

Abstract
One of the hallmark features of HIV-associated neurological disease is increased activation and migration of microglia. HIV transactivator of transcription (Tat) is released from infected cells and has the ability to recruit microglia. The purpose of this study was to investigate molecular mechanisms by which recombinant Tat1-72, but not heated-inactive Tat1-72,induces migration of rat primary microglia. Using primary microglia in Boyden chambers, we demonstrated the role of nonmuscle myosin light-chain kinase (nmMYLK) in Tat1-72 (14.4 nM)-mediated increased microglial migration (up to 171.85%). These findings were validated using microglia isolated from wild-type (WT) or nmMYLK−/− mice in Dunn chamber assays. Tat1-72-mediated activation of nmMYLK resulted in “inside-out” activation of β1 integrin, followed by “outside-in” activation of c-Src, Pyk2, and Cdc42-GTP (using G-LISA in primary and nmMYLK−/− microglia) and, subsequently, actin polymerization (flow cytometry and Western blot assays). In vivo corroboration of these findings demonstrated decreased migration of nmMYLK−/− microglia (2×105 cells transplanted into corpus callosum) compared with WT microglia toward microinjected Tat1-72 (2 μg/mouse) in hippocampus. Up-regulation of nmMYLK in microglia was also detected in sections of basal ganglia from humans with HIV-encephalitis compared with uninfected controls. nmMYLK is thus critical for eliciting microglial migration during the innate immune response.—Yao, H., Duan, M., Yang, L., Buch, S. Nonmuscle myosin light-chain kinase mediates microglial migration induced by HIV Tat: Involvement of β1 integrins.
Keywords: neuroinflammation, nmMYLK
Under normal CNS homeostasis, microglia manifest as ramified cells with highly branched, motile cell processes (1, 2). In response to pathological stimuli, these cells undergo phenotypic changes, elicit inflammatory responses, and migrate toward the toxic stimuli. Paradoxically, these cells, while crucial for host defense, can also contribute to neuropathology (3). During human immunodeficiency virus (HIV) infection, release of extracellular transactivator of transcription (Tat) from infected cells becomes the focal site for microglial migration and activation.
HIV Tat both is released by HIV-infected cells and can be taken up by neighboring cells. Extracellular Tat targets various cells of the CNS, causing a variety of biological effects ultimately leading to AIDS-associated pathologies (4). Five distinct functional domains of Tat have been characterized: the N-terminal, cysteine-rich, core, basic, and C-terminal regions (5). Each of these domains plays a critical role in eliciting a specific response in various cell types. For example, previous studies have demonstrated that the multiple paracrine effects of Tat are primarily due to its C-terminal domain containing an arginine-glycine-aspartic (RGD) sequence, which represents the principal cell attachment moiety recognized by the cognate integrin receptor (5). In endothelial cells, Tat, via its basic domain, phosphorylates vascular endothelial growth factor receptor type 2 (VEGF-R2; ref. 6). In cells of the monocytic lineage, Tat inhibits autophagy via its interaction with the cell surface receptors CXCR4, VEGFR, and β-integrin (7). Furthermore, in microglia, Tat-mediated induction of CCL2 involves activation of one or more receptors, including the low-density lipoprotein receptor-related protein, integrins, and the VEGF receptor (8). Despite extensive studies extant on the biological function of HIV Tat, detailed mechanisms underlying Tat-mediated microglial migration remain elusive.
Activation of cytoskeletal contractile machinery is critical for cell motility (9). The contractile force is generated on the basis of actin-myosin binding triggered by phosphorylation of myosin light chain (MLC), the major substrate of myosin light-chain kinase (MYLK). MYLK is a calcium calmodulin–dependent kinase that exists as either the short form (muscle, 108–130 kDa) or the long form (nonmuscle, 210 kDa) (9–11). Since MYLK-210, but not MYLK-130, preferentially mediates interaction with the actin cytoskeleton components (12), our studies were focused on the long form of MYLK. Consistent with its function in cell contraction, this form of MYLK contributes to endothelial cell junction opening in response to proinflammatory mediators (13, 14). Nonmuscle MYLK (nmMYLK) also participates in inflammatory processes by promoting neutrophil transendothelial migration (15, 16). The pathophysiological importance of MYLK in inflammation has been demonstrated in studies using MYLK-knockout animals. More recently, MYLK has been suggested as a new target for intervention aimed at modulating reactive astrocyte migration in glaucoma (17). The role of MYLK in regulating microglial migration, however, has never been investigated.
MYLK, via regulation of myosin II, is involved in integrin-mediated attachment of the leading edge to the matrix in T cells and fibroblasts (18, 19). MYLK also interacts with cytoskeletal components, such as actin and microtubules (12, 19, 20), which raises the possibility that MYLK regulates integrin function by organizing the cytoskeletal architecture (21). Integrins are a family of transmembrane receptors for extracellular matrix (ECM) that function as both adhesive and signaling mediators (22–25). Adhesive interactions with the ECM are critical determinants of process extension and cell migration. Intracellular signals such as those stimulated by chemokine receptors activate integrins to an intermediate active state and induce the binding of integrins to their ligands in the ECM (26). Integrin-matrix interactions, in turn, induce “outside-in” signals to activate tyrosine kinases c-Src, Syk, and Pyk2 (27, 28), resulting in actin polymerization, thereby reinforcing the integrin-cytoskeleton connection. Here, we elucidate the cellular signaling mechanisms involved in Tat-mediated migration of microglia that leads to enhanced neuroinflammation associated with HIV-associated neurocognitive disorder (HAND).
MATERIALS AND METHODS
Reagents
HIV Tat1-86, HIV Tat1-72, and mutant Tat1-72Δ31-61 were ordered from the University of Kentucky College of Medicine (Lexington, KY, USA). HIV Tat1-15 and Tat46-60 were obtained from Peptide 2.0 (Chantilly, VA, USA). HIV Tat101 was obtained from Diatheva (Fano, Italy). The specific Src kinase inhibitors PP2 and PP3, MYLK inhibitor ML-7, and myosin II inhibitor blebbistatin were purchased from Sigma-Aldrich (St. Louis, MO, USA). Leaf purified anti-mouse/rat CD29 (neutralizing β1 antibody; clone HM β1-1; cat. no. 102210), Leaf purified anti-mouse/rat CD61 (neutralizing β3 antibody; clone 2c9.G2; HM β3-1; 104310) and purified American hamster IgG isotype control HTK888 (400902) were obtained from BioLegend (San Diego, CA, USA). c(RGDfV) (H-2574) and control c(RADfV) (H-4088) were ordered from Bachem (Torrance, CA, USA). VEGF-R1 neutralizing antibody was obtained from R&D Systems (Minneapolis, MN, USA). Purified hamster anti-mouse MCP-1 and purified IgG isotype control were obtained from BD Pharmingen (Franklin Lakes, NJ, USA). Anti-integrin β1antibody activated clone HUTS-4 (MAB2079Z) was obtained from Millipore (Temecula, CA, USA). Anti-integrin β3 (phosphor Y785) antibody was purchased from AbCam (Cambridge, MA, USA).
Animals
C57BL/6N mice were purchased from Charles River Laboratories, Inc. (Wilmington, MA, USA). All the animals were housed under conditions of constant temperature and humidity on a 12-h light-dark cycle, with lights on at 7 AM. Food and water were available ad libitum. Animals were deeply anesthetized by overdose of isoflurane, followed by pneumothorax prior to perfusion. All animal procedures were performed according to the protocols approved by the Institutional Animal Care and Use Committee of the University of Nebraska Medical Center.
Isolation of primary microglia
Primary microglia cells were obtained from 1- to 3-d-old C57BL/6 or Sprague-Dawley newborn pups, as described previously (29). MYLK−/− microglia were isolated from MYLK−/− mouse brain tissue that was provided by Dr. Asrar Malik (University of Illinois College of Medicine, Chicago, IL, USA; ref. 16). After digestion and dissociation of the dissected brain cortices in Hank's buffered salt solution supplemented with trypsin (0.25%), mixed glial cultures were prepared by resuspending the cell suspension in DMEM supplemented with 10% heated inactivated fetal bovine serum, 100 U/ml penicillin, and 0.1 mg/ml streptomycin. Cells were plated at 20 × 106 cells/flask density onto 75-cm2 cell culture flasks. The cell medium was replaced every 5 d, and after the first medium change, macrophage colony-stimulating factor (M-CSF; 0.25 ng/ml; PeproTech, Rocky Hill, NJ, USA) was added to the flasks to promote microglial proliferation.
On confluence (7–10 d), mixed glial cultures were subjected to shaking at 37°C at 220 g for 2 h, to promote microglia detachment from the flasks. The cell medium, containing the released microglia cells, was collected from each flask and centrifuged at 100 g for 5 min to collect cells, then plated on cell culture plates for all subsequent experiments. The purity of microglial cultures was evaluated by immunohistochemical staining using antibodies against ionized calcium binding adaptor molecule 1 (Iba-1,1:250; Wako Pure Chemical Industries, Ltd., Osaka, Japan) and glial fibrillary acidic protein (GFAP; Santa Cruz Biotechnology, Santa Cruz, CA, USA) to identify microglia and astrocytes, respectively, and was routinely >95%.
BV-2 cell culture
BV-2 immortalized cell line was obtained from Dr. Sanjay Maggirwar (University of Rochester Medical Center, Rochester, NY, USA) and was grown and routinely maintained in DMEM (4.5 mg/ml glucose, 2% fetal bovine serum, and 50 μg/ml gentamicin) at 37°C and 5% CO2 and used up to passage 20.
