

Abstract
Background
This study investigates the association between TOMM40 poly-T length, age-at-onset, and neuropathology in Alzheimer’s disease (AD) individuals with the APOE ε3/ε3 allele.
Methods
Thirty-two PSEN1 mutation carriers with AD, 27 PSEN2 mutation carriers with AD, 59 participants with late-onset AD (LOAD), and 168 participants with autopsies from a community-based cohort were genotyped for TOMM40 intron 6 poly-T (rs10524523) length using short tandem repeat assays.
Results
Among AD patients with PSEN2 mutations, the presence of a long poly-T was associated with an earlier age-at-onset, whereas there were no such associations for patients with PSEN1 mutations or LOAD. In community-based participants, the presence of a long poly-T was associated with increased neuritic tangles and a higher likelihood of pathologically diagnosed AD.
Conclusions
TOMM40 intron 6 poly-T length may explain some of the variation in age-at-onset in PSEN2 familial AD and may be associated with AD neuropathology in persons with APOE ε3/ε3.
Keywords: Alzheimer’s disease, age-at-onset, genetic, APOE, TOMM40, PSEN1 mutation, PSEN2 mutation, neuropathology
1. Introduction
Our previous study showed that participants with familial Alzheimer’s disease (FAD) caused by a presenilin 2 (PSEN2) mutation (N141I) have a wide variation in age-at-onset that cannot be simply explained by the presence of an apolipoprotein E (APOE) ε4 allele [1–2]. A later analysis used Bayesian oligogenic segregation and linkage analysis to simultaneously estimate the effects of APOE, PSEN2, and additional loci on AD age-at-onset in nine families with PSEN2 mutations. Although this study found a small modifying effect of APOE on age-at-onset in affected participants, it also provided strong evidence that there is at least one additional modifier locus besides APOE ε4 allele. A more recent study found that the variable length of a poly-T nucleotide repeat (rs10524523) within intron 6 of the translocase of outer mitochondrial membrane 40 homolog (TOMM40) gene is associated with age-at-onset in late-onset AD (LOAD) [3]. A TOMM40 single nucleotide polymorphism (SNP), rs2075650, located within intron 2, is considered a tagging SNP for the APOE ε4 allele and has been associated with both LOAD risk [4–5] and quantitative traits of AD [6–10], such as age-at-onset and Aβ42 levels. Our previous study reported that SNPs within the TOMM40 gene, as well as other SNPs both proximal and distal to APOE, are associated with cerebrospinal fluid (CSF) ApoE levels [11] and postmortem ApoE expression in AD hippocampus [12]. These findings suggest that the TOMM40 region contributes to LOAD phenotypes and that genetic variation within the TOMM40 gene may actually play a functional role in LOAD risk.
Reports on APOE locus linkage disequilibrium (LD) patterns suggest that strong LD with APOE ε4 exists both proximally (in a region partially spanning the TOMM40 gene) and distally to APOE [8, 13–14]. Interestingly, the association between TOMM40 and LOAD risk is not fully explained by LD between TOMM40 SNPs and the APOE SNP (rs429358) that defines ε4 status, which suggests that other APOE locus SNPs contribute to this association with LOAD [13].
The TOMM40 poly-T variant that is found on the APOE ε4 chromosomal haplotype is relatively homogeneous, and it ranges between 22 and 29 T nucleotides. In contrast, there are two distinct subgroups of poly-T lengths found on the APOE ε3 chromosomal haplotype, the short poly-T length of 11 to 16 repeats and the longer poly-T of 29 to 39 repeats [3]. Thus, the haplotype of the APOE ε4 allele with either the short or the long poly-T and the haplotype of the APOE ε3 with the intermediate length poly-T are rare. Because of the low frequency of these haplotypes in the general population, the independent effect of the TOMM40 poly-T on AD risk or the gene-gene interaction could not be fully assessed using the conventional method of adjusting the APOE genotype or testing an interaction term of APOE x TOMM40 poly-T in a multiple regression model. To discern the effect of the APOE ε4 allele on age-at-onset and risk of AD, we restricted our analysis to participants with the APOE ε3/ε3 genotype only. We first compared age-at-onset in AD patients with and without long poly-T lengths (≥ 20 T nucleotides) in three patient samples: (1) participants recruited from the University of Washington (UW) Alzheimer’s Disease Research Center (ADRC) with either the presenilin 1 (PSEN1) mutation or the PSEN2 mutation (FAD cohort); (2) participants from the UW ADRC clinic with LOAD (≥ age 60 years) who may or may not have a family history of AD (LOAD cohort); and (3) participants from a Seattle-area, community-based cohort study, the Adult Changes in Thought (ACT) study, with AD (ACT cohort). We next examined the associations between the presence of a long poly-T and the time-to-onset of AD using survival analysis. Finally, we examined the association between poly-T length and neuropathological changes of AD in the ACT autopsy cohort.
2. Methods
2.1. Participants
To differentiate the effect of TOMM40 from that of APOE on age-at-onset, we selected study participants with the APOE ε3/ε3 genotype from three different cohorts.
