

Abstract
11β-Hydroxysteroid dehydrogenase type-1 (11β-HSD1) converts inert cortisone into active cortisol, amplifying intracellular glucocorticoid action. 11β-HSD1 deficiency improves cardiovascular risk factors in obesity but exacerbates acute inflammation. To determine the effects of 11β-HSD1 deficiency on atherosclerosis and its inflammation, atherosclerosis-prone apolipoprotein E-knockout (ApoE-KO) mice were treated with a selective 11β-HSD1 inhibitor or crossed with 11β-HSD1-KO mice to generate double knockouts (DKOs) and challenged with an atherogenic Western diet. 11β-HSD1 inhibition or deficiency attenuated atherosclerosis (74–76%) without deleterious effects on plaque structure. This occurred without affecting plasma lipids or glucose, suggesting independence from classical metabolic risk factors. KO plaques were not more inflamed and indeed had 36% less T-cell infiltration, associated with 38% reduced circulating monocyte chemoattractant protein-1 (MCP-1) and 36% lower lesional vascular cell adhesion molecule-1 (VCAM-1). Bone marrow (BM) cells are key to the atheroprotection, since transplantation of DKO BM to irradiated ApoE-KO mice reduced atherosclerosis by 51%. 11β-HSD1-null macrophages show 76% enhanced cholesterol ester export. Thus, 11β-HSD1 deficiency reduces atherosclerosis without exaggerated lesional inflammation independent of metabolic risk factors. Selective 11β-HSD1 inhibitors promise novel antiatherosclerosis effects over and above their benefits for metabolic risk factors via effects on BM cells, plausibly macrophages.—Kipari, T., Hadoke, P. W. F., Iqbal, J., Man, T. Y., Miller, E., Coutinho, A. E., Zhang, Z., Sullivan, K. M., Mitic, T., Livingstone, D. E. W., Schrecker, C., Samuel, K., White, C. I., Bouhlel, M. A., Chinetti-Gbaguidi, G., Staels, B., Andrew, R., Walker, B. R., Savill, J. S., Chapman, K. E., Seckl, J. R. 11β-hydroxysteroid dehydrogenase type 1 deficiency in bone marrow-derived cells reduces atherosclerosis.
Keywords: atherogenesis, glucocorticoids, inflammation
Chronic inflammation is a key process in atherogenesis, with extensive macrophage and lymphocyte invasion of lesions, promoting pathogenesis (1–6). Glucocorticoids in high doses suppress inflammation, but glucocorticoid pharmacotherapy (7, 8) or Cushing's disease (9, 10) exacerbate atherosclerosis and increase cardiovascular events, presumably via glucocorticoid exacerbation of systemic cardiovascular risk factors.
11β-Hydroxysteroid dehydrogenase type 1 (11β-HSD1) catalyzes regeneration of active glucocorticoids (cortisol, corticosterone) from inert 11-keto forms (cortisone, 11-dehydrocorticosterone), acting as an intracellular amplifier of glucocorticoid action. 11β-HSD1 is up-regulated in adipose tissue in obesity in humans (11) and rodents (12), leading to the notion of intracellular Cushing's syndrome of adipose tissue as a cause of obesity and its cardiometabolic consequences. Indeed, transgenic overexpression of 11β-HSD1 in adipose tissue produces local, but not systemic, glucocorticoid excess and causes visceral obesity and metabolic syndrome (13). Conversely, 11β-HSD1 deficiency protects mice from the adverse metabolic consequences of dietary obesity (14–16). A selective 11β-HSD1 inhibitor lowered blood glucose, glycated hemoglobin A1c (HbA1c) and cholesterol in patients with type 2 diabetes (17). These metabolic effects are presumed atheroprotective. Indeed in mice, a selective 11β-HSD1 inhibitor that reduced circulating cholesterol also reduced intra-aortic cholesterol, but this study did not address lesion structure or, crucially, inflammation (18). Another inhibitor had no effect on atherosclerotic lesion size (19). The key concern is whether or not lesions are more inflamed or structurally vulnerable.
11β-HSD1 is expressed in differentiated/activated macrophages and lymphocytes and is up-regulated during an inflammatory response (20–22) in which glucocorticoids promote macrophage phagocytosis of apoptotic neutrophils (23). 11β-HSD1 deficiency delays acquisition of phagocytic competence by macrophages and exacerbates acute inflammation, at least in some models (21, 24, 25). Glucocorticoids, albeit in high doses, reduce the response to vascular injury and its associated inflammation (26), and they attenuate migration (27) and proliferation (28) of vascular smooth muscle cells, effects contributing to plaque stability. 11β-HSD1 in the vessel wall, though without effect on the contractility of normal vessels (29), amplifies antiproliferative effects of glucocorticoids (30). Conversely, glucocorticoids reduce cholesteryl ester hydrolysis and export by macrophages (31) and inhibit formation of fibrous tissue (32, 33), processes contributing to plaque instability. Thus, the overall effects of 11β-HSD1 deficiency/inhibition on atherosclerotic plaques are uncertain, with systemic metabolic improvements potentially offset by worse lesional inflammation and changes in lesion structure. Indeed, any role for 11β-HSD1 in inflammatory/immune cells in atherogenesis is unknown. To address these key questions, we examined the effects of selective pharmacological inhibition or genetic deletion of 11β-HSD1 in apolipoprotein E-knockout (ApoE-KO) mice, a model of spontaneous atherogenesis on high cholesterol Western diet (WD).
MATERIALS AND METHODS
Animals
All animal experiments were carried out under the auspices of the UK Animals (Scientific Procedures) Act of 1986, and with approval from the University of Edinburgh Ethical Review Committee. Male, 11β-HSD1−/− mice congenic on the C57BL/6J genetic background have been described previously (16). 11β-HSD1−/− mice were crossed with ApoE−/− mice (also congenic on C57BL/6J; Charles River, Margate, Kent, UK) to produce 11β-HSD1−/−, ApoE−/− double-knockout (DKO) mice, 11β-HSD1+/−, apoE−/− heterozygote (het) mice, and apoE−/− (ApoE-KO) controls. Animals were born in the expected mendelian ratios, and DKO and het mice were indistinguishable from ApoE-KO mice at birth, weaning, and in adulthood. Genotyping using tail-tip DNA was performed as described previously (15). apoE−/− genotyping was achieved using hot start PCR with forward (exon 3, POS 285; 5′-AACTTACTCTACACAGGATGCC-3′) and reverse (exon 4 pos 869; 5′-CGTCATAGTGTCCTCCATCAGTGC-3′) primers. This amplified PCR products of 584 bp for the wild-type allele and 1500 bp for the null allele. All in vivo experiments and ex vivo analyses were performed blind to genotype.
Induction, detection, and quantification of atherosclerosis
The effects of short-term inhibition of 11β-HSD1 on atherosclerosis and lesion structure were assessed. Adult (10 wk old) male ApoE-KO mice were fed a high-cholesterol WD (D12079B; Research Diets, New Brunswick, NJ, USA) ad libitum, for 14 wk. For the last 8 wk of this period, mice were randomized (balanced design, n=6/group) to receive either vehicle or a selective 11β-HSD1 inhibitor (compound 544 [3-(1-adamantyl)-6,7,8,9-tetrahydro-5H-(1,2,4)-triazolo[4,3-α]azepine; ref. 18]; Enamine, Kiev, Ukraine). Mice consumed ∼10 mg/kg/d. This dose inhibited hepatic 11β-reductase activity by 52 ± 4% (n=3) 4 h after administration by oral gavage, consistent with previous reports (18).
To confirm specificity of action, male (4-wk or 4.5-mo-old) DKO, het, and ApoE-KO mice were similarly fed WD or normal chow diet for 14 wk. Body weight and food intake were measured weekly.
Animals were killed by asphyxiation (CO2), blood was collected, and plasma was stored at −80°C. The vascular tree was removed and cleaned of adherent tissue, and the aortic arch was used immediately for en face Sudan IV staining to identify neutral lipid incorporation (34), expressed as a percentage of the total surface area. The innominate artery was formalin fixed, paraffin embedded, cut into serial 4-μm sections, and stained with U.S. trichrome (UST; ref. 35). Luminal, lesion (neointima), and medial areas of atherosclerotic vessels were quantified by light microscopy with computerized planimetry (MCID Basic 7.0 image analysis software; Imaging Research, London, ON, Canada) and, where appropriate, expressed as a percentage of the total area within the internal elastic laminae (IEL). The section with the maximum cross-sectional narrowing was chosen to represent each artery when calculating the mean area in a group. Empty spaces within the lesion (areas of extracellular lipid or cholesterol crystal accumulation; ref. 36) were determined, and fibrous caps were counted (37). Serial sections at the point of maximal cross-sectional narrowing were stained with picrosirius red (PSR) to show collagen content; quantification of picrosirius red staining was achieved using color deconvolution (Photoshop CS3 Extended, Adobe Systems, San Francisco, CA, USA) and expressed as a fraction of the total neointimal area, as described previously (38). All image analysis was performed blind to genotype or treatment.