Cell treatments
Primary rat microglia were used in all experiments except in the studies comparing MYLK−/− microglia to wild-type (WT) cells and in MYLK-knockdown experiments. Pyk2- or Src-knockdown studies were done in BV-2 cells. Primary rat microglia was treated with varying concentrations of Tat (3.6, 7.2, and 14.4 nM). Treatment of primary rat microglia with pharmacological inhibitors (ML-7, 20 μM; blebbistatin, 10 μM; PP2, 1 μM; PP3, 1 μM; RGDfV, 50 μM; RADfV, 50 μM; anti-β1-neutralizing antibody, 10 μM; anti-β3-neutralizing antibody, 10 μM; IgG isotype, 10 μM; anti-VEGF-R1 neutralizing antibody, 1 and 10 μg/ml) involved pretreating cells with the respective inhibitors for 1 h, followed by exposure with Tat, followed by examination for cell adhesion or migration.
Short interfering RNA (siRNA) transfection
Primary mouse microglia were transfected with siRNA targeted against MYLK, and BV-2 cells were transfected with siRNA targeted against Pyk2 or Src, obtained from Thermo Scientific Dharmacon RNAi Technologies (Lafayette, CO, USA), as well as nonsense siRNA, by using the SiImporter (Millipore) according to the manufacturer's protocol. The knockdown efficiency of MYLK was determined by RT-PCR at 96 h after siRNA delivery, while the knockdown efficiency of Pyk2 or Src was determined by Western blot of extracts from transfected cultures at 48 h after siRNA delivery. Primary mouse microglial migration was tested at 96 h after siRNA delivery. Cell adhesion or migration of BV-2 cell lines was assessed at 48 h after siRNA delivery.
RT-PCR
Primers for mouse MYLK, rat VEGF-R1/R2, and mouse/rat GAPDH were as follows: 1) mouse MYLK (GenBank accession no. NM139300.3): upstream primer, 5-GTTCATCAGCAAGCCTCGTT-3; and downstream primer, 5-TTCTGGAGCAGCTCAAAGTG-3; 2) VEFG-R1 (GenBank NM019306.1): upstream primer, 5-GCCACAGCCAGTGAGTACAA-3; and downstream primer, 5-TTCCACGATCACCATCAGAG-3; 3) VEGF-R2 (GenBank NM013062.1): upstream primer, 5-TTCTGGACTCTCCCTGCCTA-3; and downstream primer, 5-AAGGACCATCCCACTGTCTG-3; 4) mouse GAPDH (GenBank NM008084.2): upstream primer, 5-TGCACCACCAACTGCTTAGC-3; and downstream primer, 5-GGCATGGACTGTGGTCATGAG-3; 5) rat GAPDH (GenBank NM017008.3): upstream primer, 5-TGCACCACCAACTGCTTAGC-3; and downstream primer, 5-GGCATGGACTGTGGTCATGAG-3; 6) integrin α1 (GenBank NM001033228.3): upstream primer, 5-GCAACAAGTGACAGTGAAGAG-3; and downstream primer, 5-TCCAGTTGGGTACAGTACAGG-3; 7) integrin α2 (GenBank NM008396.2): upstream primer, 5-GATGAGAACGCTCCTTCTGTC-3; and downstream primer, 5-AGTGGTTCTGCAGTCCAATTC-3; 8) integrin α3 (GenBank NM 013565.2): upstream primer, 5-AATCATCGGTTGCAGAGCTTC-3; and downstream primer, 5-GTGGTGTGACTCCAGGGTCAG-3; 9) integrin α4 (GenBank NM010576.3): upstream primer, 5-CCTGCAGCCTAGGTTACATAT-3; and downstream primer, 5-GGAAAGTGAGGTTCAGCTTCT-3; 10) integrin α5 (GenBank NM 010577.3): upstream primer, 5-AGCTCCATCAGCCAGGGTGTG-3; and downstream primer, 5-CAGCTGCAGACTACGGCTCTC-3; 11) integrin α6 (GenBank NM 008397.3): upstream primer, 5-CCTTGAATATACAGTGGCCCA-3; and downstream primer, 5-GAGAGGCCTTGCTGTCCAGCC-3. The mixtures were annealed at 50°C (1 min), extended at 72°C (2 min), and denatured at 94°C (1 min) for 35 cycles. The PCR product was visualized by electrophoresis on 1.2% agarose gels.
Western blotting
Treated cells were lysed using the Mammalian Cell Lysis kit (Sigma-Aldrich). Equal amounts of the proteins were electrophoresed in a sodium dodecyl sulfate-polyacrylamide gel (12%) under reducing conditions followed by transfer to PVDF membranes. The blots were blocked with 5% nonfat dry milk in PBS. The Western blots were then probed with antibodies recognizing the phosphorylated forms of MYLK (1:10,000; Sigma-Aldrich), integrin β1, integrin β3, p-Src, Src, p-Pyk2, Pyk2, p-MLC, MLC (1:200; Cell Signaling Technologies, Danvers, MA, USA), and β-actin (1:4000; Sigma-Aldrich). The secondary antibodies were alkaline phosphatase conjugated to goat anti-mouse/rabbit IgG (1:5000) as described previously (30).
Immunochemistry
For immunocytochemistry, primary rat microglia were plated on coverslips. After 2 d, cells were fixed with 4% paraformaldehyde for 15 min at room temperature, followed by permeabilization with 0.3% Triton X-100 in PBS. Cells were then incubated with a blocking buffer containing 10% NGS in PBS for 1 h at room temperature, followed by addition of rabbit anti-MYLK (1:100; Sigma-Aldrich) and mouse anti-p-MLC (1:500; Cell Signaling Technologies) antibodies and incubated overnight at 4°C. Finally, the secondary AlexaFluor 488 goat anti-rabbit IgG and AlexaFluor 569 goat anti-mouse IgG were added at a 1:500 dilution for 2 h to detect MYLK and p-MLC. Cells were washed 3 times in buffer and mounted with prolong gold antifade reagent with 4′,6-diamidino-2-phenylindole (DAPI; Invitrogen, Eugene, OR, USA).
Immunoprecipitation
The procedure for immunoprecipitation was performed as described previously (31). Briefly, primary microglia were treated with Tat or MYLK inhibitor-ML-7 or respective siRNAs, followed by lysis in radioimmunoprecipitation assay (RIPA) buffer (50 mm Tris, pH 8.0; 150 mM NaCl; 0.1% SDS; 1.0% Nonidet P-40; and 0.5% sodium deoxycholate) containing proteinase and phosphatase inhibitors. For each sample, 200 μg of protein was used for immunoprecipitation. The sample protein was incubated with 2 μg diluted anti-4-G-10 (Millipore), active β1 integrin, clone HUTS-4 (Millipore), MYLK (Sigma-Aldrich), β1 integrin, Pyk2, and Src antibody (Cell Signaling Technologies) overnight at 4°C, followed by incubation with 20 μl of protein A-Sepharose for 3 h at 4°C The mixture was then centrifuged (6000 g for 30 s), and the cell pellets were rinsed twice with RIPA buffer, followed by boiling in 2× Western blot loading buffer for 4 min. After spinning (6000 g for 30 s), the supernatants were subjected to Western blot as described above for detection of VEGF-R1 phosphorylation; β1 integrin activation; and Pyk2, Src, β1, β3, MYLK, Talin, and α-actinin (Cell Signaling Technologies).
F-actin content of microglia
F-actin was measured in primary rat microglia as described previously (16), with some modifications. Cells were stimulated for various times at 37°C with Tat and immediately fixed for 30 min at 25°C with 3.7% (v/v) paraformaldehyde in PBS, followed by washes in PBS. Cells were simultaneously permeabilized and stained for 10 min at 37°C in the dark with a fresh mixture of l-α-lysophosphatidylcholine palmitoyl (0.5 mg/ml; Sigma-Aldrich), Alexa Fluor-594 phalloidin (4×10−7 M; Invitrogen), and 18% formaldehyde in PBS. Following staining, samples were washed and suspended in 0.5ml of 1% paraformaldehyde solution for fluorescence acquisition, and data were analyzed with FACSDiva software (BD Biosciences, San Jose, CA, USA).
Microglial adhesion assay
Plates (96 wells) were coated with 50 μl of fibronectin (Millipore) diluted with PBS (10 μg/ml) and were incubated at 4°C overnight. To block any remaining protein binding sites on the plates, BSA (1% in PBS; 150 μl) was added to each well after removing the coating solution, followed by incubation for 30 min at room temperature. Plates were washed 3 times with 150 μl of PBS before addition of the cells. The wells were washed 3 times with serum-free DMEM as reported previously (32). Microglia (2–3×104 cells) labeled with 10 μM cell tracker green (Invitrogen) for 10 min were suspended in 200 μl of DMEM, followed by various treatments. Microglia were then rinsed 3 times with PBS to eliminate the nonadherent cells. Fluorescence intensity of adherent microglia was measured using a Synergy Mx fluorescence plate reader (BioTek Instruments, Winooski, VT, USA).
Microglial migration
Migration of microglia in vitro was determined using a Boyden chamber (Corning Costar; Corning, Inc., Corning, NY, USA) as described previously (33, 34). Cells were washed with PBS, then fluorescently labeled with 10 μM cell tracker green (Invitrogen) for 10 min at room temperature. Labeled cells (2×105 cells) were added to the upper compartment of transwell inserts in serum-free medium and treated (1 h at 37°C) with various inhibitors (ML-7, blebbistatin, PP2, and PP3), RGDfV, RADfV, anti-β1, or β3 neutralizing antibody or control IgG. Tat was placed in the lower chamber. The transwell plates were incubated for 3 h at 37°C, followed by quantification of microglial migration by measuring the number of migrated cells following detachment of cells from the insert using a Synergy Mx fluorescence plate reader (BioTek Instruments).
