

Key Points
The increased expression of PI3K p110α in mantle cell lymphoma, particularly at relapse, suggests a role for p110α in disease progression.
A high PIK3CA/PIK3CD ratio identifies patients unlikely to respond to p110δ inhibitors and supports use of dual p110α/p110δ inhibitors in MCL.
Abstract
Phosphoinositide-3 kinase (PI3K) pathway activation contributes to mantle cell lymphoma (MCL) pathogenesis, but early-phase studies of the PI3K p110δ inhibitor GS-1101 have reported inferior responses in MCL compared with other non-Hodgkin lymphomas. Because the relative importance of the class IA PI3K isoforms p110α, p110β, and p110δ in MCL is not clear, we studied expression of these isoforms and assessed their contribution to PI3K signaling in this disease. We found that although p110δ was highly expressed in MCL, p110α showed wide variation and expression increased significantly with relapse. Loss of phosphatase and tensin homolog expression was found in 16% (22/138) of cases, whereas PIK3CA and PIK3R1 mutations were absent. Although p110δ inhibition was sufficient to block B-cell receptor–mediated PI3K activation, combined p110α and p110δ inhibition was necessary to abolish constitutive PI3K activation. In addition, GDC-0941, a predominantly p110α/δ inhibitor, was significantly more active compared with GS-1101 against MCL cell lines and primary samples. We found that a high PIK3CA/PIK3CD ratio identified a subset of primary MCLs resistant to GS-1101 and this ratio increased significantly with relapse. These findings support the use of dual p110α/p110δ inhibitors in MCL and suggest a role for p110α in disease progression.
Introduction
Mantle cell lymphoma (MCL) is an aggressive disease in the vast majority of patients and is incurable with conventional therapy. Although there has been an improvement in median overall survival (OS), from the 2- to 4-year range cited in earlier series to between 5 and 7 years more recently,1 outcome is still one of the poorest among B-cell lymphomas. MCL is characterized by t(11;14), which results in juxtaposition of the IgH enhancer on chromosome 14 to the cyclin D1 locus on chromosome 11, leading to the characteristic overexpression of cyclin D1. Secondary hits primarily leading to defective DNA damage repair and cell -cycle dysregulation occur in MCL,2 and a number of studies have implicated activation of the phosphoinositide-3 kinase (PI3K) pathway, one of the most commonly dysregulated pathways in human cancer, in the pathogenesis of this disease.3-5 The serine-threonine kinase AKT, which is the major downstream target of PI3K, is thought to be important in MCL survival through its role in stabilizing cyclin D1 messenger RNA (mRNA), preventing nuclear export of cyclin D1 by phosphorylation of GSK-3b and increasing cyclin D1 translation through mammalian target of the rapamycin (mTOR) activation.6-8
PI3Ks are heterodimeric lipid kinases that have a regulatory and a catalytic subunit. Class IA PI3Ks primarily signal downstream of the B-cell receptor (BCR) and tyrosine kinase receptors to mediate downstream effects that lead to increased cell metabolism, proliferation, and survival. They have 3 catalytic subunit isoforms—p110α, p110β, and p110δ (encoded by PIK3CA, PIK3CB, and PIK3CD, respectively)—that dimerize with a p85 regulatory subunit. The p85 subunits (p85α, p85β, p55γ, p55α, and p50α) recognize phosphorylated tyrosine motifs.9 These motifs are found in the intracellular domains of CD19 and BCAP (the B-cell adaptor for PI3K) and are phosphorylated upon BCR stimulation via phosphorylation of Syk and Lyn.10 On binding to these sites, the p110 catalytic subunits are activated and convert cell membrane–bound PIP2 (phosphatidylinositol 4,5 biphosphate) to the important second messenger PIP3 (phosphatidylinositol 3,4,5 triphosphate). PIP3 binds and activates proteins that have a pleckstrin homology domain, such as AKT and PDK1. Full activation of Akt requires PI3K-induced phosphorylation at threonine 308 and mTOR complex 2–induced phosphorylation at serine 473. PTEN (phosphatase and tensin homolog) is a lipid phosphatase that opposes activation of this pathway by converting PIP3 back to PIP2.11
The p110δ isoform is a key messenger in BCR signaling and is highly enriched in leukocytes,12,13 making it an attractive target in B-cell malignancies. However, in addition to quantitative isoform expression, the mechanism of PI3K activation may predict sensitivity to isoform selective inhibition. P110α (PIK3CA) is the only PI3K catalytic unit isoform that harbors cancer-associated somatic mutations and these occur at a high frequency in solid tumors.14 PIK3CA mutations have not been found in 2 separate studies of MCL primary samples, but interestingly, gene amplification of PIK3CA related to increased copy number has been described in this disease.3,15 Loss of PTEN expression is another mechanism leading to constitutive activation of the PI3K pathway, and studies in solid tumors have demonstrated a key role for p110β in PTEN-deficient tumors.16-18 Loss of PTEN expression has been described in approximately 15% of MCLs.15 Other mechanisms of PTEN inactivation that have been suggested in MCL include phosphorylation of PTEN and negative regulation by the microRNA-17–92 cluster.4,19 More recently, activation of all 3 class IA isoforms has been described in association with somatic mutations in the gene encoding the regulatory p85α subunit (PIK3R1) in solid tumors.20 These mutations have not been studied in MCL.
Preclinical studies have demonstrated the potential of inhibiting the PI3K pathway in MCL,3,4,15,21 but early-phase studies of the first p110δ selective inhibitor, GS-1101, demonstrated more modest responses in patients with MCL compared with the impressive results seen in indolent non-Hodgkin lymphoma (NHL) and chronic lymphocytic leukemia.22 Because loss of PTEN expression (∼15%) and gene amplification of p110α (∼68%) have been described in MCL,15 understanding the relative contribution of the class IA isoforms in this disease may help in targeting this important oncogenic pathway more effectively.
