

Abstract
Long-range migrations and the resulting admixtures between populations have been important forces shaping human genetic diversity. Most existing methods for detecting and reconstructing historical admixture events are based on allele frequency divergences or patterns of ancestry segments in chromosomes of admixed individuals. An emerging new approach harnesses the exponential decay of admixture-induced linkage disequilibrium (LD) as a function of genetic distance. Here, we comprehensively develop LD-based inference into a versatile tool for investigating admixture. We present a new weighted LD statistic that can be used to infer mixture proportions as well as dates with fewer constraints on reference populations than previous methods. We define an LD-based three-population test for admixture and identify scenarios in which it can detect admixture events that previous formal tests cannot. We further show that we can uncover phylogenetic relationships among populations by comparing weighted LD curves obtained using a suite of references. Finally, we describe several improvements to the computation and fitting of weighted LD curves that greatly increase the robustness and speed of the calculations. We implement all of these advances in a software package, ALDER, which we validate in simulations and apply to test for admixture among all populations from the Human Genome Diversity Project (HGDP), highlighting insights into the admixture history of Central African Pygmies, Sardinians, and Japanese.
Keywords: admixture, linkage disequilibrium
ADMIXTURE between previously diverged populations has been a common feature throughout the evolution of modern humans and has left significant genetic traces in contemporary populations (Li et al. 2008; Wall et al. 2009; Reich et al. 2009; Green et al. 2010; Gravel et al. 2011; Pugach et al. 2011; Patterson et al. 2012). Resulting patterns of variation can provide information about migrations, demographic histories, and natural selection and can also be a valuable tool for association mapping of disease genes in admixed populations (Patterson et al. 2004).
Recently, a variety of methods have been developed to harness large-scale genotype data to infer admixture events in the history of sampled populations, as well as to estimate a range of gene flow parameters, including ages, proportions, and sources. Some of the most popular approaches, such as STRUCTURE (Pritchard et al. 2000) and principal component analysis (PCA) (Patterson et al. 2006), use clustering algorithms to identify admixed populations as intermediates in relation to surrogate ancestral populations. In a somewhat similar vein, local ancestry inference methods (Tang et al. 2006; Sankararaman et al. 2008; Price et al. 2009; Lawson et al. 2012) analyze chromosomes of admixed individuals with the goal of recovering continuous blocks inherited directly from each ancestral population. Because recombination breaks down ancestry tracts through successive generations, the time of admixture can be inferred from the tract length distribution (Pool and Nielsen 2009; Pugach et al. 2011; Gravel 2012), with the caveat that accurate local ancestry inference becomes difficult when tracts are short or the reference populations used are highly diverged from the true mixing populations.
A third class of methods makes use of allele frequency differentiation among populations to deduce the presence of admixture and estimate parameters, either with likelihood-based models (Chikhi et al. 2001; Wang 2003; Sousa et al. 2009; Wall et al. 2009; Laval et al. 2010; Gravel et al. 2011) or with phylogenetic trees built by taking moments of the site-frequency spectrum over large sets of SNPs (Reich et al. 2009; Green et al. 2010; Patterson et al. 2012; Pickrell and Pritchard 2012; Lipson et al. 2012). For example, f-statistic-based three- and four-population tests for admixture (Reich et al. 2009; Green et al. 2010; Patterson et al. 2012) are highly sensitive in the proper parameter regimes and when the set of sampled populations sufficiently represents the phylogeny. One disadvantage of drift-based statistics, however, is that because the rate of genetic drift depends on population size, these methods do not allow for inference of the time that has elapsed since admixture events.
Finally, Moorjani et al. (2011) recently proposed a fourth approach, using associations between pairs of loci to make inference about admixture, which we further develop in this article. In general, linkage disequilibrium (LD) in a population can be generated by selection, genetic drift, or population structure, and it is eroded by recombination. Within a homogeneous population, steady-state neutral LD is maintained by the balance of drift and recombination, typically becoming negligible in humans at distances of more than a few hundred kilobases (Reich et al. 2001; International HapMap Consortium 2007). Even if a population is currently well mixed, however, it can retain longer-range admixture LD (ALD) from admixture events in its history involving previously separated populations. ALD is caused by associations between nearby loci co-inherited on an intact chromosomal block from one of the ancestral mixing populations (Chakraborty and Weiss 1988). Recombination breaks down these associations, leaving a signature of the time elapsed since admixture that can be probed by aggregating pairwise LD measurements through an appropriate weighting scheme; the resulting weighted LD curve (as a function of genetic distance) exhibits an exponential decay with rate constant giving the age of admixture (Moorjani et al. 2011; Patterson et al. 2012). This approach to admixture dating is similar in spirit to strategies based on local ancestry, but LD statistics have the advantage of a simple mathematical form that facilitates error analysis.
In this article, we comprehensively develop LD-based admixture inference, extending the methodology to several novel applications that constitute a versatile set of tools for investigating admixture. We first propose a cleaner functional form of the underlying weighted LD statistic and provide a precise mathematical development of its properties. As an immediate result of this theory, we observe that our new weighted LD statistic can be used to infer mixture proportions as well as dates, extending the results of Pickrell et al. (2012). Moreover, such inference can still be performed (albeit with reduced power) when data are available from only the admixed population and one surrogate ancestral population, whereas all previous techniques require at least two such reference populations. As a second application, we present an LD-based three-population test for admixture with sensitivity complementary to the three-population f-statistic test (Reich et al. 2009; Patterson et al. 2012) and characterize the scenarios in which each is advantageous. We further show that phylogenetic relationships between true mixing populations and present-day references can be inferred by comparing weighted LD curves using weights derived from a suite of reference populations. Finally, we describe several improvements to the computation and fitting of weighted LD curves: we show how to detect confounding LD from sources other than admixture, improving the robustness of our methods in the presence of such effects, and we present a novel fast Fourier transform-based algorithm for weighted LD computation that reduces typical run times from hours to seconds. We implement all of these advances in a software package, ALDER (Admixture-induced Linkage Disequilibrium for Evolutionary Relationships).
We demonstrate the performance of ALDER by using it to test for admixture among all HGDP populations (Li et al. 2008) and compare its results to those of the three-population test, highlighting the sensitivity trade-offs of each approach. We further illustrate our methodology with case studies of Central African Pygmies, Sardinians, and Japanese, revealing new details that add to our understanding of admixture events in the history of each population.
Methods
Properties of weighted admixture LD
In this section we introduce a weighted LD statistic that uses the decay of LD to detect signals of admixture given SNP data from an admixed population and reference populations. This statistic is similar to, but has an important difference from, the weighted LD statistic used in ROLLOFF (Moorjani et al. 2011; Patterson et al. 2012). The formulation of our statistic is particularly important in allowing us to use the amplitude (i.e., y-intercept) of the weighted LD curve to make inferences about history. We begin by deriving quantitative mathematical properties of this statistic that can be used to infer admixture parameters.
Basic model and notation:
We will primarily consider a point-admixture model in which a population C′ descends from a mixture of populations A and B to form C, n generations ago, in proportions α + β = 1, followed by random mating (Figure 1). As we discuss later, we can assume for our purposes that the genetic drift between C and C′ is negligible, and hence we will simply refer to the descendant population as C as well; we will state whether we mean the population immediately after admixture vs. n generations later when there is any risk of ambiguity. We are interested in the properties of the LD in population C induced by admixture. Consider two biallelic, neutrally evolving SNPs x and y, and for each SNP call one allele ‘0’ and the other ‘1’ (this assignment is arbitrary; ‘0’ and ‘1’ do not need to be oriented with regard to ancestral state via an outgroup). Denote by pA(x), pB(x), pA(y), and pB(y) the frequencies of the ‘1’ alleles at x and y in the mixing populations A and B (at the time of admixture), and let δ(x) := pA(x) − pB(x) and δ(y) := pA(y) − pB(y) be the allele frequency differences.
Figure 1.

Notational diagram of phylogeny containing admixed population and references. Population C′ is descended from an admixture between A and B to form C; populations A′ and B′ are present-day references. In practice, we assume that postadmixture drift is negligible; i.e., the C–C′ branch is extremely short and C′ and C have identical allele frequencies. The branch points of A′ and B′ from the A–B lineage are marked A″ and B″; note that in a rooted phylogeny, these need not be most recent common ancestors (as in panel B; compare to panel A).
Let d denote the genetic distance between x and y and assume that x and y were in linkage equilibrium in populations A and B. Then the LD in population C immediately after admixture is
where D is the standard haploid measure of linkage disequilibrium as the covariance of alleles at x and y (Chakraborty and Weiss 1988). After n generations of random mating, the LD decays to
assuming infinite population size (Chakraborty and Weiss 1988). For a finite population, the above formula holds in expectation with respect to random drift, with a small adjustment factor caused by post-admixture drift (Ohta and Kimura 1971),
where Ne is the effective population size. In most applications the adjustment factor is negligible, so we will omit it in what follows (Moorjani et al. 2012, Note S1).
In practice, our data consist of unphased diploid genotypes, so we expand our notation accordingly. Consider sampling a random individual from population C (n generations after admixture). We use a pair of {0, 1} random variables X1 and X2 to refer to the two alleles at x and define random variables Y1 and Y2 likewise. Our unphased SNP data represent observations of the {0, 1, 2} random variables X := X1 + X2 and Y := Y1 + Y2.
Define z(x, y) to be the covariance
| (1) |
which can be decomposed into a sum of four haplotype covariances:
| (2) |
The first two terms measure D for the separate chromosomes, while the third and fourth terms vanish, since they represent covariances between variables for different chromosomes, which are independent. Thus, the expectation (again with respect to random drift) of the total diploid covariance is simply
| (3) |
Relating weighted LD to admixture parameters:
Moorjani et al. (2011) first observed that pairwise LD measurements across a panel of SNPs can be combined to enable accurate inference of the age of admixture, n. The crux of their approach was to harness the fact that the ALD between two sites x and y scales as e−nd multiplied by the product of allele frequency differences δ(x)δ(y) in the mixing populations. While the allele frequency differences δ(⋅) are usually not directly computable, they can often be approximated. Thus, Moorjani et al. (2011) formulated a method, ROLLOFF, that dates admixture by fitting an exponential decay e−nd to correlation coefficients between LD measurements and surrogates for δ(x)δ(y). Note that Moorjani et al. (2011) define z(x, y) as a sample correlation coefficient, analogous to the classical LD measure r, as opposed to the sample covariance (Equation 1) we use here; we find the latter more mathematically convenient.
We build upon these previous results by deriving exact formulas for weighted sums of ALD under a variety of weighting schemes that serve as useful surrogates for δ(x)δ(y) in practice. These calculations will allow us to interpret the magnitude of weighted ALD to obtain additional information about admixture parameters. Additionally, the theoretical development will generally elucidate the behavior of weighted ALD and its applicability in various phylogenetic scenarios.
