface regions (e.g. residues 121–133, 328–354 and 449–453) were released to allow the refinement of real differences between the two copies of VP55 in the structure. The final coordinates were validated using the RCSB validation server before deposition. A simulated-annealing OMIT 2F o − F c electron-density map was calculated using the composite_omit_map routine in CNS with a starting temperature of 500 K. The Ramachandran plot created by MolProbity (Chen et al., 2010 ▶) reported only four outliers on the borderline of the allowed regions out of 906 residues evaluated. 810 residues (89.4%) were in favored regions and 902 residues (99.6%) were in allowed regions. The PDB code for this structure is 3owg. Crystallographic data-collection and structure-refinement statistics are summarized in Table 1 ▶.
Table 1. Crystallographic data-collection and refinement statistics for VP55 (PDB entry 3owg).
Values in parentheses are for the last shell.
| Data collection | |
| Space group | P21212 |
| Unit-cell parameters () | a = 80.13, b = 161.29, c = 97.67 |
| Resolution () | 422.86 (2.902.86) |
| R merge | 0.060 (0.580) |
| I/(I) | 22.6 (2.0) |
| No. of unique reflections | 29731 (1448) |
| Completeness (%) | 99.9 (100.0) |
| Multiplicity | 3.7 (3.8) |
| Refinement | |
| Resolution () | 2.86 |
| No. of reflections | 29693 (4153) |
| R work/R free † | 0.253/0.316 |
| No. of atoms | |
| Protein | 7436 |
| Water | 0 |
| Wilson B factor (2) | 90.0 |
| Mean B factor (2) | 97.9 |
| R.m.s. deviations | |
| Bond lengths () | 0.012 |
| Bond angles () | 1.5 |
R free was calculated with 5% of reflections that were set aside randomly.
3. Results and discussion
3.1. Crystal structure of monomeric VP55: overview
Rod-like crystals of monomeric VP55 of dimensions ∼20 × 30 × 300 µm formed over an ∼5 d period at 277 K as described in §2. The resulting crystals belonged to space group P21212. The X-ray crystallographic structure of monomeric VP55 was solved and refined to 2.86 Å resolution (Table 1 ▶) by molecular replacement. The electron density clearly showed the presence of two molecules of VP55 (A and B) in the asymmetric unit (Fig. 1 ▶). Refinement was aided by the imposition of noncrystallographic symmetry (NCS) restraints between molecules A and B, with relaxation of NCS restraints during the later stages of refinement. A cis-proline was introduced at Pro332 to improve the fitting of the model to the density.
Figure 1.

Two VP55 molecules in the asymmetric unit. Green, chain A; orange, chain B. The previously invisible loop (residues 118–129, blue) was visible in both chains. The N- and C-terminal domains are apparent from the residue numbering.
Residues 125–129 were involved in unique and critical crystal-packing contacts between molecules A and B in the asymmetric unit (Fig. 1 ▶) as well as between the space-group symmetry mates. As a result of these contacts, density was visible for the first time for a surface loop (118–129; Fig. 1 ▶) that had been missing from all previous structural models of VP55, namely those of the crystals of the VP55–VP39 heterodimer (Moure et al., 2006 ▶; Li et al., 2009 ▶). This loop, which was visible in both molecules A and B, extends from the N-terminal domain (defined here as residues 12–117) right across the span of the central domain (residues 161–278) and places the final helix of the N-terminal domain of VP55 (residues 130–149, termed the ‘crossover’ helix) against the C-terminal domain of VP55 (Moure et al., 2006 ▶). The portion of the backbone returning from the C-terminal end of the crossover helix to the start of the central domain (residues 150–160) remained missing from the density.