We therefore studied the expression of class IA isoforms in MCL and evaluated their contribution to PI3K signaling in relation to BCR activation, loss of PTEN expression, and increased PIK3CA expression. We demonstrate that although p110δ remains highly expressed in MCL, tumor cells with increased PIK3CA expression can sustain constitutive PI3K signaling despite p110δ inhibition. Further, a PIK3CA/PIK3CD ratio greater than twice that in healthy B-cell controls identified primary MCL cases that were resistant to p110δ inhibition but significantly more sensitive to GDC-0941, a p110α/δ inhibitor in vitro. We also demonstrate a significant increase in both p110α expression and the PIK3CA/PIK3CD ratio with MCL progression.
Materials and methods
Cell lines
Granta519 and Jeko-1 MCL cell lines were used after confirmation of their identity by short tandem repeat profiling (LGC standards, Teddington, UK). Jeko-1 was cultured in RPMI (Sigma, St. Louis, MO) and Granta519 in Dulbecco’s modified Eagle medium (Sigma). Both were supplemented with 10% heat-inactivated FCS (Sigma) and 1% gentamicin (GIBCO, Life Technologies, Paisley, UK).
Patient samples
In accordance with the updated Declaration of Helsinki, all samples were obtained following ethical approval, and after informed consent from patients treated at St Bartholomew’s hospital. Solid tissue used in tissue microarray construction was fixed in formalin-fixed paraffin-embedded (FFPE) tissue. Snap-frozen tissue was evaluated for tumor content using CD20 staining of sections and homogenized using the Qiagen TissueLyserII (Qiagen, Hilden, Germany) for DNA and RNA extraction. Mononuclear cells from peripheral blood (PBMCs), bone marrow, and spleen-derived cell suspensions were isolated using Ficoll-paque density gradient centrifugation, and 22 primary MCL cell suspensions confirmed to have greater than 85% CD20-positive cells by flow cytometry were used in experiments. Clinical details of these primary samples are listed in supplemental Table 1. Cell suspensions were cultured in Iscove modified Dulbecco medium (Sigma) supplemented with 10% human serum, 1% gentamicin, 5 µg/mL bovine insulin, 50 µg/mL human transferrin, and 1 mM sodium pyruvate. PBMCs for control B cells were obtained from healthy volunteers and tonsil controls were obtained from tonsillectomies performed for nonmalignant pathology.
Antibodies and reagents
Primary antibodies for western blotting against p110α (#4249), glyceraldehyde-3-phosphate dehydrogenase (GAPDH, #2118), total Syk (#2712), phospho-Syk thr525/526 (#2710), total Akt (#9272), phospho-Akt ser473 and thr308 (#9271, #2965), phospho-GSK3b ser9 (#9323), total ribosomal S6 (#2217), and phospho-S6 ser235/ser236 (#2211) were purchased from Cell Signaling Technologies (Danvers, MA). Antibodies against p110β (sc-602) and p110δ (sc-55589) were purchased from Santa Cruz Biotechnology (Santa Cruz, CA) and anti-human PTEN (ABM-2052) from Cascade Bioscience (Winchester, MA). Goat anti-human immunoglobulin (Ig)M F(ab′)2 fragments were purchased from Southern Biotech (Cambridge, UK). GDC-0941,23 A66,24 and TGX22125 were purchased from Selleck Chem (Houston, TX) and CAL-101/GS-1101 from Active Biochem (Maplewood, NJ). For cytotoxicity studies, cells were treated in triplicate with increasing concentrations of inhibitor (0.1-10 μM) for 72 hours while an inhibitor concentration of 1 μM was used to assess downstream effects by western blotting.
Immunohistochemistry
Tissue microarrays (TMA) were constructed using triplicate 1-mm cores of FFPE tissue. Clinical details of samples included in the tissue microarray are listed in supplemental Table 2. Although antibodies against p110α, p110β, and PTEN have previously been validated on FFPE tissue,26 there was no previous literature describing the use of a p110δ-selective antibody for immunohistochemistry (IHC) at the time of performing these experiments. We therefore constructed a cell block microarray with high and low p110δ–expressing cell lines to optimize and validate this antibody (supplemental Figure 1). Details of antibodies used, concentrations, and antigen retrieval methods are listed in Table 1. All slides were scanned using an Olympus scanning microscope to obtain high-resolution images that were analyzed on Ariol SL-50 version 3.2 (Genetix, San Jose, CA) visual analysis software. Briefly, images of cores were screened individually to exclude nontumor tissue and the software was trained to calculate the percentage of positive cells and staining intensity of cores. Biopsies with only 1 evaluable core on the TMA were excluded and IHC scores for duplicates and triplicates were averaged. The IHC score was calculated as the product of percentage positive cells and mean intensity. PTEN expression was scored independently by 2 authors (S.I. and A.L.) as 2+ (strong), 1+ (moderate) or 0 (no expression) based on expression in tumor cells compared with blood vessels or nontumor cells in the TMA cores.
Table 1.
Details of primary antibodies used for immunohistochemistry
| Primary antibody | Supplier/code | Species (clone) | Antigen retrieval | Concentration | Incubation |
|---|---|---|---|---|---|
| P110α | Cell Signaling#4249 | Rabbit polyclonal(C73F8) | Citrate pH6 | 1:250 | 40 seconds |
| P110β | Abcamab55593 | Mouse monoclonal | Citrate pH6 | 1:250 | 40 seconds |
| P110δ | Santa Cruzsc-55589 | Mouse monoclonal (A-8) | Citrate pH6 | 1:500 | 40 seconds |
| PTEN | CascadeABM-2052 | Mouse monoclonal (6H2.1) | Citrate pH6 | 1:25 | 60 seconds |
Western blotting
Briefly, cell lysates were prepared by resuspending cell pellets in lysis buffer containing protease and phosphatase inhibitors (Roche, Basel, Switzerland). Total protein was estimated using the Pierce BCA protein assay kit (Thermo Scientific, Waltham, MA). Lysates were resolved on a NuPAGE 4% to 12% gel (Invitrogen, Life Technologies, Paisley, UK) and transferred onto a polyvinylidene difluoride membrane using the iBlot dry transfer method (Invitrogen). Membranes were incubated with primary antibody either overnight at 4°C or at room temperature for 2 hours at the recommended dilutions. Membranes were then washed and incubated with horseradish peroxidase-labeled secondary antibody (Dako, Denmark) for an hour at room temperature. Electrochemiluminescence reagent (GE Health Care, Uppsala, Sweden) was applied to visualize the blots using the Fuji LAS 2000 digital imager (Fujifilm, Japan).