Peritoneal inflammation
Peritonitis was induced in 12-wk-old female ApoE-KO or DKO mice after injection of 0.3 ml of 10% sterile thioglycollate (Sigma-Aldrich, Poole, UK). Peritoneal lavage cells were analyzed by flow cytometry after peritoneal lavage with 5 ml of PBS, 72 h postinjection.
Bone marrow (BM) transplantation
Ten-week-old male recipient ApoE-KO mice were housed under specific pathogen-free conditions in individually ventilated cages and given Baytril antibiotic (2.5%) in the drinking water for 1 wk before and 4 wk after BM transplantation. The mice were lethally irradiated with 1050 rad (10.5 Gy) delivered from a GammaCell 40E (MDS Nordian, Fleuvus, Belgium) with a cesium 137 source at a dose rate of 114 rad/min. Following irradiation, the mice were tail vein injected with 5 × 107 donor BM cells prepared after flushing the femurs and tibiae from female DKO or ApoE-KO with PBS. The mice were allowed to recover for 6 wk, and BM chimerism was verified by flow cytometric staining for 11β-HSD1 and CD45 antigen on blood leukocytes. 11β-HSD1 staining was performed using 11β-HSD1 sheep-derived antibody, generated in-house (39) in combination with donkey anti-sheep secondary antibody (Alexa Fluor 488; Invitrogen, Paisley, UK). Cells were fixed and permeabilized using a commercial kit (Fix and Perm; Invitrogen), according to the manufacturer's instructions, in order to allow for intracellular staining with the 11β-HSD1 antibody. Engraftment was high [flow cytometric analysis of BM transplanted from DKO donors to ApoE-KO recipients (n=5) indicated that 94.3±1.3% of circulating leukocytes were donor derived]. The mice were then placed on WD and were culled after 12 wk. The aortic arch and the abdominal aorta were formalin fixed, and optical projection tomography (OPT) analysis was used to quantify lesion and lumen volumes of the innominate artery and of the abdominal aorta. OPT scanning and quantification were performed as described previously (38). The cross-sectional area of innominate atherosclerotic lesions was also assessed, as above.
Immunohistochemistry
Immunohistochemistry was performed in serial sections taken at the point of maximal cross-sectional narrowing in each artery. Paraffin-embedded sections (4 μm) of innominate arteries were deparaffinized, blocked with normal rabbit or goat serum, incubated overnight (4°C) with primary antibodies against Mac-2 (Cedarlane, Tyne & Wear, UK; 1:6000), CD3ε (Santa Cruz Biotechnology, Santa Cruz, CA, USA; 1:750), or α-smooth muscle actin (α-SMA; Sigma-Aldrich; 1:400) to detect macrophages, T cells, and smooth muscle cells, respectively. Antigen retrieval was performed using Borg Decloaker (Biocare Medical, Concord, CA, USA) as recommended by the manufacturer for CD3. Sections were visualized with DAB substrate (Vector Laboratories, Peterborough, UK) and counterstained with hematoxylin. Controls included a nonspecific IgG raised in the same species as the primary antibody. Immunoreactivity for α-SMA was quantified using color deconvolution (as described for collagen) and expressed as a fraction of the total neointimal area, as described previously (38). Mac-2 and CD3 staining were quantified by manually counting nuclei in areas of immunoreactivity and expressing the data as number of cells per square millimeter of lesion. All image analysis was performed blind to genotype.
Aortic function
Aortic ring functional responses to a vasoconstrictor (serotonin) and endothelium-dependent (acetylcholine) and -independent [sodium nitroprusside (SNP)] vasorelaxants were measured by isometric myography (29).
Plasma and serum measurements
Total plasma cholesterol and triglyceride levels were measured with assay kits (Infinity, Fisher Scientific, Loughborough, UK) and nonesterified fatty acid (NEFA) levels using the Wako estar C kit (Alpha Laboratories, Eastleigh, UK), according to the manufacturer's instructions. Insulin levels were measured by ultrasensitive mouse insulin ELISA kit (Crystal Chem, Downers Grove, IL, USA) and glucose levels by the glucose hexokinase reagent (Infinity; Thermo Electron, Waltham, MA, USA). Monocyte chemoattractant protein 1 (MCP-1) ELISA on serum samples was performed as recommended (R&D Systems, Abingdon, UK). Plasma corticosterone levels were measured using an in-house radioimmunoassay, as described previously (40).
RNA extraction and real-time PCR
Total RNA was extracted from macrophages (21), and levels of specific mRNAs were measured as described previously (41). Total RNA from mesenteric adipose tissue (without lymph nodes) was extracted by homogenization in TRIzol (Invitrogen). RNA was isolated from ascending aortas following homogenization under liquid nitrogen, then TRIzol RNA was purified using the RNeasy kit (Qiagen, Crawley, UK) and reverse transcripted, and real-time quantitative RT-PCR (qRT-PCR) was performed on a Roche Light Cycler 480 (Roche Applied Science, Burgess Hill, UK). Primers (Invitrogen) were designed to match intron-spanning probes within the Roche Universal Probe Library for vascular cell adhesion molecule 1 (VCAM-1), intercellular adhesion molecule-1 (ICAM-1), chemokine (C-C motif) receptor 2 (CCR2), fractalkine receptor (CX3CR1), β-actin, leptin, inducible nitric oxide synthase (iNOS), resistin-like molecule-α (Relm-α), and mannose receptor (Table 1). The primer/probe set for MCP-1 (Mm00441242_m1) assay was commercially designed (TaqMan Gene Expression Assays; Applied BioSystems, Cheshire, UK). Each sample was run in triplicate. β-Actin (Table 1) or TATA-box binding protein (TBP; Mm00446973_m1; Applied Biosystems) were used as internal references.
Table 1.
Primer sequences for RT-PCR primers
| Accession number | Gene | Forward primer | Reverse primer | UPL probe |
|---|---|---|---|---|
| NM_010493.2 | Icam1 | cccacgctacctctgctc | gatggatacctgagcatcacc | 81 |
| NM_011693.3 | Vcam1 | tggtgaaatggaatctgaacc | cccagatggtggtttcctt | 34 |
| NM_009915.2 | Ccr2 | acctgtaaatgccatgcaagt | tgtcttccatttcctttgatttg | 27 |
| NM_013684.2 | Tbp | ggcggtttggctaggttt | gggttatcttcacacaccatga | 107 |
| NM_011146.3 | PPAR-γ | tgctgttatgggtgaaactctg | ctgtgtcaaccatggtaatttctt | 2 |
| NM_011333.3 | Ccl2 | catccacgtgttggctca | gatcatcttgctggtgaatgagt | 62 |
| NM_009987.3 | Cx3cr1 | aagttcccttcccatctgct | caaaattctctagatccagttcagg | 10 |
| NM_008493.3 | Lep | caggatcaatgacatttcacaca | gctggtgaggacctgttgat | 93 |
| NM_007393.3 | Actb | ctaaggccaaccgtgaaaag | accagaggcatacagggaca | 64 |
| NM_010927.3 | Nos2 | ctttgccacggacgagac | tcattgtactctgagggctgac | 13 |
| NM_020509.3 | Retnla | ccctccactgtaacgaagactc | cacacccagtagcagtcatcc | 51 |
| NM_008625.1 | Mrc1 | ggacgagcaggtgcagtt | caacacatcccgcctttc | 47 |
Flow cytometry
BM cells were obtained after flushing the femurs with 5 ml of PBS. Tail-vein blood was collected into 3.9% sodium citrate. Spleens were pressed through a 40-μm cell strainer (BD Biosciences, Oxford, UK) to produce a single-cell suspension. Red blood cells (RBCs) in the BM and blood were lysed with BD lysis buffer (BD Biosciences, Oxford, Oxfordshire, UK), and RBCs from spleens were lysed with Sigma lysis buffer (Sigma-Aldrich). Cell suspensions were enumerated using a NucleoCassette and a NucleoCounter NC-100 (ChemoMetec, Allerød, Denmark). Nonspecific binding was blocked by incubating the cells with 10% mouse serum (MS; Sigma-Aldrich) for 10 min (4°C). Cells were stained in PBS with 10% MS using conjugated antibodies at concentrations suggested by the supplier, for 30 min at 4°C. After washing, flow cytometric analyses were performed using BD FACS Calibur or LSRFortessa Cell Analyzer (BD Biosciences). Data analysis was performed using FlowJo 8.2 software (TreeStar, Ashland, OR, USA). Fluorescent flow-check fluorospheres (Beckman and Coulter, High Wycombe, UK) were added to blood, BM, and peritoneal lavage samples prior to analysis in order to calculate the absolute number of cells. Antibodies used were as follows. Anti-CD11b-peridinin chlorophyll protein cyanin 5.5 (PerCPCy5.5) or -fluorescein isothiocyanate (FITC) (clone M1/70), anti-F4/80-PerCPCy5.5 (clone BM8), anti-CD45-pacific blue (clone 30-F11), and anti-Ly6G-phycoerythrin-cyanine 7 (PECy7) or -FITC (clone 1A8) were all from Biolegend (San Diego, CA, USA). Anti-CD115 phycoerythrin (PE; clone AFS98), anti-CD3-allophycocyanin (APC) or -PE (clone 17A2), and anti-CD45-PerCPCy5.5 (clone 30-F11) were all from BD Biosciences. Anti-CD4-FITC or -PerCPCy5.5 (clone RM4-5), anti-B220-PE or -FITC (clone RA3-6B2), anti-CD25-APC (clone PC61.5), and anti-CD8a-APC (clone 53-6.7) were all from eBiosciences (Hatfield, UK). Anti-7/4-Alexa Fluor 647 (clone 7/4; AbD Serotec, Oxford, UK), anti-CD11c-APC (clone N418), and anti-B220-Pacific Orange (clone RA3-6B2) were from Invitrogen. Negative controls were performed using isotype control antibodies. The gating scheme was used in flow cytometric analysis of mouse blood and BM was performed as described previously (42).