3.2. VP55 conformational flexibility
Considering the three known states for VP55 and VP39, namely the VP39 monomer, the VP55–VP39 heterodimer and the VP55 monomer, the backbone conformation of VP39 has previously been shown to be indistinguishable in liganded or unliganded VP39 monomers (Hodel et al., 1996 ▶, 1997 ▶, 1998 ▶; Hu et al., 2002 ▶; Quiocho et al., 2000 ▶) and in the primer-free or primer-bound forms of the VP55–VP39 heterodimer (Moure et al., 2006 ▶; Li et al., 2009 ▶). Similarly, the backbone conformation of VP55 was indistinguishable in the primer-free (Moure et al., 2006 ▶) and primer-bound (Li et al., 2009 ▶) forms of the heterodimer. However, in the current study, in which the conformation of VP55 in the monomer versus the VP55–VP39 heterodimer could be addressed for the first time, substantial conformational differences were observed. An initial alignment of VP55 monomer molecules A and B on the basis of secondary-structural elements (using the SSM protocol in Coot; Emsley et al., 2010 ▶) highlighted significant differences in the backbone conformation in the N- and C-terminal domains (Fig. 2 ▶). In the central domain, which contains the polymerase catalytic center, molecules A and B exhibited a low r.m.s. deviation (r.m.s.d.; Fig. 2 ▶ b). The largest spikes in r.m.s.d. (Fig. 2 ▶ b) corresponded to the previously missing loop (residues 118–129, above), indicating flexibility in this loop.
Figure 2.

Backbone structural comparison for the two VP55 monomer molecules in the asymmetric unit. (a) Structural overlay: molecule A, cyan; molecule B, magenta. The N-terminal domain lies on the right-hand side and the C-terminal domain lies on the left. The 118–129 loop is at the bottom. (b) R.m.s. deviations for the two molecules. Domain boundaries are indicated by blue vertical lines (the region between the N-terminal and central domains encompasses the crossover helix and two adjoining loops).
Figs. 3 ▶(a) and 3 ▶(b) show least-squares backbone (Cα) superpositions (SSM protocol) of VP55 monomer molecules A and B, respectively, onto the VP55–VP39 heterodimer (coordinates from PDB entry 2ga9; Moure et al., 2006 ▶). R.m.s.d. plots from these superimpositions are shown in Fig. 3 ▶(c). As between the two molecules of VP55 in the asymmetric unit (Fig. 2 ▶ c), the most intense spikes of conformational difference, irrespective of the monomer molecule compared, tended to concentrate in the N- and C-terminal domains (Fig. 3 ▶ c). The three largest spikes were (i) the N-terminal end of helix E (71–84), (ii) the cis-proline in the VP55 monomer structure (Pro332) and (iii) the loop after helix Q (362–374). The tendency for conformational differences to concentrate in the N- and C-terminal domains was consistent with in-solution hydrogen–deuterium exchange (HDX) experiments and Langevin dynamics simulations (Li et al., 2010 ▶), which showed small but widespread differences within the N-terminal and C-terminal domains of VP55 when comparing VP55 with the VP55–VP39 heterodimer.
Figure 3.

Backbone structural comparison of the VP55 monomer with VP55 from the heterodimer. (a) Overlay of VP55 chain A (magenta) on PDB entry 2ga9 (green). (b) Overlay of VP55 chain B (yellow) on PDB entry 2ga9 (green). The N-terminal domain of VP55 is shown on the right-hand side, the C-terminal domain is shown on the left and the loop (118–129) is at the bottom. (c) R.m.s. deviations: VP55 chain A versus PDB entry 2ga9, red; VP55 chain B versus PDB entry 2ga9, black.