Quantitative real-time PCR
Total RNA was extracted from cell suspensions and frozen tissue using the RNeasy mini kit (Qiagen, Hilden, Germany). A total of 2 µg RNA was reverse transcribed using the high-capacity reverse transcription kit (Applied Biosystems, Life Technologies, Paisley, UK) as per manufacturer’s recommendations. Quantitative real-time polymerase chain reaction (qRT-PCR) was performed on the ABI HT-7900 instrument (Applied Biosystems) using Taqman gene expression assays (Applied Biosystems) for PIK3CA (Hs00192399_m1), PIK3CB (Hs00927728_m1), and PIK3CD (Hs00180679_m1). Data were analyzed using SDS2.1 software and relative expression values were calculated using the ΔCt method with GAPDH (Hs99999905_m1) as endogenous control. All reactions were run in triplicate.
PIK3CA and PIK3R1 mutation analysis
Genomic DNA was extracted from frozen lymph nodes (verified to have >80% tumor content) and PBMCs of MCL patients using the DNeasy mini kit (Qiagen). Primers used for PCR amplification of PIK3CA exons 9 and 20 and PIK3R1 exons 9, 10, 11, 13, 15, and 16 are listed in supplemental Table 3. After DNA purification, bidirectional Sanger sequencing was performed on the purified PCR products.
Cytotoxicity assays
Growth inhibition was measured with the Guava ViaCount assay (Millipore, Billerica, MA). The ViaCount reagent was added to cells treated in triplicate in a 96-well plate, and cell count and viability was determined on a Guava express plus instrument (Millipore). The adenosine triphosphate (ATP) cytotoxicity assay kit (Lonza, Basel, Switzerland) was used to measure the cytotoxic effects of PI3K inhibitors on primary MCL cell suspensions. Briefly, cells were plated in triplicate in a 96-well format and treated with PI3K inhibitors at increasing concentrations before measurement of ATP luminescence in a plate reader at 72 hours.
Statistical analysis
Prism version 5.03 (GraphPad, La Jolla, CA) was used for statistical analysis. Normally distributed data sets were tested with paired or unpaired t tests, as appropriate, whereas for 3 data sets, 1-way analysis of variance followed by a Bonferroni multiple comparison post-hoc analysis was used. Data sets that were not normally distributed were analyzed using the Mann-Whitney test for unpaired samples or the Wilcoxon matched-pairs signed-rank test for paired samples. A P value below .05 was considered significant.
Results
P110α expression in MCL is significantly higher beyond first relapse
Expression of p110α, β, and δ was evaluated by IHC analysis of 144 evaluable biopsies from 109 MCL patients. P110δ was consistently expressed at high levels in MCL. P110β expression was the weakest, whereas p110α showed a wide variation in expression across biopsies (Figure 1A). Median expression of p110α (median IHC score = 44.5 vs 33.7, P = .15) and p110δ (median IHC scores = 77.4 vs 81.1, P = .2) in MCL was not different from that seen in the germinal center area of tonsil controls, whereas P110β expression was significantly lower in MCL (P = .01). As shown in Figure 1B, p110α expression was significantly higher in biopsies taken beyond first relapse compared to those taken at diagnosis (P = .04). These differences were even more striking in sequential biopsies (P = .008) with 5 of 6 lymphoma samples showing increased p110α expression beyond first relapse (Figure 1B-D). Expression of p110α beyond first relapse was also significantly higher than expression in tonsil controls (P = .024). No significant change in expression of p110β or p110δ was seen with relapse. We also did not find a significant difference in expression of the individual isoforms between blastoid and classical MCL (data not shown).
Figure 1.
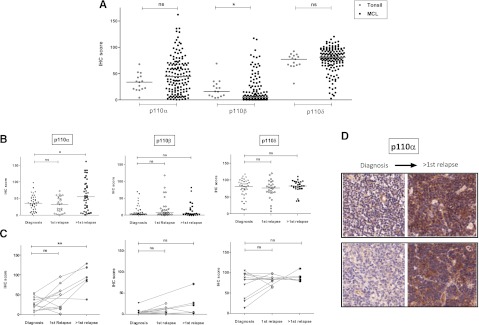
IHC expression of class IA PI3K isoforms in mantle cell lymphoma. (A) Dot plot showing expression of class IA isoforms in MCL biopsies (144 biopsies evaluable for all 3 isoforms from 109 patients) compared with tonsil controls (n = 14). Each dot represents the average IHC score of triplicate cores and bars represent median expression. P110δ is highly expressed in MCL and p110α shows a wide range of expression, whereas p110β expression is the weakest. (B) Dot plots showing significant increase in p110α expression, but not p110β or p110δ, beyond first relapse. (C) This finding is more striking in 12 sequential cases, 6 of whom had biopsies beyond first relapse (connected by gray lines). (D) Representative IHC images (original magnification ×200) of sequential biopsies from 2 patients showing a significant increase in p110α expression with relapse *P < .05, **P < .01. ns, not significant.
Loss of PTEN expression is relatively common in MCL, whereas PIK3CA and PIK3R1 mutations are rare or absent
We assessed PTEN protein expression by IHC (Figure 2). In keeping with a previous report,15 we found loss of PTEN expression in 17% of diagnostic (6/35) and 16% of all (22/138) biopsies (Figure 2C). A higher proportion of tumors exhibiting blastoid morphology had loss of PTEN expression compared to cases with classical histology, but this was not statistically significant (20% vs 15%, P = .55 by Fisher’s exact test). No significant change was found in the pattern of isoform expression, particularly for p110β, in biopsies that had loss of PTEN expression (Figure 2D). Loss of PTEN expression was seen by western blotting in 2 of 12 primary MCL samples (Figure 2E), and the results seen on western blotting corresponded to those seen by IHC. Unlike reports in solid tumors,16 we did not find significant additional cytotoxicity in these 2 samples using the combination of GS-1101 and TGX221 (p110β selective inhibitor) compared to GS-1101 alone (Figure 2F). Exons 9 and 20 of PIK3CA and exons 9, 11, 12, 13, 15, and 16 of PIK3R1 were sequenced in a total of 20 primary MCL samples and 2 cell lines (Granta519, Jeko-1); no mutations were found, suggesting these are rare or do not occur in MCL.