Measurement of cholesterol uptake/efflux in macrophages
Cholesterol efflux was measured (41) in BM-derived macrophages from 11β-HSD1-KO and C57BL/6J control mice, generated as described previously (21). Briefly, BM was flushed from femurs, and 4 × 105 cells/well were plated in 1 ml DMEM/F12 (Invitrogen) supplemented with 10% FCS, 500 U/ml penicillin, 500 U/ml streptomycin and 10% monocyte-CSF-conditioned supplement from murine fibrosarcoma cell (L929) cultures (the concentrations of cortisol and cortisone in FCS at this concentration are below the limit of detection and <0.5 nM, respectively; below the Km of the enzyme). Cells differentiated into macrophages over 7 d with medium changed every 3 d. Apolipoprotein AI (ApoAI)-mediated cholesterol efflux was measured in macrophages loaded with acetylated low-density lipoprotein (AcLDL) by incubation with 50 μg/ml [3H]-cholesterol-AcLDL for 24 h, then a further 24 h in the presence or absence of 10 μg/ml human ApoAI. [3H]-cholesterol was measured in centrifuged medium and in cells after lipids were extracted with hexane:isopropanol (3:2 v/v). Total cholesterol was measured on cellular lipids using an enzymatic assay (Boehringer, Ingelheim, Germany). The percentage of specific cholesterol efflux was calculated [medium cholesterol/(medium cholesterol + cellular cholesterol)] after subtraction of values for ApoAI-free medium.
Statistics
Values are expressed as means ± se. Statistical analysis was performed using Student's t test, 1-way ANOVA, 2-way ANOVA, or repeated-measures ANOVA, followed by Tukey post hoc test, as appropriate. Values of P < 0.05 were considered statistically significant.
RESULTS
11β-HSD1 deficiency or inhibition attenuates atherosclerosis in ApoE-KO mice
WD-fed ApoE-KO mice had extensive atherosclerotic lesions in the aortic arch and its major branches; narrowing was most severe in the innominate artery (Fig. 1Ai). Innominate artery lesions in WD-fed ApoE-KO mice were complex, with fibrous caps comprising smooth muscle, collagen, and elastin overlying areas of lipid deposition with evidence of cholesterol crystal formation (Fig. 1Aii–iv).
Figure 1.

Selective 11β-HSD1 inhibition reduces atherosclerotic lesions in WD-fed ApoE-KO mice. A) i) On gross inspection, lesions were evident on the lesser curvature of the aortic arch (black arrow), in the innominate artery (red arrow) and at the origins of the left carotid and left subclavian arteries (black arrowheads). Lesions appeared to be smaller following 11β-HSD1 inhibition. ii) UST demonstrated large complex lesions in the innominate artery. iii) Immunohistochemistry with α-SMA identified smooth muscle cells. iv) PSR staining identified collagen. B) Image analyses of these sections confirmed that short-term selective 11β-HSD1 inhibition reduced innominate artery atherosclerosis in ApoE-KO mice. C) 11β-HSD1 inhibition did not alter the proportion of smooth muscle cells in atherosclerotic lesions in the innominate artery of ApoE-KO mice. However, collagen content was increased following 11β-HSD1 inhibition, and smaller areas devoid of cells or collagen, probably reflecting extracellular lipid pools, were reduced, compared with ApoE-KO controls (n=6/group). Original view ×40. *P < 0.05 vs. vehicle control.
Administration of a selective 11β-HSD1 inhibitor to WD-fed ApoE-KO mice for 8 wk reduced atherosclerotic lesion size in all major (aortic arch, common carotid, innominate) arterial territories examined (Fig. 1Ai). The maximum cross-sectional narrowing of the innominate artery was significantly attenuated by 11β-HSD1 inhibition (Fig. 1B). Interestingly, 11β-HSD1 inhibition for 8 wk improved markers of plaque stability (Fig. 1C), with significantly more lesional collagen, smaller areas devoid of cells or collagen (probably reflecting extracellular lipid pools), and no increase in smooth muscle cells (Fig. 1C).
The atheroprotective effects of 11β-HSD1 inhibitors were recapitulated in genetic deficiency of 11β-HSD1, confirming on-target actions. Thus, in WD-fed DKO mice, the area of the innominate artery affected by atherosclerotic lesions was strikingly reduced compared to ApoE-KO controls (Fig. 2A, C). WD-fed 11β-HSD1+/− (het) mice also had reduced atherosclerotic lesions at the innominate artery, showing that partial 11β-HSD1 deficiency is also protective against severe atherosclerosis (Fig. 2A). Reduced lesion size in het and DKO mice was accompanied by an increase in luminal area (Fig. 2A).
Figure 2.
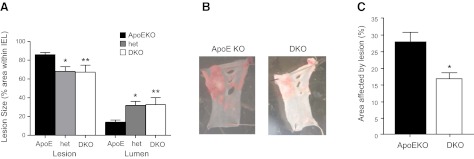
11β-HSD1 deficiency reduces atherosclerotic lesions in WD-fed ApoE-KO mice. A) Atherosclerotic lesions were smaller in the innominate arteries of 4.5-mo-old DKO mice (n=10) fed high-cholesterol WD for 14 wk. Age-matched 11β-HSD1+/−, ApoE-KO het mice (n=3) also showed significant atheroprotection compared with age-matched ApoE-KO control mice (n=11). *P < 0.05, B) Lipid incorporation was also measured by en face Sudan IV staining of sections of the aortic arch. C) Quantification of Sudan IV staining showed that DKO mice had less lipid incorporation than ApoE-KO controls. (n=10–11/group). *P < 0.05, **P < 0.01 vs. ApoE-KO.
Reduced lesion size with 11β-HSD1 deficiency is not due to improvements in metabolic cardiovascular risk factors or endothelial function
Systemic risk factors contribute to atherosclerosis, and 11β-HSD1 inhibition/deletion reduces several of these (obesity, glucose intolerance, and dyslipidemia) in high-fat dietary obesity in mice (14–16) and humans with type 2 diabetes (17). However, although WD feeding caused modest weight gain, there were no major differences between DKO and ApoE-KO mice in food intake or body weight (Table 2); if anything, DKO mice were slightly heavier. Notably, 11β-HSD1 deficiency on the ApoE-KO background did not affect plasma cholesterol and triglyceride levels in chow-fed mice, or the marked elevation of cholesterol on WD, although the WD-induced elevation of plasma triglycerides was attenuated in DKO mice (Table 2). Moreover, short-term (8 wk) 11β-HSD1 inhibition in WD-fed ApoE-KO mice did not alter fasting plasma cholesterol, triglyceride, or NEFA levels (Table 3), while reduced lesion size in WD-fed 11β-HSD1 hets was associated with higher cholesterol (34.4±2.5 mM) and triglyceride (2.90±0.30 mM) levels. Thus, the development of smaller lesions in mice with 11β-HSD1 deletion or inhibition is independent of lipid lowering. Similarly, 11β-HSD1 inhibition did not affect fasting plasma glucose or insulin levels in ApoE-KO mice (Table 3), suggesting that the protection against lesion formation is not due to improved glycemia on this strain background and diet.
Table 2.
