

Abstract
Exogenous glucagon-like peptide-2 receptor (GLP-2R) activation elicits proliferative and cytoprotective responses in the gastrointestinal mucosa and ameliorates experimental small and large bowel gut injury. Nevertheless, the essential physiological role(s) of the endogenous GLP-2R remain poorly understood. We studied the importance of the GLP-2R for gut growth, epithelial cell lineage allocation, the response to mucosal injury, and host-bacterial interactions in Glp2r−/− and littermate control Glp2r+/+ mice. Glp2r−/− mice exhibit normal somatic growth and preserved small and large bowel responses to IGF-I and keratinocyte growth factor. However, Glp2r−/− mice failed to up-regulate intestinal epithelial c-fos expression in response to acute GLP-2 administration and do not exhibit changes in small bowel conductance or small or large bowel growth after administration of GLP-2R agonists. The crypt and villus compartment and the numbers and localization of Paneth, enteroendocrine, and goblet cells were comparable in Glp2r+/+ vs. Glp2r−/− mice. Although the severity and extent of colonic mucosal injury in response to 3% oral dextran sulfate was similar across Glp2r genotypes, Glp2r−/− mice exhibited significantly increased morbidity and mortality and increased bacterial translocation after induction of enteritis with indomethacin and enhanced mucosal injury in response to irinotecan. Moreover, bacterial colonization of the small bowel was significantly increased, expression of Paneth cell antimicrobial gene products was reduced, and mucosal bactericidal activity was impaired in Glp2r−/− mice. Although the Glp2r is dispensable for gut development and the response to colonic injury, Glp2r−/− mice exhibit enhanced sensitivity to small bowel injury, and abnormal host-bacterial interactions in the small bowel.
Glucagon-like peptide-2 (GLP-2) is a member of the proglucagon-derived peptide family produced in gut endocrine L cells. GLP-2 is secreted at low basal levels in the fasting state and its circulating levels rise rapidly after nutrient ingestion (1). Exogenous administration of GLP-2 expands the small and large bowel mucosa through stimulation of crypt cell proliferation and inhibition of cell death (2). These actions of GLP-2 are conserved in the setting of experimental intestinal injury, leading to preservation and/or regeneration of damaged mucosal epithelium.
GLP-2 also increases intestinal blood flow (3, 4) and gut barrier function (5) and enhances nutrient absorption via stimulation of glucose transport activity (6, 7). Moreover, GLP-2 infusion enhances lipid absorption and chylomicron secretion in mice (8) and increases postprandial levels of triglycerides and free fatty acids in normal human subjects (9). The ability of GLP-2 to enhance nutrient absorption has led to its evaluation for the treatment of short bowel syndrome. Once- or twice-daily administration of a GLP-2 analog, teduglutide, increased the nutrient absorption and reduced energy malabsorption in human subjects with short bowel syndrome (10, 11).
Despite the delineation of multiple actions arising after exogenous pharmacological GLP-2 administration, the endogenous physiological action(s) of GLP-2 remain less well defined. Acute administration of a GLP-2 antagonist GLP-2 (3–33) partially attenuates the normal adaptive response of the small bowel mucosa to fasting/refeeding through modulation of crypt cell proliferation and apoptosis (12, 13). Similarly, attenuation of circulating GLP-2 bioactivity using immunoneutralizing antibodies reduced the extent of adaptive mucosal hyperplasia in rats with streptozotocin-induced diabetes (14).
Elucidation of mechanisms underlying endogenous GLP-2 action has been challenging in part due to the limitations of available GLP-2 receptor antagonists and the complexity of the cellular localization of GLP-2 receptor signaling. The antagonist GLP-2 (3–33) is also a weak partial agonist (12, 15), limiting its suitability for complete sustained blockade of GLP-2 action in vivo. Moreover, transient selective reduction of GLP-2 receptor (GLP-2R) expression is challenging because the GLP-2R is expressed on rare subsets of enteric neurons, enteroendocrine cells, and myofibroblasts and exerts its actions indirectly, through activation of incompletely characterized downstream mediators (16 –18).
To delineate the importance of the endogenous GLP-2R, we generated mice with targeted inactivation of the gene encoding the GLP-2R. Glp2r−/− mice fail to up-regulate intestinal growth factor expression in response to acute GLP-2 administration (18), manifest defective up-regulation of crypt cell proliferation in response to fasting-refeeding (19), but do not exhibit defects in the intestinal adaptive response to experimental streptozotocin-induced diabetes (20). We now report that germline loss of the Glp2r does not result in defects in cell lineage allocation within the gut epithelium, susceptibility to colonic injury, or defective control of intestinal permeability. However Glp2r−/− mice demonstrate increased susceptibility to mucosal injury induced by indomethacin or irinotecan in the small bowel. Furthermore, Glp2r−/− mice exhibit defective Paneth cell function as evidenced by reduced expression of defensins and impaired bactericidal activity ex vivo. Moreover, the small bowel of uninjured Glp2r−/− mice exhibit significantly increased numbers of bacteria, and microbiome analysis demonstrates a significant shift in the proportion of Firmicutes, Bacteroidetes, and Actinobacteria phyla in the feces. These findings reveal that basal GLP-2R activity is essential for the prevention of mucosal injury in the small bowel and the maintenance of normal host-bacteria interactions along the gastrointestinal tract.
Materials and Methods
Animals
Glp2r−/− mice were generated in the C57BL/6 background by replacing exons 7–9 of the Glp2r with a neomycin resistance cassette inserted in the opposite direction to that of Glp2r transcription (Supplemental Fig. 1, published on The Endocrine Society’s Journals Online web site at http://endo.endojournal-s.org). The genotyping strategy is shown in Supplemental Fig. 2. The wild-type Glp2r+/+ littermates were used as controls for all studies involving Glp2r−/− mice. Studies were performed on either male or female mice, as indicated in the figure legends, aged 8 –12 wk, that were bred at the Toronto Centre for Phenogenomics Animal facility. All animal experiments were approved by the Animal Care Committee of the Mount Sinai Hospital.