Figure 2.

Loss of PTEN expression in MCL. (A) Core from a PTEN-null adenocarcinoma control showing PTEN-negative tumor islands surrounded by PTEN-positive stroma by IHC (original magnifications ×50 and ×200). (B) Representative images of MCL cores with and without loss of PTEN expression (original magnifications ×50 and ×200). Macrophages and blood vessels were used as internal controls. (C) Pie chart showing the proportion of all biopsies with PTEN loss and blastoid morphology accompanied by a bar graph of distribution of PTEN loss in blastoid and nonblastoid MCL. (D) Dot plot comparing p110β expression levels between cores with and without loss of PTEN expression showing no difference among the 3 groups (PTEN−, PTEN 1+, and PTEN2+). There was also no significant difference in expression of p110α and δ between these groups (data not shown). (E) Western blot for PTEN expression in 12 MCL cell suspensions showing loss of expression in 2 samples (indicated by arrows). (F) Results of ATP cytotoxicity assay after 72 hours’ treatment showing no benefit from addition of a p110β-selective inhibitor to GS-1101 in 2 MCL suspensions exhibiting loss of PTEN expression.
P110δ inhibition prevents BCR-induced PI3K activation in MCL, whereas additional p110α inhibition is required to abolish constitutive PI3K activation
The Jeko-1 MCL cell line exhibits low or undetectable levels of p-Akt and was therefore used to study the effect of isoform selective inhibition on agonist-induced BCR signaling. Whereas the p110δ inhibitor GS-1101 prevented phosphorylation of Akt (thr308) in response to BCR stimulation with IgM F(ab′)2 fragments, the p110α inhibitor A6624 and the p110β inhibitor TGX22125 had minimal effects on p-Akt (thr308) (Figure 3A). We then studied the effect of isoform-selective inhibition on the Granta519 MCL cell line, which exhibits constitutive Akt phosphorylation in association with increased p110α expression as a result of increased PIK3CA copy number and gene expression15 (Figure 3B). Interestingly, GS-1101 (1 µM) was unable to abolish Akt phosphorylation in Granta519, whereas at equimolar concentrations GDC-0941, a predominantly p110α/δ inhibitor, blocked Akt phosphorylation in a sustained manner over 24 hours. This was reflected in reduced downstream phosphorylation of ribosomal S6 as a result of mTOR inhibition (Figure 3C). To establish that this effect on constitutive PI3K activation was related to activity of the p110α isoform, we combined GS-1101 separately with the p110α inhibitor A66 and a p110β inhibitor TGX221 and tested these combinations along with GS-1101 and GDC-0941 alone in serum-starved Granta519 cells. Combining GS-1101 with A66 had a similar effect as GDC-0941, whereas combining GS-1101 with TGX221 made no significant difference, confirming that residual constitutive PI3K signaling in GS-1101–treated cells is attributable to p110α (Figure 3D).
Figure 3.

Role of class IA isoforms in BCR-induced and constitutive PI3K signaling. (A) Jeko-1 cells were pretreated for an hour with isoform-selective inhibitors (1 µM) as indicated, followed by IgM stimulation with 10 µg/mL anti-human IgM F(ab') fragments for 10 minutes. Phospho-Akt (thr308) levels were compared by western blotting with nonstimulated and IgM-stimulated Jeko-1 controls. P-Syk (tyr525/526) was used as a marker of BCR activation. GS-1101 (p110δ-selective) blocked p-Akt production, whereas A66 (p110α-selective) and TGX-221 (p110β-selective) did not. (B) Real-time PCR and western blotting confirming increased expression of PIK3CA and p110α in Granta519 MCL cell lines compared with the lymphoblastoid cell line NcNc. (C) Western blot comparing downstream effects of GS-1101 (1 μM) and GDC-0941 (1 μM), at 2 time points demonstrating incomplete and nonsustained effect of GS-1101 on p-Akt, p-GSK3β, and p-S6. (D) Western blots were performed with serum-starved (4 hours) Granta519 cells treated with GS-1101, GDC-0941, and combinations of GS-1101 with A66 or TGX-221 (all 1 µM), for 2 hours, confirming the effect of p110α on constitutive PI3K activation. (E) Comparison of 50% inhibition/inhibitory concentration of GS-1101 and GDC-0941 for the 4 class I isoforms. GS-1101 is highly p110δ-selective, whereas p110α is predominantly p110α/δ-selective. The activity of both inhibitors against p110γ is comparable. (F) Western blot showing expression of p-Akt (t308) and the class IA catalytic unit isoforms in Jeko-1 and Granta519. (G) Greater growth inhibition is seen in Granta519 and Jeko-1 with GDC-0941 compared with GS-1101, whereas A66 has a minimal effect. (H) Dot plots showing greater cytotoxicity (ATP assay) with GDC-0941 compared with GS-1101 in 12 primary MCL samples with significant toxicity at and above 0.1 µM. (I) Western blot showing p-Akt (thr308) expression in the same 12 MCL cell suspensions. (J) Comparative cytotoxicity (ATP assay) of GDC-0941 and GS-1101 in 3 healthy B cells showing somewhat greater, but not statistically significant, cytotoxicity at all concentrations with GDC-0941. *P < .05, **P < .01.