Chemotaxis assay
Chemotaxis was assessed using the Dunn chemotaxis chamber (Weber Scientific International Ltd., Teddington, UK), which allows direct observation of cell movement. This assay was performed as described previously (35–37). Briefly, microglia were plated onto square coverslips coated with collagen for 1 h, washed 3 times with serum-free DMEM, and kept for 4–16 h until the chemotaxis assay was performed. The coverslip was first placed over the chamber, the outer and inner wells of which were filled with DMEM, and then sealed with a 1:1 mixture of molten paraffin wax and Vaseline around the three edges to leave a slit for exchange of the medium in the outer well. To observe the chemically directed cell migration, the medium in the outer well was exchanged through the slit for DMEM containing 14.4 nM Tat. The open edge of the coverslip was then sealed immediately, and the chamber was set on the stage of a microscope, which was maintained at 37°C, 5% CO2. Control experiments were performed under the condition in which both outer and inner wells were filled with DMEM. A region of the bridge was viewed via a Carl Zeiss confocal laser scanning microscope, and the phase-contrast images were recorded every 20 s during 1 h of observation using LSM 510 software (v. 4.0 SP1; Carl Zeiss MicroImaging GmbH, Cologne, Germany). The distance and direction were shown as x, y coordinates on scatter diagrams with the x axis positioned at a 45° angle to the outer edge of the bridge. Distance and direction of movement by the cell's leading edge was monitored over a 1-h period by phase-contrast time-lapse microscopy; the average distance migrated in the absence of a stimulus (from 0 to 0.8 mm) was subtracted to obtain final values. Images were processed and analyzed using ImageJ software (U.S. National Institutes of Health, Bethesda, MD, USA).
Cdc42 GTPase activation assay
Activities of Cdc42 GTPases were measured using colorimetric G-LISA assays (Cytoskeleton, Denver, CO, USA) according to the manufacturer's instructions.
Transduction of primary mouse microglia with AAV2-GFP
The rodent GFP PCR product was cloned into rAAV2-MCS-WPRE vector, which shows robust expression efficiency in the CNS. The vector was packaged in AAV-293 cells (derived from HEK293 cells to produce higher viral titers) using CaPO4 transient transfection (simultaneously) of the vector plasmid and virion production plasmids, followed by lysis of cells to recover replication-deficient virions. Virions were purified by affinity column (ViraTrap AAV purification kit; GeneMega Inc., San Diego, CA, USA). Recombinant AAV was titered for total particles by serial-dilution transfection in HEK293 and verified by quantitative-competitive PCR assay. rAAV2-GFP was transduced into microglia at an MOI of 2×105, followed by gentle swirling, incubation, and replacement of fresh feed medium. At 4 d post-transduction, GFP-labeled microglia was incubated with anti-Iba-1 antibody (1:250; Wako).
In vivo chemotaxis assay of microglia
Based on previous study about the in vivo microglial chemotaxis (34), Tat was microinjected into the hippocampus of C57 BL/6N mice (2 μg/2 μl) using the microinjection parameters (coordinates 2.06 mm behind the bregma, 2.0 mm lateral from the sagittal midline at a depth of 1.7 mm to skull surface). At 1 d after Tat injection, microglia from WT and MYLK−/− mice (2 μl, 1×105 cells/μl) were transduced with AAV2-GFP and then transplanted (at 0.5 μl/min) into the medial corpus callosum at the following coordinates: 2.06 mm behind the bregma, 0 mm lateral from the sagittal midline, at a depth of 1.5 mm to skull surface. Animals were perfused 7 d following cell transplantation, and immunohistochemical procedures were performed as described below. Sections were incubated with primary anti-Iba-1 antibody (1:250; Wako) overnight at 4°C. Secondary AlexaFluor 594 goat anti-mouse IgG was added at a 1:500 dilution for 2 h to detect Iba-1, followed by mounting of cells with prolong gold antifade reagent with DAPI (Invitrogen). Fluorescent images were acquired at room temperature on a Zeiss Observer. A Z1 inverted microscope was used; images were processed using AxioVs 40 4.8.0.0 software (Carl Zeiss MicroImaging). Photographs were acquired using an AxioCam MRm digital camera.
Tissue and immunostaining
Formalin-fixed, paraffin-embedded sections (5 μm) of basal ganglia from HIV− subjects and patients with HIV-encephalitis (HIV-E) were obtained from the National NeuroAIDS Tissue Consortium (Bethesda, MD, USA; Table 1) and stained with antibodies specific for MYLK (polyclonal goat, 1:100 dilutions; Santa Cruz Biotechnology), Iba-1 (1:250 dilution; Wako), p24 (1:10 dilutions; Dako, Carpinteria, CA, USA). Sections were washed 3 times in PBS, followed by incubation in secondary AlexaFluor 488 donkey anti-goat antibody (1:200) and AlexaFluor 488 goat anti-mouse antibody (1:200) or AlexaFluor 594 goat anti-rabbit antibody (1:200). Fluorescent images were acquired as described in the above.
Table 1.
Clinical data for human brain tissue samples
| Case no. | HIV-1 infection | Age (yr) | Gender | Viral load (plasma) | CD4 cell count | Neurocognitive assessment | Antiretroviral therapy | Pathological evidence of encephalitis |
|---|---|---|---|---|---|---|---|---|
| 1 (3015) | Seronegative control | 64 | Female | NA | 118 | Neuropsychological impairment or dementia due to other cause | None | HIV−, PML |
| 2 (ID: 7101958083) | Seronegative control | 46 | Male | NA | NA | Unable to reliably assign neurocognitive diagnosis | None | Normal |
| 3 (ID: 7102308387) | Seronegative control | 49 | Male | NA | NA | Unable to reliably assign neurocognitive diagnosis | None | Normal |
| 4 (4084) | HIV-E | 46 | Male | 40,519 | 18 | Neuropsychological impairment or dementia due to other cause | None | HIV-E, microglial nodule encephalitis |
| 5 (ID: 7100616568) | HIV-E | 32 | Male | 489,796 | 77 | Possible HAD | 3TC, ABC, EFV | HIV-E |
| 6 (ID: 7100107766) | HIV-E | 38 | Male | 1,843 | 43 | Possible HAD | EFV, NVP | HIV-E |
Statistical analysis
Statistical analysis was performed using 1-way analysis of variance with a post hoc Student's t test. Results were judged statistically significant if P < 0.05 by analysis of variance.
RESULTS
MYLK is required for microglial migration
Since a critical and early component of the inflammatory response in HAND is increased microglial mobility and chemotactic activity to the Tat (38), we sought to investigate first the effect of Tat (Tat1-72 was used in this study) on primary rat microglial migration and then to explore the molecular mechanisms involved in this process. As shown in Fig. 1A, different concentrations of Tat (3.6, 7.2, and 14.4 nM) induced microglial migration up to 135.34, 163.28, and 171.85%, respectively. Interestingly, pretreatment of microglia with the MYLK inhibitor-ML-7 significantly ameliorated microglial migration by 80%, thereby underpinning the role of MYLK in Tat-mediated migration of microglia (Fig. 1B). Since Tat in its biological form in vivo is present as the long forms (Tat1-86 or Tat1-101), we sought to test the effect of both these long forms on microglial migration. As shown in Fig. 1C, both Tat1-86 and Tat1-101 significantly increased microglial migration. There was no significant difference among the various Tat forms in inducing migration. Having demonstrated the role of MYLK in microglial migration, we next sought to validate the findings using the loss of function approach. Microglia isolated from WT and MYLK−/− mice were exposed to Tat and assessed for microglial migration using the Boyden chambers. In the WT microglia, Tat induced migration of microglia (up to 60% compared with untreated cells) but failed to do so in microglia isolated from the MYLK−/− mice (Fig. 1D). These findings were also confirmed in isolated mouse microglia transfected with siRNA MYLK to knock down the expression of MYLK (Fig. 1E).
Figure 1.

Effect of Tat on microglial migration. A) Tat induced primary rat microglial migration in a concentration-dependent manner using a Boyden chamber. B) MYLK inhibitor ML-7 ameliorated Tat-induced microglial migration. C) Effect of Tat1-72, Tat1-86, and Tat1-101 on primary rat microglial migration using Boyden chambers. Data are presented as means ± se of 4 individual experiments. *P < 0.05, **P < 0.01, ***P < 0.001 vs. control group; ###P < 0.001 vs. Tat-treated group. D) Tat-induced migration of microglia isolated from WT (solid bar) or MYLK−/− (shaded bar) mice using Boyden chamber. E) Tat-induced migration of primary mouse microglia in the presence of nonsense (non) siRNA (solid bar) or siRNA MYLK (shaded bar). RT-PCR analysis of cell lysates harvested in Trazol from primary mouse microglia transfected with siRNAs MYLK or nonsense siRNA. F) Microglia from WT or MYLK−/− mice placed in a gradient of Tat (14.4–0 nM) using Dunn chemotaxis chamber and visualized by phase-contrast time-lapse microscopy. G) Scatter plot indicating distance (μm) traveled by the leading edge of WT (solid circles) or MYLK−/− (open circles) microglia in a gradient of Tat after 40 min. Values are plotted in the x and y directions relative to location of the Tat source. Data are presented as means ± se of 4 individual experiments. **P < 0.01, ***P < 0.001 vs. control group.
Next, as a measure to assess motility of primary rat microglia in response to Tat, purified microglia were placed in a Dunn chemotaxis chamber and monitored for motility in a gradient of Tat (14.4–0 nM). Microglia isolated from the WT mice showed a clear and robust polarization or chemotaxis toward the Tat source within 40 min. Microglia isolated from MYLK−/− mice, on the other hand, showed a significant decrease in directed movement compared with microglia from WT mice (Fig. 1F, G).