Effect of 11β-HSD1 deficiency on physiologic characteristics and metabolic parameters in ApoE-KO mice fed chow or WD for 14 wk
| Parameter | Chow |
WD |
||
|---|---|---|---|---|
| ApoE KO | DKO | ApoE KO | DKO | |
| Body weight (g) | 33.2 ± 1.2 (n=9) | 33.4.0 ± 0.8 (n=7) | 35.4 ± 0.5 (n=7) | 37.0 ± 1.2 (n=8)† |
| Food intake (g/wk) | ND | ND | 22.9 ± 2.0 (n=11) | 21.1 ± 0.8 (n=11) |
| Cholesterol (mM) | 7.6 ± 0.7 (n=14) | 12.8 ± 1.2 (n=14)* | 23.1 ± 1.9 (n=8)††† | 22.3 ± 1.9 (n=14)††† |
| Triglycerides (mM) | 0.92 ± 0.10 (n=14) | 0.96 ± 0.11 (n=14) | 2.07 ± 0.21 (n=11)††† | 1.40 ± 0.14 (n = 13)**,† |
| Mesenteric adipose tissue weight (mg) | 200.2 ± 35.8 (n=9) | 300.7 ± 50.4 (n=7) | 440.0 ± 64.6 (n=7)† | 446.4 ± 87.4 (n=8) |
| Liver weight ratio (mg/g BW) | 56.5 ± 3.4 (n=9) | 53.2 ± 2.4 (n=7) | 50.9 ± 2.9 (n=7) | 47.0 ± 3.2 (n=8) |
| Spleen weight ratio (mg/g BW) | 4.19 ± 0.34 (n=9) | 3.04 ± 0.16 (n=7)* | 4.03 ± 0.51 (n=7) | 5.60 ± 0.79 (n=8)†† |
| Thymus weight ratio (mg/g BW) | 1.42 ± 0.16 (n=9) | 1.30 ± 0.12 (n=7) | 1.32 ± 0.10 (n=7) | 1.18 ± 0.12 (n=8) |
Data are means ± se. BW, body weight; ND, not determined.
P < 0.05,
P < 0.01 for effect of genotype;
P < 0.05,
P < 0.01,
P < 0.001 for effect of diet.
Table 3.
Effect of selective 11β-HSD1 inhibition in ApoE-KO mice on metabolic parameters
| Parameter | Vehicle | 11β-HSD1 inhibitor | P |
|---|---|---|---|
| Cumulative weight gain (g) | 1.9 ± .0.2 | 0.8 ± 0.4* | 0.03 |
| Food intake (g/d) | 8.0 ± 0.4 | 7.6 ± 0.2 | 0.3 |
| Fasting plasma cholesterol (mM) | 13.2 ± 1.5 | 12.0 ± 1.6 | 0.2 |
| Fasting plasma triglycerides (mM) | 2.04 ± 0.04 | 2.15 ± 0.06 | 0.2 |
| Fasting plasma NEFA (mM) | 2.7 ± 0.3 | 3.0 ± 0.5 | 0.6 |
| Fasting plasma glucose (AUC) | 54630 ± 1504 | 50100 ± 4448 | 0.4 |
| Fasting plasma insulin (AUC) | 108 ± 15 | 71.6 ± 14.7 | 0.1 |
Data are means ± se; n = 6/group. AUC, area under the curve.
P < 0.05 vs. vehicle control.
Endothelial dysfunction may contribute to atherosclerosis. Aortic rings from DKO mice and ApoE-KO controls, whether fed normal chow or WD, showed identical acetylcholine-elicited endothelium-dependent vasodilation (Table 4). There were also no differences by genotype or diet in SNP-mediated vasodilation (Table 4). Thus, changes in endothelial and vascular smooth muscle function do not underlie atheroprotection.
Table 4.
Effect of 11β-HSD1 deletion on functional responses of intact aortic rings from ApoE KO mice fed a chow or WD diet
| Diet |
Emax (% relaxation) |
Sensitivity (−log IC50) |
||
|---|---|---|---|---|
| ApoE KO | DKO | ApoE KO | DKO | |
| Chow | ||||
| ACh | 80.2 ± 8.5 | 90.1 ± 2.8 | 7.86 ± 0.21 | 7.89 ± 026 |
| SNP | 106.9 ± 1.9 | 106.0 ± 2.8 | 7.88 ± 0.18 | 7.88 ± 0.02 |
| WD | ||||
| ACh | 71.1 ± 4.6 | 69.3 ± 8.5 | 7.9 ± 0.2 | 7.7 ± 0.4 |
| SNP | 115.4 ± 5.6 | 104.7 ± 2.5 | 7.9 ± 0.1 | 8.2 ± 0.1 |
Data are expressed as means ± se. Chow data show the effect of 11β-HSD1 deletion on functional responses of intact aortic rings from ApoE-KO mice fed a chow diet; n = 5. WD data show the effect of 11β-HSD1 deletion on functional responses of aortic rings from apoE−/− mice fed a WD for 12 wk; n = 6. Emax, maximum response; ACh, acetylcholine; SNP, sodium nitroprusside.
Circulating glucocorticoid excess may contribute to atherogenesis. However, basal plasma corticosterone levels were similar in ApoE-KO (42.8±13.6 nM; n=6) and DKO (50.6±8.4 nM; n=8) mice. As reported previously (14), adrenal glands were heavier (P=0.001) in DKO (0.20±0.01 mg/g body wt; n=6) than ApoE-KO (0.14±0.01 mg/g body weight; n=7) mice, reflecting increased adrenal corticosterone production with 11β-reductase deficiency.
11β-HSD1-deficiency modestly reduces inflammatory cell infiltration of atherosclerosis
11β-HSD1, encoding an exclusive reductase, is expressed in innate (monocytes and macrophages; refs. 20, 21, 39) and adaptive (T cells; ref. 22) immune cells; both are implicated in atherosclerosis (2, 5, 43). This enzyme activity was abolished in resident peritoneal cells (macrophages and neutrophils) from mice with 11β-HSD1 deletion (11β-reductase activity: wild-type, 7.7 pmol/h/106 cells; 11β-HSD1-KO, below the limit of detection of the assay, <0.2 pmol/h/106 cells). Although 11β-HSD1 deficiency causes worsening of acute inflammation in joints and peritoneum, there was no evidence of worse chronic inflammation in atherosclerotic plaques. Indeed, in DKO atherosclerotic plaques, macrophage numbers were reduced overall (if unaltered per lesion area; Fig. 3B, C), and there was a 36% reduction in CD3+ T cells per lesion area (Fig. 3D, E). Real-time PCR detected iNOS (marker for M1), Relm-α (Fizz-1), and mannose receptor (markers for M2) in aortic lesions from ApoE−/− and DKO mice, but there was no difference by genotype (not shown), suggesting no change in macrophage polarization with 11β-HSD1 deficiency in this context. Reduced inflammatory cells in atherosclerotic plaques could be due to reduced circulating cells. In ApoE-KO controls, WD caused monocytosis, a change associated with fewer monocytes in BM and spleen, suggesting release from these stores (Fig. 4A). In contrast, in DKO mice, the WD-induced monocytosis was attenuated and WD increased the number of monocytes in spleen and BM (Fig. 4B, C). Although circulating neutrophils were unaffected by diet or genotype, these cells were also increased by WD in the BM and spleen of DKO mice, again suggesting increased proliferation and/or retention (Fig. 4). Indeed, DKO mice exhibited splenomegaly following WD, while the spleen weights in ApoE-KO mice were not increased by WD (Table 2). However, despite reduced circulating monocytes in WD-fed DKO mice, the 7/4hiCD11b+Ly6G− (corresponding to Ly6Chi) subset that accumulates in ApoE-KO atherosclerotic plaques (44, 45) was similar in the blood and BM of chow-fed DKO mice, suggesting little difference in pathogenic cell numbers. Moreover, macrophages in chow-fed DKO and ApoE-KO mice were recruited similarly to the peritoneum following injection of thioglycollate (Fig. 5), confirming their ability to be recruited to sites of local inflammation.
Figure 3.
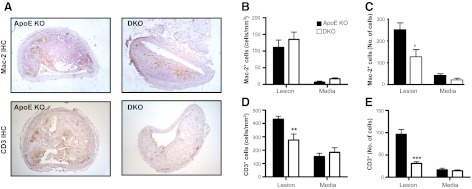
11β-HSD1 deletion attenuates macrophage and T-cell infiltration in atherosclerotic lesions. A) ApoE-KO and DKO mice (4.5 mo old) were fed WD for 14 wk, and Mac-2 or CD3 immunoreactivity was detected in the intimal lesion and the underlying media of innominate arteries. B) Cell numbers were quantified absolutely or normalized to the area of the lesion and media to give cell density (cells/mm2). Macrophage infiltration was unaltered in the lesions and media of DKO mice, when matched for lesion area. C) However, the overall numbers of macrophages were reduced in the lesions of DKO mice. D, E) T-cell infiltration (cells/mm2; D) or absolute T-cell numbers (E) were reduced into the lesions in DKO mice, but were unaltered in the media (n=5–7/group). Original view ×40. *P < 0.05, **P < 0.01, ***P < 0.001 compared with ApoE-KO.