PI3K inhibition with GDC-0941 results in greater cytotoxicity to MCL cells compared with GS-1101
We compared the cytotoxicity of GS-1101 and GDC-0941 on growth and survival of MCL cell lines and primary samples. GDC-0941 induced significantly higher growth inhibition at 72 hours in 2 MCL cell lines with intact PTEN expression (Granta519 and Jeko-1) compared with GS-1101, whereas the p110α-selective inhibitor A66 had a minimal effect on both cell lines (Figure 3F-G). Using an ATP cytotoxicity assay, a similar significant effect was observed on treating 12 primary MCL samples (Figure 3H) and GDC-0941 was significantly more cytotoxic than GS-1101 above concentrations of 0.1 µM (P = .046 at 0.1 µM, .008 at 1 µM, .005 at 5 µM, and .035 at 10 µM). Basal phospho-Akt levels were not predictive of sensitivity to either inhibitor in both cell lines and primary samples. We also treated B cells isolated from 3 healthy donors with GS-1101 and GDC-0941 and found no statistically significant difference in cytotoxicity even at higher concentrations.
A high PIK3CA/PIK3CD mRNA ratio can predict resistance to p110δ-selective inhibition
Our findings in the Granta519 MCL cell line led us to hypothesize that gene expression of the PI3K isoforms in MCL cells can predict sensitivity to isoform selective inhibition. We measured PIK3CA, PIK3CB, PIK3CD, and GAPDH mRNA levels by qRT-PCR in the same 12 MCL cell suspensions that we previously treated with GS-1101 and GDC-0941 and compared them with healthy B-cell controls. PIK3CD was highly expressed with gene expression of both PIK3CA and PIK3CD significantly up-regulated compared with healthy B-cell controls. PIK3CB showed the weakest expression (Figure 4A). Neither PIK3CA nor PIK3CD levels predicted sensitivity or resistance to the inhibitors independently. However, by plotting the ratio of PIK3CA to PIK3CD (PIK3CA/PIK3CD), we were able to separate the samples into 2 groups—1 with ratios similar to healthy B cells that had similar responses to GS-1101 and GDC-0941 and 1 with significantly higher expression. PIK3CA/PIK3CD greater than twice that seen in healthy B-cell controls identified a group that was resistant to GS-1101 and showed a significantly greater response to GDC-0941 (P = .03 at 0.1 µM, P = .008 at 1 µM) (Figure 4B). The 2 samples that were most sensitive to GS-1101 reassuringly fell in the group with the lower ratios. These differences were seen at 0.1 and 1 µM, concentrations at which the drugs are likely to be most selective for their targets. We validated our findings in a second independent cohort of 10 primary MCL samples using 1 µM GS-1101 and GDC-0941 (Figure 4C). We also treated these 10 samples with the p110α inhibitor A66 (1 µM) alone but found no significant cytotoxicity in any of the samples treated (supplemental Figure 2). As may be expected, we found some primary samples, mainly in the low-ratio group, that were relatively resistant to both GS-1101 and GDC-0941 at these lower concentrations, even when phosphorylation of Akt was blocked, suggesting alternative survival pathways were active in these tumors (data not shown).
Figure 4.

The PIK3CA/PIK3CD mRNA ratio can predict resistance to GS-1101. (A) Gene expression of class IA PI3K isoforms, relative to GAPDH, in 12 diagnostic MCL PBMCs and 3 healthy B-cell controls, showing significant increase in PIK3CA and PIK3CD compared with controls (gray) and very low levels of PIK3CB. (B) A PIK3CA/PIK3CD greater than twice the mean ratio in healthy B cells identifies a subset of primary samples (5/12) that are resistant to 0.1 µM and 1 µM GS-1101 (GS), but show significantly higher toxicity with equimolar concentrations of GDC-0941 (GDC). (C) This cutoff, when applied to an independent validation cohort (n = 10), was able to identify GS-1101–resistant samples (primary samples treated with 1 μM of GS-1101 and GDC-0941).
A high PIK3CA/PIK3CD mRNA ratio is frequently associated with MCL progression
To investigate whether disease progression was associated with a change in PIK3CA/PIK3CD, qRT-PCR was performed on RNA extracted from frozen serial biopsies from 4 MCL patients and 2 tonsil controls. Protein was extracted simultaneously for western blotting from these biopsies. Whereas PIK3CD was highly expressed and up-regulated compared with tonsil controls (Figure 5A), PIK3CA/PIK3CD was higher in all 4 cases at relapse compared with diagnosis (Figure 5C). Compared with PBMCs, PIK3CA expression, and as a result PIK3CA/PIK3CD, was relatively lower in lymph nodes. However, protein expression by western blotting showed upregulation of p110α compared with tonsil controls and also showed a marked increase with relapse in 2 of 4 serial biopsies (Figure 5B).
Figure 5.

PIK3CA/PIK3CD increases with relapse in MCL. (A) Gene expression of the PI3K class IA isoforms compared with 2 tonsil controls (gray). Serial biopsies from the same patient are in the same color (B) western blot showing very weak expression of p110β and a clear increase in MCL p110α compared with tonsil while p110δ protein expression in MCL is similar to tonsil controls. Increase in p110α with relapse is apparent in 2 out of 4 serial biopsies (C) Dot plot showing a significantly higher PIK3CA/PIK3CD ratio in all relapsed samples compared with matched diagnostic samples and tonsil controls.
To investigate whether differences existed in isoform expression between MCL and other NHL, we extracted PI3K class IA isoform expression data from publicly available gene expression profiling (www.ebi.ac.uk) from a study comparing diagnostic samples of MCL to indolent NHL.27 Even in these diagnostic samples, median expression of only the PIK3CA isoform, and therefore PIK3CA/PIK3CD, was significantly higher in MCL compared with chronic lymphocytic leukemia and indolent NHL (Figure 6A). There was no publicly available data comparing gene expression in relapsed lymphomas that we could analyze at the time of this study, but we performed immunohistochemistry for p110α in sequential biopsies from 10 follicular lymphoma patients, 4 of whom had paired biopsies beyond first relapse, and did not find evidence of increase in expression with relapse (Figure 6B-C).
Figure 6.