Myosin II is activated in the absence of MYLK
Since MLC is the only known substrate for MYLK (39, 40), we next sought to examine whether MYLK regulates the adhesion and migration of microglia secondary to activation of MLC. In WT microglia, Tat stimulation resulted in activation of MLC at serine19 (1.5-fold higher, Fig. 2A). Intriguingly, although basal phosphorylation of serine 19-MLC was slightly lower in the MYLK−/− microglia (Fig. 2A), the level of MLC activation in these cells following Tat stimulation was identical to that observed in the WT cells. These findings thus suggested that MYLK was not essential for Tat-induced phosphorylation of MLC and also that lack of migration of MYLK−/− cells was not mediated via MLC signaling. To explore the role of MYLK and MLC in Tat-mediated MLC activation, microglia were pretreated with inhibitors specific for both MYLK (ML-7) and myosin II (blebbistatin) and examined for MLC activation. There was significant inhibition of MLC activation in cells pretreated with either inhibitor (Fig. 2B).
Figure 2.

Loss of MYLK function failed to prevent myosin II activation. A) MLC phosphorylation in microglia isolated from WT or MYLK−/− mice. B) Effect of MYLK inhibitor or myosin II inhibitor on MLC phosphorylation induced by Tat in primary rat microglia. Bleb, blebbistatin. C) Tat-mediated increase in primary rat microglial adhesion was ameliorated in microglia pretreated with ML-7 (20 μM), but not with blebbistatin (10 μM). D) Myosin II failed to inhibit Tat-mediated increase in primary rat microglial migration. Data are presented as means ± se of 4 individual experiments. *P < 0.05, **P < 0.01, ***P < 0.001 vs. control group; ##P < 0.01, ###P < 0.001 vs. Tat-treated group. E) Primary rat microglia treated with Tat were double-stained using antibodies specific for MYLK (green) or p19-MLC (red). Nuclei stained with DAPI (blue). Representative pictures from 3 typical experiments. Scale bars = 20 μm. F) Actin polymerization in primary rat microglia pretreated with or without inhibitors followed by Tat treatment was measured by F-actin assay. Data are presented as means ± se of 4 individual experiments. *P < 0.05 vs. control group; #P < 0.05, ##P < 0.01 vs. respective time point in microglia treated with Tat.
Migration is a dynamic process involving the disassembly of focal adhesions at the tailing edges and the assembly of new focal adhesions at the migrating fronts (41, 42). To explore the role of MYLK and MLC in Tat-mediated microglial adhesion, primary rat microglia pretreated with ML-7 demonstrated significant inhibition of adhesion (Fig. 2C), and intriguingly, cells pretreated with blebbistatin actually resulted in enhanced adhesion (Fig. 2C). As regards migration, microglia pretreated with ML-7 demonstrated reduced migration (Fig. 1B), and those treated with blebbistatin did not exhibit any change in migration (Fig. 2D). These results indicate alternative functions for MYLK in mediating microglial adhesion and migration.
We next sought to examine the morphological and functional aspects of both myosin II and MYLK following exposure to Tat. Following exposure to Tat, primary rat microglia exhibited a polarized phenotype wherein active myosin II (stained with antibody p19-MLC) accumulated at the rear end (Fig. 2E), while MYLK localized primarily at the leading edge of the cells (Fig. 2E). Microglia treated with blebbistatin also exhibited increased actin polymerization following Tat stimulation (Fig. 2F). In contrast, F-actin content in ML-7 treated microglia was lower compared to Tat treated cells (40% less; Fig. 2F). MYLK and myosin II that appear to have distinct subcellular localizations also elicit functional differences, as evident in microglial migration.
MYLK is required for β1 integrin activation in microglia
Based on the premise that integrins are known to regulate cell migration, we rationalized that these cell surface receptors could likely be involved in Tat-mediated adhesion/migration involving MYLK. Primary rat microglial cells were pretreated with integrin inhibiting RGD peptide-RGDfV and assessed for Tat1-72-, Tat1-86-, and Tat1-101-mediated adhesion/migration. Interestingly, pretreatment of cells with RGDfV significantly suppressed adhesion/migration, while the mutated RGD peptide-RADfV (negative control) exhibited no effect (Fig. 3A, B). To further identify the integrin subtypes participating in adhesion and migration, we examined the effect of functional blocking of integrin β subtypes using neutralizing antibodies. The rationale for focusing on the integrin subtypes β1and β3 but not β2 is based on the findings that microglial migration remains unaffected in integrin-β2-deficient mice (43). Pretreatment of microglia with the neutralizing anti-β1integrin antibody resulted in significant suppression of Tat1-72-mediated adhesion (Fig. 3C) and migration (Fig. 3D). Similar to Tat1-72, cells treated with the long forms of Tat also demonstrated involvement of β1 integrin in microglial migration (Supplemental Fig. S1A). Treatment with either neutralizing anti-β3 integrin or normal IgM antibodies exhibited no effect. These results thus underscore the role of β1 integrin in Tat-mediated induction of microglial adhesion/migration. Next, we examined the activation of β1 integrin following Tat exposure in cell lysates by immunoprecipitation with the antibody against the active form of integrin β1 (clone HUTS-4). Following stimulation with Tat1-72, there was a significant time-dependent increase in the active form of β1 integrin that was significantly ameliorated in cells pretreated with ML-7 (Fig. 3E). Similar to Tat1-72, the long forms of Tat also induced activation of β1 integrin (Supplemental Fig. S1B); however, the long forms failed to activate β3 integrin (Supplemental Fig. S1C). The next step was to explore the interaction of MYLK and integrins. Immunoprecipitation of cell lysates (with and without Tat treatment) with MYLK antibody demonstrated that while both β1 and β3 integrins coprecipitated with MYLK in control cells, there was preferential interaction of MYLK with β1 compared with β3 integrin in Tat-treated cells (Fig. 3F).
Figure 3.

Involvement of integrins in Tat-mediated primary rat microglial adhesion and migration. A) Primary rat microglia pretreated with integrin-inhibiting peptide RGDfV (50 μM) or RADfV (50 μM) for 1 h were exposed to Tat and plated onto the collagen-coated plates, followed by cell adhesion assay. Cell adhesion is expressed as the ratio of the number of stimulated cells that had attached to the number of cells attached under control conditions. B) Primary rat microglia pretreated with integrin inhibiting peptide RGDfV or RADfV for 1 h were exposed to Tat followed by cell migration assay using a Boyden chamber. C, D) Primary rat microglia pretreated with neutralizing integrin β1 or β3 antibodies (10 μM) were exposed to Tat, followed by cell adhesion assay (C) and by cell migration assay (D). E) Effect of ML-7 on the Tat-induced integrin activation in primary rat microglia. F) Association of MYLK with β1 or β3 integrins in primary rat microglia stimulated with Tat. Lysates were immunoprecipitated using anti-MYLK antibody, followed by immunoblotting with anti-β1/β3 integrin antibodies or IgG. Representative immunoblots (top panel) and the densitometric analysis (lower panel) of β1/MYLK or β3/MYLK from 4 separate experiments. Data are presented as means ± se of 4 independent experiments. **P < 0.01, ***P < 0.001 vs. control group; ##P < 0.01, ###P < 0.001 vs. Tat-treated group.
Tat-induced microglial migration via β1 integrin does not involve direct binding of RGD domain
Since Tat1-72 used in our system lacks the RGD sequence, the direct involvement of RGD domain in Tat-induced adhesion/migration was ruled out (Fig. 4A). In our efforts to dissect the minimal binding domain of Tat critical for induction of primary rat microglial adhesion/migration, various Tat domains were investigated. Both Tat1–72 and Tat basic domain (Tat46–60) induced microglial adhesion/migration, while the shorter Tat1–15, or the mutant Tat72Δ31–61 peptides, failed to induce significant adhesion/migration (Fig. 4B, C).
Figure 4.

Involvement of VEGF-R1 in Tat-mediated increase of primary rat microglial adhesion/migration. A) Schematic of Tat domains. B, C) Primary rat microglia were treated with different Tat domains: Tat1-15, Tat46-60, mutant Tat1-72Δ31-61, or heated Tat for 6 h followed by microglial adhesion (B) and migration assay (C). Data are presented as means ± se of 4 independent experiments. **P < 0.01, ***P < 0.001 vs. control group. D) Detection of VEGF-R1 and R2 mRNA by RT-PCR analysis in primary rat microglia. E) Tat-mediated phosphorylation of VEGF-R1 in primary rat microglia. F, G) Effect of neutralizing VEGF-R1 antibody (1 and 10 μg/ml) on Tat-mediated MLC phosphorylation (F) and β1 integrin activation (G). H, I) Effect of neutralizing VEGF-R1 antibody on Tat-mediated primary rat microglial adhesion (H) and migration (I). Data are presented as means ± se of 4 independent experiments. ***P < 0.001 vs. control group; ##P < 0.01, ###P < 0.001 vs. Tat-treated group.
Next, we sought to examine the receptor interacting with Tat. Studies in endothelial cells have suggested that VEGF-R has the binding affinity for the basic domain of Tat (6, 44). We therefore wanted to explore the role of VEGF-R in Tat-mediated microglial migration. Primary rat microglial cells expressed VEGF-R1 mRNA, but not the VEGFR-2 isoform (Fig. 4D), and rapidly phosphorylated VEGF-R1 in response to Tat (Fig. 4E). Neutralizing VEGF-R1 antibody significantly inhibited Tat-induced MLC phosphorylation and β1 integrin activation, thereby suggesting that VEGF-R1 activation was upstream of MLC phosphorylation and β1 integrin activation (Fig. 4F, G). The role of VEGF-R1 signaling in Tat-mediated induction of adhesion/migration was further confirmed by treating cells with neutralizing antibody specific for VEGF-R1, which impaired the ability of Tat to induce adhesion/migration (Fig. 4H, I).