Peptide and drug treatments
Human [Gly2]GLP-2, hence referred to as GLP-2, was from Pepceutical Ltd. (Nottingham, UK), Long-R3-IGF-I (recombinant human IGF-I) was from Gropep Ltd. (Adelaide, Australia) and recombinant human (rHu) keratinocyte growth factor (KGF) was obtained from Amgen Inc. (Thousand Oaks, CA). GLP-2 and Long-R3-IGF-I were administered sc, whereas rHuKGF was injected into the peritoneum (ip). All peptides were administered in PBS. Experiments involving analysis of dextran sulfate colitis and indomethacin- or irinotecan-induced enteritis were carried out as previously described (21–23). To assess intestinal crypt cell proliferation 5-bromo-2′-deoxyuridine (BrdU; Sigma-Aldrich, Oakville, Ontario, Canada; 100 mg/kg) dissolved in PBS was injected ip to mice 1 h before the animals were killed.
Tissue collection and processing for morphometry and immunohistochemistry
Intestinal tissue was collected, flushed with PBS, fixed in 10% neutral buffered formalin, and paraffin embedded. Histomorphometry was performed on 5-μm sections stained with hematoxylin and eosin, using a Leica Q500MC Image Analysis System (Leica Inc., Cambridge, UK). Villus height and crypt depth were measured on 12 cross-sections with an average of 20 measurements per mouse. Immunohistochemistry was carried out using indirect immunoperoxidase detection with NovaRED substrate (Vector Laboratories, Burlington, Ontario, Canada) followed by hematoxylin counterstaining. Antilysozyme and antisynaptophysin rabbit polyclonal antibodies (DakoCytomation, Mississauga, Ontario, Canada) were used to quantify Paneth and enteroendocrine cell populations, respectively. BrdU incorporation was detected using a mouse monoclonal anti-BrdU antibody (Invitrogen Canada, Burlington, Ontario, Canada) and c-fos protein using a rabbit polyclonal anti-c-fos antibody (Sigma-Aldrich). Periodic acid-Schiff staining was used to visualize goblet cells.
Intestinal permeability
Two to three contiguous segments of proximal and midjejunum were cut along the mesenteric border, mounted in modified Ussing chambers (EasyMount chambers; Physiologic Instruments, San Diego, CA), and bathed with Kreb’s buffer [140 mM NaCl, 1.2 mM CaCl2, 1.2 mM MgCl2, 10 mM KHCO3, 0.2 mM KH2PO4, and 1.2 mM K2HPO4 (pH 7.4)] at 37 C with continuous oxygenation. Glucose (10 mM) was added to the buffer at the serosal surface balanced by 10 mM mannitol in the buffer at the mucosal surface. After equilibration for 20 min, tissues were short circuited at zero volts using an automatic multichannel voltage/current clamp (model VCC MC8; Physiologic Instruments). Tissue conductance was calculated according to Ohm’s law using potential difference and short circuit current values. Average short circuit current and conductance values were determined using Acquire and Analyze software (Physiologic Instruments).
Western blot analysis
Whole-tissue extracts were prepared by homogenization of intestinal segments in radioimmunoprecipitation assay buffer (1% Nonidet P-40, 0.5% sodium deoxycholate, and 0.1% sodium dodecyl sulfate in PBS) supplemented with protease and phosphatase inhibitors (Sigma-Aldrich), 5 mM sodium fluoride, and 200 μM sodium orthovanadate. Lectin adsorption was carried out using wheat germ agglutinin-agarose (EMD Chemicals Inc., Gibbstown, NJ) and elution with 0.3 M N-acetyl-D-glucosamine, as previously reported (24). Western blot analysis of lectin pull-down proteins was performed as described (25). GLP-2R antisera were generously provided by Professor Jens Holst (University of Copenhagen, Copenhagen, Denmark) (16).
Microbiology
Whole blood was obtained by cardiac puncture. Spleen, liver, and mesenteric lymph nodes were removed and homogenized in sterile PBS. Aliquots of whole blood and tissue homogenates were plated on blood agar plates and incubated at 37 C for 24 – 48 h under aerobic conditions before assessing the presence of bacterial colonies or quantifying the percent of the blood agar plate surface that underwent hemolysis. To determine small intestinal aerobic bacterial load, the luminal contents were collected by flushing with PBS. The number of colony-forming units per mouse small intestine was determined by plating serial dilutions of the flushed material on blood agar plates and counting colonies after incubation at 37 C for 24 h.
RNA isolation and quantitative real-time RT-PCR
Total RNA from intestinal tissue was extracted by the guanidinium thiocyanate method and cDNA synthesis performed with random hexamers and SuperScript II (Invitrogen). Real-time quantitative PCR was performed on an ABI PRISM 7900HT sequence detection system (Applied Biosystems, Foster City, CA) with TaqMan universal PCR master mix and TaqMan gene expression assays (Applied Biosystems) for glp2r (Mm01329473_ m1 for exons 2–3 and Mm01329477_m1 for exons 7–8), lysozyme P (Mm00657323_m1), defcr-rs1 (Mm00655850_m1), cryptdin 5 (Mm00651548_g1), pla2g2a (Mm00448160_m1), mmp7 (Mm00487724_m1), lgr5 (Mm00438890), epgn (Mm00504344_m1), egf (Mm00438696), erbB1 (Mm00433023), igf-1R (Mm00802831), il-6 (Mm00446190), myd88 (Mm00440338), nfkb (Mm00476361), mip-2 (Mm00436450), il-33 (Mm00505403), cxcl-10 (Mm00445235), and il-15 (Mm00434210). Relative quantification of transcript levels was performed by the 2−ΔΔCt method using the cycle threshold values obtained from the PCR amplification kinetics with the ABI PRISM SDS 2.1 software. 18S rRNA or cyclophilin was used for normalization because its intestinal expression remained unaltered, regardless of mouse genotype or treatment.
Crypt isolation, stimulation of Paneth cell secretion, and bactericidal activity assays
Small intestinal crypts were isolated after incubation of the tissue at room temperature in Ca2+- and Mg2+-free PBS containing 30 mM EDTA (26). Paneth cell secretion from the isolated crypts was induced by stimulation with the cholinergic agonist carbamylcholine (10 μM; Sigma-Aldrich) or the Toll-like receptor 4 agonist lipopolysaccharide (LPS; purified from Escherichia coli 0111:B4, 10 μg/ml; Sigma-Aldrich) for 30 min at 37 C. Crypt secretions were recovered by centrifugation and their bactericidal activity assayed against 5 × 106 colony-forming units E. coli DH5α as described (27).