2.1.1. FAD cohort
PSEN1 and PSEN2 mutation carriers were ascertained and assessed by the UW ADRC genetic core. Thirty-one PSEN2 mutation carriers were recruited from 9 pedigrees of Volga German descent [15] that ranged from 1 to 7 family members in size. Of the 31 PSEN2 mutation carriers from these families, 27 were diagnosed with AD and 4 were unaffected but at risk for AD at the time of the study (that is, at the censoring age of 46, 48, 51, and 79 years, which were later than the earliest onset age of 41). The PSEN1 mutation carriers (n = 33) were recruited from 14 pedigrees that ranged from 1 to 6 family members in size. Thirty-two PSEN1 mutation carriers were affected with FAD, and one was unaffected (age = 40 years). Unlike the PSEN2 mutations, the PSEN1 mutations differed by family. Both PSEN1 and PSEN2 mutation carriers were ascertained and assessed using the methods described in our previous report [1] Diagnosis of AD was made based on a clinical examination and/or a detailed history that was provided by informants. Study participants met criteria for “probable AD” based on the National Institute of Neurologic and Communicative Disorders and Stroke/Alzheimer’s Disease and Related Disorders Association (NINCDS-ADRDA) criteria [16], except one case with “possible AD” based on history and clinical examination. In addition, 10 of the 27 FAD patients with a PSEN2 mutation and 20 of the 32 FAD patients with a PSEN1 mutation also received a neuropathological diagnosis of AD. Age-at-onset was determined to be the age at which informant reports and medical records agreed that individuals first began showing signs of memory loss or behavioral changes.
2.1.2. LOAD cohort
Fifty-nine participants with a clinical diagnosis of probable AD and an age-at-onset of 60 years or greater were recruited from the UW ADRC. The clinical diagnosis and age-at-onset for these participants were determined using the methods described above. Of the 59 participants with a clinical diagnosis of AD, 45 were confirmed by autopsy.
2.1.3. ACT cohort
One hundred sixty-eight participants with autopsy exam were recruited from the ACT study, a community-based cohort. The ACT study design and methods have been described in detail elsewhere [17]. Beginning in 1994, cognitively normal individuals age 65 and older who resided in the Seattle area and enrolled in Group Health Cooperative (GHC), a large integrated delivery system, underwent a baseline cognitive examination using the Cognitive Abilities Screening Instrument (CASI; total score 0–100) [18] and were then followed biennially for dementia. ACT participants with poor cognition as demonstrated by a CASI score < 86 underwent a complete dementia evaluation, including a neuropsychological and clinical test battery and a physical/neurologic examination by a geriatrician or neurologist. Diagnoses were assigned at a consensus diagnosis conference using the Diagnostic and Statistical Manual of Mental Disorders-IV [19]for all-cause dementia and NINCDS-ADRDA criteria for probable and possible AD [16]. Neuropathological examinations were performed in those deceased ACT participants who consented to brain autopsy.
2.2. Neuopathological examinations
Neuropathological examinations were performed in the UW Division of Neuropathology and the UW ADRC Neuropathology Core. A detailed description of the neuropathological examination methods has been reported elsewhere [20–21]. Neuritic plaques were scored according to Consortium to Establish a Registry for Alzheimer’s Disease (CERAD) criteria [22–23], and neurofibrillary tangles were scored according to the methods of Braak and Braak [24–25]. Neuropathological AD was defined as a Braak stage ≥ V and a CERAD rating of moderate or higher. Amyloid angiopathy was scored according to the method of Vonsattel [26]. We also assessed the severity of atherosclerosis of large vessels, the number of cystic infarcts on gross inspection of the brain, and the presence of microvascular lesions of 2 mm or smaller (in their greatest dimension) that were identified by way of microscopic examination and that were defined as encephalomalacic lesions. Lewy body pathology was assessed using immunohistochemistry for α-synuclein in both subcortical and cortical regions [20–21].
2.3. Genotyping TOMM40 intron 6 poly-T and APOE
For the TOMM40 intron 6 poly-T length, we used a short tandem repeat genotyping-based assay developed in Dr. Yu’s laboratory at VA Puget Sound Health Care System, Seattle, WA. This assay used a custom PCR primer set (F: Vic-GCTGACCTCAAGCTGTCCTC, R: GGAGGGACAGGGAAAGAAAA) that has been labeled with VIC fluorescent dye to amplify a 247-bp genomic section; the assay is based on the human genome reference sequence and represents the poly-T marker. The precise length of the amplified fragments from each participant was acquired through an ABI 3130xl genetic analyzer and processed using ABI Genemapper v4.0 software (Applied Biosystems, CA). Alleles (or poly-T count) were then determined via manual inspection and cross-checking of the acquired data. Individual samples were reprocessed in instances where initial data produced unreliable reads. Poly-T length was dichotomized into short (< 20) and long (≥ 20) in the analysis based on a bimodal distribution of the number of T on the APOE ε3 chromosomal haplotype [3]. The vast majority of the alleles that were considered long TOMM40 poly-T had 34 or more T-repeats (98% of all alleles in this study); there were only two alleles with 22 T-repeats and two alleles with 29 T-repeat that were within the intermediate length which is associated with both APOE ε3 and ε4 alleles and that were classified as the long in this study.
The ε2/ε3/ε4 status of APOE was determined by the traditional approach of PCR amplification followed by digestion with the restriction enzyme HhaI [27], as reported previously [13, 28].
2.4. Statistical analysis
For continuous variables, differences by 2-category poly-T genotype (short/short vs. any long) were assessed using two sample t-tests, and differences by 3-category poly-T genotype (short/short, short/long, or long/long) were assessed using linear regression. Fisher’s exact test was used to test for differences by poly-T genotype in categorical variables. For the FAD cohort, differences in age-at-onset by poly-T genotype (short/short vs. any long) were assessed using linear regression with robust standard errors to account for clustering of participants within families. In addition, to compare mean age-at-onset by TOMM40 poly-T genotype, we also examined the relationship between TOMM40 poly-T genotype and time-to-onset of AD using survival analysis. Parametric survival models were used to model time-to-onset of AD with the assumption that the effect of having a poly-T long allele is proportional to survival time for those with a short/short genotype (i.e., an accelerated failure time assumption). Results were illustrated using Kaplan-Meier curves and presented as survival ratios and 95% confidence intervals (CIs) based on robust standard errors to account for clustering by family. Significance was assessed using likelihood ratio (LR) tests.