Following Moorjani et al. (2011), we partition all pairs of SNPs (x, y) into bins of roughly constant genetic distance,
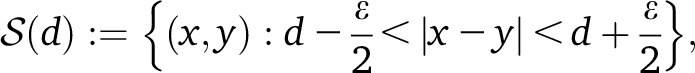 |
where ε is a discretization parameter inducing a discretization on d. Given a choice of weights w(⋅), one per SNP, we define the weighted LD at distance d as
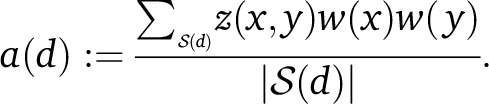 |
Assume first that our weights are the true allele frequency differences in the mixing populations, i.e., w(x) = δ(x) for all x. Applying Equation 3,
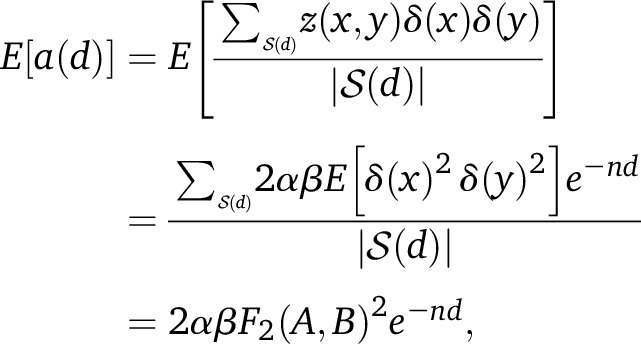 |
(4) |
where F2(A, B) is the expected squared allele frequency difference for a randomly drifting neutral allele, as defined in Reich et al. (2009) and Patterson et al. (2012). Thus, a(d) has the form of an exponential decay as a function of d, with time constant n giving the date of admixture.
In practice, we must compute an empirical estimator of a(d) from a finite number of sampled genotypes. Say we have a set of m diploid admixed samples from population C indexed by i = 1, …, m, and denote their genotypes at sites x and y by xi, yi ε {0, 1, 2}. Also assume we have some finite number of reference individuals from A and B with empirical mean allele frequencies and . Then our estimator is
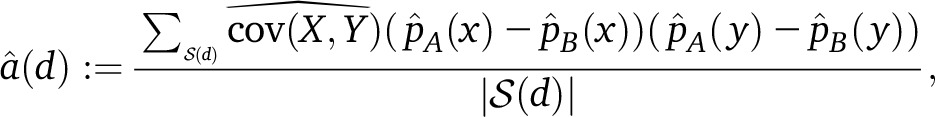 |
(5) |
where
is the usual unbiased sample covariance, so the expectation over the choice of samples satisfies (assuming no background LD, so the ALD in population C is independent of the drift processes producing the weights).
The weighted sum 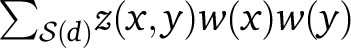 is a natural quantity to use for detecting ALD decay and is common to our weighted LD statistic and previous formulations of ROLLOFF. Indeed, for SNP pairs (x, y) at a fixed distance d, we can think of Equation 3 as providing a simple linear regression model between LD measurements z(x, y) and allele frequency divergence products δ(x)δ(y). In practice, the linear relation is made noisy by random sampling, as noted above, but the regression coefficient 2αβe−nd can be inferred by combining measurements from many SNP pairs (x, y). In fact, the weighted sum
is a natural quantity to use for detecting ALD decay and is common to our weighted LD statistic and previous formulations of ROLLOFF. Indeed, for SNP pairs (x, y) at a fixed distance d, we can think of Equation 3 as providing a simple linear regression model between LD measurements z(x, y) and allele frequency divergence products δ(x)δ(y). In practice, the linear relation is made noisy by random sampling, as noted above, but the regression coefficient 2αβe−nd can be inferred by combining measurements from many SNP pairs (x, y). In fact, the weighted sum 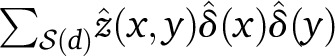 in the numerator of Equation 5 is precisely the numerator of the least-squares estimator of the regression coefficient, which is the formulation of ROLLOFF given in Moorjani et al. (2012, Note S1). Note that measurements of z(x, y) cannot be combined directly without a weighting scheme, as the sign of the LD can be either positive or negative; additionally, the weights tend to preserve signal from ALD while depleting contributions from other forms of LD.
in the numerator of Equation 5 is precisely the numerator of the least-squares estimator of the regression coefficient, which is the formulation of ROLLOFF given in Moorjani et al. (2012, Note S1). Note that measurements of z(x, y) cannot be combined directly without a weighting scheme, as the sign of the LD can be either positive or negative; additionally, the weights tend to preserve signal from ALD while depleting contributions from other forms of LD.
Up to scaling, our ALDER formulation is roughly equivalent to the regression coefficient formulation of ROLLOFF (Moorjani et al. 2012, Note S1). In contrast, the original ROLLOFF statistic (Patterson et al. 2012) computed a correlation coefficient between z(x, y) and w(x)w(y) over 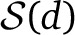 . However, the normalization term
. However, the normalization term 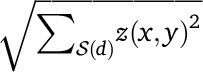 in the denominator of the correlation coefficient can exhibit an unwanted d-dependence that biases the inferred admixture date if the admixed population has undergone a strong bottleneck (Moorjani et al. 2012, Note S1) or in the case of recent admixture and large sample sizes. Beyond correcting the date bias, the curve that ALDER computes has the advantage of a simple form for its amplitude in terms of meaningful quantities, providing us additional leverage on admixture parameters. Additionally, we will show that can be computed efficiently via a new fast Fourier transform-based algorithm.
in the denominator of the correlation coefficient can exhibit an unwanted d-dependence that biases the inferred admixture date if the admixed population has undergone a strong bottleneck (Moorjani et al. 2012, Note S1) or in the case of recent admixture and large sample sizes. Beyond correcting the date bias, the curve that ALDER computes has the advantage of a simple form for its amplitude in terms of meaningful quantities, providing us additional leverage on admixture parameters. Additionally, we will show that can be computed efficiently via a new fast Fourier transform-based algorithm.
Using weights derived from diverged reference populations:
In the above development, we set the weights w(x) to equal the allele frequency differences δ(x) between the true mixing populations A and B. In practice, in the absence of DNA samples from past populations, it is impossible to measure historical allele frequencies from the time of mixture, so instead, we substitute reference populations A′ and B′ that are accessible, setting . In a given data set, the closest surrogates A′ and B′ may be somewhat diverged from A and B, so it is important to understand the consequences for the weighted LD a(d).
We show in Appendix A that with reference populations A′ and B′ in place of A and B, Equation 4 for the expected weighted LD curve changes only slightly, becoming
| (6) |
where A″ and B″ are the branch points of A′ and B′ on the A–B lineage (Figure 1). Notably, the curve still has the form of an exponential decay with time constant n (the age of admixture), albeit with its amplitude (and therefore signal-to-noise ratio) attenuated according to how far A″ and B″ are from the true ancestral mixing populations. Drift along the A′ − A″ and B′ − B″ branches likewise decreases signal-to-noise but in the reverse manner: higher drift on these branches makes the weighted LD curve noisier but does not change its expected amplitude (Supporting Information, Figure S1; see Appendix C for additional discussion). As above, given a real data set containing finite samples, we compute an estimator analogous to Equation 5 that has the same expectation (over sampling and drift) as the expectation of a(d) with respect to drift (Equation 6).
Using the admixed population as one reference:
Weighted LD can also be computed with only a single reference population by using the admixed population as the other reference (Pickrell et al. 2012, Supplement Sect. 4). Assuming first that we know the allele frequencies of the ancestral mixing population A and the admixed population C, the formula for the expected curve becomes
| (7) |
Using C itself as one reference population and R′ as the other reference (which could branch anywhere between A and B), the formula for the amplitude is slightly more complicated, but the curve retains the e−nd decay (Figure S2):
| (8) |
Derivations of these formulas are given in Appendix A.
A subtle but important technical issue arises when computing weighted LD with a single reference. In this case, the true weighted LD statistic is
where
are the mean allele frequencies of the admixed population (ignoring drift) and p(⋅) denotes allele frequencies of the reference population. Here a(d) cannot be estimated accurately by the naïve formula
which is the natural analog of (5). The difficulty is that the covariance term and the weights both involve the allele frequencies μx and μy; thus, while the standard estimators for each term are individually unbiased, their product is a biased estimate of the weighted LD.
Pickrell et al. (2012) circumvents this problem by partitioning the admixed samples into two groups, designating one group for use as admixed representatives and the other as a reference population; this method eliminates bias but reduces statistical power. We instead compute a polyache statistic (File S1) that provides an unbiased estimator of the weighted LD with maximal power.
Affine term in weighted LD curve from population substructure:
Weighted LD curves computed on real populations often exhibit a nonzero horizontal asymptote contrary to the exact exponential decay formulas we have derived above. Such behavior can be caused by assortative mating resulting in subpopulations structured by ancestry percentage in violation of our model. We show in Appendix A that if we instead model the admixed population as consisting of randomly mating subpopulations with heterogeneous amounts α—now a random variable—of mixed ancestry, our equations for the curves take the form
| (9) |
where M is a coefficient representing the contribution of admixture LD and K is an additional constant produced by substructure. Conveniently, however, the sum M + K/2 satisfies the same equations that the coefficient of the exponential does in the homogeneous case: adjusting Equation 6 for population substructure gives
| (10) |
for two-reference weighted LD, and in the one-reference case, modifying Equation 8 gives
| (11) |
For brevity, from here on we will take the amplitude of an exponential-plus-affine curve to mean M + K/2.
Admixture inference using weighted LD
We now describe how the theory we have developed can be used to investigate admixture. We detail novel techniques that use weighted LD to infer admixture parameters, test for admixture, and learn about phylogeny.
Inferring admixture dates and fractions using one or two reference populations:
As noted above, our ALDER formulation of weighted LD hones the original two-reference admixture dating technique of ROLLOFF (Moorjani et al. 2011), correcting a possible bias (Moorjani et al. 2012, Note S1), and the one-reference technique (Pickrell et al. 2012), improving statistical power. Pickrell et al. (2012) also observed that weighted LD can be used to estimate ancestral mixing fractions. We further develop this application now.
The main idea is to treat our expressions for the amplitude of the weighted LD curve as equations that can be solved for the ancestry fractions α and β = 1 − α. First consider two-reference weighted LD. Given samples from an admixed population C and reference populations A′ and B′, we compute the curve and fit it as an exponential decay plus affine term: . Let denote the amplitude of the curve. Then Equation 10 gives us a quadratic equation that we can solve to obtain an estimate of the mixture fraction α,
assuming we can estimate F2(A″, B″)2. Typically the branch-point populations A″ and B″ are unavailable, but their F2 distance can be computed by means of an admixture tree (Lipson et al. 2012; Patterson et al. 2012; Pickrell and Pritchard 2012). A caveat of this approach is that α and 1 − α produce the same amplitude and cannot be distinguished by this method alone; additionally, the inversion problem is ill-conditioned near α = 0.5, where the derivative of the quadratic vanishes.