Microbial genomic DNA extraction and analysis of the 16S rRNA locus
Fresh cecum and fecal pellets were collected and immediately frozen at −80 C. Samples were ground in liquid nitrogen and DNA extracted after microbial cell lysis with zirconia/silica beads as described (28). DNA fragments were sequenced by amplifying the 16S rRNA locus from each DNA sample using barcoded primers. Amplicons were pooled and carried through the standard Illumina paired-end sample prep. Six sequence reads were obtained (three from each strand); one sequence read from each strand corresponded to the sample-specific barcode (8 bp). The remaining four sequence reads (36 bp each) corresponded to the V5, V6, and V7 hypervariable regions of the 16S rRNA locus. These reads were concatenated and assigned bacterial taxonomies using the Ribosomal Database Project classifier (29), which had been trained using the Bergey database of sequences modified to have the same 36- × 4-bp read structure (rather than full length 16S rRNA sequences).
Statistics
Results are expressed as mean ± SE. Comparisons between genotype and/or treatment groups were performed by one- or two-way ANOVA followed by the Bonferroni or Fisher post hoc test. The χ2 test was used to analyze the data (see Fig. 3, A and C). Comparison between Kaplan-Meier survival curves shown (see Fig. 3B) was performed using the log-rank test. Statistical analyses were carried out using the GraphPad Prism 4 software package (GraphPad Software Inc., San Diego, CA) and Statistica 6 (StatSoft Inc., Tulsa, OK).
FIG. 3.

Indomethacin and irinotecan produce increased small bowel injury in Glp2r−/− mice. Proportion of moribund (A), percent survival (B), and percent positive (C) bacterial cultures in male Glp2r+/+, Glp2r+/−, and Glp2r−/− mice (n = 17–28 per group) after two sc injections of vehicle or indomethacin. *, P < 0.05, Glp2r−/− vs. Glp2r+/+ (for A and C). Jejunal crypt depth (D) and crypt density (E) and prevalence of hemolytic activity in blood (F), a reflection of bacterial septicemia, in male Glp2r−/− vs. Glp2r+/+ mice (n = 5– 6 per group) 24 h after two ip injections of vehicle or irinotecan (IRT). **, P < 0.01, ***, P < 0.001 IRT vs. vehicle; #, P < 0.05, Glp2r−/− vs. Glp2r+/+.
Results
Glp2r−/− mice were born at expected Mendelian frequencies and appeared healthy, and both body weight (Fig. 1A) and food intake (Supplemental Fig. 3) were comparable in Glp2r+/+ vs. Glp2r−/− mice. Glp2r mRNA transcripts corresponding to the targeted exons and immunoreactive GLP-2R protein were detected in intestinal tissues from Glp2r+/+ and Glp2r+/− but not Glp2r−/− mice (Supplemental Fig. 4, A and B). A single 5-μg injection of h[Gly2]-GLP-2 significantly increased nuclear c-fos expression in the epithelium of the jejunum (Fig. 1B and Supplemental Fig. 5) and colon (Supplemental Fig. 6) of Glp2r+/+ but not in Glp2r−/− mice. Basal levels of small and large bowel weight (Fig. 1C), jejunal villus height and crypt depth, colon crypt depth (Supplemental Figure 7), crypt cell proliferation, and jejunal conductance (Fig. 1, D and F) were comparable in Glp2r+/+ vs. Glp2r−/− mice. Sustained h[Gly2]-GLP-2 administration produced a robust intestinotrophic response in the small and large bowel of Glp2r+/+ mice but had no effect on jejunal ion conductance, small or large bowel weight, small bowel villus height, crypt depth, colonic mucosal thickness, DNA content, small bowel ion conductance, or crypt cell proliferation in Glp2r−/− mice (Fig. 1, C–F, and Supplemental Fig. 7). In contrast, administration of IGF-I or KGF increased small bowel mass (Fig. 1E) and KGF increased colonic mass (Supplemental Fig. 8) in Glp2r+/+ and Glp2r−/− mice. Cell lineage allocation (Goblet, enteroendocrine, and Paneth cells) was also comparable in Glp2r+/+ vs. Glp2r−/− mice (Supplemental Figs. 9 and 10). Taken together, these analyses demonstrate that the Glp2r−/− mouse fails to respond to acute or chronic GLP-2 treatment yet exhibits normal epithelial structure and cell composition in the gastrointestinal tract.
FIG. 1.

Characterization of Glp2r−/− mice. A, Body weights of male and female Glp2r+/+, Glp2r+/−, and Glp2r−/− littermates (n = 4 per group) over 24 wk. B, The c-fos-immunopositive cell number in the jejunal epithelium of Glp2r+/+ and Glp2r−/− male mice (n = 3– 4 per group) 1 h after GLP-2 (5 μg) administration. Small and large bowel weights (C) and jejunal crypt cell proliferation (D) after the administration of saline or GLP-2 (10 μg once daily) for 7 d in female Glp2r+/+, Glp2r+/−, and Glp2r−/− littermates (n = 7– 8 per group). E, Small bowel weight in response to GLP-2 (2.5 μg twice daily), Long-R3-IGF-I (25 μg twice daily), or rHuKGF (5 mg/kg body weight once daily) administration to female Glp2r+/+ and Glp2r−/− mice for 10 d (n = 5– 6 per group). F, The jejunal (Jej) transmural ion conductance from female Glp2r+/+ and Glp2r−/− mice (n = 3– 6 per group) administered saline or GLP-2 (5 μg twice daily) for 9 d. *, P < 0.05, ** or ##, P < 0.01, *** or ###, P < 0.001, treatment vs. vehicle control.
Because germline loss of the Glp2r was not associated with a demonstrable intestinal phenotype in nonstressed mice, we subjected Glp2r+/+, Glp2r+/−, and Glp2r−/− mice to experimental dextran sulfate (DS)-induced colitis, a controlled inflammatory insult responsive to exogenous GLP-2 administration (22, 30). Oral administration of 3% DS for 9 d produced comparable weight loss and similar changes in colon weight and length in Glp2r+/+ vs. Glp2r+/− and Glp2r−/− mice (Fig. 2, A–C). Furthermore, the extent of colonic mucosal injury was similar across Glp2r genotypes (Fig. 2, D and E). Body weight gain after recovery from 3% DS for 5– 8 d was also unaffected by Glp2r genotype (data not shown). Hence, loss of the Glp2r does not influence the severity of or recovery from DS-induced colonic injury.
FIG. 2.

Body weight (A), large bowel weight (B) and length (C), histology (D), and percentage intact mucosa (E) from male Glp2r+/+, Glp2r+/−, and Glp2r−/− mice treated with water alone (0% DS) or oral dextran sulfate (3% DS) in the drinking water for 9 d (n = 4 – 8 per group). ***, P < 0.001 0% vs. 3% DS-treated mice. Similar results were obtained in studies of female mice of the same genotypes (data not shown).