Figure 4.

Effect of 11β-HSD1 deficiency on inflammatory monocytes (7/4hiLy6G−CD11b+) and neutrophils (7/4+Ly6G+CD11b+) in blood, BM, and spleen. DKO and ApoE-KO mice (4 wk old) were fed chow diet or WD for 14 wk, and then monocyte and neutrophil numbers in the blood (A), BM (B), and spleen (C) were quantified by flow cytometric analysis (n=5–21/group). *P < 0.05, **P < 0.01 for effects of genotype; †P < 0.05 for effects of diet.
Figure 5.

DKO monocytes and neutrophils are recruited to sites of inflammation during sterile peritonitis induced in chow-fed mice by thioglycollate. At 72 h postinjection of thioglycollate, peritoneal cells were lavaged and analyzed by flow cytometry. Quantitative analysis of blood monocyte subsets (inflammatory 7/4hiLy6G−CD11b+ monocytes, resident 7/4loLy6G−CD11b+ monocytes, and total monocytes; A); blood neutrophils (PMNs; 7/4+Ly6G+CD11b+; B), monocyte subsets in the peritoneum (C); peritoneal PMNs, macrophages (macs), and neutrophil:macrophage aggregate (F4/80+Ly6G+CD11b+) monocytes (monos; D); and PMNs in the BM (E); n = 9–11/group. *P < 0.05 vs. ApoE-KO.
MCP-1 is the major macrophage chemoattractant in atherosclerosis (46). On a chow diet, serum MCP-1 levels were similar in DKO and ApoE-KO mice (Fig. 6A). WD increased serum MCP-1 in ApoE-KO mice, but DKO mice resisted this rise (Fig. 6A), affording an explanation for the apparent retention of cells in DKO mouse BM and spleen. Reduced elaboration of MCP-1 in adipose tissue appeared to be responsible, at least in part, because mesenteric fat MCP-1 mRNA levels were reduced (Fig. 6B), whereas levels in liver were unaffected (data not shown). This was not a general change in adipose-derived chemokines since leptin mRNA did not differ (DKO, 1.27±0.38; ApoE-KO, 1.24±0.23 arbitrary units). Despite lower MCP-1 mRNA levels in DKO adipose tissue, aortic MCP-1 mRNA levels were elevated similarly by WD in DKO and control mice (Fig. 6C). MCP-1 acts largely via CCR2, but CCR2 mRNA levels in aorta were unaltered by genotype (Fig. 6D), as were aortic levels of mRNA encoding CX3CR1 (Fig. 6E), which is also key to monocyte chemotaxis into atherosclerotic lesions (42). Thus, differences in the local vascular MCP-1/chemokine receptor system do not appear to underpin reduced inflammatory cell infiltration in DKO atherosclerosis; rather this appears likely to be due, in part, to reduced availability of MCP-1.
Figure 6.

Effect of 11β-HSD1 deficiency on circulating MCP-1 and mesenteric adipose tissue MCP-1 mRNA expression and mRNA expression of MCP-1, CCR2, CX3CR1, and VCAM-1 in the ascending aortas. ApoE-KO and DKO mice (4 wk old) were fed chow diet or WD for 14 wk. A, B) Serum MCP-1 level (A) and MCP-1 mRNA expression levels in MesAT (B) were elevated by WD in ApoE-KO control mice, but not in DKO mice. C, D) Furthermore, although aortic MCP-1 (C), and CCR2 (D) mRNA levels were elevated by WD, there was no difference by genotype. E) CX3CR1 mRNA transcript levels in the aorta were unaltered by genotype and diet. F) Aortic VCAM-1 mRNA levels were similar in ApoE-KO and DKO mice fed chow diet. However, while WD increased VCAM-1 mRNA in ApoE-KO mice, there was no change in DKO tissue (n=5–8/group). *P < 0.05, **P < 0.01 for effects of genotype; †P < 0.05, ††P < 0.01 for effects of diet.
Cell adhesion molecules, notably VCAM-1, are increased in atherosclerotic vessels and play a key role in mediating infiltration of both monocytes and lymphocytes into the vasculature (47). Aortic VCAM-1 mRNA levels were similar on chow diet (Fig. 6F) but, while WD increased aortic VCAM-1 mRNA in ApoE-KO mice, this rise did not occur in DKO mice (Fig. 6F). This was specific to VCAM-1, since aortic ICAM-1 mRNA levels were unaffected by 11β-HSD1-deficiency or WD (data not shown). This suggests that, over and above any effects on inflammatory cell availability, the host vessel is less effective in recruiting inflammatory cells in 11β-HSD1 deficiency.
11β-HSD1 deficiency increases macrophage cholesterol efflux
Of course, 11β-HSD1-deficient macrophages in DKO atherosclerotic lesions might be more atherogenic, especially as high doses of glucocorticoids reduce macrophage cholesteryl ester hydrolysis and export (31), so relative glucocorticoid deficiency might be anticipated to promote cholesterol accumulation. However, uptake of AcLDL (Fig. 7A), or oxidized LDL (data not shown), was similar in 11β-HSD1-KO and wild-type macrophages, and 11β-HSD1 deficiency had no effect on expression of scavenger receptor A (SR-A) mRNA, the major mediator of cholesterol uptake (Fig. 7C), though 11β-HSD1-KO macrophages had modestly elevated levels of CD36 mRNA, which also mediates macrophage-cholesterol influx. In contrast, 11β-HSD1-KO macrophages showed greater ATP-binding cassette transporter A1 (ABCA1)-dependent cholesterol efflux (Fig. 7B), with significantly increased levels of mRNAs encoding the major cholesterol efflux transporters and acceptor, ABCA1, ATP-binding cassette transporter G1 (ABCG1), and ApoE. Thus, 11β-HSD1-deficient macrophages show an intrinsic antiatherosclerotic balance of cholesterol trafficking.
Figure 7.
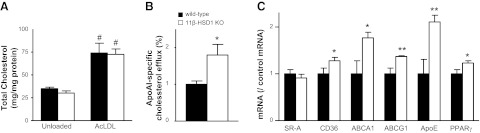
11β-HSD1 deficiency increases macrophage cholesterol efflux. A) BM-derived macrophages from 11β-HSD1-KO and C57BL/6J (wild-type) controls showed no difference in cholesterol content either basally (unloaded) or following loading with 50 mg/ml AcLDL for 24 h. Data are representative of 2 independent experiments each performed in triplicate. B) ApoAI-stimulated cholesterol efflux was increased in AcLDL-loaded macrophages from 11β-HSD1-KO mice. C) expression of mRNAs encoding cholesterol efflux transporters ApoE, ABCA1, and ABCG1 was increased in 11β-HSD1-KO macrophages, plausibly because of increased peroxisome proliferator-activated receptor γ (PPARγ). In contrast, SR-A mRNA was unaltered, though CD36 antigen was also increased. *P < 0.05, **P < 0.01 vs. wild type; #P < 0.05 vs. unloaded cells.
Atheroprotection occurs with 11β-HSD1−/− BM transplantation
To determine whether 11β-HSD1-deficient BM-derived cells are also intrinsically atheroprotective, ApoE-KO recipient mice were lethally irradiated, and BM was transplanted from either ApoE-KO or DKO mice. Following reconstitution, body and organ weights, plasma lipids, and circulating blood leukocyte numbers were not different between the groups, nor did hematopoietic 11β-HSD1 deficiency affect monocyte and neutrophil numbers in the BM and spleen (Table 5). However in ApoE-KO mice, innominate artery atherosclerotic lesion volume, assessed by OPT (38), and lesion cross-sectional area were significantly reduced by transplantation with DKO BM (Fig. 8), compared to lesions in ApoE-KO mice transplanted with autologous ApoE-KO BM. Macrophage infiltration into ApoE-KO atherosclerotic lesions was unaffected by deletion of 11β-HSD1 in BM cells (data not shown), similar to global 11β-HSD1-deficiency. Thus, atheroprotection is mediated by 11β-HSD1-deficient BM-derived cells.
Table 5.