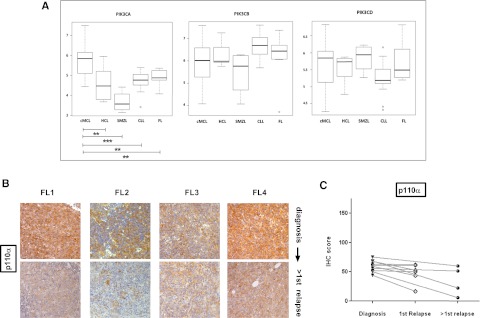
P110α expression does not increase with relapse in follicular lymphoma and gene expression of PIK3CA is significantly higher in MCL compared with indolent NHL and CLL. (A) Gene expression of class IA PI3K isoforms at diagnosis in MCL was compared with indolent NHL using publicly available Affymetrix data (European Bioinformatics Institute, www.ebi.ac.uk ref: E-GEOD-16455) with the web-based software O-miner. cMCL, conventional MCL, CLL, chronic lymphocytic leukemia, FL, follicular lymphoma; HCL, hairy cell leukemia, SMZL, splenic marginal zone lymphoma. PIK3CA expression is significantly higher in MCL compared with indolent NHLs (Benjamini-Hochberg multiple testing adjusted P values: cMCL vs HCL = .005, cMCL vs SMZL = .0001 cMCL vs CLL = .003, cMCl vs FL = .05) in this study, whereas PIK3CB and PIK3CD expression is not. (*P < .05, **P < .01, ***P < .001). (B) IHC images of 4 follicular lymphoma diagnosis: second-relapse pairs showing no increase in p110α expression with relapse (original magnification ×200). (C) Dot plot comparing expression of p110α in sequential biopsies from follicular lymphoma patients (n = 10). No evidence of increased p110α expression is seen with relapse. Pairs are connected by straight lines.
Discussion
Several studies have described the importance of the PI3K pathway in the pathogenesis of MCL.3-5 The relatively inferior effects seen in MCL in early-phase trials of the PI3K p110δ selective inhibitor GS-1101 emphasize the need to study the contribution of the other class IA PI3K isoforms to MCL survival.
In keeping with previous observations,3,15 we did not find PIK3CA mutations in 20 MCL primaries, and PTEN loss occurred at a frequency of about 16%, with an apparently, but not significantly, higher incidence in blastoid MCL. In addition, activating PIK3R1 mutations were shown to be rare or absent in MCL. Although gene amplification of PIK3CA related to increased copy number has been previously described in MCL, we demonstrate for the first time that a high PIK3CA/PIK3CD mRNA ratio can predict resistance to p110δ-selective inhibition and is frequent with relapse. The mechanism for this increase of expression with relapse is unclear. Our observation that the increase in p110α expression is particularly significant beyond first relapse may imply a chemotherapy-induced phenomenon or selection and expansion of a chemotherapy-resistant clone with high PIK3CA expression, but this needs further study.
We demonstrate that although p110δ continues to be the predominant isoform in terms of expression and agonist-induced BCR signaling in MCL, an increase in p110α expression can maintain constitutive PI3K signaling in MCL cells despite p110δ inhibition. We explored the option of an small interfering RNA study to demonstrate the reliance of Granta519 on p110α but it has been found that knocking down the expression of any 1 of the class I PI3K isoforms can result in a compensatory increase or decrease in activity of other isoforms and lead to artifacts.28-31 We therefore elected to use a highly selective p110α inhibitor, A66, for our studies.24 Studies in healthy mouse lymphocytes have demonstrated that although PI3K p110δ plays a predominant role in both agonist-dependent and agonist-independent BCR signaling, p110α contributes to agonist-independent signaling.32 The increased expression of p110α in MCL, particularly with relapse, may therefore allow tumor cells to survive independent of agonist-induced activation of the PI3K pathway. Of note, a similar phenomenon has been observed in immortalized macrophages, where p110α takes on a more prominent role in PI3K signaling, in contrast to primary macrophages where p110δ is the predominantly active isoform.33 It is possible that PI3K inhibitors may also have an effect on the tumor microenvironment; this issue is not explored here. However, the clear effect in MCL cell lines and primary MCL cells in the absence of the microenvironment, together with the relative increase of PIK3CA in MCL cells themselves, strongly suggest that this is likely to result in a differential effect with greater toxicity to the tumor cells compared with an effect on the microenvironment.
P110β shows the weakest expression at both the RNA and protein level in MCL and we found no additional effect on PI3K-induced Akt phosphorylation on combining a highly selective p110β inhibitor (TGX-221) with GS-1101. We also demonstrate that, unlike observations in solid tumors,16 there is a lack of additional cytotoxicity from combining TGX-221 and GS-1101 in MCL samples that exhibit loss of PTEN expression, but this requires confirmation on a larger number of samples. GDC-0941, an inhibitor with high selectivity for p110α and p110δ, was significantly more cytotoxic in both MCL cell lines and primary samples. Although inhibiting additional PI3K isoforms is likely to be associated with greater toxicity, this inhibitor has shown favorable toxicity profiles in early-phase clinical trials in solid tumors.34 Our findings are unlikely to be influenced by p110γ, the only class IB catalytic unit isoform, because both GS-1101 and GDC-0941 have similar efficacy against p110γ (Figure 3E). The greater contribution of p110α to PI3K signaling with MCL relapse can explain the relatively modest effects seen in relapsed/refractory MCL patients recruited into early-phase trials of GS-1101.
We propose that PIK3CA/PIK3CD is a potential biomarker that can identify patients who are resistant to p110δ inhibition and may benefit most from combined p110α/δ inhibitor therapy. This ratio may identify tumors that can use p110α-mediated constitutive PI3K signaling for microenvironment-independent survival. Our data also suggest that Akt phosphorylation, when present, is a useful marker to study responses to PI3K inhibition but absence of phosphorylation does not preclude sensitivity to PI3K inhibition. Also, we did not find higher Akt phosphorylation in MCL exhibiting increased PIK3CA/PIK3CD. This is not entirely surprising because, for instance, Akt-independent PI3K signaling has been found in cancers harboring PIK3CA mutations35,36 and a similar mechanism could operate in MCLs that have a high PIK3CA/PIK3CD ratio.
In summary, our findings suggest that MCL requires blockade of both p110α and p110δ, particularly at relapse, for effective PI3K inhibition. This will require testing in a clinical trial, but the assessment of the relative gene expression of these isoforms may help identify those patients that are most likely to respond to these exciting agents.