Logistic regression was used to determine the association between neuropathology and poly-T genotype in the ACT cohort adjusting for age-at-death and gender. Analyses were conducted using R.2.11.1 [29], and more specifically, the Design [30] and Survival packages [31] were used to carry out the survival analyses.
3. Results
As shown in Table 1, the distribution of TOMM40 intron 6 poly-T genotype among APOE ε3/ε3 AD patients with PSEN1 mutations, AD patients with PSEN2 mutation, and LOAD patients was similar. The average age-at-onset of AD patients with the PSEN1 mutation was almost a decade earlier than the age-at-onset of AD patients with the PSEN2 mutation.
Table 1.
Characteristics of participants with AD from the UW ADRC
| PSEN1 mutation carriers (N=32) | PSEN2 mutation carriers (n=27) | LOAD Participants (n=59) | |
|---|---|---|---|
| Gender, male (%) | 20 (62.5) | 15 (55.6) | 28 (47.5) |
| Race, white (%) | 26 (81.3) | 27 (100.0) | 55 (94.8)a |
| TOMM40 intron 6 poly-T genotype, n (%) | |||
| Short/short | 9 (28.1) | 9 (33.3) | 14 (23.7) |
| Short/long | 11(34.4) | 15 (55.6) | 31 (52.5) |
| Long/long | 12 (37.5) | 3 (11.1) | 14 (23.7) |
| Age-at-onset, years, mean (SD, range) | 44.9 (9.6, 31–79) | 54.1 (8.8, 41–75) | 74.1 (7.9, 60–88) |
| Short/short | 45.6 (7.5, 36–56) | 59.1 (9.5, 45–72) | 76.8 (7.8, 67–88) |
| Short/long or long/long | 44.7 (10.5, 31–79) | 51.7 (7.5, 41–75) | 73.3 (7.9, 60–85) |
1 case race unknown.
Abbreviations: AD, Alzheimer’s disease; LOAD, late-onset Alzheimer’s disease; PSEN1, presenilin 1; PSEN2, presenilin 2; SD, standard deviation; TOMM40, translocase of outer mitochondrial membrane 40 homolog; UW ADRC, University of Washington Alzheimer’s Disease Research Center
3.1. Association between TOMM40 intron 6 poly-T genotype and age-at-onset in AD
The age-at-onset of AD was an average of seven years earlier for PSEN2 mutation carriers with a long poly-T allele than for those with a short/short poly-T genotype (t = 1.94, df = 25, p = .06). The age-at-onset of AD did not differ between PSEN1 mutation carriers with a short/short poly-T genotype and those with a long poly-T genotype (Table 1; t = −0.25, df = 30, p = .81). In the ADRC LOAD cases, the average age-at-onset was 3 years earlier for cases with a long poly-T than for those with a short/short poly-T, but the difference did not reach statistical significance (t = 1.45, df = 57, p = .15).
3.2. Time-to-onset of AD and TOMM40 intron 6 poly-T
Parametric survival models were used to compare the time-to-onset of AD for PSEN2 or PSEN1 mutation carriers with at least one long poly-T allele to the time-to-onset of AD for those carriers with a short/short poly-T genotype. These models included four PSEN2 mutation carriers who remained unaffected by clinical AD at ages (46, 48, 51, and 79 years) that were later than the earliest age-at-onset (age 41 years) and one PSEN1 mutation carrier who was unaffected by clinical AD (age 40). Survival time was modeled as log-logistic. As shown in the left panel of Figure 1, PSEN2 mutation carriers with a long poly-T allele developed AD earlier than those with a short/short poly-T genotype (survival ratio 0.84, 95% CI [0.72, 0.97], LR test p < .01). In other words, on average, time-to-onset for PSEN2 mutation carriers with a poly-T long allele was shortened by 16%. The median age-at-onset for PSEN2 mutation carriers with a poly-T short/short genotype was 60.8 years; the median for those with a poly-T long allele was 50.9 years. There was no evidence that time to AD differed by poly-T genotype in AD patients who carried the PSEN1 mutation (Figure 1 middle panel, survival ratio 0.95, 95% CI [0.77, 1.17], LR test p = .51) or in patients from the LOAD sample (Figure 1 right panel, survival ratio 0.96, 95% CI [0.90, 1.06], LR test p = .25).
Figure 1.
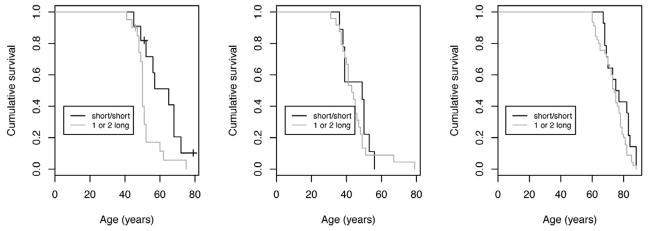
Kaplan-Meier survival curve estimates of AD by TOMM40 intron 6 poly-T genotype for the FAD participants with PSEN2 (left panel) and PSEN1 (middle panel) mutations, and for the LOAD participants (right panel)
3.3. TOMM40 intron 6 poly-T, age-at-onset and risk of AD in the ACT cohort
Of the 168 autopsied ACT participants with APOE ε3/ε3, 29% (n = 49) had a TOMM40 intron 6 poly-T short/short genotype, 51% (n = 85) had a poly-T short/long genotype, and 20% (n =34) had a poly-T long/long genotype. The TOMM40 poly-T genotype did not differ by gender, age-at-enrollment to ACT, age-at-death, or follow-up time (Table 2). During an average 7.1 year follow-up, 51 participants (30.4%) developed dementia, with a mean age-at-onset of 85.5 years. Out of these 51 participants who developed dementia, 42 were classified as probable AD (n = 25) and possible AD (n = 17) based on NINCDS-ADRDA criteria. The age-at-onset of dementia or AD did not differ by presence of a long poly-T (Table 2). The survival curves of clinically diagnosed AD did not differ by presence of a long poly-T (LR test = 0.90, p = .34).