Conformational differences were best illustrated by aligning just the N-terminal domains (residues 12–117) of VP55 molecules A and B with coordinates from the 3.21 Å resolution structure of the heterodimer (PDB entry 3erc, chain C; Li et al., 2009 ▶; Fig. 3 ▶, Table 2 ▶). Molecule B was a relatively close structural mimic of VP55 in the heterodimer. Molecule A, however, showed deviations as large as 6.5 Å at C-terminal domain residue Ser253 (Table 2 ▶). While the r.m.s.d. between N-terminally aligned molecules A and B was ∼4.1–4.2 Å greater at either of two selected points in the C-terminal domain than at two points in the N-terminal domain (Table 2 ▶), indicating a conformational difference, their direct intramolecular N-terminal to C-terminal domain distances (Met33–Asp372) differed by just 1.6 Å (see note in Table 2 ▶), indicating that the C-terminal domain does not hinge directly towards the N-terminal domain. Rather, the C-terminal domain ‘rocks’ in and out of the page in the orientation shown in Fig. 4 ▶(a) (left and right in the orientation shown in Fig. 4 ▶ b). Docking of VP39 creates a series of protein–protein contacts from a helical turn of VP39 (residues 52–55) to the loop around Asp376 of the C-terminal domain of VP55 (Fig. 4 ▶). These contacts tighten the ‘closed’ conformation of VP55 (‘molecule B-like’). In the absence of these VP39–VP55 contacts, VP55 seems to be more able to adopt alternative conformations.
Table 2. N-terminal domain alignment of the VP55VP39 heterodimer (RNA-bound form, PDB entry 3erc, chain C; Li et al., 2009 ▶) and monomeric VP55 molecules A or B: r.m.s.d. at two selected points in the C-terminal domains.
By subtraction of the tabulated intermolecular values, the r.m.s.d. between VP55 molecules A and B was 4.14.2 for the C-terminal domain. The intramolecular N-terminal to C-terminal domain distances, Met33Asp372, were 43.3 (3erc, chain C), 44.9 (VP55 chain A) and 43.9 (VP55 chain B).
Figure 4.

Overlay of chain A in the VP55 monomer (magenta) and chain C in the VP39–VP55 heterodimer (PDB entry 3erc; cyan) based on a superposition of the N-terminal domain (residues 12–117) only. VP39, as it would be docked in the heterodimer, is shown as a gray space-filling model. In (a) the N-terminal domain is on the right while the C-terminal domain is on the left. (b) is a view rotated 90° with respect to (a) such that the C-terminal domain of VP55 is facing the viewer. The residues discussed in the text are labeled. The ‘rocking’ motion of the C-terminal domain of VP55 is exemplified by the large r.m.s.d. in Cα positions of Ser353 and Asp393 (Table 2 ▶). Although molecules A and B are both shown in the context of VP39 to highlight changes to the VP39-binding site, we have no evidence that VP39 can bind VP55 in the conformation of molecule A; in fact, we suggest that it does not.
The conclusion that VP55 is relatively flexible in the absence of VP39, enabling a rocking motion of the C-terminal domain relative to the N-terminal domain, would be prima facie consistent with the experimentally observed saltatory motion of the VP55 monomer during ∼30 nt oligo(A) tail addition (Yoshizawa et al., 2007 ▶; Johnson et al., 2004 ▶; Deng & Gershon, 1997 ▶; Gershon, 1998 ▶). However, with all known primer-binding determinants being located in the central and N-terminal domains (Li et al., 2009 ▶) and no known functional determinants yet detected in the C-terminal domain of VP55, an inchworming translocation mechanism based upon the rocking motion would not be straightforward to visualize. Moreover, the magnitude of the rocking motion (accommodating an ∼2–3 nucleotide maximum step size) would be inadequate to account for the 10–13-nucleotide distances between the binding determinants in the primer that participate in the major protein–primer isomerization steps accompanying the synthesis of an ∼30 nt oligo(A) tail (Yoshizawa et al., 2007 ▶).