Acknowledgments
The authors thank Bart Vanhaesebroeck, Ezra Aksoy, the European Hematology Association (EHA)-ASH Translational Research Training in Hematology faculty, particularly Donna Neuberg, David Williams, and Linda Burns, for their valuable advice and suggestions. The authors also thank Jacek Marzec for bioinformatics, John Riches and Eleni Kiotsiu for providing healthy B cells, and all the patients at Barts Cancer Centre who kindly consented to provide samples for this research.
This work was supported by grants from the David Pitblado Foundation, National Cancer Institute (P01 CA81538) (J.G.), Cancer Research UK (C1574/A6806), and an EHA Partner Fellowship awarded by the European Hematology Association (2009/01) (C.B.).
Footnotes
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Authorship
Contribution: S.I., J.G., and S.J. designed experiments; S.I., A.C., and C.B. performed the experiments; R.A., L.M., S.I., and J.M. provided essential reagents, clinical samples, and related clinical information; M.C. and A.L. assisted with tissue microarray construction and immunohistochemistry scoring. S.I. prepared the manuscript with input from all authors.
Conflict-of-interest disclosure: J.G. and R.A. have received honoraria from Gilead for attendance at advisory boards. J.G. has received honoraria from Roche for attendance at advisory boards. The remaining authors declare no competing financial interests.
Correspondence: John Gribben, Centre for Haemato-Oncology, Barts Cancer Institute, London EC1M 6BQ, UK; e-mail: j.gribben@qmul.ac.uk.
References
- 1.Martin P, Coleman M, Leonard JP. Progress in mantle-cell lymphoma. J Clin Oncol. 2009;27(4):481-483. [DOI] [PubMed]
- 2.Fernandez V, Hartmann E, Ott G, Campo E, Rosenwald A. Pathogenesis of mantle-cell lymphoma: all oncogenic roads lead to dysregulation of cell cycle and DNA damage response pathways. J Clin Oncol. 2005;23(26):6364-6369. [DOI] [PubMed]
- 3.Rudelius M, Pittaluga S, Nishizuka S, et al. Constitutive activation of Akt contributes to the pathogenesis and survival of mantle cell lymphoma. Blood. 2006;108(5):1668–1676. doi: 10.1182/blood-2006-04-015586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Dal Col J, Zancai P, Terrin L, et al. Distinct functional significance of Akt and mTOR constitutive activation in mantle cell lymphoma. Blood. 2008;111(10):5142–5151. doi: 10.1182/blood-2007-07-103481. [DOI] [PubMed] [Google Scholar]
- 5.Rizzatti EG, Falcão RP, Panepucci RA, et al. Gene expression profiling of mantle cell lymphoma cells reveals aberrant expression of genes from the PI3K-AKT, WNT and TGFbeta signalling pathways. Br J Haematol. 2005;130(4):516–526. doi: 10.1111/j.1365-2141.2005.05630.x. [DOI] [PubMed] [Google Scholar]
- 6.Prasad A, Park IW, Allen H, et al. Styryl sulfonyl compounds inhibit translation of cyclin D1 in mantle cell lymphoma cells. Oncogene. 2009;28(12):1518–1528. doi: 10.1038/onc.2008.502. [DOI] [PubMed] [Google Scholar]
- 7.Dal Col J, Dolcetti R. GSK-3beta inhibition: at the crossroad between Akt and mTOR constitutive activation to enhance cyclin D1 protein stability in mantle cell lymphoma. Cell Cycle. 2008;7(18):2813–2816. doi: 10.4161/cc.7.18.6733. [DOI] [PubMed] [Google Scholar]
- 8.Pérez-Galán P, Dreyling M, Wiestner A. Mantle cell lymphoma: biology, pathogenesis, and the molecular basis of treatment in the genomic era. Blood. 2011;117(1):26–38. doi: 10.1182/blood-2010-04-189977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Vanhaesebroeck B, Waterfield MD. Signaling by distinct classes of phosphoinositide 3-kinases. Exp Cell Res. 1999;253(1):239–254. doi: 10.1006/excr.1999.4701. [DOI] [PubMed] [Google Scholar]
- 10.Okkenhaug K, Vanhaesebroeck B. PI3K in lymphocyte development, differentiation and activation. Nat Rev Immunol. 2003;3(4):317–330. doi: 10.1038/nri1056. [DOI] [PubMed] [Google Scholar]
- 11.Vanhaesebroeck B, Stephens L, Hawkins P. PI3K signalling: the path to discovery and understanding. Nat Rev Mol Cell Biol. 2012;13(3):195–203. doi: 10.1038/nrm3290. [DOI] [PubMed] [Google Scholar]
- 12.Vanhaesebroeck B, Welham MJ, Kotani K, et al. P110delta, a novel phosphoinositide 3-kinase in leukocytes. Proc Natl Acad Sci USA. 1997;94(9):4330–4335. doi: 10.1073/pnas.94.9.4330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bilancio A, Okkenhaug K, Camps M, et al. Key role of the p110delta isoform of PI3K in B-cell antigen and IL-4 receptor signaling: comparative analysis of genetic and pharmacologic interference with p110delta function in B cells. Blood. 2006;107(2):642–650. doi: 10.1182/blood-2005-07-3041. [DOI] [PubMed] [Google Scholar]
- 14.Samuels Y, Wang Z, Bardelli A, et al. High frequency of mutations of the PIK3CA gene in human cancers. Science. 2004;304(5670):554. doi: 10.1126/science.1096502. [DOI] [PubMed] [Google Scholar]
- 15.Psyrri A, Papageorgiou S, Liakata E, et al. Phosphatidylinositol 3′-kinase catalytic subunit alpha gene amplification contributes to the pathogenesis of mantle cell lymphoma. Clin Cancer Res. 2009;15(18):5724-5732. [DOI] [PubMed]
