

SUMMARY
Muramyl dipeptide (MDP), a product of bacterial cell-wall peptidoglycan, activatesinnate immune cells by stimulating nucleotide-binding oligomerization domain containing 2 (NOD2) -dependent activation of the transcription factor NFκB and transcription of proinflammatory genes. A20 is a ubiquitin-modifying enzyme that restricts tumor necrosis factor (TNF) receptor and Toll-like receptor (TLR) -induced signals. We now show that MDP induces ubiquitylation of receptor- interacting protein 2 (RIP2) in primary macrophages. A20-deficient cells exhibit dramatically am-plified responses to MDP, including increased RIP2 ubiquitylation, prolonged NFκB signaling, and increased production of proinflammatory cytokines. In addition, in vivo responses to MDP are exaggerated in A20-deficient mice and in chimeric mice bearing A20-deficient hematopoietic cells. These exaggerated responses occur independently of the TLR adaptors MyD88 and TRIF as well as TNF signals. These findings indicate that A20 directly restricts NOD2 induced signals in vitro and in vivo, and provide new insights into how these signals are physiologically restricted.
INTRODUCTION
Innate immune cells use a variety of molecules to sense the presence of microbes and trigger protective innate and adaptive immune responses. Hence, the regulation of signals induced by these molecules is central both to the regulation of inflammatory responses and immune homeostasis. Toll-like receptors (TLRs) are the best characterized family of cell-surface microbial sensors, and recent studies have revealed signaling proteins that are essential for activation of TLR signaling as well as those that are required for restricting TLR signals (Akira and Takeda, 2004; Liew et al., 2005). More recent studies have revealed a family of proteins called NOD (nuclear-binding and oligomeri-zation domain) like proteins or CATERPILLER (Caspase recruitment domain, transcription enhancer, R [purine]-binding, pyrin, lots of leucine repeats) proteins, which sense intracellular microbial products such as L-Ala-D-Glu-meso-diaminopimelic acid (Tri-DAP) and muramyl dipeptide (MDP) (Girardin et al., 2003a, 2003b; Inohara et al., 2003; Ting et al., 2006; Fritz et al., 2006).
MDP stimulates NFκB signaling and the activation of innate immune cells through a series of signaling events that require NOD2 (Kobayashi et al., 2005), RIP2, or RICK (Inohara et al., 1998; Kobayashi et al., 2002; Chin et al., 2002; Park et al., 2007) and IkappaB kinase gamma (IKKγ). Activation of NOD2 by MDP is thought to lead to recruitment of RIP2 via homotypic interactions between caspase recruiting domains (CARDs) of NOD2 and RIP2 (Inohara et al., 2000; Manon et al., 2007). IKKγ may subsequently be ubiquitylated and recruited to this signaling complex, after which it phosphorylates IkappaBα (IκBα) to release NFκB and induce NFκB-dependent transcriptional activity (Abbott et al., 2004). Although these studies have begun to define the biochemical mechanisms by which NOD2 activates NFκB, very little is known about how MDP- and NOD2-induced signals are restrained. As with TLR signaling, negative regulators of NOD2 signaling likely play important roles in regulating innate immunity and immune homeostasis (Liew et al., 2005).
The regulation of intracellular signaling pathways involves carefully coordinated series of rapid and reversible posttransla-tional modifications of proteins. Polyubiquitin chains can be built on target proteins via linkages through any of the seven lysine residues on ubiquitin, resulting in polyubiquitin chains of distinct conformations (Weissmann, 2001; Pickart, 2004). These distinct types of ubiquitin chains can target proteins for proteolysis via proteosomal complexes and can also recruit additional signaling proteins to active complexes or influence their localization to subcellular compartments (Chen, 2005; Chen et al., 1995; Liu et al., 2005; Deng et al., 2000). Protein ubiquitylation is enzymatically regulated by the concerted actions of E1, E2, and E3 ligases, which add ubiquitin molecules to target proteins, and deubiquitinating enzymes (DUBs), which deconjugate ubiquitin chains. The physiological importance of ubiquitylation events in regulating cellular signaling and homeostasis has recently been highlighted by studies in which enzymes responsible for adding or removing ubiquitin chains have been genetically deleted. The elimination of E3 ligases or DUBs from mice can result in either hypomorphic or excessive cell-signaling phenotypes (Lee et al., 2000; Naka et al., 1998; Chiang et al., 2000; Massoumi et al., 2006). Hence, these enzymes play critical roles in regulating signal-transduction pathways.
The A20 protein is critical for preventing inflammation in vivo (Opipari et al., 1990; Krikos et al., 1992; Lee et al., 2000). A20-or Tnfaip3-deficient (Tnfaip3−/−) mice spontaneously develop severe inflammation, cachexia, and die prematurely, even in the absence of adaptive T and B lymphocytes (Lee et al., 2000). The physiological functions of A20 in regulating innate immune homeostasis are likely related to A20’s essential roles in restricting both tumor necrosis factor (TNF) and Toll-like receptor (TLR) -induced NFκB signaling (Lee et al., 2000; Boone et al., 2004). A20 may perform these critical functions by both deubiquitylating K63 linked polyubiquitin chains and adding K48 linked polyubiquitin chains to the signaling proteins RIP1 and TRAF6 (Lee et al., 2000; Boone et al., 2004; Wertz et al., 2004). In addition to its profound biological roles in experimental models, A20 may also play important roles in human autoim-mune diseases. Recent genetic studies suggest that polymorphisms in or near the human tnfaip3 (A20) gene are associated with rheumatoid arthritis and possibly Crohn’s disease (Well-come Trust Case Control Consortium, 2007; Plenge et al., 2007; Thomson et al., 2007). Given the ability of A20 to restrict TNF- and TLR-induced NFκB signals and the genetic association of polymorphisms in the TNFAIP3 (A20) and NOD2 genes with human inflammatory diseases, we have examined whether and how A20 may regulate NOD2-induced signaling.
RESULTS
A20 Can Inhibit MDP-Induced NFκB Activity
MDP activates innate immune cells by activating NFκB-dependent transcription of proinflammatory genes. As previously reported, transfection of NOD2 into 293T cells confers responsiveness to MDP, as reflected by NFκB-dependent luciferase activity (Girardin et al., 2003b; Inohara et al., 2003). To begin to examine whether A20 inhibits MDP-induced NFκB transcriptional activity, we cotransfected NOD2 expression and NFκB-luciferase reporter plasmids into 293T cells. These experiments indicated that increasing amounts of NOD2 plasmid led to increasing amounts of NFκB activity. Additional cotransfection of an A20 expression construct dramatically inhibited this MDP- and NOD2-dependent NFκB activity, indicating that A20 can inhibit this signaling pathway (Figure 1A). A20 also inhibited NFκB signaling induced by Tri-DAP and NOD1, suggesting that A20 may restrict signaling events common to NOD1- and NOD2-dependent NFκB signaling (Figure 1B). A20 did not inhibit forskolin induced cyclic AMP response element (CRE) -dependent transcription, indicating that A20 does not globally inhibit transcriptional activation or luciferase production in these assays (Figure 1C). Taken together, these results suggest that A20 inhibits MDP and NOD-dependent NFκB activity.
Figure 1. A20 Inhibits MDP- and DAP-Dependent NFκB Transcriptional Activity.

(A–C) NFκB-dependent luciferase reporter assays of NOD2 or NOD1 transfected 293T cells. Indicated amounts of (A) NOD2, (B) NOD1, or (C) cAMP response element (CRE) plasmid trans-fected 293T cells were stimulated with MDP, Tri-DAP, or forskolin, respectively. Cells were also cotransfected with either 0 ng (black columns), 5 ng (gray columns), or 25 ng (white columns) of an A20 expression plasmid. Note the induction of NOD2-mediated NFκB transcriptional activity by MDP, the induction of NOD1-mediated NFκB by Tri-DAP, and the dose-dependent inhibition of these inductions by A20. By contrast, forskolin induced cAMP response element driven luciferase activity is not suppressed by A20. Data are reported in relative luciferase units (RLU) and are representative of three independent experiments.