MYLK is required for activation of downstream Src and Pyk2
Integrin activation can be divided into two stages (27, 28) involving the inside-out signals activating the integrins and promoting integrin-ligand binding, and the outside-in signals activating downstream kinases (Src, Fak and Pyk2). Following integrin activation, the downstream-transduced signal manifests as actin polymerization with reinforced integrin-cytoskeleton interaction (27, 28).
To explore the signaling mediators involved in MYLK-induced activation of integrins, we sought to examine the crosstalk of MYLK with the kinases regulating actin polymerization. Tat-mediated activation of Src and Pyk2 was readily observed in the WT mouse microglia (Fig. 5A, B) and, to a lesser extent, in cells isolated from MYLK−/− mice. Further validation of these findings was done at a functional level, wherein pretreatment of cells with Src inhibitor-PP2, but not its inactive anolog-PP3, resulted in reduced microglial adhesion (Fig. 5C). Corroboration of these findings was also done using the siRNA approach, wherein BV-2 cells transfected with either the Src or Pyk2 siRNAs exhibited reduced adhesion/migration of microglia in response to Tat. Tat-mediated increased adhesion/migration was attenuated by siRNA Src (Fig. 5D, E) and Pyk2 (Fig. 4F, G). Collectively, these findings underscored the role of Src and Pyk2 in Tat-mediated microglial adhesion/migration. Our findings indicate an association of MYLK with Src and Pyk2 (Fig. 5H, I).
Figure 5.

MYLK is required for activation of Src and Pyk2. A, B) Src (A) and Pyk2 (B) phosphorylation in microglia isolated from WT or MYLK−/− mice. C) Tat-mediated increase in microglial adhesion was ameliorated in primary rat microglia pretreated with PP2 (1 μM), but not PP3 (1 μM). D) Right panel: effect of Tat on the cell adhesion of BV-2 cells in the presence of Src siRNA. Left panel: Western blot analysis of whole-cell lysates from BV-2 cells transfected with siRNAs Src or nonsense (non) siRNA using antibodies specific for Src. E) Effect of Tat on the cell migration of BV-2 cells in the presence of Src siRNA. F) Right panel: effect of Tat on the cell adhesion of BV-2 cells in the presence of Pyk2 siRNA. Left panel: Western blot analysis of whole cell lysates from BV-2 cells transfected with siRNAs Pyk2 or nonsense siRNA using antibodies specific for Pyk2. G) Effect of Tat on the cell migration of BV-2 cells in the presence of Pyk2 siRNA. H) Association of MYLK with Pyk2 or Src in primary rat microglia stimulated with Tat. I) Association of Pyk2 with MYLK or Src in primary rat microglia stimulated with Tat. Lysates were immunoprecipitated using anti-MYLK/Pyk2 antibody or followed by immunoblotting with anti Pyk2/Src/MYLK or IgG antibodies. Data are presented as means ± se of 4 independent experiments. *P < 0.05, **P < 0.01, ***P < 0.001 vs. control group; ###P < 0.001 vs. Tat-treated group.
MYLK regulates the interaction of β1 integrin with Src/Pyk2
We next sought to explore the interaction of β1 integrin with both Src and Pyk2. β1, but not β3, integrin demonstrated increased interaction with Pyk2 (Fig. 6A) and Src (Fig. 6B). Further validation of the role of MYLK in this process was examined using the WT and the MYLK−/− mouse microglia. There was reduced binding of Src/Pyk2 with β1 integrin in the latter cells (Fig. 6B). Next, a set of experiments was performed to examine whether β1 integrin regulated Src and Pyk2 activation. Pretreatment of primary rat microglia with RGD peptide-RGDfV significantly suppressed Tat-induced Pyk2 and Src phosphorylation, further confirming that β1 integrin-ECM binding is involved in Pyk2 and Src activation (Fig. 6C).
Figure 6.

MYLK regulates β1 integrin-Pyk2/Src interaction. A) Association of Pyk2 with integrin β1 or β3 in primary rat microglia stimulated with Tat. B) Top panel: association of integrin β1 with Pyk2 or Src in microglia isolated from WT or MYLK−/− mice stimulated with Tat. Lysates were immunoprecipitated using anti-Pyk2/β1 integrin antibodies, followed by immunoblotting with anti β1/β3/Pyk2/Src antibodies or IgG. Bottom panel: densitometric analyses of Pyk2 or Src from 4 separate experiments. C) Top panel: effect of integrin-inhibiting peptide RGDfV or RADfV on Tat-mediated phosphorylation of Pyk2/Src. Image is representative of 4 independent experiments. Bottom panel: densitometric analyses of pPk2/Pyk2 and pSrc/Src from 4 separate experiments. Data are presented as means ± se of 4 independent experiments. **P < 0.01, ***P < 0.001 vs. control group; #P < 0.05, ##P < 0.01, ###P < 0.001 vs. Tat-treated group.
MYLK regulates β1 integrin-cytoskeleton interaction
Pyk2 has been shown to activate the small Rho GTPase Cdc42, thus promoting actin polymerization (45). Exposure of primary rat microglia to Tat resulted in time-dependent activation of Cdc42, which was ameliorated in cells pretreated with ML-7 (Fig. 7A, B). Furthermore, there was reduced Cdc42 activation in microglia from MYLK−/− compared with the WT mice (Fig. 7C), thereby suggesting that Pyk2-mediated β1 integrin-cytoskeleton interactions are critical downstream pathways in MYLK-mediated microglial migration.
Figure 7.

MYLK regulates β1 integrin-cytoskeleton interaction. A) Tat-induced Cdc42-GTP activation in a time-dependent manner in primary rat microglia. B) Effect of MYLK inhibitor-ML-7 on the Cdc42-GTP activation induced by Tat in primary rat microglia. C) Effect of Tat on Cdc42-GTP activation in microglia isolated from WT or MYLK−/− mice. D) Left top panel: effect of ML-7 on the association of β1 integrin with Talin or α-actinin in microglia stimulated with Tat in primary rat microglia. Right top panel: association of β1 integrin with Talin or α-actinin in microglia isolated from WT or MYLK−/− mice stimulated with Tat. Lysates were immunoprecipitated using anti-β1 integrin antibody. followed by immunoblotting with anti-Talin/α-actinin antibodies or IgG. Bottom panels: densitometric analyses of Talin or α-actinin from 4 separate experiment. Data are presented as means ± se of 4 independent experiments. *P < 0.05, **P < 0.01, ***P < 0.001 vs. control group; #P < 0.05, ##P < 0.01 vs. Tat-treated group.
We next examined whether MYLK participated in mediating β1 integrin-cytoskeleton assembly. Tat treatment resulted in increased interaction between β1 integrins and talin or actinin in primary rat microglia (Fig. 7D, left panel), an effect that was ameliorated in cells pretreated with ML-7. Furthermore, there was increased binding of β1 integrins to talin in the WT cells as expected and this was reduced in the MYLK−/− mouse microglia (Fig. 7D, right panel).
MYLK deficiency attenuated microglial migration induced by Tat
Based on the in vitro, we next sought to validate in vivo whether intrahippocampal injection of Tat could lead to microglial migration to the Tat-injected site (Fig. 8A). Microglia were first isolated from mouse brain, followed by infection with an AAV-expressing GFP. Figure 8B shows representative in vitro staining for microglial marker Iba-1 prior to injection. Colocalization of GFP with Iba-1 was evident for all the cells examined. GFP+ microglia were stereotactically transplanted into the medial corpus callosum (1 d after Tat injection) at the location shown (Fig. 8A, asterisk). Three areas were designated for counting GFP+ microglia (labeled 1–3 in Fig. 8A), at sites between injected GFP-microglia and Tat. At 7 d following Tat microinjection, brains of mice were stained for Iba-1. Tat-treated mice demonstrated increased Iba-1-positivity (Fig. 8C, bottom panels) with concomitant increased GFP+ microglia compared with saline-injected animals (Fig. 8C, top panels).
Figure 8.

Chemotactic responses of GFP-labeled microglia in vivo. A) Schematic diagram showing in vivo locations of GFP-labeled microglial transplantation (asterisk) and Tat injection (arrow). The 3 areas chosen for analysis of GFP+ microglial migration are indicated. B) Representative images of immunostaining for Iba-1 and GFP-labeled cells in vitro. C) Effect of Tat on microgliosis in the hippocampus. Representative image following 7 d intrahippocampal injection of saline or Tat immunostained for Iba-1 and GFP-labeled microglia (left panel) and the merged staining for GFP and Iba-1 (right panel). D) Representative GFP+ microglia in the 3 areas. Top panel: Tat and WT microglia. Bottom panel: Tat microglia from MYLK−/− mice. E) Quantification of GFP+ microglia in areas 1–3; n = 6 animals/group. Scale bars = 20 μm. **P < 0.01, ***P < 0.001 vs. WT group.
The next step was to identify the role of MYLK in microglial migration. Two groups of animals were injected with Tat, with one group receiving microglia isolated from WT (Fig. 8D, top panels) and the other group receiving microglia from MYLK−/− mice. Mice receiving WT cells showed high levels of GFP+ microglia in the Tat-injected area, with considerable levels of GFP+ microglia evident in both areas 1 (adjacent to transplantation site; Fig. 8D) and 2–3 (proximal to Tat-injected site; Fig. 8D). Although mice transplanted with cells from MYLK−/− mice exhibited similar levels of GFP+ cells (Fig. 8D, area 1), there were decreased numbers of migrating microglia in areas 2–3 (Fig. 8D, bottom panels). Quantification for in vivo chemotaxis of GFP+ microglia is presented in Fig. 8E.