Because Glp2r mRNA transcripts are also present in the small bowel (31, 32), we assessed the consequences of non-steroidal antiinflammatory drug-induced small bowel enteritis in Glp2r−/− mice (21). The proportion of moribund Glp2r−/− mice was significantly higher compared with Glp2r+/+ littermate controls 24 h after indomethacin administration (Fig. 3A), and more Glp2r−/− mice succumbed after indomethacin administration (Fig. 3B). We next assessed whether enhanced bacterial infection might underlie the increased morbidity of Glp2r−/− mice. The proportion of in-domethacin-treatedGlp2r−/− mice exhibiting positive bacterial cultures in liver, spleen, and blood trended significantly higher (P < 0.01, χ2 test for trend) relative to levels in Glp2r+/+ mice, and significantly more Glp2r−/− mice exhibited positive mesenteric lymph node bacterial cultures (Fig. 3C).
To determine whether Glp2r−/− mice exhibited enhanced sensitivity to small bowel mucosal injury in a second independent experimental model, we assessed the acute effects of irinotecan, a chemotherapeutic agent with significant gastrointestinal toxicity (23). After irinotecan treatment, jejunal crypt depth was similarly decreased in both Glp2r+/+ and Glp2r−/− mice; however, only Glp2r−/− mice exhibited a significant reduction in crypt density (Fig. 3, D and E). Furthermore, hemolysis on blood agar plates, an indirect measure of systemic bacteremia, was increased to a significantly greater extent in Glp2r−/− vs. Glp2r+/+ mice after irinotecan (Fig. 3F). To explore the basis for the enhanced mucosal damage in response to irinotecan, we assessed expression of genes important for small bowel growth, survival, and inflammation. Irinotecan-treated Glp2r−/− mice exhibited significantly reduced levels of mRNA transcripts for the stem cell marker lgr5, the erythroblastic leukemia viral oncogene homolog (ErbB) ligand EGF, and the Igf1r. Conversely, levels of mRNA transcripts for epgn and ErbB1 were not as up-regulated in irinotecan-treated Glp2r−/− vs. Glp2r+/+ mice, whereas levels of the mRNA for the cytokine Il-6 were significantly up-regulated in Glp2r−/− compared with Glp2r+/+ small bowel (Fig. 4).
FIG. 4.

Defective gene expression in response to irinotecan (IRT) in the small bowel of Glp2r−/− mice. Relative jejunal mRNA levels of lgr5 (leucine rich repeat-containing G protein coupled receptor 5) (A), epgn (epigen) (B), egf (epidermal growth factor) (C), erbB1 (epidermal growth factor receptor) (D), igf-1r (IGF-I receptor), (E) and il-6 (IL-6) (F) from Glp2r+/+ and Glp2r−/− male mice (n = 4 – 6 per group) treated with IRT. *, P < 0.05, IRT vs. vehicle; #, P < 0.05, Glp2r−/− vs. Glp2r+/+.
Because increased bacterial infection may reflect altered gut barrier function and microbial colonization, we assessed intestinal bacterial colonization in Glp2r−/− mice. Glp2r−/− mice exhibited a significantly greater bacterial load in the small bowel relative to age and sex-matched littermate controls (Fig. 5A). To identify mechanisms underlying increased small bowel bacterial colonization in Glp2r−/− mice, we determined the expression of Paneth cell genes known to be important for host-bacterial interactions. Although levels of lysozyme and the defensin-related Defcr-rs1 (crypt1) mRNA transcripts were comparable in the jejunum and ileum of Glp2r−/− vs. Glp2r+/+ mice, levels of ileal matrix metalloproteinase 7 (Mmp7) mRNA, the key enzyme responsible for activation of defensin activity, and the abundance of cryptdin 5 and secretory phospholipase A2 (Pla2g2a) mRNA transcripts were lower in both jejunum and ileum of Glp2r−/− mice (Fig. 5B). Furthermore, we detected significantly reduced intestinal expression of multiple RNA transcripts encoding proteins involved in the host response to infection in Glp2r−/− mice, including myd88, nfkb, mip2, il-6, il-33, cxcl10, and il-15 (Fig. 5C).
FIG. 5.
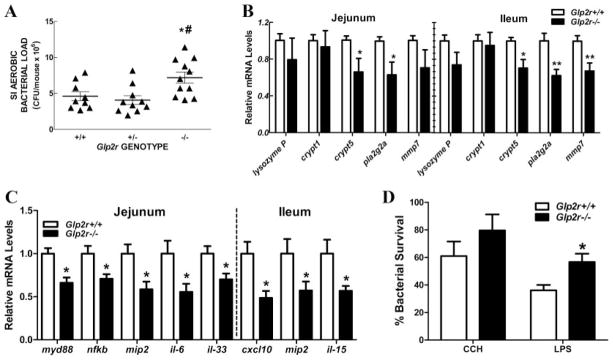
Intestinal bacterial load, gene expression profiles, and intestinal bactericidal activity in Glp2r−/− mice. A, Small intestinal aerobic bacterial load from Glp2r+/+, Glp2r+/−, and Glp2r−/− male mice (n = 11–13 per group). Each data point corresponds to one mouse. *, P < 0.05, and #, P < 0.01 for Glp2r−/− vs. Glp2r+/+ and Glp2r+/−, respectively. Relative mRNA levels of Paneth cell markers (B) and inflammation-related genes (C) in jejunum and ileum of male Glp2r−/− mice and wild-type littermate controls (n = 7–9 per group). *, P < 0.05, **, P < 0.01 Glp2r−/− vs. Glp2r+/+. D, Small intestinal crypts isolated from Glp2r+/+ and Glp2r−/− male mice (n = 4 –10 per group) were stimulated with carbamylcholine (CCH) or LPS and the supernatants assayed for bactericidal activity. *, P < 0.05, Glp2r−/− vs. Glp2r+/+.
The increased bacterial load, enhanced bacterial translocation, and altered Paneth gene expression signature suggested the presence of a functional Paneth cell defect. Consistent with this hypothesis, Paneth cell secretions isolated from Glp2r−/− mice exhibited reduced bactericidal activity after stimulation with carbamylcholine and LPS ex vivo (Fig. 5D).