Among the best-characterized protein monomers that translocate along ssNA are the ATP-dependent molecular motors of the SF1/SF2 helicase/translocase superfamilies (Singleton et al., 2007 ▶, and references therein). These ATP-dependent ratchets have been proposed to move along ssNA in an inchworming fashion with step sizes of ∼1–5 nt for SF1 motors, proposed on the basis of kinetic studies (Ali & Lohman, 1997 ▶; Dillingham et al., 2000 ▶; Singleton et al., 2007 ▶), and step sizes as large as 18 nt for the SF2 helicase NS3 (Dumont et al., 2006 ▶; Levin et al., 2004 ▶; Serebrov et al., 2009 ▶). However, the apparent absence of a crystal structure for any single SF1 or SF2 helicase core in both open and closed conformations renders more precise estimation of their step sizes difficult (Singleton et al., 2007 ▶), not to mention a more precise definition of conformational states matching their proposed tracking movements on ssNA. While rigid-body domain-level conformational differences have been observed between crystallographic structures of either the same or structurally related helicases (Singleton et al., 2007 ▶), these differences seem to be comparatively small and insufficient in size to allow multi-nucleotide steps of ∼10 Å or greater. Indeed, rigid-body domain movements of 10 Å or greater have only rarely been observed in the crystal structure of any nucleic acid-binding protein (Håkansson et al., 1997 ▶). It therefore seems to some extent to be a matter of speculation how monomeric helicases can incur large step sizes via the mechanisms proposed.
For VP55, inferences can be drawn: the central domain of VP55, in which the catalytic center is located (and from which the primer 3′ end is extended), is likely to move in lockstep with the C-terminal domain, with hinging primarily affecting the N-terminal domain alone. The rocking motion observed in the current study could provide a partial correlate to the major isomerization steps known to occur during the translocation of VP55 (Yoshizawa et al., 2007 ▶). Assuming that this rocking motion represents the maximum dynamic range of VP55 and assuming that it is involved in translocation, then one possibility might be that the ‘closing’ motions of the N-terminal domain represent a ‘shuffling’ of nascent nucleic acid in small (∼1–3 nt) steps from the catalytic center to an as-yet undetected (sequence-nonspecific) site on the N-terminal domain closer to the catalytic center than the U-binding site that initially interacts with position −10 in the primer. As nascent nucleic acid accumulates immediately upstream of the putative sequence-nonspecific site, larger and less frequent steps would occur at the U-binding sites, corresponding to the previously characterized periodic isomerizations of the VP55–primer complex (Yoshizawa et al., 2007 ▶). Understanding the precise role of the rocking motion of VP55 awaits further experimentation.
The flexibility of the VP55 monomer may be consistent with the observation of crystal cracking during soaks of VP55 monomer crystals with either ATP alone or ATP plus metal ions, hinting that ATP binding may be one of the causes of conformational changes that are not tolerated within the packed apo crystals generated here. An alternative explanation might be slow unprimed poly(A) synthesis within the crystal. The relative inflexibility of monomeric VP39, and of both VP55 and VP39 within the heterodimer (above), suggest a role for VP39 in the ‘rigidification’ of VP55 during the processive generation of tails hundreds of adenylates in length (see §1).
The schematic in Fig. 5 ▶ depicts secondary-structural elements that ‘cross over’ between domains, which may play a role in controlling the conformational flexibility of VP55. One of these is the ‘crossover helix’ of the N-terminal domain (above), the motions of which are likely to match those of the C-terminal domain. The two loops flanking the crossover helix may guide the rocking motion of the C-terminal domain, much like the reins of a horse. One of the two loops (118–129), which is visible for the first time in the monomer structure (the other, 150–160, remained invisible), may have been insufficiently ‘tensioned’ in the heterodimer crystals for visualization. An additional ‘crossover’ element of VP55 that may play a role in regulating the conformational register of the N-terminal and C-terminal domains of VP55 is the extreme C-terminal β-hairpin (residues 469–475), which crosses back to the N-terminal domain (12–117; Fig. 5 ▶). Nonetheless, no dramatic differences in the relationship of the β-hairpin to the N-terminal domain were observed between molecules A and B or between these molecules and the heterodimer conformation: residue Asp475 within the β-hairpin forms a weak salt bridge with the N-terminal domain residue Lys84 in both the monomer and heterodimer structures, and the main-chain C=O of residue Lys474 forms a weak hydrogen bond to the side chain of Lys93 in all structures.
Figure 5.