Table 2.
Characteristics of participants by TOMM40 intron 6 poly-T genotype in the ACT cohort
| TOMM40 intron 6 poly-T genotype | |||||
|---|---|---|---|---|---|
| Short/short (n = 49) | Short/long (n = 85) | Long/long (n = 34) | Total (n = 168) | p-value | |
| Gender, male, n (%) | 24 (49.0) | 36 (42.4) | 10 (29.4) | 70 (41.7) | .20 |
| Race, Caucasian, n (%) | 48 (98.0) | 81 (95.3) | 34 (100) | 163 (97.0) | .60 |
| Clinical diagnosis of dementia, n (%) | 16 (32.7) | 22 (25.9) | 13 (38.2) | 51 (30.4) | .38 |
| Clinical AD (probable + possible), n (%) | 15 (30.6) | 15 (17.6) | 12 (35.3) | 42 (25.0) | .07 |
| Age-at-baseline, mean (SD, range) | 79.4 (5.3, 68–93) | 78.5 (65, 66–92) | 80.6 (7.6, 65–90) | 79.2 (6.4, 65–93) | .26 |
| Age-at-death, mean (SD, range) | 88.1 (5.3, 78–99) | 87.3 (7.1, 68–102) | 88.1 (7.7, 72–100) | 87.7 (6.7, 68–102) | .72 |
| Year of follow-up, mean (SD, range) a | 7.3 (3.0, 2.0–13.3) | 7.3 (3.2, 1.8–14.2) | 6.2 (3.3, 1.8–13.0) | 7.1 (3.2, 1.8–14.2) | .28 |
| Age-at-onset, years (SD, range) | |||||
| Dementia (n = 51) | 84.8 (3.7, 74–90) | 84.5 (6.2, 73–96) | 88.2 (4.6, 80–94) | 85.5 (5.3, 73–96) | .10 |
| Clinical AD (n = 42) | 85.1 (3.7, 74–90) | 84.1 (7.1, 73–96) | 88.8 (4.4, 80–94) | 85.8 (5.6, 73–96) | .08 |
1 case excluded with no clinical follow-up before death.
Abbreviations: ACT, Adult Changes in Thought; AD, Alzheimer’s disease; SD, standard deviation; TOMM40, translocase of outer mitochondrial membrane 40 homolog
3.4. TOMM40 intron 6 poly-T and brain pathologies
As shown in Table 3, individuals with a higher number of poly-T alleles (0 vs. 1 vs. 2) were found to exhibit a high level of neuritic tangle (Braak stage of V–VI) and a higher frequency of pathologically defined AD (Braak stage ≥ V and CERAD ≥ moderate). In a logistic regression model of pathologically diagnosed AD that adjusted for age-at-death and gender, pathologically diagnosed AD was associated with an increasing number of poly-T long alleles (LR test p = .04). Using no long poly-T (short/short) as the reference group, the odds ratios (OR) for 1 and 2 alleles were 2.96, 95% CI (0.80, 11.0), and 5.66, 95% CI (1.40, 22.9), respectively. However, there were no significant differences between participants with and without a long poly-T for CERAD score of neuritic plaques or the presence of amyloid angiopathy (Table 3). There also were no associations between the presence of a long poly-T and cerebrovascular damage or Lewy body pathology in the exploratory analyses.
Table 3.
Brain pathologies by TOMM40 intron 6 poly-T genotype in ACT study participants with APOE ε3/ε3
|
TOMM40 intron 6 poly-T
|
|||||
|---|---|---|---|---|---|
| Short/short n = 49 | Short/long n = 85 | Long/long n = 34 | Total n = 168 | p-value | |
| Brain weight, mean (SD) | 1209.7 (158.3) | 1218.5 (123.8) | 1202.6 (158.6) | 1212.8 (140.9) | .85 |
| AD pathology, n (%) | |||||
| Braak stage ≥ V–VI | 3 (6.1) | 17 (20.0) | 10 (29.4) | 30 (17.9) | .01 |
| CERAD score ≥ moderate | 22 (44.9) | 33 (38.8) | 14 (41.2) | 69 (41.1) | .80 |
| Pathologically diagnosed AD | 3 (6.1) | 14 (16.5) | 10 (29.4) | 27 (16.1) | .02 |
| Amyloid angiopathy (≥ moderate) | 2 (4.2) | 9 (10.6) | 2 (5.9) | 13 (7.8) | .44 |
| Cerebrovascular pathology, n (%) | |||||
| Any microvascular lesions | 17 (35.4) | 42 (49.4) | 18 (52.9) | 77 (46.1) | .21 |
| Any cystic infarcts | 13 (27.7) | 20 (24.4) | 10 (29.4) | 43 (26.4) | .82 |
| Atherosclerosis (≥ moderate) | 31 (66.0) | 49 (59.0) | 14 (43.8) | 94 (58.0) | .14 |
| Lewy body pathology, n (%) | |||||
| In cortex | 6 (12.2) | 4 (4.7) | 2 (5.9) | 12 (7.1) | .29 |
| In any region | 12 (26.1) | 9 (11.4) | 5 (16.7) | 26 (16.8) | .11 |
NOTE. Number of missing cases: 2 in the brain weight row, 1 in the microvascular lesions row, 1 in the amyloid angiopathy row, 5 in the cystic infarcts row, 6 in the atherosclerosis row, and 13 in Lewy body pathology in any region row.