To further explore whether loss of the Glp2r impaired host-microbiome interactions, we assessed the proportion of key bacterial phyla in the cecum and feces of Glp2r−/− mice. A number of significant differences were detected in the representation of specific fecal bacterial populations, including significant reductions in the proportions of Firmicutes and Actinobacteria and a significant increase in Bacteroidetes and Clostridiales in Glp2r−/− mice (Fig. 6).
FIG. 6.
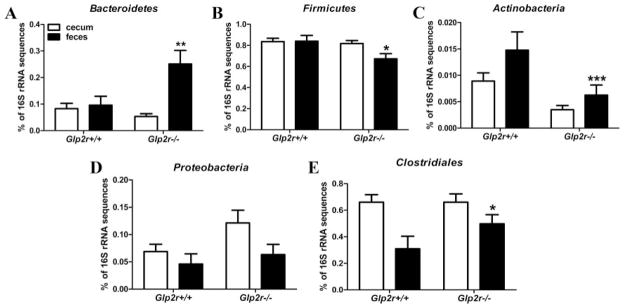
16S rRNA sequencing reveals differential expression of the gut microbiome in the feces of Glp2r−/− mice. Relative abundance of Bacteroidetes (A), Firmicutes (B), Actinobacteria (C), Proteobacteria (D), and Clostridiales (E) in the cecum and feces of male Glp2r−/− mice and wild-type littermates (n = 5– 6 per group). *, P < 0.05, **, P < 0.01, ***, P < 0.001, Glp2r−/− mice vs. wild-type littermates.
Discussion
Current concepts of GLP-2 action have evolved primarily from studies using pharmacological administration of GLP-2R agonists, which produce expansion of the small bowel mucosal epithelium through stimulation of crypt cell proliferation and inhibition of enterocyte apoptosis (2, 33). The identification of GLP-2 (3–33) as a GLP-2R antagonist (15) has enabled experiments delineating the importance of endogenous GLP-2 action in vivo. These studies demonstrated that GLP-2 (3–33) attenuates the adaptive mucosal response to refeeding in mice (12) and rats (13). Nevertheless, as GLP-2 (3–33) is also a partial agonist and augments intestinal mucosal growth (15), we generated Glp2r−/− mice as an alternative model for assessing complete loss of endogenous GLP-2 action in vivo (8, 18, 19). Surprisingly, despite the potent intestinotrophic actions of exogenous GLP-2, crypt cell proliferation, mucosal histology, and cell lineage allocation within the gut epithelium are comparable in Glp2r+/+ vs. Glp2r−/− mice. These findings are consistent with observations that the growth-promoting actions of GLP-2 occur predominantly at pharmacological levels of circulating GLP-2 in the parenterally fed neonatal pig (34). Nevertheless, these results do not exclude the possibility that locally produced GLP-2, acting in a paracrine manner, contributes to the control of mucosal growth.
An alternative explanation for the normal development of the Glp2r−/− gastrointestinal tract invokes the possibility of a second functional GLP-2 receptor capable of responding to endogenously produced GLP-2. Indeed, treatment of Caco-2 and T84 intestinal cell lines with GLP-2 activates growth-related signal transduction pathways and stimulates cell proliferation despite the absence of expression of the classical GLP-2R in these two cell lines (35–37). Similarly GLP-2 stimulates cell migration in multiple intestinal cell lines, despite the absence of Glp2r mRNA transcripts encoding the known cloned GLP-2R (38, 39). Although the existence of a second receptor responsive to exogenous GLP-2 cannot be excluded by our data, the Glp2r−/− mouse fails to up-regulate immediate early gene expression, crypt cell proliferation, or small and large bowel growth in response to exogenous GLP-2 administration. In contrast, Glp2r−/− mice exhibit a robust intestinotrophic response to exogenous IGF-I or KGF, implying preservation of growth factor-dependent pathways in both the small and large bowel. Hence, the available data suggest that the Glp2r gene functionally disrupted in Glp2r−/− mice encodes for the major GLP-2-responsive receptor transducing classical GLP-2 actions in the murine gastrointestinal tract.
A second explanation for the surprisingly normal phenotype of Glp2r−/− mice involves the up-regulation of related pathways compensating for the lack of GLP-2R action in the gastrointestinal tract. Our current understanding of GLP-2 action involves recruitment of downstream mediators, including IGF-I, KGF, vasoactive intestinal polypeptide , endothelial nitric oxide synthase, and ErbB ligands (16 –19, 40 – 42). Although we have not detected evidence for up-regulation of components of the IGF-I, KGF, or ErbB pathways in the gut of the Glp2r−/− mice, our analyses have involved a limited examination of basal levels of mRNA transcripts encoding key mediators in these pathways (18, 19), and we cannot exclude the possibility that the activity of one or more downstream targets of GLP-2 action is significantly up-regulated in a compensatory manner in Glp2r−/− mice.
Despite previous studies demonstrating proliferative and therapeutic actions of GLP-2 in the normal (16, 43, 44) and diseased (22, 30) colon, the severity of weight loss and colonic mucosal damage was comparable in the presence or absence of the Glp2r. Hence, GLP-2R signaling appears dispensable for the adaptive colonic response to mucosal injury in our model of DS-induced colitis. In contrast, Glp2r−/− mice exhibit greater susceptibility to indomethacin-induced enteritis, in association with increased evidence for bacterial infection in peripheral tissues, particularly mesenteric lymph nodes. Similarly, Glp2r−/− mice exhibited greater intestinal injury and more pronounced bacteremia after the administration of irinotecan, a potent topoisomerase inhibitor. Hence, the disruption of basal GLP-2R signaling appears to preferentially affect the small, rather than the large, bowel.
Consistent with findings of enhanced bacterial translocation in Glp2r−/− mice with experimental small bowel injury, levels of mRNA transcripts for the Paneth cell-derived defensin cryptdin 5, the antimicrobial enzyme Pla2g2a and associated enzymatic activators of defensin activity such as Mmp7, were significantly reduced in the Glp2r−/− small bowel. Furthermore, the Glp2r−/− small bowel exhibited reduced expression of multiple genes important for different components of the inflammatory response and increased bacterial content. Collectively, these observations point to one or more defects in the regulation of the epithelial-microbiota relationship in Glp2r−/− mice, findings supported by the demonstration that Glp2r−/− Paneth cell secretions exhibit reduced bactericidal activity ex vivo. Furthermore, significant alterations in the proportion of major bacterial microbiome constituents were detected in feces of uninjured Glp2r−/− mice. At present, we cannot ascertain whether the Paneth cell defects observed reflect loss of GLP-2 action indirectly on Paneth cells or whether these defects are secondary to other cellular phenotypes arising in Glp2r−/− mice.