- 16.Wee S, Wiederschain D, Maira SM, et al. PTEN-deficient cancers depend on PIK3CB. Proc Natl Acad Sci USA. 2008;105(35):13057–13062. doi: 10.1073/pnas.0802655105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Edgar KA, Wallin JJ, Berry M, et al. Isoform-specific phosphoinositide 3-kinase inhibitors exert distinct effects in solid tumors. Cancer Res. 2010;70(3):1164–1172. doi: 10.1158/0008-5472.CAN-09-2525. [DOI] [PubMed] [Google Scholar]
- 18.Jia S, Liu Z, Zhang S, et al. Essential roles of PI(3)K-p110beta in cell growth, metabolism and tumorigenesis. Nature. 2008;454(7205):776–779. doi: 10.1038/nature07091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Rao E, Jiang C, Ji M, et al. The miRNA-17 approximately 92 cluster mediates chemoresistance and enhances tumor growth in mantle cell lymphoma via PI3K/AKT pathway activation. Leukemia. 2012;26(5):1064-1072. [DOI] [PubMed]
- 20.Jaiswal BS, Janakiraman V, Kljavin NM, et al. Somatic mutations in p85alpha promote tumorigenesis through class IA PI3K activation. Cancer Cell. 2009;16(6):463–474. doi: 10.1016/j.ccr.2009.10.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lannutti BJ, Meadows SA, Herman SE, et al. CAL-101, a p110delta selective phosphatidylinositol-3-kinase inhibitor for the treatment of B-cell malignancies, inhibits PI3K signaling and cellular viability. Blood. 2011;117(2):591–594. doi: 10.1182/blood-2010-03-275305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kahl B, Byrd JC, Flinn IW, et al. Clinical safety and activity in a phase 1 study of CAL-101, an isoform-selective inhibitor of phosphatidylinositol 3-kinase P110δ, in patients with relapsed or refractory non-Hodgkin lymphoma [abstract]. Blood (ASH Annual Meeting Abstracts). 2010;116(21):1777. [Google Scholar]
- 23.Folkes AJ, Ahmadi K, Alderton WK, et al. The identification of 2-(1H-indazol-4-yl)-6-(4-methanesulfonyl-piperazin-1-ylmethyl)-4-morpholin-4-yl-thieno[3,2-d]pyrimidine (GDC-0941) as a potent, selective, orally bioavailable inhibitor of class I PI3 kinase for the treatment of cancer. J Med Chem. 2008;51(18):5522–5532. doi: 10.1021/jm800295d. [DOI] [PubMed] [Google Scholar]
- 24.Jamieson S, Flanagan JU, Kolekar S, et al. A drug targeting only p110α can block phosphoinositide 3-kinase signalling and tumour growth in certain cell types. Biochem J. 2011;438(1):53–62. doi: 10.1042/BJ20110502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jackson SP, Schoenwaelder SM, Goncalves I, et al. PI 3-kinase p110beta: a new target for antithrombotic therapy. Nat Med. 2005;11(5):507–514. doi: 10.1038/nm1232. [DOI] [PubMed] [Google Scholar]
- 26.Carvalho S, Milanezi F, Costa JL. Amendoeira I, Schmitt F. PIKing the right isoform: the emergent role of the p110beta subunit in breast cancer. Virchows Arch. 2010;456(3):235-243. [DOI] [PubMed]
- 27.Fernàndez V, Salamero O, Espinet B, et al. Genomic and gene expression profiling defines indolent forms of mantle cell lymphoma. Cancer Res. 2010;70(4):1408–1418. doi: 10.1158/0008-5472.CAN-09-3419. [DOI] [PubMed] [Google Scholar]
- 28.Vanhaesebroeck B, Ali K, Bilancio A, et al. Signalling by PI3K isoforms: insights from gene-targeted mice. Trends Biochem Sci. 2005;30(4):194–204. doi: 10.1016/j.tibs.2005.02.008. [DOI] [PubMed] [Google Scholar]
- 29.Vanhaesebroeck B, Rohn JL, Waterfield MD. Gene targeting: attention to detail. Cell. 2004;118(3):274–276. doi: 10.1016/j.cell.2004.07.018. [DOI] [PubMed] [Google Scholar]
- 30.Vanhaesebroeck B, Guillermet-Guibert J, Graupera M, et al. The emerging mechanisms of isoform-specific PI3K signalling. Nat Rev Mol Cell Biol. 2010;11(5):329–341. doi: 10.1038/nrm2882. [DOI] [PubMed] [Google Scholar]
- 31.Geering B, Cutillas PR, Nock G, et al. Class IA phosphoinositide 3-kinases are obligate p85-p110 heterodimers. Proc Natl Acad Sci USA. 2007;104(19):7809–7814. doi: 10.1073/pnas.0700373104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ramadani F, Bolland DJ, Garcon F, et al. The PI3K isoforms p110alpha and p110delta are essential for pre-B cell receptor signaling and B cell development. Sci Signal. 2010;3(134):ra60. doi: 10.1126/scisignal.2001104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Papakonstanti EA, Zwaenepoel O, Bilancio A, et al. Distinct roles of class IA PI3K isoforms in primary and immortalised macrophages. J Cell Sci. 2008;121(Pt 24):4124–4133. doi: 10.1242/jcs.032763. [DOI] [PubMed] [Google Scholar]
- 34.Wagner AJ, Von Hoff DH, LoRusso PM, et al. A first-in-human phase I study to evaluate the pan-PI3K inhibitor GDC-0941 administered QD or BID in patients with advanced solid tumors [abstract]. J Clin Oncol. 2010;28(15s) suppl. Abstract 2541. [Google Scholar]
- 35.Vasudevan KM, Barbie DA, Davies MA, et al. AKT-independent signaling downstream of oncogenic PIK3CA mutations in human cancer. Cancer Cell. 2009;16(1):21–32. doi: 10.1016/j.ccr.2009.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Morrow CJ, Gray A, Dive C. Comparison of phosphatidylinositol-3-kinase signalling within a panel of human colorectal cancer cell lines with mutant or wild-type PIK3CA. FEBS Lett. 2005;579(23):5123–5128. doi: 10.1016/j.febslet.2005.07.096. [DOI] [PubMed] [Google Scholar]