Abbreviations: ACT, Adult Changes in Thought; AD, Alzheimer’s disease; APOE, apolipoprotein E; CERAD, Consortium to Establish a Registry for Alzheimer’s Disease; SD, standard deviation; TOMM40, translocase of outer mitochondrial membrane 40 homolog.
4. Discussion
Our study found an association between the TOMM40 intron 6 poly-T variant and an earlier age-at-onset in FAD patients with the APOE ε3/ε3 genotype who carry PSEN2a mutation, but we observed no significant association between the TOMM40 poly-T and age-at-onset in families with a PSEN1 mutation or in participants with LOAD. In a community-based autopsy cohort who had the APOE ε3/ε3 genotype and who were initially free of dementia, we found that the presence of a long TOMM40 intron 6 poly-T was associated with a higher level of neuritic tangles and with increased risk of pathologically diagnosed AD but not with increased risk of clinical AD during life. The association between long poly-T and earlier penetrance of AD in the PSEN2 mutation carriers, as well as with increased neuropathological changes in AD, suggests that the TOMM40 intron 6 poly-T locus is either an effect modifier itself or a surrogate marker for effect modifier of AD.
The association between the TOMM40 poly-T variant and an earlier age-at-onset has been inconsistent. The association between the TOMM40 poly-T variant and an earlier age-at-onset in FAD patients with the APOE ε3/ε3 genotype and PSEN2 mutation is in line with finding from Roses and colleagues who found that the TOMM40 long poly-T polymorphism was associated with an earlier age-at-onset of LOAD [3]. However, in a much larger sample of research participants with LOAD, Cruchaga and colleagues were unable to replicate this finding [32]; moreover, they found that the poly-T polymorphism was associated with a decreased risk of LOAD. In another study, Johnson and colleagues found that homozygosity of the long poly-T allele was associated with poorer cognitive functioning and a decrease in the volume of gray matter in the ventral precuneus, a brain region that is affected early in AD, in middle-aged nondemented adults with the APOE ε3/ε3 genotype [33]. Despite these controversial findings, several lines of additional evidence have suggested that TOMM40 may have an independent role in the AD disease process. The TOMM40 gene encodes a translocase of the outer mitochondrial membrane protein, and mitochondrial function plays an important role in the aging processes and has been implicated in the pathogenesis of AD [34–36]. In a pathway analysis using genome-wide association data from a large French population, Hong and colleagues [37] found a highly significant enrichment of genes involved in intracellular protein transmembrane transport, including several mitochondrial proteins and nucleoporins that are associated with AD. When they further analyzed how the identified protein transport pathway was related to the APOE locus, which contains four genes that cannot be readily discerned due to LD (i.e., APOE, TOMM40, PVRL2, and BCL3), only TOMM40 was closely connected to the set of 18 protein transport genes but not to APOE. In addition, our group has recently reported that in contrast to the TOMM40 short poly-T, the TOMM40 long poly-T is associated with a decrease in TOMM40 promoter activity [38]. This finding suggests that poly-T length may functionally contribute to TOMM40 expression, which implicates a decrease in TOMM40 expression as a contributing factor in AD pathogenesis through the disruption of mitochondrial function [38]. Together with our observations in the current study, this suggests that it is plausible that accelerated onset in persons with a long TOMM40 poly-T might be due to the decreased mitochondrial function that has been associated with advancing age [37].
In this study we found no relationship between the TOMM40 poly-T and age-at-onset of AD in families with the PSEN1 mutation. The reason for this lack of a relationship is not entirely clear. However, in contrast to the single PSEN2 mutation (N1411) that appears to cause AD in our sample of PSEN2 mutation carriers, there are multiple PSEN1 mutations that are known to cause AD. Therefore, it is plausible that the heterogeneity of PSEN1 mutations results in a larger variation in age-at-onset and that this variation decreases precision in detecting modifying effects of TOMM40. Because of our relatively small sample size, the possibility of a poly-T effect on age-at-onset in PSEN1 cases cannot be excluded, but if such an effect exists, its magnitude seems to be much smaller than the magnitude of the effect that was observed in our PSEN2 cases. A large family study with multiple affected AD cases who share the same PSEN1 mutation will be useful to answer this question. The lack of association between the poly-T and age-at-onset in PSEN1 families could also be related to age-at-onset itself; compared to PSEN2 cases, PSEN1 cases had a much earlier age-at-onset, with a mean age of around 44, and this may suggest that PSEN1 mutations had a stronger effect in determining age-at-onset than the PSEN2 mutation. It is also possible that TOMM40 functionally interacts with PSEN1 and PSEN2 differently. Presenilin mutations have been shown to sensitize cells to apoptosis by mechanisms that impair mitochondrial function [39]. A study that investigated the impact of presenilin/γ-secretase on mitochondrial function using mouse embryonal fibroblasts derived from wild-type, PS1−/−, PS2−/−, and PS double knock-out embryos showed a specific role of presenilin2/γ-secretase activity for proper mitochondrial function but no role for PS1/γ-secretase [40].