Complementary evidence linking GLP-2 action to modulation of gut inflammation derives from observations that probiotic administration increased levels of GLP-2 and reduced gut inflammation in obese mice, whereas reduction of GLP-2 activity with the antagonist GLP-2 (3–33) reversed many of the beneficial antiinflammatory actions of GLP-2 (45). Similarly, GLP-2R activation suppressed intestinal leukocyte infiltration and reduced the expression of genes regulating inflammation and barrier function in a murine model of surgical ileus (46) and in mice with experimental ileitis and colitis (40). Taken together, our findings establish the importance of the intestinal Glp2r for the regulation of host bacterial interactions and reveal a new role for GLP-2 in Paneth cell-derived enteric defense, and control of gut microbial communities. It is important to recognize that Paneth cell activity represents only one component of an integrated intestinal and immune network that defends the mucosal epithelium from bacterial invasion. Furthermore, our data indicate that loss of the Glp2r clearly impairs the small bowel inflammatory response to mucosal injury. Hence, we cannot rule out other subtle abnormalities in distinct arms of the local innate immune response that contribute to the phenotypes observed in Glp2r−/− mice. Taken together, these findings establish the Glp2r as an essential mediator of the small bowel mucosal response to pathogens and inflammation. Whether enhancement of GLP-2 activity may be beneficial in human intestinal diseases characterized by enhanced inflammation (47) requires more careful investigation.
Supplementary Material
Acknowledgments
S.-J.L., J.L., K.K.L., D.H., H.G., D.S.G., and B.Y. carried out the experiments, analyzed the data, and wrote the manuscript. B.Y. and D.J.D. designed the experiments, and D.J.D. also wrote the manuscript. We thank Jennifer Estall for technical assistance with antisera characterization.
This work was supported by Canadian Institutes for Health Research Grant MOP-14799.
Abbreviations
- BrdU
5-Bromo-2′-deoxyuridine
- DS
dextran sulfate
- ErbB
erythroblastic leukemia viral oncogene homolog
- GLP-2
glucagon-like peptide-2
- GLP-2R
GLP-2 receptor
- KGF
keratinocyte growth factor
- LPS
lipopolysaccharide
- rHu
recombinant human
Footnotes
Disclosure Summary: None of the authors have any disclosures directly related to this manuscript. D.J.D., NPS Pharmaceuticals, the University of Toronto, and the University Health Network are parties to an agreement for licensing of GLP-2 patents.
References
- 1.Brubaker PL, Crivici A, Izzo A, Ehrlich P, Tsai CH, Drucker DJ. Circulating and tissue forms of the intestinal growth factor, glucagon-like peptide-2. Endocrinology. 1997;138:4837– 4843. doi: 10.1210/endo.138.11.5482. [DOI] [PubMed] [Google Scholar]
- 2.Drucker DJ, Erlich P, Asa SL, Brubaker PL. Induction of intestinal epithelial proliferation by glucagon-like peptide 2. Proc Natl Acad Sci USA. 1996;93:7911–7916. doi: 10.1073/pnas.93.15.7911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Guan X, Stoll B, Lu X, Tappenden KA, Holst JJ, Hartmann B, Burrin DG. GLP-2-mediated up-regulation of intestinal blood flow and glucose uptake is nitric oxide-dependent in TPN-fed piglets 1. Gastroenterology. 2003;125:136–147. doi: 10.1016/s0016-5085(03)00667-x. [DOI] [PubMed] [Google Scholar]
- 4.Bremholm L, Hornum M, Henriksen BM, Larsen S, Holst JJ. Glucagon-like peptide-2 increases mesenteric blood flow in humans. Scand J Gastroenterol. 2009;44:314–319. doi: 10.1080/00365520802538195. [DOI] [PubMed] [Google Scholar]
- 5.Benjamin MA, McKay DM, Yang PC, Cameron H, Perdue MH. Glucagon-like peptide-2 enhances intestinal epithelial barrier function of both transcellular and paracellular pathways in the mouse. Gut. 2000;47:112–119. doi: 10.1136/gut.47.1.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cheeseman CI. Upregulation of SGLT-1 transport activity in rat jejunum induced by GLP-2 infusion in vivo. Am J Physiol. 1997;273:R1965–R1971. doi: 10.1152/ajpregu.1997.273.6.R1965. [DOI] [PubMed] [Google Scholar]
- 7.Cottrell JJ, Stoll B, Buddington RK, Stephens JE, Cui L, Chang X, Burrin DG. Glucagon-like peptide-2 protects against TPN-induced intestinal hexose malabsorption in enterally refed piglets. Am J Physiol Gastrointest Liver Physiol. 2006;290:G293–G300. doi: 10.1152/ajpgi.00275.2005. [DOI] [PubMed] [Google Scholar]
- 8.Hsieh J, Longuet C, Maida A, Bahrami J, Xu E, Baker CL, Brubaker PL, Drucker DJ, Adeli K. Glucagon-like peptide-2 increases intestinal lipid absorption and chylomicron production via CD36. Gastroenterology. 2009;137:997–1005. 1005.e1–4. doi: 10.1053/j.gastro.2009.05.051. [DOI] [PubMed] [Google Scholar]
- 9.Meier JJ, Nauck MA, Pott A, Heinze K, Goetze O, Bulut K, Schmidt WE, Gallwitz B, Holst JJ. Glucagon-like peptide 2 stimulates glucagon secretion, enhances lipid absorption, and inhibits gastric acid secretion in humans. Gastroenterology. 2006;130:44–54. doi: 10.1053/j.gastro.2005.10.004. [DOI] [PubMed] [Google Scholar]