The situation is more complicated when using the admixed population as one reference. First, the amplitude relation from Equation 11 gives a quartic equation in :
Second, the F2 distances involved are in general not possible to calculate by solving allele frequency moment equations (Lipson et al. 2012; Patterson et al. 2012). In the special case that one of the true mixing populations is available as a reference, however—i.e., R′ = A—Pickrell et al. (2012) demonstrated that mixture fractions can be estimated much more easily. From Equation 7, the expected amplitude of the curve is 2αβ3F2(A, B)2. On the other hand, assuming no drift in C since the admixture, allele frequencies in C are given by weighted averages of allele frequencies in A and B with weights α and β; thus, the squared allele frequency differences from A to B and C satisfy
and F2(A, C) is estimable directly from the sample data. Combining these relations, we can obtain our estimate by solving the equation
| (12) |
In practice, the true mixing population A is not available for sampling, but a closely related population A′ may be. In this case, the value of given by Equation 12 with A′ in place of A is a lower bound on the true mixture fraction α (see Appendix A for theoretical development and Results for simulations exploring the tightness of the bound). This bounding technique is the most compelling of the above mixture fraction inference approaches, as prior methods cannot perform such inference with only one reference population. In contrast, when more references are available, moment-based admixture tree-fitting methods, for example, readily estimate mixture fractions (Lipson et al. 2012; Patterson et al. 2012; Pickrell and Pritchard 2012). In such cases we believe that existing methods are more robust than LD-based inference, which suffers from the degeneracy of solutions noted above; however, the weighted LD approach can provide confirmation based on a different genetic mechanism.
Testing for admixture:
Thus far, we have taken it as given that the population C of interest is admixed and developed methods for inferring admixture parameters by fitting weighted LD curves. Now we consider the question of whether weighted LD can be used to determine whether admixture occurred in the first place. We develop a weighted LD-based formal test for admixture that is broadly analogous to the drift-based three-population test (Reich et al. 2009; Patterson et al. 2012) but sensitive in different scenarios.
A complication of interpreting weighted LD is that certain demographic events other than admixture can also produce positive weighted LD that decays with genetic distance, particularly in the one-reference case. Specifically, if population C has experienced a recent bottleneck or an extended period of low population size, it may contain long-range LD. Furthermore, this LD typically has some correlation with allele frequencies in C; consequently, using C as one reference in the weighting scheme produces a spurious weighted LD signal.
In the two-reference case, LD from reduced population size in C is generally washed out by the weighting scheme assuming the reference populations A′ and B′ are not too genetically similar to C. The reason is that the weights arise from drift between A′ and B′ that is independent of demographic events producing LD in C (beyond genetic distances that are so short that the populations share haplotypes descended without recombination from their common ancestral haplotype). Thus, observing a two-reference weighted LD decay curve is generally good evidence that population C is admixed. There is still a caveat, however. If C and one of the references, say A′, share a recent population bottleneck, then the bottleneck-induced LD in C can be correlated to the allele frequencies of A′, resulting once again in spurious weighted LD. In fact, the one-reference example mentioned above is the limiting case A′ = C of this situation.
With these considerations in mind, we propose an LD-based three-population test for admixture that includes a series of pre-tests safeguarding against the pathological demographies that can produce a non-admixture weighted LD signal. We outline the test now; details are in Appendix B. Given a population C to test for admixture and references A′ and B′, the main test is whether the observed weighted LD using A′ − B′ weights is positive and well-fit by an exponential decay curve. We estimate a jackknife-based p-value by leaving out each chromosome in turn and refitting the weighted LD as an exponential decay; the jackknife then gives us a standard error on the fitting parameters—namely, the amplitude and the decay constant—that we use to measure the significance of the observed curve.
The above procedure allows us to determine whether there is sufficient signal in the weighted LD curve to reject the null hypothesis (under which is random “colored” noise in the sense that it contains autocorrelation). However, in order to conclude that the curve is the result of admixture, we must rule out the possibility that it is being produced by demography unrelated to admixture. We therefore apply the following pre-test procedure. First, we determine the distance to which LD in C is significantly correlated with LD in either A′ or B′; to minimize signal from shared demography, we subsequently ignore data from SNP pairs at distances smaller than this correlation threshold. Then, we compute one-reference weighted LD curves for population C with A′–C and B′–C weights and check that the curves are well-fit as exponential decays. In the case that C is actually admixed between populations related to A′ and B′, the one-reference A′–C and B′–C curves pick up the same e−nd admixture LD decay signal. If C is not admixed but has experienced a shared bottleneck with A′ (producing false-positive admixture signals from the A′ – B′ and B′–C curves), however, the A′–C weighting scheme is unlikely to produce a weighted LD curve, especially when fitting beyond the LD correlation threshold.
This test procedure is intended to be conservative, so that a population C identified as admixed can strongly be assumed to be so, whereas if C is not identified as admixed, we are less confident in claiming that C has experienced no admixture whatsoever. In situations where distinguishing admixture from other demography is particularly difficult, the test will err on the side of caution; for example, even if C is admixed, the test may fail to identify C as admixed if it has also experienced a bottleneck. Also, if a reference A′ shares some of the same admixture history as C or is simply very closely related to C, the pre-test will typically identify long-range correlated LD and deem A′ an unsuitable reference to use for testing admixture. The behavior of the test and pre-test criteria are explored in detail with coalescent simulations in Appendix C.
Learning about phylogeny:
Given a triple of populations (C; A′, B′), our test can identify admixture in the test population C, but what does this imply about the relationship of populations A′ and B′ to C? As with the drift-based three-population test, test results must be interpreted carefully: even if C is admixed, this does not necessarily mean that the reference populations A′ and B′ are closely related to the true mixing populations. However, computing weighted LD curves with a suite of different references can elucidate the phylogeny of the populations involved, since our amplitude Equations 10 and 11 provide information about the locations on the phylogeny at which the references diverge from the true mixing populations.
More precisely, in the notation of Figure 1, the amplitude of the two-reference weighted LD curve is 2αβF2(A″, B″)2, which is maximized when A″ = A and B″ = B and is minimized when A″ = B″. So, for example, we can fix A′ and compute curves for a variety of references B′; the larger the resulting amplitude, the closer the branch point B″ is to B. In the one-reference case, as the reference R′ is varied, the amplitude 2αβ(αF2(A, R″) − βF2(B, R″))2 traces out a parabola that starts at 2αβ3F2(A, B)2 when R″ = A, decreases to a minimum value of 0, and increases again to 2α3βF2(A, B)2 when R″ = B (Figure S2). Here, the procedure is more qualitative because the branches F2(A, R″) and F2(B, R″) are less directly useful and the mixture proportions α and β may not be known.
Implementation of ALDER
We now describe some more technical details of the ALDER software package in which we have implemented our weighted LD methods.
Fast Fourier transform algorithm for computing weighted LD:
We developed a novel algorithm that algebraically manipulates the weighted LD statistic into a form that can be computed using a fast Fourier transform (FFT), dramatically speeding up the computation (File S2). The algebraic transformation is made possible by the simple form (Equation 5) of our weighted LD statistic along with a genetic distance discretization procedure that is similar spirit to ROLLOFF (Moorjani et al. 2011) but subtly different: instead of binning the contributions of SNP pairs (x, y) by discretizing the genetic distance |x − y| = d, we discretize the genetic map positions x and y themselves (using a default resolution of 0.05 cM) (Figure S3). For two-reference weighted LD, the resulting FFT-based algorithm that we implemented in ALDER has computational cost that is approximately linear in the data size; in practice, it ran three orders of magnitude faster than ROLLOFF on typical data sets we analyzed.
Curve fitting:
We fit discretized weighted LD curves as from Equation 9, using least-squares to find best-fit parameters. This procedure is similar to ROLLOFF, but ALDER makes two important technical advances that significantly improve the robustness of the fitting. First, ALDER directly estimates the affine term K that arises from the presence of subpopulations with differing ancestry percentages by using interchromosome SNP pairs that are effectively at infinite genetic distance (Appendix A). The algorithmic advances we implement in ALDER enable efficient computation of the average weighted LD over all pairs of SNPs on different chromosomes, giving and, importantly, eliminating one parameter from the exponential fitting. In practice, we have observed that ROLLOFF fits are sometimes sensitive to the maximum inter-SNP distance d to which the weighted LD curve is computed and fit; ALDER eliminates this sensitivity.
Second, because background LD is present in real populations at short genetic distances and confounds the ALD signal (interfering with parameter estimates or producing spurious signal entirely), it is important to fit weighted LD curves starting only at a distance beyond which background LD is negligible. ROLLOFF used a fixed threshold of d > 0.5 cM, but some populations have longer-range background LD (e.g., from bottlenecks), and moreover, if a reference population is closely related to the test population, it can produce a spurious weighted LD signal due to recent shared demography. ALDER therefore estimates the extent to which the test population shares correlated LD with the reference(s) and fits only the weighted LD curve beyond this minimum distance as in our test for admixture (Appendix B).
We estimate standard errors on parameter estimates by performing a jackknife over the autosomes used in the analysis, leaving out each in turn. Note that the weighted LD measurements from individual pairs of SNPs that go into the computed curve are not independent of each other; however, the contributions of different chromosomes can reasonably be assumed to be independent.
Data sets
We primarily applied our weighted LD techniques to a data set of 940 individuals in 53 populations from the CEPH–Human Genome Diversity Cell Line Panel (HGDP) (Rosenberg et al. 2002) genotyped on an Illumina 650K SNP array (Li et al. 2008). To study the effect of SNP ascertainment, we also analyzed the same HGDP populations genotyped on the Affymetrix Human Origins Array (Patterson et al. 2012). For some analyses we also included HapMap Phase 3 data (International HapMap Consortium 2010) merged either with the Illumina HGDP data set, leaving ∼600,000 SNPs, or with the Indian data set of Reich et al. (2009) including 16 Andaman Islanders (9 Onge and 7 Great Andamanese), leaving ∼500,000 SNPs.
We also constructed simulated admixed chromosomes from 112 CEU and 113 YRI phased HapMap individuals using the following procedure, described in Moorjani et al. (2011). Given desired ancestry proportions α and β, the age n of the point admixture, and the number m of admixed individuals to simulate, we built each admixed chromosome as a composite of chromosomal segments from the source populations, choosing breakpoints via a Poisson process with rate constant n, and sampling blocks at random according to the specified mixture fractions. We stipulated that no individual haplotype could be reused at a given locus among the m simulated individuals, preventing unnaturally long identical-by-descent segments but effectively eliminating postadmixture genetic drift. For the short time scales we study (admixture occurring 200 or fewer generations ago), this approximation has little impact. We used this method to maintain some of the complications inherent in real data.
Results
Simulations
First, we demonstrate the accuracy of several forms of inference from ALDER on simulated data. We generated simulated genomes for mixture fractions of 75% YRI/25% CEU and 90% YRI/10% CEU and admixture dates of 10, 20, 50, 100, and 200 generations ago. For each mixture scenario we simulated 40 admixed individuals according to the procedure above.