(D and E) A20 is required for restricting MDP induced responses. ELISA analyses of secreted IL-1β and IL-6 from BMDCs. BMDCs from Tnfaip3+/+ (white columns) and Tnfaip3−/− (black columns) mice were stimulated with the indicated doses of MDP for 12, 24, or 36 hr (for IL-6 analyses) or 24 hr (for IL-1β analyses), after which supernatants were harvested and analyzed by ELISA for (D) IL-6 and (E) IL-1β secretion. Data are representative of three independent experiments, with standard deviation (bars) indicated.
A20 Is Required for Restricting MDP-Induced Signals
To determine whether the ability of A20 to inhibit MDP triggered NFκB transcription correlates with a physiological requirement for A20 to restrict MDP-stimulated signals, we tested the responses of Tnfaip3−/− and Tnfaip3+/+ bone marrow-derived dendritic cells (BMDCs) to MDP. Notably, Tnfaip3−/− and Tnfaip3+/+ BMDCs were phenotypically similar, as judged by the expression of CD11c, CD40, and MHC class II molecules, after 10 days of culture in GM-CSF containing media (Figure S1). Stimulation of these Tnfaip3−/− and Tnfaip3+/+ BMDCs with MDP at day 10 of culture revealed thatTnfaip3−/− BMDCs secrete more IL-6 and more IL-1β than Tnfaip3+/+ BMDCs, indicating that A20 is required for restricting MDP-triggered inflammatory responses (Figures 1D and 1E).
A20 Is Required for Restricting MDP-Induced NFκB-Signaling Activity
MDP stimulates NFκB-signaling activity, which in turn leads to the production of NFκB-dependent genes such as IL-6 and IL-1β. We thus investigated whether A20 is directly required for restricting MDP-induced NFκB-signaling activity. Bone marrow-derived macrophages (BMDMs) from Tnfaip3+/+ and Tnfaip3−/− mice were stimulated with MDP, after which cell lysates were acutely analyzed by immunoblotting for IκBα and phospho-IκBα protein expression as indicators of NFκB-signaling activity. As compared to Tnfaip3+/+ BMDMs, Tnfaip3−/− cells express elevated amounts of phospho-IκBα protein, suggesting that A20 is physiologically required for restricting MDP-induced NFκB signaling (Figure 2A). To confirm that A20 was required for proximate NFκB-signaling activity, we analyzed the activity of the IKK signalosome by performing IKK kinase assays on lysates from MDP stimulated Tnfaip3+/+ and Tnfaip3−/− cells. These experiments revealed that Tnfaip3−/− cells displayed exaggerated IKK kinase activity when compared to Tnfaip3+/+ cells (Figure 2B). As these assays measure acute signaling events, these findings suggest that A20 plays a direct role in terminating MDP-induced NFκB signaling.
Figure 2. A20 Is Required for Restricting MDP-Induced Signaling Activity.

(A) Immunoblotting analyses of phospho-IκBα, IκBα, A20, NOD2, RIP2, and actin protein expression in Tnfaip3−/− and Tnfaip3+/+ BMDMs after MDP stimulation. Tnfaip3−/− and Tnfaip3+/+ BMDMs were stimulated with 10 μg/ml of MDP, and cell lysates were harvested at the indicated time points for immunoblotting analysis. Note increased phospho-IκBα protein expression in Tnfaip3−/− cells at later time points, indicating prolonged NFκB-signaling activity. Note similar amounts of IκBα, NOD2, and RIP2 in Tnfaip3+/+ and Tnfaip3−/− cells after MDP stimulation. Note constant A20 protein expression in Tnfaip3+/+ cells (and not in Tnfaip3−/− cells) after MDP stimulation. Actin protein expression is shown as a control.
(B) IKK kinase assay of Tnfaip3−/− and Tnfaip3+/+ BMDMs after MDP stimulation. Tnfaip3−/− and Tnfaip3+/+ BMDMs were stimulated with MDP, and cell ly-sates were harvested at the indicated time points, immunoprecipitated with anti-IKKγ antibody, incubated with GST-IκBα substrate, and analyzed by immunoblotting for phospho-GST-IκBα levels. Note increased phospho-GST-IκBα expression in Tnfaip3−/− BMDMs indicating prolonged IKK kinase activity. IKKβ protein expression in the immunoprecipitates are shown as a control.
As A20 protein is induced by LPS in macrophages, we examined whether A20 is also induced by MDP in these cells (Boone et al., 2004). MDP treatment did not substantially change the basal level of A20 protein expression in BMDMs, consistent with the fact that MDP causes much weaker stimulation of NFκB signaling in macrophages than LPS (Figure 2A). MDP responses in macrophages are thought to depend upon NOD2 and RIP2. We thus examined whether expression of these proteins are affected by A20 deficiency. These studies revealed that both NOD2 and RIP2 proteins were expressed at similar levels in Tnfaip3+/+ and Tnfaip3−/− cells and did not change significantly after MDP stimulation (Figure 2A). Considered together with the findings above, A20 appears to directly regulate MDP-induced NFκB signaling at or above the level of the IKK signalsome.
A20 Directly Restricts MDP-Induced NFκB Signaling Activity, Independently of LPS or TNF Signals
Some sources of MDP can be contaminated with LPS, and as A20 restricts LPS-induced NFκB signaling, we sought to rule out the possibility that Tnfaip3−/− cells were aberrantly responding to contaminating LPS, rather than MDP, in our assays. First, we pretested batches of MDP using a sensitive limulus endo-toxin assay and utilized endotoxin-free MDP for our signaling studies. Second, we obtained an MDP isomer that does not activate NOD2 from the same commercial source as the active MDP. We tested the ability of this MDP isomer to stimulate NFκB signals in wild-type BMDMs. This experiment revealed that the inactive MDP isomer fails to induce phospho-IκBα, consistent with the absence of contaminating LPS from this source (Figure 3A). Third, as the adaptor protein MyD88 is required for most TLR-induced NFκB signaling and is not thought to be involved in NOD-induced signals, we tested the responses of BMDMs from Tnfaip3+/+ Myd88−/− and Tnfaip3−/− Myd88−/− double-mutant mice to MDP. These experiments showed that Tnfaip3−/− Myd88−/− BMDMs exhibited exaggerated NFκB-signaling responses to MDP when compared with Tnfaip3+/+ Myd88−/−cells (Figure 3B). Thus, A20 regulates NOD2 responses independently of MyD88-dependent TLR signals. Finally, MyD88-independent TLR signals are mediated by an alternative adaptor, TRIF (Hoebe et al., 2003; Yamamoto et al., 2003). To examine A20s role in regulating MDP signaling in the absence of all TLR signals, we interbred Tnfaip3−/− Myd88−/− mice with Ticam1lps2/lps2 mutant mice that lack TRIF-dependent signaling. We then tested MDP responses in Tnfaip3−/− Myd88−/− Ticam1lps2/lps2 and Tnfaip3+/+ Myd88−/− Ticam1lps2/lps2 BMDMs. This experiment revealed that MDP induces exaggerated NFκB signaling in Tnfaip3−/− Myd88−/− Ticam1lps2/lps2 cells when compared to Tnfaip3+/+ Myd88−/− Ticam1lps2/lps2 cells (Figure 3C). Together, these results show that A20 regulates MDP-induced signals independently of TLR signals.
Figure 3. A20 Directly Regulates MDP-Induced NFκB Signaling.

(A) Immunoblotting analyses of MDP versus MDP isomer-stimulated BMDMs. Wild-type BMDMs were stimulated with either MDP or an MDP isomer from the same commercial source that does not activate NOD2 for the indicated time points, after which lysates were analyzed for expression of phospho-IκBα. Note that the MDP isomer fails to elicit NFκB signaling in BMDMs.