MYLK expression in microglia from HIV-infected patients
Since the degree of neurological deficit in HIV-infected individuals strongly correlates with the number of activated microglia within the basal ganglia, we wanted to assess whether MYLK expression colocalized with microglia in the postmortem brain tissues from HIV− or HIV-E subjects. HIV-1− controls demonstrated very few MYLK+ cells, while MYLK expression was increased in sections of brains from patients with HIV-E (Fig. 9A). Intriguingly, increased MYLK immunostaining seen in subjects with HIV-E colocalized with Iba-1 microglia. In addition, Iba-1 microglia were also positive for virus p24 (Fig. 9B).
Figure 9.

Microglial MYLK expression in the basal ganglia sections of HIV− subjects and subjects with HIV-E. A) Left panels: double immunolabeling and confocal image analyses demonstrating increased MYLK expression, localized primarily in Iba-1-labeled microglia (green, MYLK; red, Iba-1). Right panel: semiquantitative analysis of the signal intensity from 3-μm z stack in the images in left panels. B) Left panels: double immunolabeling and confocal image analyses demonstrating HIV p24 expression, exclusively in Iba-1-labeled microglia (green, p24; red, Iba-1). Right panel: semiquantitative analysis of the signal intensity from 3-μm z stack in the images in left panels. Nuclei were stained with DAPI (blue). Scale bars = 20 μm. Images are representative from 4 experiments with 3 HIV− subjects and 3 patients with HIV-E.
DISCUSSION
Chronic inflammation is a hallmark feature of HIV disease in the CNS involving glial migration and activation. In the CNS, HIV Tat released from infected cells serves as a chemoattractive agent, as well as a focal site for recruitment of microglia, leading to expansion of the neuroinflammatory response. Herein we demonstrate that Tat mediates microglial adhesion/migration via activation of VEGF-R1. These findings are consistent with the previous reports in endothelial cells (6). This study provides direct evidence for the involvement of Tat-mediated activation of MYLK in microglial migration. While MYLK has been implicated in neutrophil migration (15, 16) and endothelial dysfunction (46), its role in microglial migration has never been reported. The long form of MYLK is the predominant isoform that triggers actomyosin contraction (47–50). Inhibition of MYLK kinase activity or genetic deletion of MYLK has resulted in beneficial effects against acute lung injury (51), endotoxic shock (52), and burn-associated injuries (53). In line with these findings, our results demonstrated that MYLK−/− or inhibition attenuated microglial migration induced by Tat protein.
Our findings about the effect of Tat on microglial migration were consistent with the previous report (54); however, the extent of microglial migration observed by Eugenin et al. (54) and by us was not as robust as that reported by Fraga et al. (55). One possible explanation for this discrepancy could be the differing amounts of Tat used in the two studies (14 nM compared with ≥25 nM used by Fraga et al.). The rationale for choosing 14 nM Tat concentration is in keeping with the physiological levels of Tat found in the cerebrospinal fluid and in the serum of HIV-infected patients, as reported previously (56–60).
While MYLK activation is known to phosphorylate its well-recognized substrate MLC, emerging evidence also suggests an alternative MLC-phosphorylation-uncoupled mechanism in MYLK-mediated events (16). For example, MYLK is found to promote neutrophil infiltration in septic lung injury, an effect involving Pyk2 activation that is independent of MLC phosphorylation (16). Similarly, in endothelial cells, MYLK deficiency attenuated thrombin-induced hyperpermeability without altering its substrate activity (46). Our findings demonstrated that microglial cells treated with the pharmacological inhibitor of MYLK-ML-7 exhibited reduced Tat-mediated MLC phosphorylation and chemotaxis. Validation of these findings in MYLK−/− cells also demonstrated reduced microglial migration to Tat, however, with no effect on MLC phosphorylation. It can thus be envisioned that in addition to MYLK mediated MLC phosphorylation, MYLK also mediates other alternative cellular pathways that could be critical for Tat-mediated cell migration. Similar findings of a MLC phosphorylation-independent role of MYLK have been reported by others (16).
To further assess the role of chemokines in Tat-mediated migration of microglia, Luminex analysis was performed on supernatants from Tat-treated cultures, and among the various chemokines examined, MCP-1 levels were found to be dramatically increased, as shown previously by Eugenin et al. (54). It was therefore a logical choice to examine the effect of neutralizing MCP-1 antibody on Tat-mediated microglial migration. As shown in Supplemental Fig. S2, neutralizing MCP-1 antibody significantly ameliorated Tat-mediated microglial migration, thereby implicating its role in Tat-mediated migration of microglia.
This study identifies the role of MYLK signaling pathways in Tat-mediated microglial migration that are independent of MLC phosphorylation. Our findings implicate β1 integrins downstream of MYLK as the key targets responsible for regulating Tat-mediated adhesion/migration of microglia. Previous studies have demonstrated that Tat, through its ability to bind α5β1 and αvβ3 integrins via its RGD rich domain, regulates the endothelial cell cycle progression (5, 61). In contrast, our studies with Tat1–72, lacking the RGD domain, demonstrate Tat-mediated inside-out activation of β1 integrins. This leads then to an outside-in signal that results in the activation of Src and Pyk2. Kinase activation then leads to actin polymerization, which reinforces the integrin-cytoskeleton interaction and sustains β1 integrins in the high-affinity and high-avidity state, reflecting the full activation for integrins. Integrin β1, also known as CD29, is a transmembrane glycoprotein that forms noncovalent complexes with various integrin α subunits (α1–6). Using immunoprecipitation assay, we identified integrin α4–6 as the putative binding partners of integrin β1 that were critical for microglial migration (Supplemental Fig. S3). This is consistent with the previous reports demonstrating the involvement of α4β1, α5β1, and α6β1 integrins in microglial adhesion (62, 63).
Our findings shed light on the adhesive activity of the basic domain of Tat that is MYLK dependent. However, the RGD sequence that is present in the longer forms of Tat is a known mediator of integrin activation and adhesion. To understand the role of MYLK in Tat1-101-mediated downstream activation of Src and FAK, cells were exposed to Tat1–101 in the presence or absence of MYLK inhibitor ML-7. Intriguingly, pretreatment of cells with ML-7 failed to completely inhibit Tat1–101-mediated activation of Src and FAK, suggesting thereby that Tat1–101 induced Src and FAK activation via an alternative MYLK/MLC-independent pathway (data not shown). Interestingly, Src phosphorylation was attenuated in microglia isolated from MYLK−/− mice, thereby lending support that MYLK-mediated Src activation is independent of its kinase activity or MLC phosphorylation. While it is known that Src can directly phosphorylate MYLK (64), in our findings it appears that, reciprocally, MYLK can also regulate Src through a possible feedback loop.
MYLK, in addition to possessing a kinase domain, also has binding sites for cytoskeletal proteins and tyrosine kinases, implying its function as an adaptor protein. Our findings indicate that MYLK bound to both Src and Pyk2. MYLK was shown to interact directly with Pyk2 while phosphorylating it, and ML-7 partially blocked Pyk2 activation, thereby implying that the kinase activity of MYLK and MYLK-Pyk2 interaction may be both critical events for Pyk2 activation. Similar to published findings (45), our results also demonstrate that inhibition of MYLK kinase resulted in mitigation of Cdc42 activation and actin polymerization induced by Tat. MYLK is thus essential for Tat-mediated activation of a tyrosine-Cdc42-actin signaling pathway leading to microglial adhesion and migration.
These findings were also validated in vivo wherein intrahippocampal injection of Tat followed by administration of AAV2 GFP-infected WT microglia in the corpus callosum resulted in microgliosis and microglial migration. Intriguingly, injection of microglia isolated from MYLK−/− mice into Tat administered mice failed to demonstrate microglial migration. These findings are in agreement with previous reports describing the role of MYLK in inducing neutrophil and astrocyte migration (16, 17).
The next step was to validate our findings in a clinical setting. HIV-E is known to correlate with chronic neuroinflammation, activated microglia, and presence of Tat. There was increased expression of MYLK that colocalized with Iba-1 microglial cells in the brains of patients with HIV-E. In addition, Iba-1+ cells in HIV-E sections were also positive for virus antigen p24. In contrast, HIV− subjects failed to demonstrate MYLK and p24 staining.
In summary, this study implies a pathophysiological role of MYLK in HIV-E. MYLK participates in microgliosis by augmenting microglial migration in response to Tat protein. The mechanism involved in this process includes Tat-mediated activation of MYLK, followed by inside-out activation of β1 integrin, subsequent outside-in signaling of β1 integrin involving activation of the downstream kinases Src and Pyk2 and activation of Cdc42-GTP, culminating ultimately into actin polymerization (Fig. 10). MYLK thus mediates a regulatory pathway critical for eliciting innate immune responses in microglia. Specific blocking of the long form of MYLK could be considered as a potential therapeutic target for treating neuroinflammatory conditions involving microgliosis.
Figure 10.