We first investigated the admixture dates estimated by ALDER using a variety of reference populations drawn from the HGDP with varying levels of divergence from the true mixing populations. On the African side, we used HGDP Yoruba (21 samples; essentially the same population as HapMap YRI) and San (5 samples); on the European side, we used French (28 samples; very close to CEU), Han (34 samples), and Papuan (17 samples). We computed two-reference weighted LD curves using pairs of references, one from each group, as well as one-reference curves using the simulated population as one reference and each of the above HGDP populations as the other.
For the 75% YRI mixture, estimated dates are nearly all accurate to within 10% (Table S1). The noise levels of the fitted dates (estimated by ALDER using the jackknife) are the lowest for the Yoruba–French curve, as expected, followed by the one-reference curve with French, consistent with the admixed population being mostly Yoruba. The situation is similar but noisier for the 90% YRI mixture (Table S2); in this case, the one-reference signal is quite weak with Yoruba and undetectable with San as the reference, due to the scaling of the amplitude (Equation 11) with the cube of the CEU mixture fraction.
We also compared fitted amplitudes of the weighted LD curves for the same scenarios to those predicted by Equations 10 and 11; the accuracy trends are similar (Table S3 and Table S4). Finally, we tested Equation 12 for bounding mixture proportions using one-reference weighted LD amplitudes. We computed lower bounds on the European ancestry fraction using French, Russian, Sardinian, and Kalash as successively more diverged references. As expected, the bounds are tight for the French reference and grow successively weaker (Table S5 and Table S6). We also tried lower-bounding the African ancestry using one-reference curves with an African reference. In general, we expect lower bounds computed for the major ancestry proportion to be much weaker (Appendix A), and indeed we find this to be the case, with the only slightly diverged Mandenka population producing extremely weak bounds. An added complication is that the Mandenka are an admixed population with a small amount of West Eurasian ancestry (Price et al. 2009), which is not accounted for in the amplitude formulas we use here.
Another notable feature of ALDER is that, to a much greater extent than f-statistic methods, its inference quality improves with more samples from the admixed test population. As a demonstration of this, we simulated a larger set of 100 admixed individuals as above, for both 75% YRI/25% CEU and 90% YRI/10% CEU scenarios, and compared the date estimates obtained on subsets of 5–100 of these individuals with two different reference pairs (Table S7 and Table S8). With larger sample sizes, the estimates become almost uniformly more accurate, with smaller standard errors. By contrast, we observed that while using a very small sample size (say 5) for the reference populations does create noticeable noise, using 20 samples already gives allele frequency estimates accurate enough that adding more reference samples has only minimal effects on the performance of ALDER. This is similar to the phenomenon that the precision of f-statistics does not improve appreciably with more than a moderate number of samples and is due to the inherent variability in genetic drift among different loci.
Robustness
A challenge of weighted LD analysis is that owing to various kinds of model violation, the parameters of the exponential fit of an observed curve may depend on the starting distance d0 from which the curve is fit. We therefore explored the robustness of the fitting parameters to the choice of d0 in a few scenarios (Figure 2). First, in a simulated 75%/25% YRI–CEU admixture 50 generations ago, we find that the decay constant and amplitude are both highly robust to varying d0 from 0.5 to 2.0 cM (Figure 2, top). This result is not surprising because our simulated example represents a true point admixture with minimal background LD in the admixed population.
Figure 2.

Dependence of date estimates and weighted LD amplitudes on fitting start point. Rows correspond to three test scenarios: simulated 75% YRI/25% CEU mixture 50 generations ago with Yoruba–French weights (A–C); Uygur with Han–French weights (D–F); HapMap Maasai with Yoruba–French weights (G–I). (A, D, and G) The weighted LD curve (blue) with best-fit exponential decay curve (red), fit starting from d0 = 0.5 cM. The middle and right columns show the date estimate (B, E, H) and amplitude (C, F, I) as a function of d0. (We note that our date estimates for Uygur are somewhat more recent than those in Patterson et al. 2012, most likely because of our direct estimate of the affine term in the weighted LD curve.)
In practice, we expect some dependence on d0 due to background LD or longer-term admixture (either continuously over a stretch of time or in multiple waves). Both of these tend to increase the weighted LD for smaller values of d relative to an exact exponential curve, so that estimates of the decay constant and amplitude decrease as we increase the fitting start point d0; the extent to which this effect occurs depends on the extent of the model violation. We studied the d0 dependence for two example admixed populations, HGDP Uygur and HapMap Maasai (MKK). For Uygur, the estimated decay constants and amplitudes are fairly robust to the start point of the fitting, varying roughly by ±10% (Figure 2, middle). In contrast, the estimates for Maasai vary dramatically, decreasing by a factor of >2 as d0 is increased from 0.5 to 2.0 cM (Figure 2, bottom). This behavior is likely due to multiple-wave admixture in the genetic history of the Maasai; indeed, it is visually evident that the weighted LD curve for Maasai deviates from an exponential fit (Figure 2) and is in fact better fit as a sum of exponentials. (See Figure S4 and Appendix C for further simulations exploring continuous admixture.)
It is also important to consider the possibility of SNP ascertainment bias, as in any study based on allele frequencies. We believe that for weighted LD, ascertainment bias could have modest effects on the amplitude, which depends on F2 distances (Lipson et al. 2012; Patterson et al. 2012), but does not affect the estimated date. Running ALDER on a suite of admixed populations in the HGDP under a variety of ascertainment schemes suggests that admixture date estimates are indeed quite stable to ascertainment (Table S9). Meanwhile, the amplitudes of the LD curves can scale substantially when computed under different SNP ascertainments, but their relative values are different only for extreme cases of African vs. non-African test populations under African vs. non-African ascertainment (Table S10; cf. Patterson et al. 2012, Table 2).
Admixture test results for HGDP populations
To compare the sensitivity of our LD-based test for admixture to the f-statistic-based three-population test, we ran both ALDER and the three-population test on all triples of populations in the HGDP. Interestingly, while the tests concur on the majority of the populations they identify as admixed, each also identifies several populations as admixed that the other does not (Table 1), showing that the tests have differing sensitivity to different admixture scenarios.
Table 1. Results of ALDER and three-population tests for admixture on HGDP populations.
| Both | No. LD | No. f3 | Only LD | No. | Only f3 | No. | Neither |
|---|---|---|---|---|---|---|---|
| Adygei | 205 | 139 | BiakaPygmy | 81 | French | 99 | Basque |
| Balochi | 123 | 204 | Colombian | 5 | Han | 13 | Dai |
| BantuKenya | 30 | 182 | Druze | 128 | Italian | 46 | Hezhen |
| BantuSouthAfrica | 27 | 11 | Japanese | 1 | Orcadian | 1 | Karitiana |
| Bedouin | 300 | 63 | Kalash | 20 | Tujia | 8 | Lahu |
| Brahui | 363 | 16 | MbutiPygmy | 77 | Tuscan | 59 | Mandenka |
| Burusho | 450 | 377 | Melanesian | 96 | Miao | ||
| Cambodian | 266 | 158 | Pima | 489 | Naxi | ||
| Daur | 29 | 8 | San | 155 | Papuan | ||
| Han-NChina | 1 | 77 | Sardinian | 45 | She | ||
| Hazara | 699 | 593 | Yakut | 435 | Surui | ||
| Makrani | 173 | 163 | Yi | ||||
| Maya | 784 | 124 | Yoruba | ||||
| Mongola | 76 | 385 | |||||
| Mozabite | 313 | 107 | |||||
| Oroqen | 68 | 5 | |||||
| Palestinian | 308 | 64 | |||||
| Pathan | 113 | 348 | |||||
| Russian | 158 | 153 | |||||
| Sindhi | 264 | 366 | |||||
| Tu | 22 | 315 | |||||
| Uygur | 428 | 616 | |||||
| Xibo | 101 | 335 |
We ran both ALDER and the three-population test for admixture on each of the 53 HGDP populations using all pairs of other populations as references. We group the populations according to whether each test methodology produced at least one test identifying them as admixed; for each population, we list the number of reference pairs with which with each method (abbreviated LD and f3) detected admixture. We used a significance threshold of P < 0.05 after multiple-hypothesis correction.
Admixture identified only by ALDER:
The three-population test loses sensitivity primarily as a result of drift subsequent to splitting from the references’ lineages. More precisely, using the notation of Figure 1, the three-population test statistic f3(C; A′, B′) estimates the sum of two directly competing terms: −αβF2(A″, B″), the negative quantity arising from admixture that we wish to detect, and α2F2(A″, A) + β2F2(B″, B) + F2(C, C″), a positive quantity from the “off-tree” drift branches. If the latter term dominates, the three-population test will fail to detect admixture regardless of the statistical power available. For example, Melanesians are only found to be admixed according to the ALDER test; the inability of the three-population test to identify them as admixed is likely due to long off-tree drift from the Papuan branch prior to admixture. The situation is similar for the Pygmies, for whom we do not have two close references available.
Small mixture fractions also diminish the size of the admixture term −αβF2(A, B) relative to the off-tree drift, and we believe this effect along with postadmixture drift may be the reason Sardinians are detected as admixed only by ALDER. In the case of the San, who have a small amount of Bantu admixture (Pickrell et al. 2012), the small mixture fraction may again play a role, along with the lack of a reference population closely related to the preadmixture San, meaning that using existing populations incurs long off-tree drift.
Admixture identified only by the three-population test:
There are also multiple reasons why the three-population test can identify admixture when ALDER does not. For the HGDP European populations in this category (Table 1), the three-population test is picking up a signal of admixture identified by Patterson et al. (2012) and interpreted as a large-scale admixture event in Europe involving Neolithic farmers closely related to present-day Sardinians and an ancient northern Eurasian population. This mixture likely began quite anciently (e.g., 7000–9000 years ago when agriculture arrived in Europe; Bramanti et al. 2009; Soares et al. 2010; Pinhasi et al. 2012), and because admixture LD breaks down as e−nd, where n is the age of admixture, there is nearly no LD left for ALDER to harness beyond the correlation threshold d0. An additional factor that may inhibit LD-based testing is that to prevent false-positive identifications of admixture, ALDER typically eliminates reference populations that share LD (and in particular, admixture history) with the test population, whereas the three-population test can use such references.
To summarize, the ALDER and three-population tests both analyze a test population for admixture using two references, but they detect signal based on different “genetic clocks.” The three-population test uses signal from genetic drift, which can detect quite old admixture but must overcome a counteracting contribution from postadmixture and off-tree drift. The LD-based test uses recombination, which is relatively unaffected by small population size-induced long drift and has no directly competing effect, but has limited power to detect chronologically old admixtures because of the rapid decay of the LD curve. Additionally, as discussed above in the context of simulation results, the LD-based test may be better suited for large data sets, since its power is enhanced more by the availability of many samples. The tests are thus complementary and both valuable. (See Figure S5 and Appendix C for further exploration.)
Case studies
We now present detailed results for several human populations, all of which ALDER identifies as admixed but are not found by the three-population test (Table 1). We infer dates of admixture and in some cases gain additional historical insights.