- 10.Jeppesen PB, Sanguinetti EL, Buchman A, Howard L, Scolapio JS, Ziegler TR, Gregory J, Tappenden KA, Holst J, Mortensen PB. Teduglutide (ALX-0600), a dipeptidyl peptidase IV resistant glucagon-like peptide 2 analogue, improves intestinal function in short bowel syndrome patients. Gut. 2005;54:1224–1231. doi: 10.1136/gut.2004.061440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jeppesen PB, Gilroy R, Pertkiewicz M, Allard JP, Messing B, O’Keefe SJ. Randomised placebo-controlled trial of teduglutide in reducing parenteral nutrition and/or intravenous fluid requirements in patients with short bowel syndrome. Gut. 2011;60:902–914. doi: 10.1136/gut.2010.218271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Shin ED, Estall JL, Izzo A, Drucker DJ, Brubaker PL. Mucosal adaptation to enteral nutrients is dependent on the physiologic actions of glucagon-like peptide-2 in mice. Gastroenterology. 2005;128:1340–1353. doi: 10.1053/j.gastro.2005.02.033. [DOI] [PubMed] [Google Scholar]
- 13.Nelson DW, Murali SG, Liu X, Koopmann MC, Holst JJ, Ney DM. Insulin-like growth factor I and glucagon-like peptide-2 responses to fasting followed by controlled or ad libitum refeeding in rats. Am J Physiol Regul Integr Comp Physiol. 2008;294:R1175–R1184. doi: 10.1152/ajpregu.00238.2007. [DOI] [PubMed] [Google Scholar]
- 14.Hartmann B, Thulesen J, Hare KJ, Kissow H, Orskov C, Poulsen SS, Holst JJ. Immunoneutralization of endogenous glucagon-like peptide-2 reduces adaptive intestinal growth in diabetic rats. Regul Pept. 2002;105:173–179. doi: 10.1016/s0167-0115(02)00013-7. [DOI] [PubMed] [Google Scholar]
- 15.Thulesen J, Knudsen LB, Hartmann B, Hastrup S, Kissow H, Jeppesen PB, Ørskov C, Holst JJ, Poulsen SS. The truncated metabolite GLP-2 (3–33) interacts with the GLP-2 receptor as a partial agonist. Regul Pept. 2002;103:9–15. doi: 10.1016/s0167-0115(01)00316-0. [DOI] [PubMed] [Google Scholar]
- 16.Ørskov C, Hartmann B, Poulsen SS, Thulesen J, Hare KJ, Holst JJ. GLP-2 stimulates colonic growth via KGF, released by sub-epithelial myofibroblasts with GLP-2 receptors. Regul Pept. 2005;124:105–112. doi: 10.1016/j.regpep.2004.07.009. [DOI] [PubMed] [Google Scholar]
- 17.Dubé PE, Forse CL, Bahrami J, Brubaker PL. The essential role of insulin-like growth factor-1 in the intestinal tropic effects of glucagon-like peptide-2 in mice. Gastroenterology. 2006;131:589– 605. doi: 10.1053/j.gastro.2006.05.055. [DOI] [PubMed] [Google Scholar]
- 18.Yusta B, Holland D, Koehler JA, Maziarz M, Estall JL, Higgins R, Drucker DJ. ErbB signaling is required for the proliferative actions of GLP-2 in the murine gut. Gastroenterology. 2009;137:986–996. doi: 10.1053/j.gastro.2009.05.057. [DOI] [PubMed] [Google Scholar]
- 19.Bahrami J, Yusta B, Drucker DJ. ErbB activity links the glucagon-like peptide-2 receptor to refeeding-induced adaptation in the murine small bowel. Gastroenterology. 2010;138:2447–2456. doi: 10.1053/j.gastro.2010.03.006. [DOI] [PubMed] [Google Scholar]
- 20.Bahrami J, Longuet C, Baggio LL, Li K, Drucker DJ. The glucagon-like peptide-2 receptor modulates islet adaptation to metabolic stress in the ob/ob mouse. Gastroenterology. 2010;139:857– 868. doi: 10.1053/j.gastro.2010.05.006. [DOI] [PubMed] [Google Scholar]
- 21.Boushey RP, Yusta B, Drucker DJ. Glucagon-like peptide 2 decreases mortality and reduces the severity of indomethacin-induced murine enteritis. Am J Physiol. 1999;277:E937–E947. doi: 10.1152/ajpendo.1999.277.5.E937. [DOI] [PubMed] [Google Scholar]
- 22.Drucker DJ, Yusta B, Boushey RP, DeForest L, Brubaker PL. Human [Gly2]-GLP-2 reduces the severity of colonic injury in a murine model of experimental colitis. Am J Physiol. 1999;276:G79–G91. doi: 10.1152/ajpgi.1999.276.1.G79. [DOI] [PubMed] [Google Scholar]
- 23.Boushey RP, Yusta B, Drucker DJ. Glucagon-like peptide (GLP)-2 reduces chemotherapy-associated mortality and enhances cell survival in cells expressing a transfected GLP-2 receptor. Cancer Res. 2001;61:687– 693. [PubMed] [Google Scholar]
- 24.Hedo JA, Kahn CR. Radioactive labeling and turnover studies of the insulin receptor subunits. Methods Enzymol. 1985;109:593– 609. doi: 10.1016/0076-6879(85)09117-0. [DOI] [PubMed] [Google Scholar]
- 25.Koehler JA, Yusta B, Drucker DJ. The HeLa cell glucagon-like peptide-2 receptor is coupled to regulation of apoptosis and ERK1/2 activation through divergent signaling pathways. Mol Endocrinol. 2005;19:459– 473. doi: 10.1210/me.2004-0196. [DOI] [PubMed] [Google Scholar]
- 26.Bjerknes M, Cheng H. Methods for the isolation of intact epithelium from the mouse intestine. Anat Rec. 1981;199:565–574. doi: 10.1002/ar.1091990412. [DOI] [PubMed] [Google Scholar]
- 27.Ayabe T, Satchell DP, Wilson CL, Parks WC, Selsted ME, Ouellette AJ. Secretion of microbicidal alpha-defensins by intestinal Paneth cells in response to bacteria. Nat Immunol. 2000;1:113–118. doi: 10.1038/77783. [DOI] [PubMed] [Google Scholar]
- 28.Ley RE, Bäckhed F, Turnbaugh P, Lozupone CA, Knight RD, Gordon JI. Obesity alters gut microbial ecology. Proc Natl Acad Sci USA. 2005;102:11070–11075. doi: 10.1073/pnas.0504978102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wang Q, Garrity GM, Tiedje JM, Cole JR. Naive Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Appl Environ Microbiol. 2007;73:5261–5267. doi: 10.1128/AEM.00062-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.L’Heureux MC, Brubaker PL. Glucagon-like peptide-2 and common therapeutics in a murine model of ulcerative colitis. J Pharmacol Exp Ther. 2003;306:347–354. doi: 10.1124/jpet.103.051771. [DOI] [PubMed] [Google Scholar]