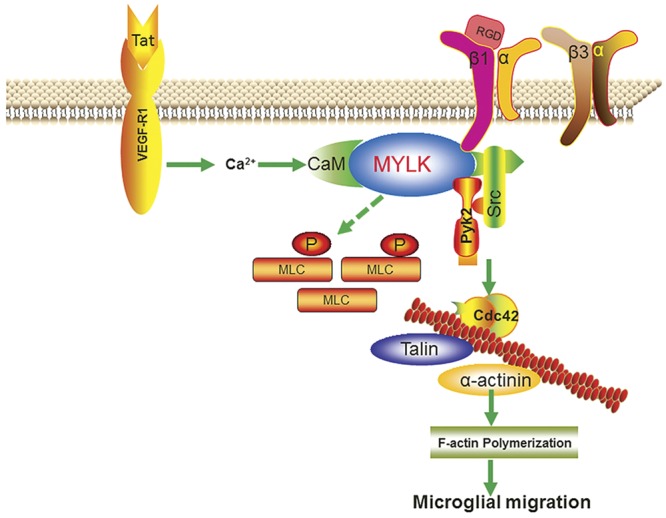
Schematic illustration demonstrating signaling pathways involved in Tat-mediated microglial migration. Tat-mediated engagement of the VEGFR1 stimulated activation of MYLK, followed by inside-out activation of β1 integrin, and subsequent outside-in signaling of β1 integrin involving activation of the downstream kinases c-Src, Pyk2, and Cdc42-GTP, culminating ultimately in actin polymerization, leading to microglial migration.
Acknowledgments
The authors thank Yaman Lu for technical support.
This work was supported by grants MH-068212, DA020392, DA023397, and DA024442 (S.B.), and DA030285 (H.Y.) from the U.S. National Institutes of Health.
The authors declare no conflicts of interest.
This article includes supplemental data. Please visit http://www.fasebj.org to obtain this information.
- DAPI
- 4′,6-diamidino-2-phenylindole
- ECM
- extracellular matrix
- GFAP
- glial fibrillary acidic protein
- HAND
- HIV-associated neurocognitive disorder
- HIV
- human immunodeficiency virus
- HIV-E
- HIV-encephalitis
- Iba-1
- ionized calcium binding adaptor molecule 1
- MLC
- myosin light chain
- MYLK
- myosin light-chain kinase
- nmMYLK
- nonmuscle myosin light-chain kinase
- RGD
- Arg-Gly-Asp
- RIPA
- radioimmunoprecipitation assay
- siRNA
- short interfering RNA
- Tat
- transactivator of transcription
- VEGF-R
- vascular endothelial growth factor receptor
- WT
- wild type
REFERENCES
- 1. Davalos D., Grutzendler J., Yang G., Kim J. V., Zuo Y., Jung S., Littman D. R., Dustin M. L., Gan W. B. (2005) ATP mediates rapid microglial response to local brain injury in vivo. Nat. Neurosci. 8, 752–758 [DOI] [PubMed] [Google Scholar]
- 2. Nimmerjahn A., Kirchhoff F., Helmchen F. (2005) Resting microglial cells are highly dynamic surveillants of brain parenchyma in vivo. Science 308, 1314–1318 [DOI] [PubMed] [Google Scholar]
- 3. Rostasy K. M. (2005) Inflammation and neuroaxonal injury in multiple sclerosis and AIDS dementia complex: implications for neuroprotective treatment. Neuropediatrics 36, 230–239 [DOI] [PubMed] [Google Scholar]
- 4. Urbinati C., Nicoli S., Giacca M., David G., Fiorentini S., Caruso A., Alfano M., Cassetta L., Presta M., Rusnati M. (2009) HIV-1 Tat and heparan sulfate proteoglycan interaction: a novel mechanism of lymphocyte adhesion and migration across the endothelium. Blood 114, 3335–3342 [DOI] [PubMed] [Google Scholar]
- 5. Toschi E., Bacigalupo I., Strippoli R., Chiozzini C., Cereseto A., Falchi M., Nappi F., Sgadari C., Barillari G., Mainiero F., Ensoli B. (2006) HIV-1 Tat regulates endothelial cell cycle progression via activation of the Ras/ERK MAPK signaling pathway. Mol. Biol. Cell 17, 1985–1994 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Albini A., Soldi R., Giunciuglio D., Giraudo E., Benelli R., Primo L., Noonan D., Salio M., Camussi G., Rockl W., Bussolino F. (1996) The angiogenesis induced by HIV-1 Tat protein is mediated by the Flk-1/KDR receptor on vascular endothelial cells. Nat. Med. 2, 1371–1375 [DOI] [PubMed] [Google Scholar]
- 7. Van Grol J., Subauste C., Andrade R. M., Fujinaga K., Nelson J., Subauste C. S. (2010) HIV-1 inhibits autophagy in bystander macrophage/monocytic cells through Src-Akt and STAT3. PLoS One 5, e11733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Minghetti L., Visentin S., Patrizio M., Franchini L., Ajmone-Cat M. A., Levi G. (2004) Multiple actions of the human immunodeficiency virus type-1 Tat protein on microglial cell functions. Neurochem. Res. 29, 965–978 [DOI] [PubMed] [Google Scholar]
- 9. Gallagher P. J., Herring B. P., Stull J. T. (1997) Myosin light chain kinases. J. Muscle Res. Cell Motil. 18, 1–16 [DOI] [PubMed] [Google Scholar]
- 10. Taylor D. A., Stull J. T. (1988) Calcium dependence of myosin light chain phosphorylation in smooth muscle cells. J. Biol. Chem. 263, 14456–14462 [PubMed] [Google Scholar]
- 11. Stull J. T., Kamm K. E., Taylor D. A. (1988) Calcium control of smooth muscle contractility. Am. J. Med. Sci. 296, 241–245 [DOI] [PubMed] [Google Scholar]
- 12. Smith L., Parizi-Robinson M., Zhu M. S., Zhi G., Fukui R., Kamm K. E., Stull J. T. (2002) Properties of long myosin light chain kinase binding to F-actin in vitro and in vivo. J. Biol. Chem. 277, 35597–35604 [DOI] [PubMed] [Google Scholar]
- 13. Graham W. V., Wang F., Clayburgh D. R., Cheng J. X., Yoon B., Wang Y., Lin A., Turner J. R. (2006) Tumor necrosis factor-induced long myosin light chain kinase transcription is regulated by differentiation-dependent signaling events. Characterization of the human long myosin light chain kinase promoter. J. Biol. Chem. 281, 26205–26215 [DOI] [PubMed] [Google Scholar]
- 14. Ma T. Y., Boivin M. A., Ye D., Pedram A., Said H. M. (2005) Mechanism of TNF-α modulation of Caco-2 intestinal epithelial tight junction barrier: role of myosin light-chain kinase protein expression. Am. J. Physiol. Gastrointest. Liver Physiol. 288, G422–G430 [DOI] [PubMed] [Google Scholar]
- 15. Saito H., Minamiya Y., Kitamura M., Saito S., Enomoto K., Terada K., Ogawa J. (1998) Endothelial myosin light chain kinase regulates neutrophil migration across human umbilical vein endothelial cell monolayer. J. Immunol. 161, 1533–1540 [PubMed] [Google Scholar]
- 16. Xu J., Gao X. P., Ramchandran R., Zhao Y. Y., Vogel S. M., Malik A. B. (2008) Nonmuscle myosin light-chain kinase mediates neutrophil transmigration in sepsis-induced lung inflammation by activating beta2 integrins. Nat. Immunol. 9, 880–886 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Miao H., Crabb A. W., Hernandez M. R., Lukas T. J. (2010) Modulation of factors affecting optic nerve head astrocyte migration. Invest. Ophthalmol. Vis. Sci. 51, 4096–4103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Smith A., Bracke M., Leitinger B., Porter J. C., Hogg N. (2003) LFA-1-induced T cell migration on ICAM-1 involves regulation of MLCK-mediated attachment and ROCK-dependent detachment. J. Cell Sci. 116, 3123–3133 [DOI] [PubMed] [Google Scholar]
- 19. Webb D. J., Donais K., Whitmore L. A., Thomas S. M., Turner C. E., Parsons J. T., Horwitz A. F. (2004) FAK-Src signalling through paxillin, ERK and MLCK regulates adhesion disassembly. Nat. Cell Biol. 6, 154–161 [DOI] [PubMed] [Google Scholar]
- 20. Dudek S. M., Birukov K. G., Zhan X., Garcia J. G. (2002) Novel interaction of cortactin with endothelial cell myosin light chain kinase. Biochem. Biophys. Res. Commun. 298, 511–519 [DOI] [PubMed] [Google Scholar]
- 21. Kudryashov D. S., Chibalina M. V., Birukov K. G., Lukas T. J., Sellers J. R., Van Eldik L. J., Watterson D. M., Shirinsky V. P. (1999) Unique sequence of a high molecular weight myosin light chain kinase is involved in interaction with actin cytoskeleton. FEBS Lett. 463, 67–71 [DOI] [PubMed] [Google Scholar]
- 22. Ginsberg M. H., Partridge A., Shattil S. J. (2005) Integrin regulation. Curr. Opin. Cell Biol. 17, 509–516 [DOI] [PubMed] [Google Scholar]
- 23. Hynes R. O. (2002) Integrins: bidirectional, allosteric signaling machines. Cell 110, 673–687 [DOI] [PubMed] [Google Scholar]
- 24. Lowell C. A., Berton G. (1999) Integrin signal transduction in myeloid leukocytes. J. Leukoc. Biol. 65, 313–320 [DOI] [PubMed] [Google Scholar]
- 25. Luo B. H., Carman C. V., Springer T. A. (2007) Structural basis of integrin regulation and signaling. Annu. Rev. Immunol. 25, 619–647 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Ley K. (2002) Integration of inflammatory signals by rolling neutrophils. Immunol. Rev. 186, 8–18 [DOI] [PubMed] [Google Scholar]
- 27. Calderwood D. A., Shattil S. J., Ginsberg M. H. (2000) Integrins and actin filaments: reciprocal regulation of cell adhesion and signaling. J. Biol. Chem. 275, 22607–22610 [DOI] [PubMed] [Google Scholar]