Pygmies:
Both Central African Pygmy populations in the HGDP, the Mbuti and Biaka, show evidence of admixture (Table 1), about 28 ± 4 generations (800 years) ago for Mbuti and 38 ± 4 generations (1100 years) ago for Biaka, estimated using San and Yoruba as reference populations (Figure 3, A and C). The intrapopulation heterogeneity is low, as demonstrated by the negligible affine terms. In each case, we also generated weighted LD curves with the Pygmy population itself as one reference and a variety of second references. We found that using French, Han, or Yoruba as the second reference gave very similar amplitudes, but the amplitude was significantly smaller with the other Pygmy population or San as the second reference (Figure 3, B and D). Using the amplitudes with Yoruba, we estimated mixture fractions of at least 15.9 ± 0.9% and 28.8 ± 1.4% Yoruba-related ancestry (lower bounds) for Mbuti and Biaka, respectively.
Figure 3.

Weighted LD curves for Mbuti using San and Yoruba as reference populations (A) and using Mbuti itself as one reference and several different second references (B), and analogous curves for Biaka (C and D). Genetic distances are discretized into bins at 0.05 cM resolution. Data for each curve are plotted and fit starting from the corresponding ALDER-computed LD correlation thresholds. Different amplitudes of one-reference curves (B and D) imply different phylogenetic positions of the references relative to the true mixing populations (i.e., different split points ), suggesting a sketch of a putative admixture graph (E). Relative branch lengths are qualitative, and the true root is not necessarily as depicted.
The phylogenetic interpretation of the relative amplitudes is complicated by the fact that the Pygmy populations, used as references, are themselves admixed, but a plausible coherent explanation is as follows (see Figure 3E). We surmise that a proportion β (bounds given above) of Bantu-related gene flow reached the native Pygmy populations on the order of 1000 years ago. The common ancestors of Yoruba or non-Africans with the Bantu population are genetically not very different from Bantu, due to high historical population sizes (branching at positions X1 and X2 in Figure 3E). Thus, the weighted LD amplitudes using Yoruba or non-Africans as second references are nearly 2α3βF2(A, B)2, where B denotes the admixing Bantu population. Meanwhile, San and Western (resp. Eastern) Pygmies split from the Bantu–Mbuti (resp. Biaka) branch toward the middle or the opposite side from Bantu (X3 and X4), giving a smaller amplitude (Figure S2).
Our results are in agreement with previous studies that have found evidence of gene flow from agriculturalists to Pygmies (Quintana-Murci et al. 2008; Verdu et al. 2009; Patin et al. 2009; Jarvis et al. 2012). Quintana-Murci et al. (2008) suggested based on mtDNA evidence in Mbuti that gene flow ceased several thousand years ago, but more recently, Jarvis et al. (2012) found evidence of admixture in Western Pygmies, with a local-ancestry-inferred block-length distribution of 3.1 ± 4.6 Mb (mean and standard deviation), consistent with our estimated dates.
Sardinians:
We detect a very small proportion of sub-Saharan African ancestry in Sardinians, which our ALDER tests identified as admixed (Table 1 and Figure 4A). To investigate further, we computed weighted LD curves with Sardinian as the test population and all pairs of the HapMap CEU, YRI, and CHB populations as references (Table 2). We observed an abnormally large amount of shared long-range LD in chromosome 8, likely because of an extended inversion segregating in Europeans (Price et al. 2008), so we omitted it from these analyses. The CEU–YRI curve has the largest amplitude, suggesting both that the LD present is due to admixture and that the small non-European ancestry component, for which we estimated a lower bound of 0.6 ± 0.2%, is from Africa. (For this computation we used single-reference weighted LD with YRI as the reference, fitting the curve after 1.2 cM to reduce confounding effects from correlated LD that ALDER detected between Sardinian and CEU. Changing the starting point of the fit does not qualitatively affect the results.) The existence of a weighted LD decay curve with CHB and YRI as references provides further evidence that the LD is not simply due to a population bottleneck or other nonadmixture sources, as does the fact that our estimated dates from all three reference pairs are roughly consistent at about 40 generations (1200 years) ago. Our findings thus confirm the signal of African ancestry in Sardinians reported in Moorjani et al. (2011). The date, small mixture proportion, and geography are consistent with a small influx of migrants from North Africa, who themselves traced only a fraction of their ancestry ultimately to sub-Saharan Africa, consistent with the findings of Dupanloup et al. (2004).
Figure 4.

Weighted LD curves for HGDP Sardinian using Italian–Yoruba weights (A) and HapMap Japanese (JPT) using JPT itself as one reference and HapMap Han Chinese in Beijing (CHB) as the second reference (B). The exponential fits are performed starting at 1 cM and 1.2 cM, respectively, as selected by ALDER based on detected correlated LD.
Table 2. Amplitudes and dates from weighted LD curves for Sardinian using various reference pairs.
| Ref 1 | Ref 2 | Weighted LD amplitude | Date estimate |
|---|---|---|---|
| CEU | YRI | 0.00003192 ± 0.00000903 | 48 ± 10 |
| CHB | YRI | 0.00001738 ± 0.00000679 | 34 ± 8 |
| CEU | CHB | 0.00000873 ± 0.00000454 | 52 ± 21 |
Data are shown from ALDER fits to weighted LD curves computed using Sardinian as the test population and pairs of HapMap CEU, YRI, and CHB as the references. Date estimates are in generations. We omitted chromosome 8 from the analysis because of anomalous long-range LD. Curves were fit for d > 1.2 cM, the extent of LD correlation between Sardinian and CEU computed by ALDER.
Japanese:
Genetic studies have suggested that present-day Japanese are descended from admixture between two waves of settlers, responsible for the Jomon and Yayoi cultures (Hammer and Horai 1995; Hammer et al. 2006; Rasteiro and Chikhi 2009). We also observed evidence of admixture in Japanese, and while our ability to learn about the history was limited by the absence of a close surrogate for the original Paleolithic mixing population, we were able to take advantage of the one-reference inference capabilities of ALDER. More precisely, among our tests using all pairs of HGDP populations as references (Table 1), one reference pair, Basque and Yakut, produced a passing test for Japanese. However, as we have noted, the reference populations need not be closely related to the true mixing populations, and we believe that in this case this seemingly odd reference pair arises as the only passing test because the data set lacks a close surrogate for Jomon.
In the absence of a reference on the Jomon side, we computed single-reference weighted LD using HapMap JPT as the test population and JPT–CHB weights, which confer the advantage of larger sample sizes (Figure 4B). The weighted LD curve displays a clear decay, yielding an estimate of 45 ± 6 generations, or about 1300 years, as the age of admixture. To our knowledge, this is the first time genome-wide data have been used to date admixture in Japanese. As with previous estimates based on coalescence of Y-chromosome haplotypes (Hammer et al. 2006), our date is consistent with the archeologically attested arrival of the Yayoi in Japan ∼2300 years ago (we suspect that our estimate is from later than the initial arrival because admixture may not have happened immediately or may have taken place over an extended period of time). Based on the amplitude of the curve, we also obtain a (likely very conservative) genome-wide lower bound of 41 ± 3% “Yayoi” ancestry using Equation 12 (under the reasonable assumption that Han Chinese are fairly similar to the Yayoi population). It is important to note that the observation of a single-reference weighted LD curve is not sufficient evidence to prove that a population is admixed, but the existence of a pair of references with which the ALDER test identified Japanese as admixed, combined with previous work and the lack of any signal of reduced population size, makes us confident that our inferences are based on true historical admixture.
Onge:
Finally, we provide a cautionary example of weighted LD decay curves arising from demography and not admixture. We observed distinct weighted LD curves when analyzing the Onge, an indigenous population of the Andaman Islands. However, this curve is present only when using Onge themselves as one reference; moreover, the amplitude is independent of whether CEU, CHB, YRI, GIH (HapMap Gujarati), or Great Andamanese is used as the second reference (Figure 5), as expected if the weighted LD is due to correlation between LD and allele frequencies in the test population alone (and independent of the reference allele frequencies). Correspondingly, ALDER’s LD-based test does not identify Onge as admixed using any pair of these references. Thus, while we cannot definitively rule out admixture, the evidence points toward internal demography (low population size) as the cause of the elevated LD, consistent with the current census of <100 Onge individuals.
Figure 5.

Weighted LD curves for Onge using Onge itself as one reference and several different second references.
Discussion
Strengths of weighted LD for admixture inference
The statistics underlying weighted LD are quite simple, making the formula for the expectation of , as well as the noise and other errors from our inference procedure, relatively easy to understand. By contrast, local ancestry-based admixture dating methods (e.g., Pool and Nielsen (2009) and Gravel (2012)) are sensitive to imperfect ancestry inference, and it is difficult to trace the error propagation to understand the ultimate effect on inferred admixture parameters. Similarly, the wavelet method of Pugach et al. (2011) uses reference populations to perform (fuzzy) ancestry assignment in windows, for which error analysis is challenging.
Another strength of our weighted LD methodology is that it has relatively low requirements on the quality and quantity of reference populations. Our theory tells us exactly how the statistic behaves for any reference populations, no matter how diverged they are from the true ancestral mixing populations. In contrast, the accuracy of results from clustering and local ancestry methods is dependent on the quality of the reference populations used in ways that are difficult to characterize. On the quantity side, previous approaches to admixture inference require a surrogate for each ancestral population, whereas as long as one is confident that the signal is truly from admixture, weighted LD can be used with only one available reference to infer times of admixture (as in our analysis of the Japanese) and bound mixing fractions (as in our Pygmy case study and Pickrell et al. (2012)), problems that were previously intractable.
Weighted LD also advances our ability to test for admixture. As discussed above, ALDER offers complementary sensitivity to the three-population test and allows the identification of additional populations as admixed. Another formal test for admixture is the four-population test (Reich et al. 2009; Patterson et al. 2012), which is quite sensitive but also has trade-offs; for example, it requires three distinctly branching references, whereas ALDER and the three-population test need only two. Additionally, the phylogeny of the populations involved must be well understood to allow interpretation of a signal of admixture from the four-population test properly (i.e., to determine which population is admixed). Using weighted LD, on the other hand, largely eliminates the problem of determining the destination or direction of gene flow, since the LD signal of admixture is intrinsic to a specified test population.
One-reference vs. two-reference curves
In practice, it is often useful to compute weighted LD curves using both the one-reference and two-reference techniques, as both can be used for inferences in different situations. Generally, we consider two-reference curves to be more reliable for parameter estimation, since using the test population as one reference is more prone to introducing unwanted signals, such as recent admixture from a different source, nonadmixture LD from reduced population size, or population structure among samples. In particular, populations with more complicated histories and additional sources of LD beyond the specifications of our model often have different estimates of admixture dates with one- and two-reference curves. There is a small chance that date disagreement can reflect a false-positive admixture signal, but this is very unlikely if both one- and two-reference curves exist beyond the correlated LD threshold (see Appendix B). Two-reference curves also allow for direct estimation of mixture fractions, although, as discussed above, we prefer instead to use the method of single-reference bounding.