(B) Immunoblotting analyses of phospho-IκBα protein levels in Tnfaip3−/− Myd88−/− and Tnfaip3+/+ Myd88−/− BMDMs after MDP stimulation. Tnfaip3−/− Myd88−/− and Tnfaip3+/+ Myd88−/− BMDMs were stimulated with 10 μg/ml of MDP, and cell lysates were harvested at the indicated time points for immunoblotting analysis. Note that increased phospho-IκBα protein expression is observed in Tnfaip3−/− Myd88−/− cells at later time points, indicating that A20 restricts MDP NFκB-signaling activity independently of MyD88.
(C) Immunoblotting analyses of phospho-IκBα protein levels in Tnfaip3−/− Myd88−/− Ticam1lps2/lps2 and Tnfaip3+/+ Myd88−/− Ticam1lps2/lps2 BMDMs after MDP stimulation. Tnfaip3−/− Myd88−/− Ticam1lps2/lps2 and Tnfaip3+/+ Myd88−/− Ticam1lps2/lps2 BMDMs were stimulated with 10 μg/ml of MDP, and cell lysates were harvested at the indicated time points for immunoblotting analysis of phospho-IκBα and IκBα. Note that increased phospho-IκBα protein levels are observed in Tnfaip3−/− Myd88−/− Ticam1lps2/lps2 BMDMs compared with Tnfaip3+/+ Myd88−/− Ticam1lps2/lps2 cells.
(D) Immunoblotting analyses of phospho-IκBα protein levels in Tnfaip3−/− Myd88−/− Tnf−/− and Tnfaip3+/+ Myd88−/− Tnf−/− BMDMs after MDP stimulation. Tnfaip3−/− Myd88−/− Tnf−/− and Tnfaip3+/+ Myd88−/− Tnf−/− BMDMs were stimulated with 10 μg/ml of MDP, and cell lysates were harvested at the indicated time points for immunoblotting analysis of phospho-IκBα and IκBα proteins. Note that increased phospho-IκBα protein levels are observed in Tnfaip3−/− Myd88−/− Tnf−/− BMDMs compared with Tnfaip3+/+ Myd88−/− Tnf−/− cells. Immunoblots for actin expression are shown as controls for all experiments. All data are representative of at least three independent experiments.
A20 regulates TNF-induced NFκB signals, and MDP stimulation of BMDMs can stimulate modest levels of TNF secretion. To ensure that prolonged NFκB-signaling activity in Tnfaip3−/− BMDMs was not due to aberrant responses to TNF secreted by these cells, we interbred Tnfaip3−/− Myd88−/− mice with Tnf−/− mice and compared the MDP responses of Tnfaip3−/− Myd88−/− Tnf−/− BMDMs with Tnfaip3+/+ Myd88−/− Tnf−/− BMDMs. These experiments showed that Tnfaip3−/− Myd88−/− Tnf−/− BMDMs exhibit exacerbated NFκB signaling compared with Tnfaip3+/+ Myd88−/− Tnf−/− BMDMs (Figure 3D). Taken together, these experiments indicate that A20 directly restricts MDP-induced NFκB signaling independently of TLR or TNF signals.
A20 Inhibits RIP2-Induced NFκB Transcriptional Activity
We then turned to investigate the mechanism by which A20 might restrict MDP-induced NFκB signaling. Prior studies indicated that MDP-induced NFκB signaling requires RIP2 in addition to NOD2 (Kobayashi et al., 2002, 2005; Chin et al., 2002; Park et al., 2007). MDP induces NOD2 oligomerization that leads to recruitment of RIP2. Activated RIP2 is thought to recruit IKKγ to this signaling complex and phosphorylates IκBα to release NFκB and induce NFκB-dependent transcriptional activity (Inohara et al., 2000; Abbott et al., 2004). We were interested in investigating whether A20 might inhibit NOD2-induced signaling at or below the point of RIP2 activation. We thus tested whether A20 could inhibit RIP2-induced NFκB activity by cotransfecting RIP2, A20, and an NFκB-dependent luciferase reporter into 293T cells and assaying luciferase production. Transfection of RIP2 expression plasmid induces NFκB-dependent luciferase activity in a dose-dependent fashion. Cotransfec-tion of A20 expression plasmid suppressed RIP2-induced NFκB transcriptional activity (Figure 4A). These studies indicate that A20 inhibits RIP2-induced NFκB transcriptional activity in a dose-dependent fashion. This result also suggests that A20 can restrict MDP-induced NFκB signaling at or below the point of RIP2 in this pathway.
Figure 4. A20 Deubiquitylates RIP2 and Restricts RIP2-Dependent NFκB Activity.

(A) NFκB-dependent luciferase reporter assays of RIP2-transfected 293T cells. 293T cells were transfected with either 0 ng (white columns), 5 ng (light gray columns), 25 ng (dark gray columns), or 125 ng (black columns) of RIP2 expression plasmid and the indicated amounts of A20 expression plasmids. (White columns are essentially at baseline.) Cell lysates were analyzed for luciferase activity. Note the dose-dependent induction of NFκB transcriptional activity by RIP2 and the dose-dependent inhibition of this induction by A20. Data are reported in relative luciferase units and are representative of three independent experiments.
(B) Immunoblotting assay of RIP2 ubiquitylation in cells. FLAG-tagged RIP2 and Myc-tagged ubiquitin were cotransfected into 293T cells, after which cells were lysed in NP-40 buffer, and boiled in 1% SDS. Samples were then diluted 1:10 with PBS, immunoprecipitated with FLAG-specific antibody (RIP2), and immunoblotted for the presence of Myc-ubiquitin. Note that RIP2 is preferentially ubiquitylated with K48R ubiquitin compared to K63R ubiquitin in cells (compare second and third lanes of top panel). Immunoblotting of Myc-ubiquitin proteins in preimmunoprecipitates are shown in bottom panel as controls.
(C) A20 restricts RIP2 ubiquitylation in cells. Immunoblotting analyses of RIP2 ubiquitylation with A20 cotransfection. HA-RIP2, Myc-K48R ubiquitin, and either wild-type or mutant C103A A20 were cotransfected into 293T cells, after which lysates were immunoblotted for Myc (ubiquitin). Note that cotrans-fected wild-type A20 but not C103A mutant A20 reduces RIP2 ubiquitylation in cells (top panels). Immunoblotting of Myc-ubiquitin proteins is shown in bottom panel as controls.
(D) Recombinant A20 protein deubiquitylates RIP2 in a cell-free assay. Immu-noblotting analyses of RIP2 ubiquitylation after cell-free deubiquitylation. FLAG-RIP2 and myc-K48R mutant ubiquitin were cotransfected into 293T cells, after which cells were lysed in NP-40 lysis buffer. Samples were then immunoprecipitated with FLAG specific antibody (RIP2), washed, and incubated with bacterial recombinant N-terminal A20 protein or mutant C103A A20 protein for 90 min. Wild-type N-terminal A20 protein was also incubated with ubiquitylated RIP2 in the presence of the cysteine protease inhibitor N-ethyl maleimide (NEM). Reactions were then analyzed by immunoblotting for (Myc) ubiquitin. Note that wild-type A20 but not mutant C103A A20 protein reduces RIP2 ubiquitylation (compare second and third lanes). Note also that A20-mediated reduction of RIP2 ubiquitylation is inhibited by NEM (compare second and fourth lanes). Data are representative of three independent experiments.
A20 Deubiquitylates RIP2, a Critical NOD2 Signaling Protein
To better understand the molecular mechanism by which A20 restricts MDP-induced NFκB signaling, we considered that A20 is a ubiquitin-modifying enzyme that regulates RIP1 ubiqui-tylation after TNFR signaling (Wertz et al., 2004). RIP2 shares a kinase domain with RIP1. We thus hypothesized that RIP2 might undergo ubiquitylation in response to MDP and that A20, a ubiq-uitin-modifying enzyme, may regulate RIP2 ubiquitylation. To test this hypothesis, we first investigated whether RIP2 could be ubiquitylated in cells. We cotransfected FLAG-tagged RIP2 and myc-tagged forms of ubiquitin bearing either K48R or K63R mutations into 293T cells, immunoprecipitated FLAG-RIP2, and analyzed the ubiquitylation status of this protein by immunoblotting. This experiment suggested that transfected RIP2 can be ubiquitylated preferentially with K63 polyubiquitin chains, a linkage that has been associated with protein activation (Figure 4B).