Our study also found that the TOMM40 intron 6 poly-T was associated with neuropathological changes of AD but not with a clinical diagnosis of AD. We suspect that this finding is due to the heterogeneity of neuropathological changes that underlie the clinical diagnosis of AD, which in our study included both probable and possible AD by NINCDS-ADRDA criteria, and due to variability in the classification of study outcome. Although AD pathologies are the most common cause of dementia, other late-life pathological changes, such as cerebrovascular damage, cortical Lewy bodies, and hippocampal sclerosis, also contribute to cognitive impairment and dementia diagnosis [20]. For example, of the 23 cases in our study with a clinical diagnosis of AD who did not meet criteria for pathological AD, 15 (65.2%) had a Braak stage of III–IV. Of these 15 cases with intermediate Braak stages, 13 had comorbid microvascular lesions, cystic infarts, and/or Lewy bodies, and only 2 had no comorbid pathology identified. Similarly, 7 cases with a low Braak stage had comorbid pathologies. These additional dementia-causing brain pathologies likely explain the differences we observed between clinical and pathological diagnoses in this study. If these comorbid non-AD brain pathologies contribute to an accelerated clinical manifestation of AD-like symptoms or to the clinical diagnoses of AD but are not associated with TOMM40, the strength of the association between TOMM40 and clinical diagnosis of AD, or age-at-onset, would be weakened. In a previous clinicopathological study of genetic risk factors and AD [41], people with the APOE ε3/ε4 genotype were at greater risk for clinical AD than people with the APOE ε3/ε3 genotype (OR 3.4, 95% CI 3.0–3.8), and their risk for pathologically diagnosed AD was even higher (OR 5.5, 95% CI 4.2–7.2). These findings suggest that increasing diagnostic precision through the use of neuropathological phenotypes [42] and diagnostic surrogate biomarkers, such as CSF Aβ1–42, tau [9–10, 32], and neuroimaging [7], could potentially increase our statistical power to discover or test a susceptibility gene for AD, especially for those genes with moderate risk.
The major strength of this study is its use of highly homogeneous samples. These samples provide great statistical power to detect a moderate association and offer a unique opportunity to understand the potential mechanisms of TOMM40, including the way that TOMM40 interacts with PSEN1 and PSEN2. Moreover, our autopsy study enabled us to examine the relationship between TOMM40 and specific brain pathologies of AD. However, the study also has several shortcomings. First, because the sample size of each cohort is relatively small, the positive associations of TOMM40 ploy-T variant with age-at-onset in the PSEN2 families and with the increased neuropathologies of AD could be spurious and the lack of a significant association between TOMM40 and age-at-onset in the participants with PSEN1 mutations or in the participants with LOAD could be due to our limited statistical power to detect a moderate association. These findings need to be further investigated in a larger sample. Second, the definition and determination of age-at-onset may vary across studies. For example, our determination of age-at-onset in AD in the clinical case series is primarily based on the recollection of informants, and such recollections could be largely subjective and prone to recall bias. In the ACT study, which included biennial follow-up appointments, onset was determined as halfway between the visit in which participants received a diagnosis and the previous visit. Third, because of the considerable discrepancy between clinical and pathological classifications of AD and the potential selection bias associated with autopsy [43], our findings of an association between TOMM40 intron 6 poly-T length and pathological AD should be extrapolated to clinical AD with the greatest caution, if at all. Associations of TOMM40 poly-T with neuritic plaques and other brain pathologies need to be further investigated in an independent study. Finally, we should emphasize that the association between the TOMM40 intron 6 poly-T variant, age-at-onset, and AD neuropathological changes is limited to those participants with APOE ε3/ε3 and should not be extrapolated to persons with other APOE genotypes, especially the ε4 allele, because it is conceivable that APOE ε4 interacts differently with the TOMM40 intron 6 poly-T in terms of the moderation of age-at-onset or the pathogenesis of AD. Whether the TOMM40 poly-T length has an independent effect on the risk of AD or a synergistic effect with APOE ε4 needs to be further investigated. However, the interaction between APOE and TOMM40 poly-T length could not be tested by simply using joint analysis in multiple regression models because the APOE ε4 allele is almost exclusively associated with an intermediate length of the TOMM40 poly-T, whereas the ε3 allele is almost exclusively associated with either short or long poly-T but not intermediate poly-T.
In conclusion, TOMM40 intron 6 poly-T length may explain some of the variation in age-at-onset in PSEN2 familial AD and may be associated with AD neuropathology in persons with APOE ε3/ε3. If these findings are confirmed in independent studies, the risk of AD could be further stratified in those “risk-neutral” people with APOE ε3/ε3. Furthermore, our results, together with previous contrasting findings [3, 32–33], suggest that it is important for future studies to use a biological markers, such as CSF biomarkers or neuroimaging results, to increase the precision of measuring phenotypes and to thereby increase the power to detect a moderate genetic risk factor in AD.
Acknowledgments
This study is supported by grants AG033693, AG006781, AG05136, and AG034214 from the National Institute of Aging and by the US Department of Veterans Affairs, Office of Research and Development, Biomedical Laboratory Research Program. The study sponsors had no role in the design and conduct of the study; collection, management, analysis, and interpretation of the data; or preparation, review, and approval of the manuscript.
We thank Andrew David for editing the manuscript.