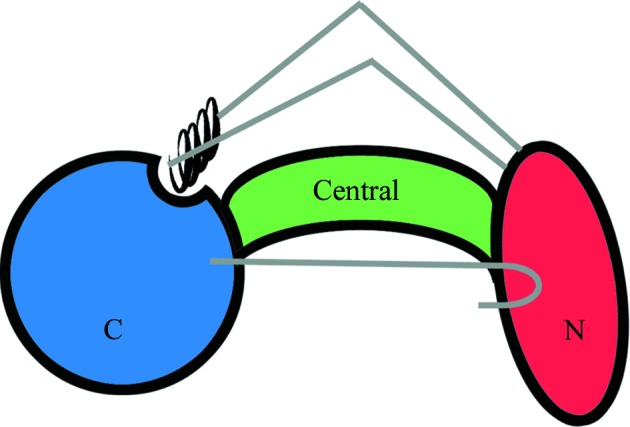
Schematic depicting the secondary-structural features of VP55 that ‘cross over’ between domains. The black coil represents the ‘crossover helix’ (see text). Loops 118–129 and 150–160, which connect the crossover helix to the N-terminal domain (see text), are colored gray. Also shown in gray (sideways ‘hockey-stick’ shape) is the extreme C-terminal β-hairpin of VP55, which makes tertiary contacts to the N-terminal domain of VP55.
3.3. Primer binding
The binding sites for U and UU within the primer, based around VP55 residues Ile476 and Phe47, respectively (Li et al., 2009 ▶), both lie within the N-terminal domain of VP55 (Li et al., 2009 ▶). The N-terminal domain was virtually identical in conformation in the VP55 monomer and heterodimer. The only tangible difference in the vicinity of the UU–Phe47 primer contact site was in the side chain of VP55 residue Lys43 (Fig. 6 ▶), which hydrogen bonds to the primer in the primer-bound heterodimer structure (Li et al., 2009 ▶). The Lys43 side chain appeared to extend further out in the structure of the unliganded monomer. The side chains of Ile51, Leu55 and Tyr108 showed possible minor conformational differences, although at 2.86 Å resolution, and given the marginal quality of electron density for these side chains in the monomer structure these differences remain unconfirmed. Despite similarities in the N-terminal domain, the VP55 monomer and the VP55–VP39 heterodimer differ profoundly in their primer-binding properties. Notably, the optimal distance between the proximal U and distal UU of the primer is 10 nt greater in the heterodimer than in the VP55 monomer (Johnson & Gershon, 1999 ▶; Deng et al., 1999 ▶). Since the maximum ‘rocking’ distance in VP55 is 6.5 Å (above), a distance that is inadequate to absorb 10 nt of additional ssNA, a more likely mechanism for absorption of the longer primer would seem to be primer re-routing in the heterodimer.
Figure 6.
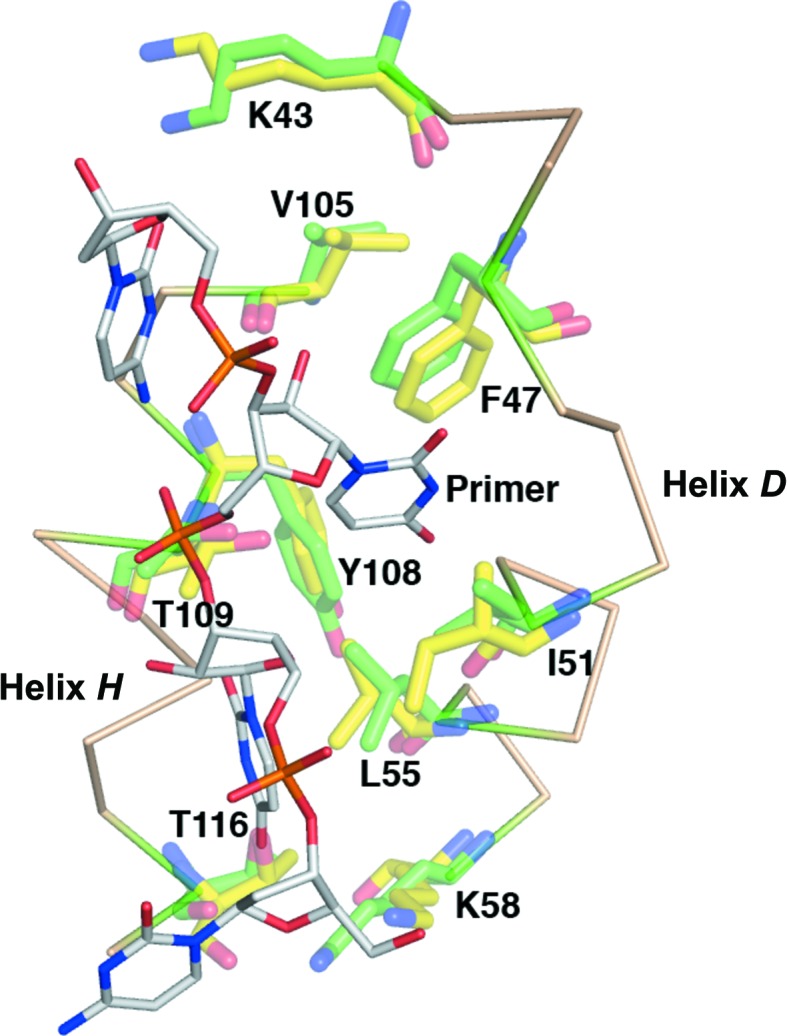
The overlaid primer UU-binding sites of the VP55 monomer (yellow) and heterodimer (green). The primer-binding site is flanked by N-terminal domain helices D and H. Primer-contacting side chains are shown.
3.4. Catalytic center