We then asked whether A20 could regulate RIP2’s signaling activity by regulating RIP2 ubiquitylation in cells. Cotransfection of HA-RIP2 and myc-K48R mutant ubiquitin with wild-type A20 revealed that A20 reduced the amount and size of polyubiquity-lated RIP2 (Figure 4C). By contrast, coexpression of a mutant form of A20 bearing a cysteine to alanine mutation at position 103 (C103A), a mutation known to eliminate deubiquitylating activity of A20, failed to reduce RIP2 ubiquitylation (Figure 4C). This finding suggests that A20 deubiquitylates non-K48-linked polyubiquitylated RIP2, thereby reducing its signaling activity.
The reduction of polyubiquitylated RIP2 by cotransfected A20 in cells could be due to A20 deubiquitylating RIP2 or to A20 preventing the ubiquitylation of RIP2. To better determine whether A20 directly removes ubiquitin chains from RIP2, we cotransfected FLAG-RIP2 and myc-K48R ubiquitin into cells, immunoprecipitated FLAG-RIP2, and tested the ability of bacterially derived recombinant N-terminal A20 protein to deubiquity-late RIP2 in a cell-free assay. Recombinant N-terminal A20 protein reduced the level of ubiquitylated RIP2, while a mutant N-terminal A20 protein bearing a cysteine to alanine (C103A) substitution exhibited reduced deubiquitylating activity (Figure 4D, compare first, second, and third lanes). In addition, N-ethylemaleimide, a cysteine protease inhibitor that inhibits A20’s deubiquitylating activity, inhibits A20’s activity toward ubiquitylated RIP2 in these assays (Figure 4D, compare first, second, and fourth lanes). These experiments indicate that A20 can directly deubiquitylate non-K48-linked ubiquitin chains from RIP2. Thus, A20 may terminate MDP-induced NFκB signaling by removing activating K63-linked polyubiquitin chains from RIP2.
A20 Is Required for Restricting MDP-Induced Ubiquitylation of Endogenous RIP2
The physiological role of A20 in regulating MDP-induced RIP2 ubiquitylation can be directly assessed by measuring the status of endogenous RIP2 protein in Tnfaip3+/+ and Tnfaip3−/− cells after MDP stimulation. We thus treated Tnfaip3+/+ and Tnfaip3−/− BMDMs with MDP, lysed the cells at various time points, boiled the lysates in 1% SDS, immunoprecipitated RIP2, and tested the ubiquitylation status of RIP2 by immuno-blotting for ubiquitin. These experiments reveal that MDP causes ubiquitylation of endogenous RIP2 in both Tnfaip3+/+ and Tnfaip3−/− BMDMs (Figure 5A). Strikingly, the levels of ubiquity-lated RIP2 are exaggerated and prolonged in Tnfaip3−/− BMDMs after MDP treatment (Figure 5A). As these lysates were boiled in 1% SDS prior to immunoprecipitation, the ubiquitylated proteins detected on the immunoblot should represent RIP2 and not RIP2-associated proteins, such as TRAF6.
Figure 5. A20 Is Required for Restricting Endogenous RIP2 and Not TRAF6 Ubiquitylation after MDP Stimulation.

(A) Immunoblotting analysis of endogenous ubiquitylated RIP2 in BMDMs after MDP stimulation. Tnfaip3−/− and Tnfaip3+/+ BMDMs were stimulated with 10 μg/ml MDP and lysed in RIPA lysis buffer at the indicated time points, boiled in 1%SDS, diluted 1:10 with RIPA buffer, and immunoprecipitated with anti-RIP2 antibody. Immunoprecipitated RIP2 was then analyzed by immuno-blotting for ubiquitin. Note the presence of prolonged RIP2 ubiquitylation in Tnfaip3−/− BMDMs after MDP stimulation. Immunoblotting of RIP2 immuno-precipitates is shown below as a control.
(B) Immunoblotting analysis of endogenous ubiquitylated TRAF6 in BMDMs after MDP stimulation. Tnfaip3−/− and Tnfaip3+/+ BMDMs were stimulated as in (A) above and immunoprecipitated with anti-TRAF6 antibody. Immuno-precipitated TRAF6 was then analyzed by immunoblotting for ubiquitin. Note similar TRAF6 ubiquitylation in Tnfaip3+/+ and Tnfaip3−/− BMDMs after MDP stimulation. Immunoprecipitates using antibody-coated beads only (no lysates) are shown as controls in right lane of each immunoblot. Immunoblot-ting for TRAF6 on TRAF6 immunoprecipitates is shown below as a control. Data are representative of seven independent experiments.
A20 can regulate TRAF6 ubiquitylation after TLR-induced signals, and TRAF6 may contribute to NOD2- induced NFκB signaling (Boone et al., 2004; Abbott et al., 2007). We thus tested whether endogenous TRAF6 ubiquitylation was regulated by A20 after MDP stimulation. We stimulated Tnfaip3+/+ and Tnfaip3−/− BMDMs with MDP, boiled lysates in 1%SDS, diluted the samples, immunoprecipitated endogenous TRAF6 (instead of RIP2), and immunoblotted for ubiquitin. These experiments revealed that MDP induces negligible TRAF6 ubiquitylation in Tnfaip3+/+ BMDMs, and no increased TRAF6 ubiquitylation is seen in Tnfaip3−/− BMDMs (Figure 5B). Thus, A20 is required for restricting MDP-induced RIP2 and not TRAF6 ubiquitylation. By contrast, stimulation of Tnfaip3+/+ and Tnfaip3−/− BMDMs with LPS readily induced TRAF6 ubiquitylation (data not shown). Therefore, A20 is physiologically required for restricting the ubiquitylation of RIP2 after MDP stimulation.
A20 Is Required for Restricting MDP Responses In Vivo
The ability of MDP to activate NOD2-expressing innate immune cells suggests that MDP would stimulate inflammatory responses in intact mice. We thus tested the responses of Tnfaip3−/− and Tnfaip3+/+ mice to MDP. Tnfaip3−/− and Tnfaip3+/+ mice were injected intraperitoneally with either MDP or H2O, after which serum IL-6 protein was measured by ELISA. These experiments revealed that MDP induced significantly higher levels of serum IL-6 in Tnfaip3−/− mice than in Tnfaip3+/+ mice (data not shown). To ensure that these responses were not mediated by contaminating TLR ligands, we repeated these studies with Tnfaip3−/− Myd88−/− and Tnfaip3+/+ Myd88−/− mice. MDP again induced serum IL-6 production in these mice, and the levels of IL-6 were dramatically higher in Tnfaip3−/− Myd88−/− mice than in Tnfaip3+/+ Myd88−/− mice (Figure 6A). Thus, A20 restricts MDP responses in vivo in the absence of MyD88-dependent TLR responses. This finding is notable, as most prior studies of in vivo MDP responses have relied upon accompanying TLR signals.
Figure 6. A20 Restricts MDP-Induced Inflammatory Responses In Vivo.