A number of practical considerations make the one-reference capabilities of ALDER desirable. Foremost is the possibility that one may not have a good surrogate available for one of the ancestral mixing populations, as in our Japanese example. Also, while our method of learning about phylogenetic relationships is best suited to two-reference curves because of the simpler form of the amplitude in terms of branch lengths, it is often useful to begin by computing a suite of single-reference curves, both because the data generated will scale linearly with the number of references available and because observing a range of different amplitudes gives an immediate signal of the presence of admixture in the test population.
Overall, then, a sample sequence for applying ALDER to a new data set might be as follows: (1) test all populations for admixture using all pairs of references from among the other populations; (2) explore admixed populations of interest by comparing single-reference weighted LD curves; (3) learn more detail by analyzing selected two-reference curves alongside the one-reference ones; and (4) estimate parameters using one- or two-reference curves as applicable. Of course, step 1 itself involves the complementary usefulness of both one- and two-reference weighted LD, since our test for admixture requires the presence of exponential decay signals in both types of curves.
Effect of multiple-wave or continuous admixture
As discussed in our section on robustness of results, in the course of our data analysis, we observed that the weighted LD date estimate almost always becomes more recent when the exponential decay curve is fit for a higher starting distance d0. Most likely, this is because admixtures in human populations have taken place over multiple generations, such that our estimated times represent intermediate dates during the process. To whatever extent an admixture event is more complicated than posited in our point-admixture model, removing low-d bins will lead the fitting to capture proportionately more of the more recent admixture. By default, ALDER sets d0 to be the smallest distance such that nonadmixture LD signals can be confidently discounted for d > d0 (see Methods, Testing for admixture, and Appendix B), but it should be noted that the selected d0 varies for different sets of populations, and in each case the true admixture signal at d < d0 is also excluded. Theoretically, this pattern could allow us to learn more about the true admixture history of a population, since the value of a(d) at each d represents a particular function of the amount of admixture that took place at each generation in the past. However, in our experience, fitting becomes difficult for any model involving more than two or three parameters. Thus, we made the decision to restrict ourselves to assuming a single-point admixture, fit for a principled threshold d > d0, accepting that the inferred date n represents some form of average value over the true history.
Other possible complications
In our derivations, we have assumed implicitly that the mixing populations and the reference populations are related through a simple tree. However, it may be that their history is more complicated, for example, involving additional admixtures. In this case, our formulas for the amplitude of the ALD curve will be inaccurate if, for example, A and A′ have different admixture histories. However, if our assumptions are violated only by events occurring before the divergences between the mixing populations and the corresponding references, then the amplitude will be unaffected. Moreover, no matter what the population history is, as long as A and B are free of measurable LD (so that our assumption of independence of alleles conditional on a single ancestry is valid), there will be no effect on the estimated date of admixture.
Conclusions and future directions
In this study, we have shown how LD generated by population admixture can be a powerful tool for learning about history, extending previous work that showed how it can be used for estimating dates of mixture (Moorjani et al. 2011; Patterson et al. 2012). We have developed a new suite of tools, implemented in the ALDER software package, that substantially increases the speed of admixture LD analysis, improves the robustness of admixture date inference, and exploits the amplitude of LD as a novel source of information about history. In particular, (a) we show how admixture LD can be leveraged into a formal test for mixture that can sometimes find evidence of admixture not detectable by other methods, (b) we show how to estimate mixture proportions, and (c) we show that we can even use this information to infer phylogenetic relationships. A limitation of ALDER at present, however, is that it is designed for a model of pulse admixture between two ancestral populations. Important directions for future work will be to generalize these ideas to make inferences about the time course of admixture in the case that it took place over a longer period of time (Pool and Nielsen 2009; Gravel 2012) and to study multiway admixture. In addition, it would be valuable to be able to use the information from admixture LD to constrain models of history for multiple populations simultaneously, either by extending ALDER itself or by using LD-based test results in conjunction with methods for fitting phylogenies incorporating admixture (Lipson et al. 2012; Patterson et al. 2012; Pickrell and Pritchard, 2012).
Software
Executable and C++ source files for our ALDER software package are available online at the Berger and Reich Lab websites: http://groups.csail.mit.edu/cb/alder/, http://genetics.med.harvard.edu/reich/Reich_Lab/Software.html.
Acknowledgments
We are grateful to the reviewer for many suggestions that substantially increased the quality of this manuscript. M.L. and P.L. acknowledge National Science Foundation (NSF) Graduate Research Fellowship support. P.L. was also partially supported by National Institutes of Health (NIH) training grant 5T32HG004947-04 and the Simons Foundation. N.P., J.P. and D.R. are grateful for support from NSF HOMINID grant 1032255 and NIH grant GM100233.
Appendix A: Derivations of Weighted LD Formulas
Expected Weighted LD Using Two Diverged Reference Populations
We now derive Equation 6 for the expected weighted LD (with respect to random drift) using references A′ and B′ in place of A and B, retaining the notation of Figure 1. Let A′ and B′ have allele frequencies and , and let denote the allele frequency divergences with which we weight the LD z(x, y), giving the two-site statistic
[For brevity, we drop the binning procedure of averaging over SNP pairs (x, y) at distance |x − y| ≈ d here.] The value of the random variable z(x, y) is affected by sampling noise as well as genetic drift between A and B, while the random variables δ′(x) and δ′(y) are outcomes of genetic drift between A′ and B′. These random variables are uncorrelated conditional on the allele frequencies of x and y in A″ and B″. We also assume that x and y are distant enough to have negligible background LD and hence the drifts at the two sites are independent. We then have
where in the last step the relation E[δ(x)δ′(x)] = E[δ(y)δ′(y)] = F2(A″, B″) follows from the fact that the intersection of the drift paths δ(⋅) and δ′(⋅) is the branch between A″ and B″ (Reich et al. 2009).
Expected Weighted LD Using One Diverged Reference Population
Using the admixed population C as one reference and a population A′ as the other, we have pC(⋅) = αpA(⋅) + βpB(⋅) (assuming negligible postadmixture drift), giving weights
where δPQ denotes the allele frequency difference between populations P and Q. Arguing as above, the expected weighted LD is given by
To complete the calculation, we compute
For the first term, the intersection of the A–B and A′–A drift paths is the A–A″ branch, so with the negative sign arising because the paths traverse this branch in opposite directions. For the second term, the intersection of the A–B and A′–B drift paths is the A″–B branch (traversed in the same direction), so . Combining these results gives Equation 8. (Note that a slight subtlety arises now that we are using population C in our weights: sites x and y can exhibit admixture LD at appreciable distances, so and are not independent. However, only the portions of and arising from postadmixture drift are correlated, and this drift is negligible for typical scenarios we study in which admixture occurred 200 or fewer generations ago.)
Bounding Mixture Fractions Using One Reference
We now establish our claim in the main text that the estimator given in Equation 12 for the mixture fraction α is a lower bound when the reference population A′ is diverged from A. Equation 12 gives a correct estimate when A′ = A but becomes an approximation when there is genetic drift between A and A′ or between C and C′. (For accuracy, in this section we relax our usual assumption of negligible drift from C to C′.)
Rearranging Equation 12, we have by definition
| (A1) |
From Equation 7, the amplitude is in truth given by
where we have included the postadmixture drift multiplier from the C–C′ branch. It follows that
| (A2) |
We claim that F2(A′, C′)2 > (−αβF2(A, A″) + β2F2(B, A″))2, in which case combining (A1) and (A2) gives and hence . Indeed, we have
Squaring both sides appears to give our claim, but we must be careful because it is possible for the final expression to be negative. We assume that A′ is closer to A than B, i.e., F2(A, A″) < F2(B, A″). Then, if α < β, the final expression is clearly positive. If α > β, we have α2F2(A, A″) > αβF2(A, A″) and so
Thus, squaring the inequality is valid in either case, establishing our bound. From the above we also see that the accuracy of the bound depends on the sizes of the terms that are lost in the approximation—αF2(A, A″), F2(A′, A″), and F2(C, C′)—relative to the term that is kept, β2F2(B, A″). In particular, aside from the bound being tighter the closer A′ is to A, it is also more useful when the reference A′ comes from the minor side α < 0.5.
Affine Term from Population Substructure
In the above, we have assumed that population C is homogeneously admixed; i.e., an allele in any random admixed individual from C has a fixed probability α of having ancestry from A and β of having ancestry from B. In practice, many admixed populations experience assortative mating such that subgroups within the population have varying amounts of each ancestry. Heterogeneous admixture among subpopulations creates LD that is independent of genetic distance and not broken down by recombination: intuitively, knowing the value of an allele in one individual changes the prior on the ancestry proportions of that individual, thereby providing information about all other alleles (even those on other chromosomes). This phenomenon causes weighted LD curves to exhibit a nonzero horizontal asymptote, the form of which we now derive.
We model assortative mating by taking α to be a random variable rather than a fixed probability, representing the fact that individuals from different subpopulations of C have different priors on their A ancestry. As before we set β := 1 − α and we now denote by and the population-wide mean ancestry proportions; thus, . We compute the expected diploid covariance E[z(x, y)], which we saw in Equation 2 splits into four terms corresponding to the LD between each copy of the x allele and each copy of the y allele.
Previously, the cross-terms cov(X1, Y2) and cov(X2, Y1) vanished because a homogeneously mixed population does not exhibit interchromosome LD. Now, however, writing cov(X1, Y2) = E[(X1 − μx)(Y2 − μy)] as an expectation over individuals from C in the usual way, we find if we condition on the prior α for A ancestry,
That is, subpopulations with different amounts of A ancestry make nonzero contributions to the covariance. We can now compute cov(X1, Y2) by taking the expectation of the above over the whole population (i.e., over the random variable α)
| (A3) |
and likewise for cov(X2, Y1).
To compute the same-chromosome covariance terms, we split into two cases according to whether or not recombination has occurred between x and y since admixture. In the case that recombination has not occurred—i.e., the ancestry of the chromosomal region between x and y can be traced back as one single chunk to the time of admixture, which occurs with probability e−nd—the region from x to y has ancestry from A with probability α and from B with probability β. Thus,
Taking the expectation over the whole population,
| (A4) |
as without assortative mating.
In the case where there has been a recombination, the loci are independent conditioned upon the ancestry proportion α, as in our calculation of the cross-terms; hence,
| (A5) |
and this occurs with probability 1 − e−nd.
Combining Equations A3, A4, and A5, we obtain
Importantly, our final expression for E[z(x, y)] still factors as the product of a d-dependent term—now an exponential decay plus a constant—and the allele frequency divergences δ(x)δ(y). Because it is the product δ(x)δ(y) that interacts with our various weighting schemes, the formulas that we have derived for the weighted LD curve E[a(d)]—Equations 4, 6, 7, and 8—retain the same factors involving F2 distances and change only in the replacement of 2αβe−nd with .
Appendix B: Testing for Admixture
Here we provide details of the weighted LD-based test for admixture we implement in ALDER. The test procedure is summarized in the main text; we focus here on technical aspects not given explicitly in Methods.