Footnotes
Conflict of Interest Disclosure: We have no conflicts of interest or financial disclosures to report.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Bird TD, Levy-Lahad E, Poorkaj P, Sharma V, Nemens E, Lahad A, et al. Wide range in age of onset for chromosome 1--related familial Alzheimer’s disease. Annals of neurology. 1996;40:932–936. doi: 10.1002/ana.410400619. [DOI] [PubMed] [Google Scholar]
- 2.Wijsman EM, Daw EW, Yu X, Steinbart EJ, Nochlin D, Bird TD, et al. APOE and other loci affect age-at-onset in Alzheimer’s disease families with PS2 mutation. Am J Med Genet B Neuropsychiatr Genet. 2005;132B:14–20. doi: 10.1002/ajmg.b.30087. [DOI] [PubMed] [Google Scholar]
- 3.Roses AD, Lutz MW, Amrine-Madsen H, Saunders AM, Crenshaw DG, Sundseth SS, et al. A TOMM40 variable-length polymorphism predicts the age of late-onset Alzheimer’s disease. Pharmacogenomics J. 2010;10:375–384. doi: 10.1038/tpj.2009.69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Harold D, Abraham R, Hollingworth P, Sims R, Gerrish A, Hamshere ML, et al. Genome-wide association study identifies variants at CLU and PICALM associated with Alzheimer’s disease. Nat Genet. 2009;41:1088–1093. doi: 10.1038/ng.440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Seshadri S, Fitzpatrick AL, Ikram MA, DeStefano AL, Gudnason V, Boada M, et al. Genome-wide analysis of genetic loci associated with Alzheimer disease. JAMA. 2010;303:1832–1840. doi: 10.1001/jama.2010.574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Potkin SG, Turner JA, Guffanti G, Lakatos A, Torri F, Keator DB, et al. Genome-wide strategies for discovering genetic influences on cognition and cognitive disorders: methodological considerations. Cogn Neuropsychiatry. 2009;14:391–418. doi: 10.1080/13546800903059829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Shen L, Kim S, Risacher SL, Nho K, Swaminathan S, West JD, et al. Whole genome association study of brain-wide imaging phenotypes for identifying quantitative trait loci in MCI and AD: A study of the ADNI cohort. Neuroimage. 2010;53:1051–1063. doi: 10.1016/j.neuroimage.2010.01.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Han MR, Schellenberg GD, Wang LS. Genome-wide association reveals genetic effects on human Abeta42 and tau protein levels in cerebrospinal fluids: a case control study. BMC Neurol. 2010;10:90. doi: 10.1186/1471-2377-10-90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kim S, Swaminathan S, Shen L, Risacher SL, Nho K, Foroud T, et al. Genome-wide association study of CSF biomarkers Abeta1–42, t-tau, and p-tau181p in the ADNI cohort. Neurology. 2011;76:69–79. doi: 10.1212/WNL.0b013e318204a397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Schott JM. Using CSF biomarkers to replicate genetic associations in Alzheimer’s disease. Neurobiol Aging. 2011 doi: 10.1016/j.neurobiolaging.2011.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bekris LM, Millard SP, Galloway NM, Vuletic S, Albers JJ, Li G, et al. Multiple SNPs within and surrounding the apolipoprotein E gene influence cerebrospinal fluid apolipoprotein E protein levels. J Alzheimers Dis. 2008;13:255–266. doi: 10.3233/jad-2008-13303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bekris LM, Galloway NM, Montine TJ, Schellenberg GD, Yu CE. APOE mRNA and protein expression in postmortem brain are modulated by an extended haplotype structure. Am J Med Genet B Neuropsychiatr Genet. 2010;153B:409–417. doi: 10.1002/ajmg.b.30993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yu CE, Seltman H, Peskind ER, Galloway N, Zhou PX, Rosenthal E, et al. Comprehensive analysis of APOE and selected proximate markers for late-onset Alzheimer’s disease: patterns of linkage disequilibrium and disease/marker association. Genomics. 2007;89:655–665. doi: 10.1016/j.ygeno.2007.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chu SH, Roeder K, Ferrell RE, Devlin B, DeMichele-Sweet MA, Kamboh MI, et al. TOMM40 poly-T repeat lengths, age of onset and psychosis risk in Alzheimer disease. Neurobiol Aging. 2011;32:2328 e2321–2329. doi: 10.1016/j.neurobiolaging.2011.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bird TD, Lampe TH, Nemens EJ, Miner GW, Sumi SM, Schellenberg GD. Familial Alzheimer’s disease in American descendants of the Volga Germans: probable genetic founder effect. Annals of neurology. 1988;23:25–31. doi: 10.1002/ana.410230106. [DOI] [PubMed] [Google Scholar]
- 16.McKhann G, Drachman D, Folstein M, Katzman R, Price D, Stadlan EM. Clinical diagnosis of Alzheimer’s disease: report of the NINCDS-ADRDA Work Group under the auspices of Department of Health and Human Services Task Force on Alzheimer’s Disease. Neurology. 1984;34:939–944. doi: 10.1212/wnl.34.7.939. [DOI] [PubMed] [Google Scholar]
- 17.Kukull WA, Higdon R, Bowen JD, McCormick WC, Teri L, Schellenberg GD, et al. Dementia and Alzheimer disease incidence: a prospective cohort study. Arch Neurol. 2002;59:1737–1746. doi: 10.1001/archneur.59.11.1737. [DOI] [PubMed] [Google Scholar]
- 18.Teng EL, Hasegawa K, Homma A, Imai Y, Larson E, Graves A, et al. The Cognitive Abilities Screening Instrument (CASI): a practical test for cross-cultural epidemiological studies of dementia. Int Psychogeriatr. 1994;6:45–62. doi: 10.1017/s1041610294001602. [DOI] [PubMed] [Google Scholar]
- 19.American Psychiatric Association. Diagnostic and statistical manual of mental disorders: DSM-IV. Wshington DC: American Psychiatric Association; 1994. [Google Scholar]