The current structures are the first not only of monomeric VP55, but also of VP55 in the absence of bound ATP or analogs thereof. The polymerase catalytic center of VP55 is located in a narrow cleft between the C-terminal domain and the β-sheet of the central domain, on the opposite side of the central β-sheet from the VP39 docking cleft described above. In overlays of the VP55 monomer with the 3′-dATP-bound VP55–VP39 heterodimer (PDB entry 3erc, chain C) no backbone conformational changes larger than 1.0 Å were detected within or immediately adjacent to the catalytic center. The relative stability of the central domain is also reflected in the B factors, which are lower for the central domain of VP55 than for the N- and C-terminal domains (Fig. 7 ▶). Nonetheless, the cleft that binds the extreme 3′ end of the primer as it extends away from the catalytic center appears to be slightly narrower in the ATP-bound heterodimer owing to side-chain adjustments and slight backbone changes on the C-terminal side of the cleft (Fig. 8 ▶). Correspondingly, small but significant backbone differences are clearly manifest in the Cα positions of catalytic center residues lying to the C-terminal side of the central domain boundary (e.g. Arg294, Lys304 and Arg308; Fig. 8 ▶). Adjacent to the triphosphate moiety of ATP, the side chains of Arg294 and Lys304 were swung away in the absence of ATP, while the side chains of both Ser188 and Arg308 protruded into the ATP-binding site. These differences are considered to be meaningful even in the context of an averaged coordinate error of as high as ∼0.5 Å for this 2.86 Å resolution structure. Additional slight rotations of a few aromatic side chains (e.g. Tyr186, Tyr240 and Phe255) were also noticeable along the primer-binding cleft at the catalytic center (Fig. 8 ▶). The side-chain orientation and position of Tyr236 and His467 in the primer-free VP55 monomer (Fig. 8 ▶) would need to make substantial adjustments in order to achieve the stacking interactions observed in the heterodimer structure with primer present (Li et al., 2009 ▶). Of the three Asp residues that are responsible for metal binding (Asp202, Asp204 and Asp253), Asp202 adopted a different rotamer in the VP55 monomer in the absence of metal ions. Overall, whether or not ATP and primer binding trigger significant backbone conformational change in VP55, side-chain adjustments appear to be necessary for optimal ligand contacts.
Figure 7.