- 31.Munroe DG, Gupta AK, Kooshesh F, Vyas TB, Rizkalla G, Wang H, Demchyshyn L, Yang ZJ, Kamboj RK, Chen H, McCallum K, Sumner-Smith M, Drucker DJ, Crivici A. Prototypic G protein-coupled receptor for the intestinotrophic factor glucagon-like peptide 2. Proc Natl Acad Sci USA. 1999;96:1569–1573. doi: 10.1073/pnas.96.4.1569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yusta B, Huang L, Munroe D, Wolff G, Fantaske R, Sharma S, Demchyshyn L, Asa SL, Drucker DJ. Enteroendocrine localization of GLP-2 receptor expression. Gastroenterology. 2000;119:744–755. doi: 10.1053/gast.2000.16489. [DOI] [PubMed] [Google Scholar]
- 33.Tsai CH, Hill M, Asa SL, Brubaker PL, Drucker DJ. Intestinal growth-promoting properties of glucagon-like peptide 2 in mice. Am J Physiol. 1997;273:E77–E84. doi: 10.1152/ajpendo.1997.273.1.E77. [DOI] [PubMed] [Google Scholar]
- 34.Burrin DG, Stoll B, Guan X, Cui L, Chang X, Holst JJ. Glucagon-like peptide 2 dose-dependently activates intestinal cell survival and proliferation in neonatal piglets. Endocrinology. 2005;146:22–32. doi: 10.1210/en.2004-1119. [DOI] [PubMed] [Google Scholar]
- 35.Jasleen J, Shimoda N, Shen ER, Tavakkolizadeh A, Whang EE, Jacobs DO, Zinner MJ, Ashley SW. Signaling mechanisms of glucagon-like peptide 2-induced intestinal epithelial cell proliferation. J Surg Res. 2000;90:13–18. doi: 10.1006/jsre.2000.5818. [DOI] [PubMed] [Google Scholar]
- 36.Jasleen J, Ashley SW, Shimoda N, Zinner MJ, Whang EE. Glucagon-like peptide 2 stimulates intestinal epithelial proliferation in vitro. Dig Dis Sci. 2002;47:1135–1140. doi: 10.1023/a:1015062712767. [DOI] [PubMed] [Google Scholar]
- 37.Rocha FG, Shen KR, Jasleen J, Tavakkolizadeh A, Zinner MJ, Whang EE, Ashley SW. Glucagon-like peptide-2: divergent signaling pathways. J Surg Res. 2004;121:5–12. doi: 10.1016/j.jss.2004.04.009. [DOI] [PubMed] [Google Scholar]
- 38.Bulut K, Meier JJ, Ansorge N, Felderbauer P, Schmitz F, Hoffmann P, Schmidt WE, Gallwitz B. Glucagon-like peptide 2 improves intestinal wound healing through induction of epithelial cell migration in vitro evidence for a TGF-β-mediated effect. Regul Pept. 2004;121:137–143. doi: 10.1016/j.regpep.2004.04.014. [DOI] [PubMed] [Google Scholar]
- 39.Masur K, Schwartz F, Entschladen F, Niggemann B, Zaenker KS. DPPIV inhibitors extend GLP-2 mediated tumour promoting effects on intestinal cancer cells. Regul Pept. 2006;137:147–155. doi: 10.1016/j.regpep.2006.07.003. [DOI] [PubMed] [Google Scholar]
- 40.Sigalet DL, Wallace LE, Holst JJ, Martin GR, Kaji T, Tanaka H, Sharkey KA. Enteric neural pathways mediate the anti-inflammatory actions of glucagon-like peptide 2. Am J Physiol Gastrointest Liver Physiol. 2007;293:G211–G221. doi: 10.1152/ajpgi.00530.2006. [DOI] [PubMed] [Google Scholar]
- 41.Amato A, Baldassano S, Serio R, Mulè F. Glucagon-like peptide-2 relaxes mouse stomach through vasoactive intestinal peptide release. Am J Physiol Gastrointest Liver Physiol. 2009;296:G678–G684. doi: 10.1152/ajpgi.90587.2008. [DOI] [PubMed] [Google Scholar]
- 42.Guan X, Karpen HE, Stephens J, Bukowski JT, Niu S, Zhang G, Stoll B, Finegold MJ, Holst JJ, Hadsell D, Hadsell DL, Nichols BL, Burrin DG. GLP-2 receptor localizes to enteric neurons and endocrine cells expressing vasoactive peptides and mediates increased blood flow. Gastroenterology. 2006;130:150–164. doi: 10.1053/j.gastro.2005.11.005. [DOI] [PubMed] [Google Scholar]
- 43.Litvak DA, Hellmich MR, Evers BM, Banker NA, Townsend CM., Jr Glucagon-like peptide 2 is a potent growth factor for small intestine and colon. J Gastrointest Surg. 1998;2:146–150. doi: 10.1016/s1091-255x(98)80005-x. [DOI] [PubMed] [Google Scholar]
- 44.Ghatei MA, Goodlad RA, Taheri S, Mandir N, Brynes AE, Jordinson M, Bloom SR. Proglucagon-derived peptides in intestinal epithelial proliferation: glucagon-like peptide-2 is a major mediator of intestinal epithelial proliferation in rats. Dig Dis Sci. 2001;46:1255–1263. doi: 10.1023/a:1010615429639. [DOI] [PubMed] [Google Scholar]
- 45.Cani PD, Possemiers S, Van de Wiele T, Guiot Y, Everard A, Rottier O, Geurts L, Naslain D, Neyrinck A, Lambert DM, Muccioli GG, Delzenne NM. Changes in gut microbiota control inflammation in obese mice through a mechanism involving GLP-2-driven improvement of gut permeability. Gut. 2009;58:1091–1103. doi: 10.1136/gut.2008.165886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Moore BA, Peffer N, Pirone A, Bassiri A, Sague S, Palmer JM, Johnson DL, Nesspor T, Kliwinski C, Hornby PJ. GLP-2 receptor agonism ameliorates inflammation and gastrointestinal stasis in murine postoperative ileus. J Pharmacol Exp Ther. 2010;333:574–583. doi: 10.1124/jpet.109.161497. [DOI] [PubMed] [Google Scholar]
- 47.Buchman AL, Katz S, Fang JC, Bernstein CN, Abou-Assi SG. Teduglutide, a novel mucosally active analog of glucagon-like peptide-2 (GLP-2) for the treatment of moderate to severe Crohn’s disease. Inflamm Bowel Dis. 2010;16:962–973. doi: 10.1002/ibd.21117. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.