Physiological characteristics, metabolic parameters, and immune cell phenotyping in ApoE-KO mice transplanted with ApoE-KO or DKO BM cells
| Parameter | ApoE-KO BM → ApoE-KO | DKO BM → ApoE-KO |
|---|---|---|
| Body weight (g) | 32.4 ± 2.2 | 36.4 ± 2.6 |
| Subcutaneous adipose tissue weight, absolute weight (mg) | 809.7 ± 249.5 | 1106.4 ± 218.2 |
| Epididymal adipose tissue weight, absolute weight (mg) | 1091.3 ± 317.2 | 1481.3 ± 285.7 |
| Mesenteric adipose tissue weight, absolute weight (mg) | 364.1.3 ± 70.9 | 606.1 ± 135.0 |
| Spleen weight ratio (mg/g BW) | 4.56 ± 0.41 | 4.51 ± 0.73 |
| Thymus weight ratio (mg/g BW) | 1.12 ± 0.17 | 0.92 ± 0.16 |
| Heart weight ratio (mg/g BW) | 4.88 ± 0.44 | 4.68 ± 0.38 |
| Left kidney weight ratio (mg/g BW) | 6.19 ± 0.41 | 5.65 ± 0.23 |
| Monocytes in blood (105 cells/ml) | 10.8 ± 1.7 | 10.9 ± 1.7 |
| Neutrophils in blood (105 cells/ml) | 22.3 ± 6.5 | 16.2 ± 2.9 |
| T lymphocytes in blood (105 cells/ml) | 16.6 ± 1.8 | 14.7 ± 1.0 |
| B lymphocytes in blood (105 cells/ml) | 69.4 ± 4.2 | 58.7 ± 7.8 |
| CD45+11β-HSD1+ in blood (105 cells/ml) | 60.5 ± 10.6 | 3.2 ± 0.7*** |
| BM monocytes (105 cells/femur) | 7.1 ± 2.3 | 8.3 ± 1.9 |
| BM neutrophils (105 cells/femur) | 35.8 ± 11.8 | 53.2.±14.4 |
| BM CD45+11β-HSD1+ (105 cells/femur) | 50.0 ± 16.2 | 1.8 ± 0.4* |
| Splenic monocytes (105 cells/femur) | 5.2 ± 1.7 | 9.2 ± 3.2 |
| Splenic neutrophils (105 cells/femur) | 6.2 ± 1.4 | 9.6 ± 2.6 |
| Splenic T lymphocytes (105 cells/ml) | 10.0 ± 2.8 | 12.1 ± 4.9 |
| Splenic B lymphocytes (105 cells/ml) | 22.5 ± 7.3 | 24.2 ± 8.8 |
Data are expressed as means ± se; n = 5/group. BW, body weight.
P < 0.001 vs. ApoE-KO BM → ApoE-KO.
Figure 8.

11β-HSD1 deletion in BM cells reduces atherosclerotic lesion size. Lethally irradiated ApoE-KO recipient mice were reconstituted with BM from ApoE-KO mice or DKO mice and fed WD for 12 wk. Formalin-fixed innominate arteries underwent OPT scanning. Analysis of lesion volumes (A) and lesion cross-sectional area (B) in the innominate arteries demonstrated that ApoE-KO recipient mice reconstituted with DKO BM cells exhibited significantly smaller lesions when compared to ApoE-KO mice reconstituted with ApoE-KO BM cells (n=5/group). *P < 0.05, **P < 0.01 vs. ApoE-KO mice reconstituted with ApoE-KO BM cells.
DISCUSSION
11β-HSD1 deficiency or inhibition substantially reduces atherosclerosis in ApoE-KO mice. The effect was clearcut, was similar with short-term selective inhibition or genetic deficiency of 11β-HSD1, and involved both a reduction in atherosclerotic lesion volume and, crucially, an increase in the vessel lumen. Partial deficiency of 11β-HSD1 was also atheroprotective. Despite theoretical concerns of worse lesional inflammation, 11β-HSD1 deficiency was accompanied by no increase in macrophages and reduced T-cell infiltration. There was also an improvement in histological lesional stability with increased fibrosis, plausibly reflecting attenuated glucocorticoid-mediated inhibition of collagen biosynthesis (48). Atheroprotection did not require improvements in metabolic cardiovascular risk factors. Instead, 11β-HSD1-deficient BM cells were responsible for atheroprotection, plausibly at least in part because they exhibit an atheroprotective phenotype of enhanced cholesterol export.
The anticipated increase in lesional inflammation with 11β-HSD1 deficiency was not seen, with fewer T cells and no increase in macrophages per lesional area, plausibly due to lower circulating MCP-1 and less aortic VCAM-1. Physiological glucocorticoids increase VCAM-1 via stimulation of mineralocorticoid receptors (which are high-affinity sites for corticosterone in the absence of high expression of 11β-HSD type 2) in mouse aortic endothelial cells (49), affording a possible explanation to contrast with the effects of high doses of synthetic glucocorticoids which act via glucocorticoid receptors to reduce endothelial VCAM-1 transcription in vitro (50), while glucocorticoid receptor antagonists elevate VCAM-1 mRNA (51).
Perhaps surprisingly, there was little alteration in circulating lipids to explain 11β-HSD1 deficiency/inhibition-mediated atheroprotection on the ApoE-KO background. This may reflect the already markedly hyperlipidemic state of this strain and/or the only marginally obesogenic effect of WD—the beneficial effects of 11β-HSD1 deficiency/inhibition on cardiovascular risk factors predominantly occur in obesity (16, 17). 11β-HSD1 inhibition/deficiency also ameliorates a number of cardiovascular risk factors that are likely to add to the atheroprotective benefits in wild-type rodents and perhaps humans.
11β-HSD1-deficient macrophages were able more efficiently to export cholesterol, an impact expected to be even greater when ApoE is present (ApoE mRNA levels were higher in 11β-HSD1-KO macrophages), since ApoE is a key atheroprotective VLDL cholesterol transporter with anti-inflammatory effects (52, 53). This suggests that 11β-HSD1 inhibitor-mediated atheroprotection may extend to models of atherosclerosis expressing ApoE and perhaps to humans. Notably, the atheroprotective effect was seen with 11β-HSD1 deficiency restricted in transplanted BM, implying a key role for 11β-HSD1-deficiency in BM-derived cells over and beyond any effects on host metabolism or the aortic milieu per se. Overall, this study localizes the atheroprotective effects of 11β-HSD1 deficiency/inhibition to 11β-HSD1 deficiency in BM. This is very likely to be mediated by leukocytes, e.g., changes in macrophages, but further work is required to characterize which BM-derived leukocytes are responsible. These results support the clinical development of selective inhibitors of 11β-HSD1 as a novel multimodal treatment for atherosclerosis and lesional inflammation over and above their role in metabolic disease, from which the major cause of mortality is atherosclerotic disease.
Acknowledgments
The authors thank Dr. Valerie S. Densmore for generating the original 11β-HSD1, ApoE double-knockout line, and Dr. Nicholas Kirkby for help with optical projection tomography.
This work was supported by grants from the Wellcome Trust (program grant 083184/Z/07/Z), the Medical Research Council (project grant 86642), the Society for Endocrinology, the British Heart Foundation (BHF), Agence Nationale de la Recherche (AlMHA project), and the Carnegie Trust.
The authors acknowledge the support of the BHF Centre of Excellence. Conflict of interest: B.R.W. and J.R.S. hold intellectual property on the concept of 11β-HSD1 inhibitors in metabolic and cardiovascular disease.
This article includes supplemental data. Please visit http://www.fasebj.org to obtain this information.