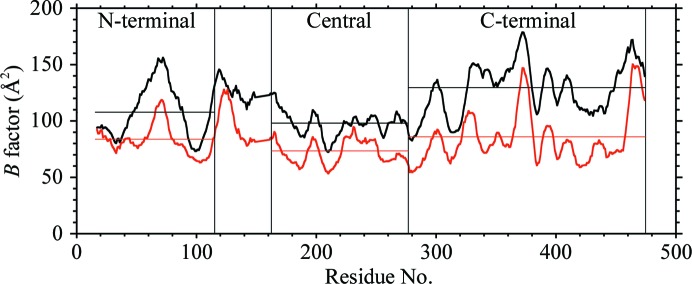
11-residue rolling average B factor for VP55 molecules A (black) and B (red). Molecule B shows less thermal motion than molecule A. The N-terminal, central and C-terminal domains of VP55 are indicated. The region between the N-terminal and central domains encompasses the crossover helix and two adjoining loops.
Figure 8.

The overlaid catalytic center of VP55 in the VP55 monomer and the VP39–VP55 heterodimer based on a superposition of the central domain (residues 161–278). The residues (as well as 3′-dATP and the RNA primer) from the 3′-dATP/RNA-bound heterodimer (PDB entry 3erc, chain C) are colored gray, while those from chain B in the VP55 monomer are green. The 2F o − F c OMIT map for the VP55 monomer is shown at the 1σ contour level. Residues discussed in the text are labeled.
In conclusion, an X-ray crystallographic structure has been generated for unliganded VP55 monomer, showing the first domain-level structural isoforms among either VP55 poly(A) polymerase, its processivity factor (VP39), or the VP55-VP39 heterodimer. The occurrence of domain-level motion specifically in monomeric VP55 is consistent with the finding that the monomeric protein undergoes saltatory translocation whereas the heterodimer does not.
Supplementary Material
PDB reference: VP55, 3owg
Acknowledgments
This work was supported by NIH grant GM51953.
References
- Ali, J. A. & Lohman, T. M. (1997). Science, 275, 377–380. [DOI] [PubMed]
- Brünger, A. T., Adams, P. D., Clore, G. M., DeLano, W. L., Gros, P., Grosse-Kunstleve, R. W., Jiang, J.-S., Kuszewski, J., Nilges, M., Pannu, N. S., Read, R. J., Rice, L. M., Simonson, T. & Warren, G. L. (1998). Acta Cryst. D54, 905–921. [DOI] [PubMed]
- Chen, V. B., Arendall, W. B., Headd, J. J., Keedy, D. A., Immormino, R. M., Kapral, G. J., Murray, L. W., Richardson, J. S. & Richardson, D. C. (2010). Acta Cryst. D66, 12–21. [DOI] [PMC free article] [PubMed]
- Deng, L., Beigelman, L., Matulic-Adamic, J., Karpeisky, A. & Gershon, P. D. (1997). J. Biol. Chem. 272, 31542–31552. [DOI] [PubMed]
- Deng, L. & Gershon, P. D. (1997). EMBO J. 16, 1103–1113. [DOI] [PMC free article] [PubMed]
- Deng, L., Johnson, L., Neveu, J. M., Hardin, S., Wang, S.-M., Lane, W. S. & Gershon, P. D. (1999). J. Mol. Biol. 285, 1417–1427. [DOI] [PubMed]
- Dillingham, M. S., Wigley, D. B. & Webb, M. R. (2000). Biochemistry, 39, 205–212. [DOI] [PubMed]
- Dumont, S., Cheng, W., Serebrov, V., Beran, R. K., Tinoco, I., Pyle, A. M. & Bustamante, C. (2006). Nature (London), 439, 105–108. [DOI] [PMC free article] [PubMed]
- Edmonds, M. (1982). The Enzymes, edited by P. D. Boyer, pp. 217–224. New York: Academic Press.