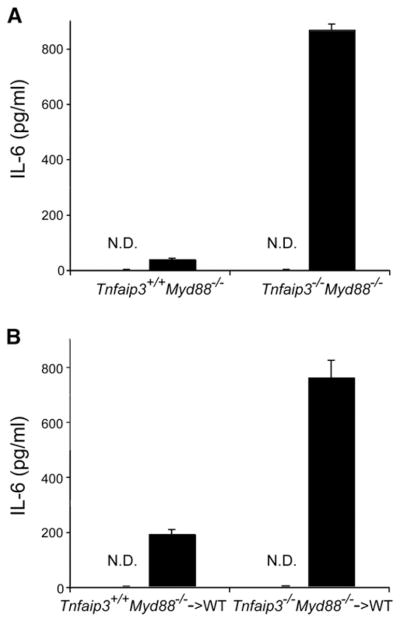
(A and B) ELISA analyses of serum from MDP-injected mice. Twenty-five milligrams over kilograms of MDP (or H2O) was injected into (A) intact Tnfaip3+/+ Myd88−/− and Tnfaip3−/− Myd88−/− mice, and (B) chimeric mice reconstituted with HSCs from Tnfaip3+/+ Myd88−/− and Tnfaip3−/− Myd88−/− mice. Serum was harvested 4 hr after MDP injection and analyzed for IL-6 levels by ELISA. White columns indicate samples from mice injected with H2O, and indicate that no IL-6 above baseline was detected (ND). Black columns indicate samples from MDP-injected mice. Note increased levels of serum IL-6 in (A) intact Tnfaip3−/− Myd88−/− mice compared with Tnfaip3+/+ Myd88−/− mice and in (B) chimeric mice bearing Tnfaip3−/− Myd88−/− HSCs compared with those bearing Tnfaip3+/+ Myd88−/− cells. Data were obtained from three sets of paired mice.
NOD2 is expressed in radiation-sensitive hematopoietic cells such as macrophages and dendritic cells as well as in radiation resistant cells such as Paneth cells. We thus investigated whether A20 restricts MDP-induced IL-6 production specifically in hematopoietic cells in vivo. We used either Tnfaip3−/− Myd88−/− or Tnfaip3+/+ Myd88−/− fetal liver hematopoietic stem cells (HSCs) to reconstitute irradiated wild-type mice, allowed 6 weeks for hematopoietic reconstitution, and then tested the responses of these radiation chimera to MDP. These experiments revealed that Tnfaip3−/− Myd88−/− HSC reconstituted chimera elaborated greater amounts of serum IL-6 than Tnfaip3+/+ Myd88−/− HSC reconstituted chimera after MDP injection (Figure 6B). Thus, consistent with our in vitro studies, A20 expression in hematopoietic cells is required for restricting MDP-induced NOD2 signaling in vivo.
DISCUSSION
We have identified a unique mechanism by which NOD2-induced signaling and innate immune responses are restricted. Our results suggest that ubiquitylation of RIP2 is a physiological event in macrophages in response to MDP- and NOD2-induced signals, and that the ubiquitin-modifying enzyme A20 is essential for deubiquitylating RIP2 and negatively regulating these signals. These findings provide new insights into how innate immune responses are regulated by non-TLR ligands and signaling.
The recognition of microbial products through non-TLR host proteins such as NOD or CATERPILLER proteins has emerged as an important aspect of innate immunity. The requirement for A20 in restricting MDP- and NOD2-induced signals demonstrates that these signals, like TLR signals, are physiologically restricted by endogenous proteins. A recent study suggests that a short isoform of NOD2, NOD2-S, may also restrict NOD2-induced signals (Rosenstiel et al., 2006). In addition, certain members of the CATERPILLER family appear to negatively regulate cellular activation of immune cells (Ting et al., 2006). Hence, the proper regulation of these signaling pathways requires both positive and negative regulators.
Prior studies suggested that both NOD2 and RIP2 are essential for MDP-induced NFκB responses (Girardin et al., 2003b; Kobayashi et al., 2002, 2005; Chin et al., 2002; Park et al., 2007). MDP was thought to activate NFκB signaling by inducing NOD2 oligomerization, RIP2 recruitment, IKKγ recruitment, and IKKγ ubiquitylation (Inohara et al., 2000; Abbott et al., 2004). How RIP2 becomes biochemically activated was unknown. Our current studies suggest that RIP2 is physiologically ubiquitylated in primary cells after ligand stimulation. RIP2 may be ubiquity-lated with non-K48-linked polyubiquitin chains that help recruit downstream signaling molecules such as IKKγ. As IKKγ binds polyubiquitylated RIP1 during TNF receptor-induced NFκB signaling, it is possible that IKKγ binds to polyubiquitylated RIP2 in a similar fashion during MDP-induced NFκB signaling (Ea et al., 2006; Wu et al., 2006). Recruitment of IKKγ would then lead to IκBα phosphorylation and NFκB transcriptional activity. The identification of RIP2 ubiquitylation has also been shown by others during the review of this study (Hasegawa et al., 2008; Yang et al., 2007). Together, these studies provide an important new insight for understanding MDP- and NOD2-induced NFκB signaling.
We have shown that A20 is required for restricting MDP-induced cytokine production and NFκB signaling in primary innate immune cells. These findings demonstrate that NOD2-induced signals are physiologically restricted. Thus, it appears that NOD2 signaling, like TLR signaling, requires negative as well as positive regulation. In addition, we have elucidated a biochemical mechanism by which A20 may restrict NOD2 signals. A20 can directly deubiquitylate non-K48-linked chains on RIP2, and A20 is required for physiologically restricting endogenous RIP2 ubiquitylation after MDP stimulation. As RIP2 may become conjugated with K63-linked polyubiquitin chains after MDP stimulation, A20’s deubiquitylating activity may serve to “deactivate” RIP2. It is also possible that A20 may utilize its E3 ligase activity to remove ubiquitylated RIP2 from active signaling complexes. Both of these activities would serve to terminate NOD2-induced NFκB signaling. These RIP2-targeted activities could also explain A20’s apparent role in restricting NOD1-induced NFκB signaling, which also requires RIP2 (Girardin et al., 2003a). Although A20 does not stably associate with RIP2 in cells (data not shown), it is not uncommon for ubiquitin-modifying enzymes to associate only transiently with their substrates. The combination of our findings that A20 can directly deubiquitylate RIP2 in cell-free assays and that endogenous RIP2 ubiquitylation is exaggerated in MDP stimulated Tnfaip3−/− cells provides compelling evidence that RIP2 is the predominant physiological substrate for A20 in the MDP-/NOD2-signaling pathway.
Our current studies show that purified MDP elicit inflammatory responses in vivo independently of TLR signaling. We have used Tnfaip3−/− Myd88−/− and Tnfaip3−/− Myd88−/− Ticam1lps2/lps2 mice to establish that MDP elicits proinflammatory responses in the absence of MyD88- and TRIF-dependent TLR signaling and to show that A20 restricts NOD2 signaling independently of its roles in inhibiting TLR signaling. The genetic dissection of TLR- and NOD2-induced signaling is important since NOD2 signals have been suggested to both enhance and restrict MyD88-dependent TLR signals (Netea et al., 2005; Wolfert et al., 2002; Watanabe et al., 2004). Our finding thus provides new avenues for genetically dissecting the physiological roles of A20 in regulating NOD2 versus TLR signaling as well as for understanding the interactions between TLR and NOD2 signaling. Moreover, the ability to elicit MDP responses in intact mice should facilitate further studies interrogating the roles of NOD2-induced signaling in vivo. For example, dissecting the physiological roles of NOD2-induced signals in distinct cell types such as epithelial cells and macrophages can be addressed in chimeric mice or mice bearing lineage-specific gene deletions of NOD2-signaling proteins.
NOD2 signaling is likely to be important to immune homeosta-sis, because polymorphisms in the human NOD2 gene are associated with both Blau’s syndrome and Crohn’s disease in human patients (Miceli-Richard et al., 2001; Hugot et al., 2001; Ogura et al., 2001). However, multiple questions remain in understanding this association. NOD2 polymorphisms have been proposed to exhibit both loss and gain of NOD2 function (Miceli-Richard et al., 2001; Watanabe et al., 2004; Maeda et al., 2005), and it is unclear precisely how hypomorphic or hypermorphic mutations in NOD2 predispose patients to intestinal inflammation. Nevertheless, the clearest physiological function for NOD2 is mediating cellular responses to MDP by activating NFκB signaling. Hence, understanding the molecular mechanisms by which NOD2 activates NFκB signaling and how this process is regulated will likely provide critical insights into how perturbations in NOD2 function affect cellular and ultimately organismal responses. In this context, our discovery that A20 is directly responsible for restricting MDP-triggered signals provides both an important molecular insight into physiological NOD2 function, but also a potential therapeutic target for manipulating or correcting NOD2 functions. Greater understanding of how NOD2-mediated signals regulate immune responses and homeostasis is likely to be biomedically as well as immunologically important.