Determining the Extent of LD Correlation
The first step of ALDER estimates the distance to which LD in the test population is correlated with LD in each reference population. Such correlation suggests shared demographic history that can confound the ALD signal, so it is important to determine the distance to which LD correlation extends and analyze weighted LD curves only for d greater than this threshold. Our procedure is as follows. We successively compute LD correlation for SNP pairs (x, y) within distance bins dk < |x − y| < dk+1, where dk = kr for some bin resolution r (0.05 cM by default). For each SNP pair (x, y) within a bin, we estimate the LD (i.e., sample covariance between allele counts at x and y) in the test population and the LD in the reference population. We then form the correlation coefficient between the test LD estimates and reference LD estimates over all SNP pairs in the bin. We jackknife over chromosomes to estimate a standard error on the correlation, and we set our threshold after the second bin for which the correlation is insignificant (P > 0.05). To reduce dependence on sample size, we then repeat this procedure with successively increasing resolutions up to 0.1 cM and set the final threshold as the maximum of the cutoffs obtained.
Determining Significance of a Weighted LD Curve
To define a formal test for admixture based on weighted LD, we need to estimate the significance of an observed weighted LD curve . This question is statistically subtle for several reasons. First, the null distribution of the curve is complex. Clearly the test population C should not be admixed under the null hypothesis, but as we have discussed, shared demography—particularly bottlenecks—can also produce weighted LD. We circumvent this issue by using the pretests described in the next section and assume that if the test triple (C; A′, B′) passes the pretests, then under the null hypothesis, nonadmixture demographic events have negligible effect on weighted LD beyond the correlation threshold computed above. Even so, the curve still cannot be modeled as random white noise: because SNPs contribute to multiple bins, the curve typically exhibits noticeable autocorrelation. Finally, even if we ignore the issue of colored noise, the question of distinguishing a curve of any type—in our case, an exponential decay—from noise is technically subtle: the difficulty is that a singularity arises in the likelihood surface when the amplitude vanishes, which is precisely the hypothesis that we wish to test (Davies 1977).
In light of these considerations, we estimate a P-value using the following procedure, which we feel is well justified despite not being entirely theoretically rigorous. We perform jackknife replicates of the curve computation and fitting, leaving out one chromosome in each replicate, and estimate a standard error for the amplitude and decay constant of the curve using the usual jackknife procedure. We obtain a “z-score” for the amplitude and the decay constant by dividing each by its estimated standard error. Finally, we take the minimum (i.e., less significant) of these z-scores and convert it to a P-value assuming it comes from a standard normal; we report this P-value as our final significance estimate.
Our intuition for this procedure is that checking the z-score of the decay constant essentially tells us whether or not the exponential decay is well determined: if the curve is actually just noise, then the fitting of jackknife replicates should fluctuate substantially. On the other hand, if the curve has a stable exponential decay constant, then we have good evidence that is actually well fit by an exponential—and in particular, the amplitude of the exponential is nonzero, meaning we are away from the singularity. In this case the technical difficulty is no longer an issue and the jackknife estimate of the amplitude should in fact give us a good estimate of a z-score that is approximately normal under the null. The z-score for the decay constant certainly is not normally distributed—in particular, it is always positive—but taking the minimum of these two scores makes the test only more conservative.
Perhaps most importantly, we have compelling empirical evidence that our z-scores are well behaved under the null. We applied our test to nine HGDP populations that neither ALDER nor the three-population test identified as admixed; for each test population, we used as references all populations with correlated LD detectable to no more than 0.5 cM. These test triples thus comprise a suite of approximately null tests. We computed Q–Q plots for the reported z-scores and observed that for z > 0 (our region of interest), our reported z-scores follow the normal distribution reasonably well, generally erring slightly on the conservative side (Figure S6). These findings give strong evidence that our significance calculation is sufficiently accurate for practical purposes; in reality, model violation is likely to exert stronger effects than the approximation error in our P-values, and although our empirical tests cannot probe the tail behavior of our statistic, for practical purposes the precise values of P-values less than, say, 10−6 are generally inconsequential.
Pretest Thresholds
To ensure that our test is applicable to a given triple (C; A′, B′), we need to rule out the possibility of demography producing non-admixture-related weighted LD. We do so by computing weighted LD curves for C with weights A′–B′, A′–C, and B′–C and fitting an exponential to each curve. To eliminate the possibility of a shared ancestral bottleneck between C and one of the references, we check that the three estimated amplitudes and decay constants are well determined; explicitly, we compute a jackknife-based standard error for each parameter and require the implied P-value for the parameter being positive to be <0.05. If so, we conclude that whatever LD is present is due to admixture, not other demography, and we report the P-value estimate defined above for the significance of the A′–B′ curve as the P-value of our test.
We are aware of one demographic scenario in which the ALDER test could potentially return a finding of admixture when the test population is not in fact admixed. As illustrated in Figure S7, this would occur when A′ and C have experienced a shared bottleneck and C has subsequently had a further period of low population size. We do not believe that we have ever encountered such a false-positive admixture signal, but to guard against it, we note that if it were to occur, the three decay time constants for the reference pairs A′–B′, A′–C, and B′–C would disagree. Thus, along with the test results, ALDER returns a warning whenever the three best-fit values of the decay constant do not agree to within 25%.
Multiple-Hypothesis Correction
In determining statistical significance of test results when testing a population using many pairs of references, we apply a multiple-hypothesis correction that takes into account the number of tests being run. Because some populations in the reference set may be very similar, however, the tests may not be independent. We therefore compute an effective number nr of distinct references by running PCA on the allele frequency matrix of the reference populations; we take nr to be the number of singular values required to account for 90% of the total variance. Finally, we apply a Bonferroni correction to the P-values from each test using the effective number of reference pairs.
Appendix C: Coalescent Simulations
Here we further validate and explore the properties of weighted LD with entirely in silico simulations using the Markovian coalescent simulator MaCS (Chen et al. 2009). These simulations complement the exposition in the main text in which we constructed simulated admixed chromosomes by piecing together haplotype fragments from real HapMap individuals.
Effect of Divergence and Drift on Weighted LD Amplitude
To illustrate the effect of using reference populations with varying evolutionary distances from true mixing populations, we performed a set of four simulations in which we varied one reference population in a pair of dimensions: (1) time depth of divergence from the true ancestor and (2) drift since divergence. In each case, we simulated individuals from three populations A′, C′, and B′, with 22% of C′s ancestry derived from a pulse of admixture 40 generations ago from B, where A′ and B′ diverged 1000 generations ago. We simulated five chromosomes of 100 Mb each for 20 diploid individuals from each of A′ and B′ and 30 individuals from C′, with diploid genotypes produced by randomly combining pairs of haploid chromosomes. We assumed an effective population size of 10,000 and set the recombination rate to 10−8. We set the mutation rate parameter to 10−9 to have the same effect as using a mutation rate of 10−8 and then thinning the data by a factor of 10 (as it would otherwise have produced an unnecessarily large number of SNPs). Finally, we set the MaCS history parameter (the Markovian order of the simulation, i.e., the distance to which the full ancestral recombination graph is maintained) to 104 bases.
For the first simulation (Figure S1A), we set the divergence of A′ and C′ to be immediately prior to the gene flow event, altogether resulting in the following MaCS command:
For the second simulation (Figure S1B), we increased the drift along the A′ terminal branch by reducing the population size by a factor of 20 for the past 40 generations:
For the third and fourth simulations (Figure S1, C and D), we changed the divergence time of A′ and C′ from 41 to 520 generations, half the distance to the root:
We computed weighted LD curves using A′–B′ references (Figure S1), and the results corroborate our derivation and discussion of Equation 6. In all cases, the estimated date of admixture is within statistical error of the simulated 40-generation age. The amplitude of the weighted LD curve is unaffected by drift in A′ but is substantially reduced by the shorter distance F2(A″, B″) in the latter two simulations. Increased drift to A′ does, however, make the weighted LD curves in the right side somewhat noisier than those on the left.
Validation of Pretest Criteria in Test for Admixture
To understand the effects of the pretest criteria stipulated in our LD-based test for admixture, we simulated a variety of population histories with and without mixture. In each case we used the same basic parameter settings as above, except we set the root of each tree to be 4000 generations ago and we simulated 10 chromosomes for each individual instead of 5.
Scenario 1: True admixture 40 generations ago; reference A′ diverged 400 generations ago (similar to Figure S1C). All pretests pass and our test correctly identifies admixture.
Scenario 2: True admixture 40 generations ago; reference A′ diverged 41 generations ago (similar to Figure S1A). Because of the proximity of the admixed population C′ and the reference A′, the test detects long-range correlated LD and concludes that using A′ as a reference may produce unreliable results.
Scenario 3: True admixture 40 generations ago; contemporaneous gene flow (of half the magnitude) to the lineage of the reference population A′ as well. Again, the pretest detects long-range correlated LD and concludes that A′ is an unsuitable reference.
Scenario 4: No admixture; A and C simply form a clade diverging at half the distance to the root (similar to Figure S1C without the gene flow). The test finds no evidence for admixture; weighted LD measurements do not exhibit a decay curve.
Scenario 5: No admixture; A and C diverged 40 generations ago. As above, the test finds no decay in weighted LD. In this scenario the pretest does detect substantial correlated LD to 1.95 cM because of the proximity of A and C.
Scenario 6: No admixture; same setup as scenario 4 with addition of recent bottleneck in population C (100-fold reduced population size for the past 40 generations). Here, the test finds no weighted LD decay in the two-reference curve and concludes that there is no evidence for admixture. It does, however, detect decay curves in both one-reference curves (with A–C and B–C weights); these arise because of the strong bottleneck-induced LD within population C.
Scenario 7: No admixture; shared bottleneck: A and C diverged 40 generations ago and their common ancestor underwent a bottleneck of 100-fold reduced population size for the preceding 40 generations. In this case the pretest detects an enormous amount of correlated LD between A and C and deems A an unsuitable reference.
Sensitivity Comparison of Three-Population Test and LD-Based Test for Admixture
Here we compare the sensitivities of the allele frequency moment-based three-population test (Reich et al. 2009; Patterson et al. 2012) and our LD-based test for admixture. We simulated a total of 450 admixture scenarios in which we varied three parameters: the age of the branch point A″ (1000, 2000, and 3000 generations), the date n of gene flow (20–300 in increments of 20), and the fraction α of A ancestry (50–95% in increments of 5%), as depicted in Figure S5. In each case we simulated 40 admixed individuals, otherwise using the same parameter settings as in the scenarios above. Explicitly, we ran the commands:
where tMix and tSplit correspond to n and the age of A″, while migRate and tMixStop produce a pulse of gene flow from the B′ branch giving C′ a fraction α of A ancestry.
We then ran both the three-population test (f3) and the ALDER test on C′ using A′ and B′ as references (Figure S5). The results of these simulations show clearly that the two tests do indeed have complementary parameter ranges of sensitivity. We first observe that the f3 test is essentially unaffected by the age of admixture (up to the 300 generations we investigate here). As discussed in the main text, its sensitivity is constrained by competition between the admixture signal of magnitude αβF2(A″, B″) and the “off-tree drift” arising from branches off the lineage connecting A′ and B′ (Reich et al. 2009)—in this case, essentially the quantity α2F2(A″, C′). Thus, as the divergence point A″ moves up the lineage, the threshold value of α below which the f3 test can detect mixture decreases.