- 11β-HSD1
- 11β-hydroxysteroid dehydrogenase type 1
- α-SMA
- α-smooth muscle actin
- ABCA1
- ATP-binding cassette transporter A1
- ABCG1
- ATP-binding cassette transporter G1
- AcLDL
- acetylated low-density lipoprotein
- APC
- allophycocyanin
- ApoAI
- apolipoprotein AI
- ApoE
- apolipoprotein E
- BM
- bone marrow
- CCR2
- chemokine (C-C motif) receptor 2
- CCR2
- chemokine (C-C motif) receptor 2
- CX3CR1
- fractalkine receptor
- DKO
- double knockout
- FITC
- fluorescein isothiocyanate
- HbA1c
- glycated hemoglobin A1c
- het
- heterozygote
- ICAM-1
- intercellular adhesion molecule-1
- IEL
- internal elastic laminae
- iNOS
- inducible nitric oxide synthase
- KO
- knockout
- MCP-1
- monocyte chemoattractant protein 1
- MS
- mouse serum
- NEFA
- nonesterified fatty acids
- OPT
- optical projection tomography
- PE
- phycoerythrin
- PECy7
- phycoerythrin-cyanine 7
- PerCPCy5.5
- peridinin chlorophyll protein cyanin 5.5
- PSR
- picrosirius red
- RBC
- red blood cell
- Relm-α
- resistin like molecule-α
- SNP
- sodium nitroprusside
- SR-A
- scavenger receptor A
- TBP
- TATA-box binding protein
- UST
- U.S. trichrome
- VCAM-1
- vascular cell adhesion molecule 1
- WD
- Western diet
REFERENCES
- 1. Libby P. (2002) Inflammation in atherosclerosis. Nature 420, 868–874 [DOI] [PubMed] [Google Scholar]
- 2. Hansson G. K., Libby P. (2006) The immune response in atherosclerosis: a double-edged sword. Nat. Rev. Immunol. 6, 508–519 [DOI] [PubMed] [Google Scholar]
- 3. Hansson G. K., Hermansson A. The immune system in atherosclerosis. Nat. Immunol. 12, 204–212 [DOI] [PubMed] [Google Scholar]
- 4. Taleb S., Tedgui A., Mallat Z. Adaptive T cell immune responses and atherogenesis. Curr. Opin. Pharmacol. 10, 197–202 [DOI] [PubMed] [Google Scholar]
- 5. Packard R. R., Lichtman A. H., Libby P. (2009) Innate and adaptive immunity in atherosclerosis. Semin. Immunopathol. 31, 5–22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Galkina E., Ley K. (2009) Immune and inflammatory mechanisms of atherosclerosis. Annu. Rev. Immunol. 27, 165–197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Alevizaki M., Cimponeriu A., Lekakis J., Papamichael C., Chrousos G. P. (2007) High anticipatory stress plasma cortisol levels and sensitivity to glucocorticoids predict severity of coronary artery disease in subjects undergoing coronary angiography. Metabolism 56, 222–226 [DOI] [PubMed] [Google Scholar]
- 8. Souverein P. C., Berard A., Van Staa T. P., Cooper C., Egberts A. C., Leufkens H. G., Walker B. R. (2004) Use of oral glucocorticoids and risk of cardiovascular and cerebrovascular disease in a population based case-control study. Heart 90, 859–865 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Etxabe J., Vazquez J. A. (1994) Morbidity and mortality in Cushing's disease: an epidemiological approach. Clin. Endocrinol. (Oxf.) 40, 479–484 [DOI] [PubMed] [Google Scholar]
- 10. Faggiano A., Pivonello R., Spiezia S., De Martino M. C., Filippella M., Di Somma C., Lombardi G., Colao A. (2003) Cardiovascular risk factors and common carotid artery caliber and stiffness in patients with Cushing's disease during active disease and 1 year after disease remission. J. Clin. Endocrinol. Metab. 88, 2527–2533 [DOI] [PubMed] [Google Scholar]
- 11. Rask E., Olsson T., Soderberg S., Andrew R., Livingstone D. E., Johnson O., Walker B. R. (2001) Tissue-specific dysregulation of cortisol metabolism in human obesity. J. Clin. Endocrinol. Metab. 86, 1418–1421 [DOI] [PubMed] [Google Scholar]
- 12. Livingstone D. E., Jones G. C., Smith K., Jamieson P. M., Andrew R., Kenyon C. J., Walker B. R. (2000) Understanding the role of glucocorticoids in obesity: tissue-specific alterations of corticosterone metabolism in obese Zucker rats. Endocrinology 141, 560–563 [DOI] [PubMed] [Google Scholar]
- 13. Masuzaki H., Paterson J., Shinyama H., Morton N. M., Mullins J. J., Seckl J. R., Flier J. S. (2001) A transgenic model of visceral obesity and the metabolic syndrome. Science 294, 2166–2170 [DOI] [PubMed] [Google Scholar]
- 14. Kotelevtsev Y., Holmes M. C., Burchell A., Houston P. M., Schmoll D., Jamieson P., Best R., Brown R., Edwards C. R., Seckl J. R., Mullins J. J. (1997) 11β-hydroxysteroid dehydrogenase type 1 knockout mice show attenuated glucocorticoid-inducible responses and resist hyperglycemia on obesity or stress. Proc. Natl. Acad. Sci. U. S. A. 94, 14924–14929 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Morton N. M., Holmes M. C., Fievet C., Staels B., Tailleux A., Mullins J. J., Seckl J. R. (2001) Improved lipid and lipoprotein profile, hepatic insulin sensitivity, and glucose tolerance in 11β-hydroxysteroid dehydrogenase type 1 null mice. J. Biol. Chem. 276, 41293–41300 [DOI] [PubMed] [Google Scholar]
- 16. Morton N. M., Paterson J. M., Masuzaki H., Holmes M. C., Staels B., Fievet C., Walker B. R., Flier J. S., Mullins J. J., Seckl J. R. (2004) Novel adipose tissue-mediated resistance to diet-induced visceral obesity in 11β-hydroxysteroid dehydrogenase type 1-deficient mice. Diabetes 53, 931–938 [DOI] [PubMed] [Google Scholar]
- 17. Rosenstock J., Banarer S., Fonseca V. A., Inzucchi S. E., Sun W., Yao W., Hollis G., Flores R., Levy R., Williams W. V., Seckl J. R., Huber R. The 11-β-hydroxysteroid dehydrogenase type 1 inhibitor INCB13739 improves hyperglycemia in patients with type 2 diabetes inadequately controlled by metformin monotherapy. Diabetes Care 33, 1516–1522 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Hermanowski-Vosatka A., Balkovec J. M., Cheng K., Chen H. Y., Hernandez M., Koo G. C., Le Grand C. B., Li Z., Metzger J. M., Mundt S. S., Noonan H., Nunes C. N., Olson S. H., Pikounis B., Ren N., Robertson N., Schaeffer J. M., Shah K., Springer M. S., Strack A. M., Strowski M., Wu K., Wu T., Xiao J., Zhang B. B., Wright S. D., Thieringer R. (2005) 11β-HSD1 inhibition ameliorates metabolic syndrome and prevents progression of atherosclerosis in mice. J. Exp. Med. 202, 517–527 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Lloyd D. J., Helmering J., Cordover D., Bowsman M., Chen M., Hale C., Fordstrom P., Zhou M., Wang M., Kaufman S. A., Veniant M. M. (2009) Antidiabetic effects of 11β-HSD1 inhibition in a mouse model of combined diabetes, dyslipidaemia and atherosclerosis. Diabetes Obes. Metab. 11, 688–699 [DOI] [PubMed] [Google Scholar]
- 20. Thieringer R., Le Grand C. B., Carbin L., Cai T. Q., Wong B., Wright S. D., Hermanowski-Vosatka A. (2001) 11-β-hydroxysteroid dehydrogenase type 1 is induced in human monocytes upon differentiation to macrophages. J. Immunol. 167, 30–35 [DOI] [PubMed] [Google Scholar]
- 21. Gilmour J. S., Coutinho A. E., Cailhier J. F., Man T. Y., Clay M., Thomas G., Harris H. J., Mullins J. J., Seckl J. R., Savill J. S., Chapman K. E. (2006) Local amplification of glucocorticoids by 11-β-hydroxysteroid dehydrogenase type 1 promotes macrophage phagocytosis of apoptotic leukocytes. J. Immunol. 176, 7605–7611 [DOI] [PubMed] [Google Scholar]
- 22. Zhang T. Y., Ding X., Daynes R. A. (2005) The expression of 11β-hydroxysteroid dehydrogenase type I by lymphocytes provides a novel means for intracrine regulation of glucocorticoid activities. J. Immunol. 174, 879–889 [DOI] [PubMed] [Google Scholar]
- 23. Liu Y., Cousin J. M., Hughes J., Van Damme J., Seckl J. R., Haslett C., Dransfield I., Savill J., Rossi A. G. (1999) Glucocorticoids promote nonphlogistic phagocytosis of apoptotic leukocytes. J. Immunol. 162, 3639–3646 [PubMed] [Google Scholar]
- 24. Chapman K. E., Coutinho A. E., Gray M., Gilmour J. S., Savill J. S., Seckl J. R. (2009) The role and regulation of 11β-hydroxysteroid dehydrogenase type 1 in the inflammatory response. Mol. Cell. Endocrinol. 301, 123–131 [DOI] [PubMed] [Google Scholar]