- Edmonds, M. (1990). Methods Enzymol. 181, 161–170. [DOI] [PubMed]
- Edmonds, M. (2002). Prog. Nucleic Acid Res. Mol. Biol. 71, 285–389. [DOI] [PubMed]
- Emsley, P., Lohkamp, B., Scott, W. G. & Cowtan, K. (2010). Acta Cryst. D66, 486–501. [DOI] [PMC free article] [PubMed]
- Gershon, P. D. (1998). Semin. Virol. 8, 343–350.
- Gershon, P. D., Ahn, B.-Y., Garfield, M. & Moss, B. (1991). Cell, 66, 1269–1278. [DOI] [PubMed]
- Gershon, P. D. & Moss, B. (1992). Genes Dev. 6, 1575–1586. [DOI] [PubMed]
- Gershon, P. D. & Moss, B. (1993). J. Biol. Chem. 268, 2203–2210. [PubMed]
- Håkansson, K., Doherty, A. J., Shuman, S. & Wigley, D. B. (1997). Cell, 89, 545–553. [DOI] [PubMed]
- Hodel, A. E., Gershon, P. D. & Quiocho, F. A. (1998). Mol. Cell, 1, 443–447. [DOI] [PubMed]
- Hodel, A. E., Gershon, P. D., Shi, X. & Quiocho, F. A. (1996). Cell, 85, 247–256. [DOI] [PubMed]
- Hodel, A. E., Gershon, P. D., Shi, X., Wang, S.-M. & Quiocho, F. A. (1997). Nature Struct. Biol. 4, 350–354. [DOI] [PubMed]
- Hu, G., Oguro, A., Li, C., Gershon, P. D. & Quiocho, F. A. (2002). Biochemistry, 41, 7677–7687. [DOI] [PubMed]
- Johnson, L. & Gershon, P. D. (1999). Nucleic Acids Res. 27, 2708–2721. [DOI] [PMC free article] [PubMed]
- Johnson, L., Liu, S. & Gershon, P. D. (2004). J. Mol. Biol. 337, 843–856. [DOI] [PubMed]
- Jones, T. A., Zou, J.-Y., Cowan, S. W. & Kjeldgaard, M. (1991). Acta Cryst. A47, 110–119. [DOI] [PubMed]
- Levin, M. K., Wang, Y.-H. & Patel, S. S. (2004). J. Biol. Chem. 279, 26005–26012. [DOI] [PubMed]
- Li, C.-Z., Koter, M., Ye, X., Zhou, S.-F., Chou, W., Luo, R. & Gershon, P. D. (2010). Biochemistry, 49, 6247–6262. [DOI] [PubMed]
- Li, C.-Z., Li, H., Zhou, S.-F., Sun, E., Yoshizawa, J., Poulos, T. L. & Gershon, P. D. (2009). Structure, 17, 680–689. [DOI] [PubMed]
- McCoy, A. J., Grosse-Kunstleve, R. W., Adams, P. D., Winn, M. D., Storoni, L. C. & Read, R. J. (2007). J. Appl. Cryst. 40, 658–674. [DOI] [PMC free article] [PubMed]
- Moure, C. M., Bowman, B. R., Gershon, P. D. & Quiocho, F. A. (2006). Mol. Cell, 33, 339–349. [DOI] [PubMed]
- Otwinowski, Z. & Minor, W. (1997). Methods Enzymol. 276, 307–326. [DOI] [PubMed]
- Quiocho, F. A., Hu, G. & Gershon, P. D. (2000). Curr. Opin. Struct. Biol. 10, 78–86. [DOI] [PubMed]
- Serebrov, V., Beran, R. K. & Pyle, A. M. (2009). J. Biol. Chem. 284, 2512–2521. [DOI] [PMC free article] [PubMed]
- Singleton, M. R., Dillingham, M. S. & Wigley, D. B. (2007). Annu. Rev. Biochem. 76, 23–50. [DOI] [PubMed]
- Yoshizawa, J. M., Li, C. & Gershon, P. D. (2007). J. Biol. Chem. 282, 19144–19151. [DOI] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
PDB reference: VP55, 3owg