We have previously shown that A20 restricts TNF and TLR signals by regulating the ubiquitylation of RIP1 and TRAF6, respectively (Lee et al., 2000; Boone et al., 2004; Wertz et al., 2004). Our current studies suggest that A20 is also physiologically required for restricting RIP2 ubiquitylation after MDP stimulation. These findings indicate that A20 enzymatically modifies diverse ubiquitylated target proteins, including RIP1, TRAF6, and RIP2. Hence, like other E3 ligases and deubiquitylating enzymes, A20 appears to act on multiple substrates.
A20’s physiological targets may be dictated partly by the structural nature of the substrate and partly by K63-linked ubiq-uitylation of the substrate. In this scenario, signaling intermediates that share certain structural features and that become decorated with K63-linked polyubiquitin chains in response to a specific ligand may become A20’s preferential enzymatic substrates. Thus, although TRAF6 may be a dominant K63 ubiquity-lated signaling intermediate during TLR-induced NFκB signaling (Deng et al., 2000) and may also be involved in MDP-induced signals (Abbott et al., 2004), our current data suggest that RIP2 is conjugated to K63-linked polyubiquitin chains after MDP stimulation and that ubiquitylated RIP2 is the major target for A20 during MDP-triggered signaling. The ability of A20 to recognize both signaling intermediates and their ubiquitin chains is consistent with recent structural data suggesting that A20 may bind ubiquitin chains as well as exert enzymatic activity toward ubiquitylated substrates (Penengo et al., 2006; Lee et al., 2006; Komander and Barford, 2008).
Finally, A20’s multiple functions may play independent or additive (or perhaps synergistic) roles in restricting immune responses in vivo. These diverse functions suggest that even modest changes in A20 expression or A20 activity may lead to important changes in the intensity and character of immune responses. Such changes may underlie recent genetic suggestions that polymorphisms near or within the TNFAIP3 (A20) gene are associated with inflammatory diseases in human patients (Wellcome Trust Case Control Consortium, 2007; Plenge et al., 2007; Thomson et al., 2007). Therefore, A20 may be an outstanding therapeutic target for inflammatory diseases.
EXPERIMENTAL PROCEDURES
Mice and Cell Preparations
The generation of Tnfaip3−/− mice was previously described (Lee et al., 2000). Myd88−/− mice were generously provided by S. Akira and R. Medzhitov. Tnf−/− and Ticam1lps2/lps2 mutant mice were purchased from JAX laboratories (Bar Harbor, ME). All strains of mice were backbred to the C57BL/6J background for at least six generations. Myd88−/−, Tnf−/−, and Ticam1lps2/lps2 mice were interbred with Tnfaip3−/− mice in the UCSF animal-care facility, and all procedures were conducted according to IACUC approved protocols. BMDMs and BMDCs were generated as previously described (Boone et al., 2004).
Antibodies and Cellular Reagents
Antibodies for cellular phenotyping were purchased from BD Biosciences (San Jose, CA) and used at recommended dilutions. Cells were analyzed on a FACSCalibur analyzer and quantitated using FlowJo software (Tree Star, Incorporated, Ashland, OR). Antibodies for immunoprecipitation and immu-noblotting experiments were obtained from commercial sources; anti-RIP2 (for immunoprecipitation: Cayman Chemical, Ann Arbor, MI; for immunoblott-ing: Alexis biochemicals, San Diego, CA), anti-ubiquitin, anti-IKK-β, and anti-IKKγ (Santa Cruz Biotechnology, Santa Cruz, CA). ELISA analyses of cy-tokine secretion were performed using assay kits according to manufacturer’s protocols (BD Biosciences, San Jose, CA). MDP from Calbiochem (Leicester-shire, UK) and Tri-DAP (L-Ala-D-Glu-meso-DAP) (kind gift from Stephen Girar-din) were controlled for lack of endotoxin content by the Limulus amoebocyte gelation activity test (LAL) performed using the QCL-1000 from BioWhittaker (Verviers, Belgium), according to the manufacturer recommendations.
Plasmids
cDNA expression constructs for murine NOD2, RIP2, and A20 proteins were generated by RT-PCR from normal murine cells, sequenced to confirm their accuracy, and subcloned into pCMV expression constructs. cDNA expression vectors for NOD1, NOD2, and RIP2 were described elsewhere (Kufer et al., 2006).
Luciferase Reporter Assays
Human embryonic kidney cells (HEK293T) were stimulated with 20 nM MDP, 20 nM Tri-dap, or 10 μm forskolin 30 min before transient transfection with 75 ng of a NFκB or CRE luciferase reporter (Stratagene) and 25 ng of a β-ga-lactosidase construct along with NOD1, NOD2, or RIP2 and A20 expressing vectors using the Fugene 6 reagent (Roche diagnostics, Mannheim, Germany), according to the manufacturer recommendations. For each transfection point, total DNA was adjusted to 300 ng by empty vector pcDNA3.1. After 20 hr of transfection, supernatants were discarded and cells were lysed in 100 μl of lysis buffer (Tris pH 8 25 mM, MgCl2 8 mM, Triton X-100 1%, glycerol 15%, DTT 1 mM) for 5 min at room temperature. Lysed cells were analyzed for lucif-erase activity in lysis buffer complemented with luciferin, 2 mM, and ATP, 1 mM from Sigma Aldrich (St Quentin, France) using a Microlumat plus from Berthold Technologies (Bad Wildbad, Germany). For each well, relative lucifer-ase activity was normalized to β-galactosidase activity.
MDP Stimulation of Cells
Cells were pretreated 30 min before stimulation with cytochalasine D (Calbio-chem, Leicestershire, UK) to a final concentration of 1 μM, then stimulated by addition of endotoxin tested MDP (10 μg/ml) or MDP isomer in complete medium. In some experiments, cytochalasin D was omitted. After 20 hr of stimulation, supernatants were either analyzed directly or aliquoted and frozen at −20−C for subsequent cytokine assay by ELISA.
Cell-Free Deubiquitylation Assays
Cell-free deubiquitylation assays were performed as previously described (Boone et al., 2004). Briefly, FLAG-RIP2 was transfected into 293T cells, after which cells were lysed and ubiquitylated FLAG-RIP2 was immunoprecipitated with anti-FLAG (RIP2) antibody. Ubiquitylated FLAG-RIP2 was then incubated with either wild-type or mutant forms of recombinant bacterial N-terminal A20 protein and subsequently analyzed by immunoblotting for ubiquitin. Recombinant N-terminal protein was prepared as previously described, with the modification that N-terminal GST-A20 protein was cleaved with thrombin and purified prior to use in the deubiquitylation assays (Boone et al., 2004).
Assays of Endogenous Protein Ubiquitylation
BMDMs were stimulated with 10 μg/ml MDP, lysed in RIPA lysis buffer, boiled in 1%SDS, diluted 1:10 in RIPA buffer, immunoprecipitated with antibodies specific for either RIP2 (Cayman Chemical) or TRAF6 (Santa Cruz), and analyzed by immunoblotting with antibodies to ubiquitin (Santa Cruz, P4D1), RIP2 (Alexis), or TRAF6 (EMD bioscience).
In Vivo MDP Assays
Mice were injected intraperitoneally with 25 mg/kg of MDP or H2O. Four hours later, serum was obtained and analyzed by ELISA for IL-6 levels according to manufacturer’s instructions (BD Biosciences).
Supplementary Material
Acknowledgments
This work was supported by NIH RO1 grants (A.M.), a fellowship from the Crohn’s and Colitis Foundation of America (O.H.), the Rainin Foundation, and by the immunology core of the UCSF Liver Center (5P30DK026743). This paper is dedicated to the memory of Ken Rainin.
Footnotes
Supplemental Data include one figure and can be found with this article online at http://www.immunity.com/cgi/content/full/28/3/381/DC1/.