- 20.Sonnen JA, Larson EB, Crane PK, Haneuse S, Li G, Schellenberg GD, et al. Pathological correlates of dementia in a longitudinal, population-based sample of aging. Annals of neurology. 2007;62:406–413. doi: 10.1002/ana.21208. [DOI] [PubMed] [Google Scholar]
- 21.Sonnen JA, Larson EB, Haneuse S, Woltjer R, Li G, Crane PK, et al. Neuropathology in the Adult Changes in Thought Study: A Review. J Alzheimers Dis. 2009 doi: 10.3233/JAD-2009-1180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mirra SS, Heyman A, McKeel D, Sumi SM, Crain BJ, Brownlee LM, et al. The Consortium to Establish a Registry for Alzheimer’s Disease (CERAD) Part II. Standardization of the neuropathologic assessment of Alzheimer’s disease. Neurology. 1991;41:479–486. doi: 10.1212/wnl.41.4.479. [DOI] [PubMed] [Google Scholar]
- 23.Mirra SS. The CERAD neuropathology protocol and consensus recommendations for the postmortem diagnosis of Alzheimer’s disease: a commentary. Neurobiol Aging. 1997;18:S91–94. doi: 10.1016/s0197-4580(97)00058-4. [DOI] [PubMed] [Google Scholar]
- 24.Braak H, Braak E. Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol (Berl) 1991;82:239–259. doi: 10.1007/BF00308809. [DOI] [PubMed] [Google Scholar]
- 25.Braak E, Griffing K, Arai K, Bohl J, Bratzke H, Braak H. Neuropathology of Alzheimer’s disease: what is new since A Alzheimer? Eur Arch Psychiatry Clin Neurosci. 1999;249 (Suppl 3):14–22. doi: 10.1007/pl00014168. [DOI] [PubMed] [Google Scholar]
- 26.Greenberg SM, Vonsattel JP. Diagnosis of cerebral amyloid angiopathy. Sensitivity and specificity of cortical biopsy. Stroke. 1997;28:1418–1422. doi: 10.1161/01.str.28.7.1418. [DOI] [PubMed] [Google Scholar]
- 27.Hixson JE, Vernier DT. Restriction isotyping of human apolipoprotein E by gene amplification and cleavage with HhaI. J Lipid Res. 1990;31:545–548. [PubMed] [Google Scholar]
- 28.Yu CE, Payami H, Olson JM, Boehnke M, Wijsman EM, Orr HT, et al. The apolipoprotein E/CI/CII gene cluster and late-onset Alzheimer disease. Am J Hum Genet. 1994;54:631–642. [PMC free article] [PubMed] [Google Scholar]
- 29.R: A language and environment for statistical computing [computer program] Version 2.11.1. Vienna, Austria: R Foundation for Statistical Computing; 2010. [Google Scholar]
- 30.Design Package [computer program]. Version 2.3-02009.
- 31.Survival analysis, including penalised likelihood [computer program]. Version 2.35-82009.
- 32.Cruchaga C, Nowotny P, Kauwe JS, Ridge PG, Mayo K, Bertelsen S, et al. Association and expression analyses with single-nucleotide polymorphisms in TOMM40 in Alzheimer disease. Arch Neurol. 2011;68:1013–1019. doi: 10.1001/archneurol.2011.155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Johnson SC, La Rue A, Hermann BP, Xu G, Koscik RL, Jonaitis EM, et al. The effect of TOMM40 poly-T length on gray matter volume and cognition in middle-aged persons with APOE epsilon3/epsilon3 genotype. Alzheimers Dement. 2011;7:456–465. doi: 10.1016/j.jalz.2010.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Eckert A, Schulz KL, Rhein V, Gotz J. Convergence of amyloid-beta and tau pathologies on mitochondria in vivo. Mol Neurobiol. 2010;41:107–114. doi: 10.1007/s12035-010-8109-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Swerdlow RH, Burns JM, Khan SM. The Alzheimer’s disease mitochondrial cascade hypothesis. J Alzheimers Dis. 2010;20(Suppl 2):S265–279. doi: 10.3233/JAD-2010-100339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Morais VA, De Strooper B. Mitochondria dysfunction and neurodegenerative disorders: cause or consequence. J Alzheimers Dis. 2010;20 (Suppl 2):S255–263. doi: 10.3233/JAD-2010-100345. [DOI] [PubMed] [Google Scholar]
- 37.Hong MG, Alexeyenko A, Lambert JC, Amouyel P, Prince JA. Genome-wide pathway analysis implicates intracellular transmembrane protein transport in Alzheimer disease. J Hum Genet. 2010;55:707–709. doi: 10.1038/jhg.2010.92. [DOI] [PubMed] [Google Scholar]
- 38.Bekris LM, Lutz F, Yu CE. Functional analysis of APOE locus genetic variation implicates regional enhancers in the regulation of both TOMM40 and APOE. J Hum Genet. 2011 doi: 10.1038/jhg.2011.123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Keller JN, Guo Q, Holtsberg FW, Bruce-Keller AJ, Mattson MP. Increased sensitivity to mitochondrial toxin-induced apoptosis in neural cells expressing mutant presenilin-1 is linked to perturbed calcium homeostasis and enhanced oxyradical production. J Neurosci. 1998;18:4439–4450. doi: 10.1523/JNEUROSCI.18-12-04439.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Behbahani H, Shabalina IG, Wiehager B, Concha H, Hultenby K, Petrovic N, et al. Differential role of Presenilin-1 and -2 on mitochondrial membrane potential and oxygen consumption in mouse embryonic fibroblasts. J Neurosci Res. 2006;84:891–902. doi: 10.1002/jnr.20990. [DOI] [PubMed] [Google Scholar]
- 41.Corneveaux JJ, Myers AJ, Allen AN, Pruzin JJ, Ramirez M, Engel A, et al. Association of CR1, CLU and PICALM with Alzheimer’s disease in a cohort of clinically characterized and neuropathologically verified individuals. Hum Mol Genet. 2010;19:3295–3301. doi: 10.1093/hmg/ddq221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bennett DA, De Jager PL, Leurgans SE, Schneider JA. Neuropathologic intermediate phenotypes enhance association to Alzheimer susceptibility alleles. Neurology. 2009;72:1495–1503. doi: 10.1212/WNL.0b013e3181a2e87d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Tsuang D, Simpson KL, Li G, Barnhart RL, Edland SD, Bowen J, et al. Evaluation of selection bias in an incident-based dementia autopsy case series. Alzheimer Dis Assoc Disord. 2005;19:67–73. doi: 10.1097/01.wad.0000165507.67993.47. [DOI] [PMC free article] [PubMed] [Google Scholar]