The ALDER tests behave rather differently, exhibiting a drop-off in sensitivity as the age of admixture increases, with visible noise near the thresholds of sufficient sensitivity. The difference between the f3 and ALDER results is most notable in Figure S5, bottom, where the reference A′ is substantially diverged from C′. In this case, ALDER is still able to identify small amounts of admixture from the B′ branch, whereas the f3 test cannot. Also notable are the vertical swaths of failed tests centered near α = 0.9, 0.75, and 0.65 for A″ respectively located at distances 0.75, 0.5, and 0.25 along the branch from the root to A′. This feature of the results arises because the amplitude of the single-reference weighted LD curve with A′–C′ weights vanishes near those values of α (see Equation 8 and Figure S2), causing the ALDER pretest to fail. (The two-reference weighted LD exhibits a clear decay curve, but the pretest is being overly conservative in these cases.) Finally, we also observe that for the smallest choice of mixture age (20 generations), many ALDER tests fail. In these cases, the pretest detects long-range correlated LD with the reference B′ and is again overly conservative.
Effect of Protracted Admixture on Weighted LD
The admixture model that we analyze in this article treats admixture as occurring instantaneously in a single pulse of gene flow; however, in real human populations, admixture typically occurs continuously over an extended period of time. Here we explore the effect of protracted admixture on weighted LD curves by simulating scenarios involving continuous migration. We used a setup nearly identical to the simulations above for comparing the f3 and ALDER tests, except here we modified the migration rate and start and end times to correspond to 40% B ancestry that continuously mixed into population C over a period of 0–200 generations ending 40 generations ago. We varied the duration of admixture in increments of 20 generations.
For each simulation, we used ALDER to compute the two-reference weighted LD curve and fit an exponential decay. In each case the date of admixture estimated by ALDER (Figure S4A) falls within the time interval of continuous mixture, as expected (Moorjani et al. 2011). For shorter durations of admixture spanning up to 50 generations or so, the estimated date falls very near the middle of the interval, while it is downward biased for mixtures extending back to hundreds of generations. The amplitude of the fitted exponential also exhibits a downward bias as the mixture duration increases (Figure S4B). This behavior occurs because unlike the point admixture case, in which the weighted LD curve follows a simple exponential decay (Figure S4C), continuous admixture creates weighted LD that is an average of exponentials with different decay constants (Figure S4D).
Footnotes
Communicating editor: J. Wall
Literature Cited
- Bramanti B., Thomas M., Haak W., Unterlaender M., Jores P., et al. , 2009. Genetic discontinuity between local hunter–gatherers and Central Europe’s first farmers. Science 326(5949): 137–140. [DOI] [PubMed] [Google Scholar]
- Chakraborty R., Weiss K., 1988. Admixture as a tool for finding linked genes and detecting that difference from allelic association between loci. Proc. Natl. Acad. Sci. USA 85(23): 9119–9123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen G., Marjoram P., Wall J., 2009. Fast and flexible simulation of DNA sequence data. Genome Res. 19(1): 136–142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chikhi L., Bruford M., Beaumont M., 2001. Estimation of admixture proportions: a likelihood-based approach using Markov chain Monte Carlo. Genetics 158: 1347–1362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies R., 1977. Hypothesis testing when a nuisance parameter is present only under the alternative. Biometrika 64(2): 247–254. [DOI] [PubMed] [Google Scholar]
- Dupanloup I., Bertorelle G., Chikhi L., Barbujani G., 2004. Estimating the impact of prehistoric admixture on the genome of Europeans. Mol. Biol. Evol. 21(7): 1361–1372. [DOI] [PubMed] [Google Scholar]
- Gravel S., 2012. Population genetics models of local ancestry. Genetics 191: 607–619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gravel S., Henn B., Gutenkunst R., Indap A., Marth G., et al. , 2011. Demographic history and rare allele sharing among human populations. Proc. Natl. Acad. Sci. USA 108(29): 11983–11988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Green R., Krause J., Briggs A., Maricic T., Stenzel U., et al. , 2010. A draft sequence of the Neandertal genome. Science 328(5979): 710–722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hammer M., Horai S., 1995. Y chromosomal DNA variation and the peopling of Japan. Am. J. Hum. Genet. 56(4): 951–962. [PMC free article] [PubMed] [Google Scholar]
- Hammer M., Karafet T., Park H., Omoto K., Harihara S., et al. , 2006. Dual origins of the Japanese: common ground for hunter–gatherer and farmer Y chromosomes. J. Hum. Genet. 51(1): 47–58. [DOI] [PubMed] [Google Scholar]
- International HapMap Consortium , 2007. A second generation human haplotype map of over 3.1 million SNPs. Nature 449(7164): 851–861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- International HapMap Consortium , 2010. Integrating common and rare genetic variation in diverse human populations. Nature 467(7311): 52–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jarvis J., Scheinfeldt L., Soi S., Lambert C., Omberg L., et al. , 2012. Patterns of ancestry, signatures of natural selection, and genetic association with stature in western African Pygmies. PLoS Genet. 8(4): e1002641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laval G., Patin E., Barreiro L., Quintana-Murci L., 2010. Formulating a historical and demographic model of recent human evolution based on resequencing data from noncoding regions. PLoS ONE 5(4): e10284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawson D., Hellenthal G., Myers S., Falush D., 2012. Inference of population structure using dense haplotype data. PLoS Genet. 8(1): e1002453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J., Absher D., Tang H., Southwick A., Casto A., et al. , 2008. Worldwide human relationships inferred from genome-wide patterns of variation. Science 319(5866): 1100–1104. [DOI] [PubMed] [Google Scholar]
- Lipson M., Loh P., Levin A., Reich D., Patterson N., et al. , 2012. Efficient moment-based inference of admixture parameters and sources of gene flow, arXiv arXiv:1212.2555 (in press). [DOI] [PMC free article] [PubMed]
- Moorjani P., Patterson N., Hirschhorn J., Keinan A., Hao L., et al. , 2011. The history of African gene flow into Southern Europeans, Levantines, and Jews. PLoS Genet. 7(4): e1001373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moorjani P., Patterson N., Loh P., Lipson M., Kisfali P., et al. , 2012. Reconstructing Roma history from genome-wide data., arXiv arXiv:1212.1696 (in press). [DOI] [PMC free article] [PubMed]
- Ohta T., Kimura M., 1971. Linkage disequilibrium between two segregating nucleotide sites under the steady flux of mutations in a finite population. Genetics 68: 571–580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patin E., Laval G., Barreiro L., Salas A., Semino O., et al. , 2009. Inferring the demographic history of African farmers and Pygmy hunter–gatherers using a multilocus resequencing data set. PLoS Genet. 5(4): e1000448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patterson N., Hattangadi N., Lane B., Lohmueller K., Hafler D., et al. , 2004. Methods for high-density admixture mapping of disease genes. Am. J. Hum. Genet. 74(5): 979–1000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patterson N., Price A., Reich D., 2006. Population structure and eigenanalysis. PLoS Genet. 2(12): e190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patterson N., Moorjani P., Luo Y., Mallick S., Rohland N., et al. , 2012. Ancient admixture in human history. Genetics 192: 1065–1093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pickrell J., Pritchard J., 2012. Inference of population splits and mixtures from genome-wide allele frequency data. PLoS Genet. 8(11): e1002967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pickrell J., Patterson N., Barbieri C., Berthold F., Gerlach L., et al. , 2012. The genetic prehistory of southern Africa. Nat. Commun. 3(1): 1143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinhasi R., Thomas M., Hofreiter M., Currat M., Burger J., 2012. The genetic history of Europeans. Trends Genet. 28(10): 496–505. [DOI] [PubMed] [Google Scholar]
- Pool J., Nielsen R., 2009. Inference of historical changes in migration rate from the lengths of migrant tracts. Genetics 181: 711–719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Price A., Weale M., Patterson N., Myers S., Need A., et al. , 2008. Long-range LD can confound genome scans in admixed populations. Am. J. Hum. Genet. 83(1): 132–135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Price A., Tandon A., Patterson N., Barnes K., Rafaels N., et al. , 2009. Sensitive detection of chromosomal segments of distinct ancestry in admixed populations. PLoS Genet. 5(6): e1000519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pritchard J., Stephens M., Donnelly P., 2000. Inference of population structure using multilocus genotype data. Genetics 155: 945–959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pugach I., Matveyev R., Wollstein A., Kayser M., Stoneking M., 2011. Dating the age of admixture via wavelet transform analysis of genome-wide data. Genome Biol. 12(2): R19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quintana-Murci L., Quach H., Harmant C., Luca F., Massonnet B., et al. , 2008. Maternal traces of deep common ancestry and asymmetric gene flow between Pygmy hunter–gatherers and Bantu-speaking farmers. Proc. Natl. Acad. Sci. USA 105(5): 1596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rasteiro R., Chikhi L., 2009. Revisiting the peopling of Japan: an admixture perspective. J. Hum. Genet. 54(6): 349–354. [DOI] [PubMed] [Google Scholar]
- Reich D., Cargill M., Bolk S., Ireland J., Sabeti P., et al. , 2001. Linkage disequilibrium in the human genome. Nature 411(6834): 199–204. [DOI] [PubMed] [Google Scholar]
- Reich D., Thangaraj K., Patterson N., Price A., Singh L., 2009. Reconstructing Indian population history. Nature 461(7263): 489–494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenberg N., Pritchard J., Weber J., Cann H., Kidd K., et al. , 2002. Genetic structure of human populations. Science 298(5602): 2381–2385. [DOI] [PubMed] [Google Scholar]
- Sankararaman S., Sridhar S., Kimmel G., Halperin E., 2008. Estimating local ancestry in admixed populations. Am. J. Hum. Genet. 82(2): 290–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soares P., Achilli A., Semino O., Davies W., Macaulay V., et al. , 2010. The archaeogenetics of Europe. Curr. Biol. 20(4): R174–R183. [DOI] [PubMed] [Google Scholar]
- Sousa V., Fritz M., Beaumont M., Chikhi L., 2009. Approximate Bayesian computation without summary statistics: the case of admixture. Genetics 181: 1507–1519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang H., Coram M., Wang P., Zhu X., Risch N., 2006. Reconstructing genetic ancestry blocks in admixed individuals. Am. J. Hum. Genet. 79(1): 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verdu P., Austerlitz F., Estoup A., Vitalis R., Georges M., et al. , 2009. Origins and genetic diversity of Pygmy hunter–gatherers from western Central Africa. Curr. Biol. 19(4): 312–318. [DOI] [PubMed] [Google Scholar]
- Wall J., Lohmueller K., Plagnol V., 2009. Detecting ancient admixture and estimating demographic parameters in multiple human populations. Mol. Biol. Evol. 26(8): 1823–1827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J., 2003. Maximum-likelihood estimation of admixture proportions from genetic data. Genetics 164: 747–765. [DOI] [PMC free article] [PubMed] [Google Scholar]