References
- Abbott DW, Wilkins A, Asara JM, Cantley LC. The Crohn’s disease protein, NOD2, requires RIP2 in order to induce ubiquitinylation of a novel site on NEMO. Curr Biol. 2004;14:2217–2227. doi: 10.1016/j.cub.2004.12.032. [DOI] [PubMed] [Google Scholar]
- Abbott DW, Yang Y, Hutti JE, Madhavarapu S, Kelliher MA, Cantley LC. Coordinated regulation of Toll-like receptor and NOD2 signaling by K63-linked polyubiquitin chains. Mol Cell Biol. 2007;27:6012–6025. doi: 10.1128/MCB.00270-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akira S, Takeda K. Toll-like receptor signaling. Nat Rev Immunol. 2004;4:499–511. doi: 10.1038/nri1391. [DOI] [PubMed] [Google Scholar]
- Boone DL, Turer EE, Lee EG, Ahmad RC, Wheeler MT, Tsui C, Hurley P, Chien M, Chai S, Hitotsumatsu O, et al. The ubiquitin-modifying enzyme A20 is required for termination of Toll-like receptor responses. Nat Immunol. 2004;5:1052–1060. doi: 10.1038/ni1110. [DOI] [PubMed] [Google Scholar]
- Chen ZJ. Ubiquitin signalling in the NF-κB pathway. Nat Cell Biol. 2005;7:758–765. doi: 10.1038/ncb0805-758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen ZJ, Hagler J, Palombella VJ, Melandri F, Scherer D, Ballard D, Maniatis T. Signal-induced sete-specific phosphorylation targets IκBα to the ubiquitin-proteasome pathway. Genes Dev. 1995;9:1586–1597. doi: 10.1101/gad.9.13.1586. [DOI] [PubMed] [Google Scholar]
- Chiang YJ, Kole HK, Brown K, Naramura M, Fukuhara S, Hu RJ, Jang IK, Gutkind JS, Shevach E, Gu H. Cbl-b regulates the CD28 dependence of T-cell activation. Nature. 2000;403:216–220. doi: 10.1038/35003235. [DOI] [PubMed] [Google Scholar]
- Chin AI, Dempsey PW, Bruhn K, Miller JF, Xu Y, Cheng G. Involvement of receptor-interacting protein 2 in innate and adaptive immune responses. Nature. 2002;416:190–194. doi: 10.1038/416190a. [DOI] [PubMed] [Google Scholar]
- Deng L, Wang C, Spencer E, Yang L, Braun A, You J, Slaughter C, Pickart C, Chen ZJ. Activation of the IκB kinase complex by TRAF6 requires a dimeric ubiquitin-conjugateing enzyme complex and a unique polyubiquitin chain. Cell. 2000;103:351–361. doi: 10.1016/s0092-8674(00)00126-4. [DOI] [PubMed] [Google Scholar]
- Ea CK, Deng L, Xia ZP, Pineda G, Chen ZJ. Activation of IKK by TNFα requires site-specific ubiquitination of RIP1 and polyubiquitin binding by NEMO. Mol Cell. 2006;22:245–257. doi: 10.1016/j.molcel.2006.03.026. [DOI] [PubMed] [Google Scholar]
- Fritz JH, Ferrero RL, Philpott DJ, Girardin SE. Nod-like proteins in immunity, inflammation and disease. Nat Immunol. 2006;7:1250–1257. doi: 10.1038/ni1412. [DOI] [PubMed] [Google Scholar]
- Girardin SE, Boneca IG, Carneiro LA, Antignac A, Jehanno M, Viala J, Tedin K, Taha MK, Labigne A, Zahringer U, et al. Nod1 detects a unique muropeptide from gram-negative bacterial peptidoglycan. Science. 2003a;300:1584–1587. doi: 10.1126/science.1084677. [DOI] [PubMed] [Google Scholar]
- Girardin SE, Boneca IG, Viala J, Chamaillard M, Labigne A, Thomas G, Philpott DJ, Sansonetti PJ. Nod2 is a general sensor of peptidoglycan through muramyl dipeptide (MDP) detection. J Biol Chem. 2003b;278:8869–8872. doi: 10.1074/jbc.C200651200. [DOI] [PubMed] [Google Scholar]
- Hasegawa M, Fujimoto Y, Lucas PC, Nakano H, Fukase K, Núñez G, Inohara N. A critical role of RICK/RIP2 polyubiquitination in Nod-induced NF-kappaB activation. EMBO J. 2008;27:373–383. doi: 10.1038/sj.emboj.7601962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoebe K, Du X, Georgel P, Janssen E, Tabeta K, Kim SO, Goode J, Lin P, Mann N, Mudd S, et al. Identification of Lps2 as a key transducer of MyD88-independent TIR signalling. Nature. 2003;424:743–748. doi: 10.1038/nature01889. [DOI] [PubMed] [Google Scholar]
- Hugot JP, Chamaillard M, Zouali H, Lesage S, Cezard JP, Belaiche J, Almer S, Tysk C, O’Morain CA, Gassull M, et al. Association of Nod2 leucine-rich repeat variants with susceptibility to Crohn’s disease. Nature. 2001;411:599–603. doi: 10.1038/35079107. [DOI] [PubMed] [Google Scholar]
- Inohara N, del Peso L, Koseki T, Chen S, Nunez G. RICK, a novel protein kinase containing a caspase recruitment domain, interacts with CLARP and regulates CD95-mediated apoptosis. J Biol Chem. 1998;273:12296–12300. doi: 10.1074/jbc.273.20.12296. [DOI] [PubMed] [Google Scholar]
- Inohara N, Koseki T, Lin J, del Peso L, Lucas PC, Chen FF, Ogura Y, Nunez G. An induced proximity model for NF-κB activation in the Nod1/RICK and RIP signaling pathways. J Biol Chem. 2000;275:27823–27831. doi: 10.1074/jbc.M003415200. [DOI] [PubMed] [Google Scholar]
- Inohara N, Ogura Y, Fontalba A, Gutierrez O, Pons F, Crespo J, Fukase K, Inamura S, Kusumoto S, Hashimoto M, et al. Host recognition of bacterial muramyl dipeptide mediated through Nod2. J Biol Chem. 2003;278:5509–5512. doi: 10.1074/jbc.C200673200. [DOI] [PubMed] [Google Scholar]
- Kobayashi K, Inohara N, Hernandez LD, Galan JE, Nunez G, Janeway CA, Medzhitov R, Flavell RA. RICK/RIP2/CARDIAK mediates signaling for receptors of the innate and adaptive immune systems. Nature. 2002;416:194–199. doi: 10.1038/416194a. [DOI] [PubMed] [Google Scholar]
- Kobayashi K, Chamaillard M, Ogura Y, Henegariu O, Inohara N, Nunez G, Flavell RA. Nod2-dependent regulation of innate and adaptive immunity in the intestinal tract. Science. 2005;307:731–734. doi: 10.1126/science.1104911. [DOI] [PubMed] [Google Scholar]
- Komander D, Barford D. Structure of the A20 OTU domain and mechanistic insights into deubiquitination. Biochem J. 2008;409:77–85. doi: 10.1042/BJ20071399. [DOI] [PubMed] [Google Scholar]
- Krikos A, Laherty CD, Dixit VM. Transcriptional activation of the tumor necrosic factor α-inducible zinc finger protein, A20, is mediated by κB elements. J Biol Chem. 1992;267:17971–17976. [PubMed] [Google Scholar]
- Kufer TA, Kremmer E, Banks DJ, Philpott DJ. Role for erbin in bacterial activation of Nod2. Infect Immun. 2006;74:3115–3124. doi: 10.1128/IAI.00035-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee EG, Boone DL, Chai S, Libby SL, Chien M, Lodolce JP, Ma A. Failure to regulate TNF-induced NF-κB and cell death responses in A20-deficient mice. Science. 2000;289:2350–2354. doi: 10.1126/science.289.5488.2350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee S, Tsai YC, Mattera R, Smith WJ, Kostelansky MS, Weissman AM, Bonifacino JS, Hurley JH. Structural basis for ubiquitin recognition and autoubiquitination by Rabex-5. Nat Struct Mol Biol. 2006;3:264–271. doi: 10.1038/nsmb1064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu YC, Penninger J, Karin M. Immunity by ubiquitylation: a reversible process of modification. Nat Rev Immunol. 2005;5:941–952. doi: 10.1038/nri1731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liew FY, Xu D, Brint EK, O’Neill AJ. Negative regulation of toll-like receptor-mediated immune resoponses. Nat Rev Immunol. 2005;5:446–458. doi: 10.1038/nri1630. [DOI] [PubMed] [Google Scholar]