- 25. Coutinho A. E., Gray M., Seckl J. R., Chapman K. E. (2012) 11β-hydroxysteroid dehydrogenase type 1, but not type 2, deficiency worsens acute inflammation and experimental arthritis in mice. Endocrinology 153, 234–240 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Poon M., Gertz S. D., Fallon J. T., Wiegman P., Berman J. W., Sarembock I. J., Taubman M. B. (2001) Dexamethasone inhibits macrophage accumulation after balloon arterial injury in cholesterol fed rabbits. Atherosclerosis 155, 371–380 [DOI] [PubMed] [Google Scholar]
- 27. Goncharova E. A., Billington C. K., Irani C., Vorotnikov A. V., Tkachuk V. A., Penn R. B., Krymskaya V. P., Panettieri R. A., Jr. (2003) Cyclic AMP-mobilizing agents and glucocorticoids modulate human smooth muscle cell migration. Am. J. Respir. Cell Mol. Biol. 29, 19–27 [DOI] [PubMed] [Google Scholar]
- 28. Longenecker J. P., Kilty L. A., Johnson L. K. (1984) Glucocorticoid inhibition of vascular smooth muscle cell proliferation: influence of homologous extracellular matrix and serum mitogens. J. Cell Biol. 98, 534–540 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Hadoke P. W., Christy C., Kotelevtsev Y. V., Williams B. C., Kenyon C. J., Seckl J. R., Mullins J. J., Walker B. R. (2001) Endothelial cell dysfunction in mice after transgenic knockout of type 2, but not type 1, 11β-hydroxysteroid dehydrogenase. Circulation 104, 2832–2837 [DOI] [PubMed] [Google Scholar]
- 30. Small G. R., Hadoke P. W., Sharif I., Dover A. R., Armour D., Kenyon C. J., Gray G. A., Walker B. R. (2005) Preventing local regeneration of glucocorticoids by 11β-hydroxysteroid dehydrogenase type 1 enhances angiogenesis. Proc. Natl. Acad. Sci. U. S. A. 102, 12165–12170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Ayaori M., Sawada S., Yonemura A., Iwamoto N., Ogura M., Tanaka N., Nakaya K., Kusuhara M., Nakamura H., Ohsuzu F. (2006) Glucocorticoid receptor regulates ATP-binding cassette transporter-A1 expression and apolipoprotein-mediated cholesterol efflux from macrophages. Arterioscler. Thromb. Vasc. Biol. 26, 163–168 [DOI] [PubMed] [Google Scholar]
- 32. Canalis E. (1983) Effect of glucocorticoids on type I collagen synthesis, alkaline phosphatase activity, and deoxyribonucleic acid content in cultured rat calvariae. Endocrinology 112, 931–939 [DOI] [PubMed] [Google Scholar]
- 33. Cutroneo K. R., Counts D. F. (1975) Anti-inflammatory steroids and collagen metabolism: glucocorticoid-mediated alterations of prolyl hydroxylase activity and collagen synthesis. Mol. Pharmacol. 11, 632–639 [PubMed] [Google Scholar]
- 34. Holman R. L., Mc G. H., Jr., Strong J. P., Geer J. C. (1958) Technics for studying atherosclerotic lesions. Lab. Invest. 7, 42–47 [PubMed] [Google Scholar]
- 35. Hadoke P., Wainwright C. L., Wadsworth R. M., Butler K., Giddings M. J. (1995) Characterization of the morphological and functional alterations in rabbit subclavian artery subjected to balloon angioplasty. Coron. Artery Dis. 6, 403–415 [DOI] [PubMed] [Google Scholar]
- 36. Shiomi M., Ito T., Tsukada T., Yata T., Watanabe Y., Tsujita Y., Fukami M., Fukushige J., Hosokawa T., Tamura A. (1995) Reduction of serum cholesterol levels alters lesional composition of atherosclerotic plaques. Effect of pravastatin sodium on atherosclerosis in mature WHHL rabbits. Arterioscler. Thromb. Vasc. Biol. 15, 1938–1944 [DOI] [PubMed] [Google Scholar]
- 37. Williams H., Johnson J. L., Carson K. G., Jackson C. L. (2002) Characteristics of intact and ruptured atherosclerotic plaques in brachiocephalic arteries of apolipoprotein E knockout mice. Arterioscler. Thromb. Vasc. Biol. 22, 788–792 [DOI] [PubMed] [Google Scholar]
- 38. Kirkby N. S., Low L., Seckl J. R., Walker B. R., Webb D. J., Hadoke P. W. Quantitative 3-dimensional imaging of murine neointimal and atherosclerotic lesions by optical projection tomography. PLoS One 6, e16906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. De Sousa Peixoto R. A., Turban S., Battle J. H., Chapman K. E., Seckl J. R., Morton N. M. (2008) Preadipocyte 11β-hydroxysteroid dehydrogenase type 1 is a keto-reductase and contributes to diet-induced visceral obesity in vivo. Endocrinology 149, 1861–1868 [DOI] [PubMed] [Google Scholar]
- 40. Harris H. J., Kotelevtsev Y., Mullins J. J., Seckl J. R., Holmes M. C. (2001) Intracellular regeneration of glucocorticoids by 11β-hydroxysteroid dehydrogenase (11β-HSD)-1 plays a key role in regulation of the hypothalamic-pituitary-adrenal axis: analysis of 11β-HSD-1-deficient mice. Endocrinology 142, 114–120 [DOI] [PubMed] [Google Scholar]
- 41. Rigamonti E., Helin L., Lestavel S., Mutka A. L., Lepore M., Fontaine C., Bouhlel M. A., Bultel S., Fruchart J. C., Ikonen E., Clavey V., Staels B., Chinetti-Gbaguidi G. (2005) Liver X receptor activation controls intracellular cholesterol trafficking and esterification in human macrophages. Circ. Res. 97, 682–689 [DOI] [PubMed] [Google Scholar]
- 42. Combadiere C., Potteaux S., Rodero M., Simon T., Pezard A., Esposito B., Merval R., Proudfoot A., Tedgui A., Mallat Z. (2008) Combined inhibition of CCL2, CX3CR1, and CCR5 abrogates Ly6C(hi) and Ly6C(lo) monocytosis and almost abolishes atherosclerosis in hypercholesterolemic mice. Circulation 117, 1649–1657 [DOI] [PubMed] [Google Scholar]
- 43. Weber C., Zernecke A., Libby P. (2008) The multifaceted contributions of leukocyte subsets to atherosclerosis: lessons from mouse models. Nat. Rev. Immunol. 8, 802–815 [DOI] [PubMed] [Google Scholar]
- 44. Tacke F., Alvarez D., Kaplan T. J., Jakubzick C., Spanbroek R., Llodra J., Garin A., Liu J., Mack M., van Rooijen N., Lira S. A., Habenicht A. J., Randolph G. J. (2007) Monocyte subsets differentially employ CCR2, CCR5, and CX3CR1 to accumulate within atherosclerotic plaques. J. Clin. Invest. 117, 185–194 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Swirski F. K., Libby P., Aikawa E., Alcaide P., Luscinskas F. W., Weissleder R., Pittet M. J. (2007) Ly-6Chi monocytes dominate hypercholesterolemia-associated monocytosis and give rise to macrophages in atheromata. J. Clin. Invest. 117, 195–205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Charo I. F., Taubman M. B. (2004) Chemokines in the pathogenesis of vascular disease. Circ. Res. 95, 858–866 [DOI] [PubMed] [Google Scholar]
- 47. Blankenberg S., Barbaux S., Tiret L. (2003) Adhesion molecules and atherosclerosis. Atherosclerosis 170, 191–203 [DOI] [PubMed] [Google Scholar]
- 48. Meisler N., Shull S., Xie R., Long G. L., Absher M., Connolly J. P., Cutroneo K. R. (1995) Glucocorticoids coordinately regulate type I collagen pro alpha 1 promoter activity through both the glucocorticoid and transforming growth factor beta response elements: a novel mechanism of glucocorticoid regulation of eukaryotic genes. J. Cell. Biochem. 59, 376–388 [DOI] [PubMed] [Google Scholar]
- 49. Deuchar G. A., McLean D., Hadoke P. W., Brownstein D. G., Webb D. J., Mullins J. J., Chapman K., Seckl J. R., Kotelevtsev Y. V. 11β-hydroxysteroid dehydrogenase type 2 deficiency accelerates atherogenesis and causes proinflammatory changes in the endothelium in apoe−/− mice. Endocrinology 152, 236–246 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Simoncini T., Maffei S., Basta G., Barsacchi G., Genazzani A. R., Liao J. K., De Caterina R. (2000) Estrogens and glucocorticoids inhibit endothelial vascular cell adhesion molecule-1 expression by different transcriptional mechanisms. Circ. Res. 87, 19–25 [DOI] [PubMed] [Google Scholar]
- 51. Cavalcanti D. M., Lotufo C. M., Borelli P., Ferreira Z. S., Markus R. P., Farsky S. H. (2007) Endogenous glucocorticoids control neutrophil mobilization from bone marrow to blood and tissues in non-inflammatory conditions. Br. J. Pharmacol. 152, 1291–1300 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Curtiss L. K. (2000) ApoE in atherosclerosis: a protein with multiple hats. Arterioscler. Thromb. Vasc. Biol. 20, 1852–1853 [DOI] [PubMed] [Google Scholar]
- 53. Zhu Y., Kodvawala A., Hui D. Y. Apolipoprotein E inhibits Toll-like receptor (TLR)-3- and TLR-4-mediated macrophage activation through distinct mechanisms. Biochem. J. 428, 47–54 [DOI] [PMC free article] [PubMed] [Google Scholar]