- Maeda S, Hsu LC, Liu H, Bankston LA, Iimura M, Kagnoff MF, Eckmann L, Karin M. Nod2 mutation in Crohn’s disease potentiates NF-κB activity and IL-1β processing. Science. 2005;307:734–738. doi: 10.1126/science.1103685. [DOI] [PubMed] [Google Scholar]
- Manon F, Favier A, Nunez G, Simorre JP, Cusack S. Solution structure of NOD1 CARD and mutational analysis of its interaction with the CARD of downstream kinase RICK. J Mol Biol. 2007;365:160–174. doi: 10.1016/j.jmb.2006.09.067. [DOI] [PubMed] [Google Scholar]
- Massoumi R, Chmielarska K, Hennecke K, Pfeifer A, Fassler R. Cyld inhibits tumor cell proliferation by blocking Bcl-3-dependent NF-κB signaling. Cell. 2006;125:665–677. doi: 10.1016/j.cell.2006.03.041. [DOI] [PubMed] [Google Scholar]
- Miceli-Richard C, Lesage S, Rybojad M, Prieur AM, Manouvrier-Hanu S, Hafner R, Chamaillard M, Zouali H, Thomas G, Hugot JP. CARD15 mutations in Blau syndrome. Nat Genet. 2001;29:19–20. doi: 10.1038/ng720. [DOI] [PubMed] [Google Scholar]
- Naka T, Matsumoto T, Narazaki M, Fujimoto M, Morita Y, Ohsawa Y, Saito H, Nagasawa T, Uchiyama Y, Kishimoto T. Accelerated apoptosis of lymphocytes by augmented induction of Bax in SSI-1 (STAT-induced STAT inhibitor-1) deficient mice. Proc Natl Acad Sci USA. 1998;95:15577–15582. doi: 10.1073/pnas.95.26.15577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Netea MG, Ferwerda G, de Jong DJ, Jansen T, Jacobs L, Kramer M, Naber TH, Drenth JP, Girardin SE, Kullberg BJ, et al. Nucleotide-binding oligomerization domain-2 modulates specific TLR pathways for the induction of cytokine release. J Immunol. 2005;174:6518–6523. doi: 10.4049/jimmunol.174.10.6518. [DOI] [PubMed] [Google Scholar]
- Ogura Y, Bonen DK, Inohara N, Nicolae DL, Chen FF, Ramos R, Britton H, Moran T, Karaliuskas R, Duerr RH, et al. A frameshift mutation in NOD2 associated with susceptibility to Crohn’s disease. Nature. 2001;411:603–606. doi: 10.1038/35079114. [DOI] [PubMed] [Google Scholar]
- Opipari AW, Jr, Boguski MS, Dixit VM. The A20 cDNA induced by tumor necrosis factor α encodes a novel type of zinc finger protein. J Biol Chem. 1990;265:14705–14708. [PubMed] [Google Scholar]
- Park JH, Kim YG, McDonald C, Kanneganti TD, Hasegawa M, Body-Malapel M, Inohara N, Nunez G. RICK/RIP2 mediates innate immune responses induced through Nod1 and Nod2 but not TLRs. J Immunol. 2007;178:2380–2386. doi: 10.4049/jimmunol.178.4.2380. [DOI] [PubMed] [Google Scholar]
- Penengo L, Mapelli M, Murachelli AG, Confalonieri S, Magri L, Musacchio A, Di Fiore PP, Polo S, Schneider TR. Crystal structure of the ubiquitin binding domains of Rabex-5 reveals two models of interaction with ubiquitin. Cell. 2006;124:1186–1195. doi: 10.1016/j.cell.2006.02.020. [DOI] [PubMed] [Google Scholar]
- Pickart CM. Back to the future with ubiquitin. Cell. 2004;116:181–190. doi: 10.1016/s0092-8674(03)01074-2. [DOI] [PubMed] [Google Scholar]
- Plenge RM, Cotsapas C, Davies L, Price AL, de Bakker PI, Maller J, Pe’er I, Burtt NP, Blumenstiel B, DeFelice M, et al. Two independent alleles at 6q23 associated with risk of rheumatoid arthritis. Nat Genet. 2007;39:1477–1482. doi: 10.1038/ng.2007.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenstiel P, Huse K, Till A, Hampe J, Hellmig S, Sina C, Billmann S, von Kampen O, Waetzig GH, Platzer M, et al. A short isoform of NOD2/CARD15, NOD2-S, is an endogenous inhibitor of NOD2/receptor-interacting protein kinase 2-induced signaling pathways. Proc Natl Acad Sci USA. 2006;103:3280–3285. doi: 10.1073/pnas.0505423103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomson W, Barton A, Ke X, Eyre S, Hinks A, Bowes J, Donn R, Symmons D, Hider S, Bruce IN, et al. Rheumatoid arthritis association at 6q23. Nat Genet. 2007;39:1431–1433. doi: 10.1038/ng.2007.32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ting JP, Kastner DL, Hoffman HM. CATERPILLERs, pyrin and hereditary immunological disorders. Nat Rev Immunol. 2006;6:183–195. doi: 10.1038/nri1788. [DOI] [PubMed] [Google Scholar]
- Watanabe T, Kitani A, Murray PJ, Strober W. NOD2 is a negative regulator of Toll-like receptor 2-mediated T helper type 1 responses. Nat Immunol. 2004;5:800–808. doi: 10.1038/ni1092. [DOI] [PubMed] [Google Scholar]
- Weissmann AM. Themes and variations on ubiquitylation. Nat Rev Mol Cell Biol. 2001;2:169–178. doi: 10.1038/35056563. [DOI] [PubMed] [Google Scholar]
- Wellcome Trust Case Control Consortium. Genome-wide association study of 14,000 cases of seven common diseases and 3,000 shared controls. Nature. 2007;7:661–678. doi: 10.1038/nature05911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wertz IE, O’Rourke KM, Zhou H, Eby M, Aravind L, Seshagiri S, Wu P, Wiesmann C, Baker R, Boone DL, et al. De-ubiquitination and ubiquitin ligase domains of A20 downregulate NFκB signalling. Nature. 2004;430:694–699. doi: 10.1038/nature02794. [DOI] [PubMed] [Google Scholar]
- Wolfert MA, Murray TF, Boons GJ, Moore JN. The origin of the synergistic effect of muramyl dipeptide with endotoxin and peptidoglycan. J Biol Chem. 2002;277:39179–39186. doi: 10.1074/jbc.M204885200. [DOI] [PubMed] [Google Scholar]
- Wu CJ, Conze DB, Li T, Srinivasula SM, Ashwell JD. Sensing of Lys 63-linked polyubiquitination by NEMO is a key event in NF-κB activation. Nat Cell Biol. 2006;8:398–406. doi: 10.1038/ncb1384. [DOI] [PubMed] [Google Scholar]
- Yamamoto M, Sato S, Hemmi H, Hoshino K, Kaisho T, Sanjo H, Takeuchi O, Sugiyama M, Okabe M, Takeda K, Akira S. Role of adaptor TRIF in the MyD88-independent toll-like receptor signaling pathway. Science. 2003;301:640–643. doi: 10.1126/science.1087262. [DOI] [PubMed] [Google Scholar]
- Yang Y, Yin C, Pandey A, Abbott D, Sassetti C, Kelliher MA. NOD2 pathway activation by MDP or Mycobacterium tuberculosis infection involves the stable polyubiquitination of Rip2. J Biol Chem. 2007;282:36223–36229. doi: 10.1074/jbc.M703079200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.