- 28. Totani L., Piccoli A., Manarini S., Federico L., Pecce R., Martelli N., Cerletti C., Piccardoni P., Lowell C. A., Smyth S. S., Berton G., Evangelista V. (2006) Src-family kinases mediate an outside-in signal necessary for beta2 integrins to achieve full activation and sustain firm adhesion of polymorphonuclear leucocytes tethered on E-selectin. Biochem. J. 396, 89–98 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Zhou F., Yao H. H., Wu J. Y., Ding J. H., Sun T., Hu G. (2008) Opening of microglial K(ATP) channels inhibits rotenone-induced neuroinflammation. J. Cell. Mol. Med. 12, 1559–1570 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Yao H., Peng F., Dhillon N., Callen S., Bokhari S., Stehno-Bittel L., Ahmad S. O., Wang J. Q., Buch S. (2009) Involvement of TRPC channels in CCL2-mediated neuroprotection against tat toxicity. J. Neurosci. 29, 1657–1669 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Peng F., Dhillon N. K., Yao H., Zhu X., Williams R., Buch S. (2008) Mechanisms of platelet-derived growth factor-mediated neuroprotection–implications in HIV dementia. Eur. J. Neurosci. 28, 1255–1264 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Ohsawa K., Irino Y., Sanagi T., Nakamura Y., Suzuki E., Inoue K., Kohsaka S. (2010) P2Y12 receptor-mediated integrin-beta1 activation regulates microglial process extension induced by ATP. Glia 58, 790–801 [DOI] [PubMed] [Google Scholar]
- 33. Miller A. M., Stella N. (2009) Microglial cell migration stimulated by ATP and C5a involve distinct molecular mechanisms: quantification of migration by a novel near-infrared method. Glia 57, 875–883 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Ryu J. K., Cho T., Choi H. B., Wang Y. T., McLarnon J. G. (2009) Microglial VEGF receptor response is an integral chemotactic component in Alzheimer's disease pathology. J. Neurosci. 29, 3–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Webb S. E., Pollard J. W., Jones G. E. (1996) Direct observation and quantification of macrophage chemoattraction to the growth factor CSF-1. J. Cell Sci. 109(Pt. 4), 793–803 [DOI] [PubMed] [Google Scholar]
- 36. Honda S., Sasaki Y., Ohsawa K., Imai Y., Nakamura Y., Inoue K., Kohsaka S. (2001) Extracellular ATP or ADP induce chemotaxis of cultured microglia through Gi/o-coupled P2Y receptors. J. Neurosci. 21, 1975–1982 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Haynes S. E., Hollopeter G., Yang G., Kurpius D., Dailey M. E., Gan W. B., Julius D. (2006) The P2Y12 receptor regulates microglial activation by extracellular nucleotides. Nat. Neurosci. 9, 1512–1519 [DOI] [PubMed] [Google Scholar]
- 38. Lu S. M., Tremblay M. E., King I. L., Qi J., Reynolds H. M., Marker D. F., Varrone J. J., Majewska A. K., Dewhurst S., Gelbard H. A. (2011) HIV-1 Tat-induced microgliosis and synaptic damage via interactions between peripheral and central myeloid cells. PLoS One 6, e23915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Adelstein R. S. (1983) Regulation of contractile proteins by phosphorylation. J. Clin. Invest. 72, 1863–1866 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Kamm K. E., Stull J. T. (1985) The function of myosin and myosin light chain kinase phosphorylation in smooth muscle. Annu. Rev. Pharmacol. Toxicol. 25, 593–620 [DOI] [PubMed] [Google Scholar]
- 41. Lauffenburger D. A., Horwitz A. F. (1996) Cell migration: a physically integrated molecular process. Cell 84, 359–369 [DOI] [PubMed] [Google Scholar]
- 42. Caswell P. T., Vadrevu S., Norman J. C. (2009) Integrins: masters and slaves of endocytic transport. Nat. Rev. Mol. Cell Biol. 10, 843–853 [DOI] [PubMed] [Google Scholar]
- 43. Kurpius D., Wilson N., Fuller L., Hoffman A., Dailey M. E. (2006) Early activation, motility, and homing of neonatal microglia to injured neurons does not require protein synthesis. Glia 54, 58–70 [DOI] [PubMed] [Google Scholar]
- 44. Mitola S., Sozzani S., Luini W., Primo L., Borsatti A., Weich H., Bussolino F. (1997) Tat-human immunodeficiency virus-1 induces human monocyte chemotaxis by activation of vascular endothelial growth factor receptor-1. Blood 90, 1365–1372 [PubMed] [Google Scholar]
- 45. Ren X. R., Du Q. S., Huang Y. Z., Ao S. Z., Mei L., Xiong W. C. (2001) Regulation of CDC42 GTPase by proline-rich tyrosine kinase 2 interacting with PSGAP, a novel pleckstrin homology and Src homology 3 domain containing rhoGAP protein. J. Cell Biol. 152, 971–984 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Sun C., Wu M. H., Yuan S. Y. (2011) Nonmuscle myosin light-chain kinase deficiency attenuates atherosclerosis in apolipoprotein E-deficient mice via reduced endothelial barrier dysfunction and monocyte migration. Circulation 124, 48–57 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Moy A. B., Shasby S. S., Scott B. D., Shasby D. M. (1993) The effect of histamine and cyclic adenosine monophosphate on myosin light chain phosphorylation in human umbilical vein endothelial cells. J. Clin. Invest. 92, 1198–1206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Van Nieuw Amerongen G. P., Draijer R., Vermeer M. A., van Hinsbergh V. W. (1998) Transient and prolonged increase in endothelial permeability induced by histamine and thrombin: role of protein kinases, calcium, and RhoA. Circ. Res. 83, 1115–1123 [DOI] [PubMed] [Google Scholar]
- 49. Haorah J., Knipe B., Leibhart J., Ghorpade A., Persidsky Y. (2005) Alcohol-induced oxidative stress in brain endothelial cells causes blood-brain barrier dysfunction. J. Leukoc. Biol. 78, 1223–1232 [DOI] [PubMed] [Google Scholar]
- 50. Hixenbaugh E. A., Goeckeler Z. M., Papaiya N. N., Wysolmerski R. B., Silverstein S. C., Huang A. J. (1997) Stimulated neutrophils induce myosin light chain phosphorylation and isometric tension in endothelial cells. Am. J. Physiol. 273, H981–988 [DOI] [PubMed] [Google Scholar]
- 51. Wainwright M. S., Rossi J., Schavocky J., Crawford S., Steinhorn D., Velentza A. V., Zasadzki M., Shirinsky V., Jia Y., Haiech J., Van Eldik L. J., Watterson D. M. (2003) Protein kinase involved in lung injury susceptibility: evidence from enzyme isoform genetic knockout and in vivo inhibitor treatment. Proc. Natl. Acad. Sci. U. S. A. 100, 6233–6238 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Ralay Ranaivo H., Carusio N., Wangensteen R., Ohlmann P., Loichot C., Tesse A., Chalupsky K., Lobysheva I., Haiech J., Watterson D. M., Andriantsitohaina R. (2007) Protection against endotoxic shock as a consequence of reduced nitrosative stress in MLCK210-null mice. Am. J. Pathol. 170, 439–446 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Reynoso R., Perrin R. M., Breslin J. W., Daines D. A., Watson K. D., Watterson D. M., Wu M. H., Yuan S. (2007) A role for long chain myosin light chain kinase (MLCK-210) in microvascular hyperpermeability during severe burns. Shock 28, 589–595 [DOI] [PubMed] [Google Scholar]
- 54. Eugenin E. A., Dyer G., Calderon T. M., Berman J. W. (2005) HIV-1 tat protein induces a migratory phenotype in human fetal microglia by a CCL2 (MCP-1)-dependent mechanism: possible role in NeuroAIDS. Glia 49, 501–510 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Fraga D., Raborn E. S., Ferreira G. A., Cabral G. A. (2011) Cannabinoids inhibit migration of microglial-like cells to the HIV protein Tat. J. Neuroimmune Pharmacol. 6, 566–577 [DOI] [PubMed] [Google Scholar]
- 56. Xiao H., Neuveut C., Tiffany H. L., Benkirane M., Rich E. A., Murphy P. M., Jeang K. T. (2000) Selective CXCR4 antagonism by Tat: implications for in vivo expansion of coreceptor use by HIV-1. Proc. Natl. Acad. Sci. U. S. A. 97, 11466–11471 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Westendorp M. O., Frank R., Ochsenbauer C., Stricker K., Dhein J., Walczak H., Debatin K. M., Krammer P. H. (1995) Sensitization of T cells to CD95-mediated apoptosis by HIV-1 Tat and gp120. Nature 375, 497–500 [DOI] [PubMed] [Google Scholar]
- 58. Rumbaugh J., Turchan-Cholewo J., Galey D., St Hillaire C., Anderson C., Conant K., Nath A. (2006) Interaction of HIV Tat and matrix metalloproteinase in HIV neuropathogenesis: a new host defense mechanism. FASEB J. 20, 1736–1738 [DOI] [PubMed] [Google Scholar]
- 59. Toborek M., Lee Y. W., Flora G., Pu H., Andras I. E., Wylegala E., Hennig B., Nath A. (2005) Mechanisms of the blood-brain barrier disruption in HIV-1 infection. Cell. Mol. Neurobiol. 25, 181–199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Eugenin E. A., King J. E., Nath A., Calderon T. M., Zukin R. S., Bennett M. V., Berman J. W. (2007) HIV-tat induces formation of an LRP-PSD-95- NMDAR-nNOS complex that promotes apoptosis in neurons and astrocytes. Proc. Natl. Acad. Sci. U. S. A. 104, 3438–3443 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Urbinati C., Mitola S., Tanghetti E., Kumar C., Waltenberger J., Ribatti D., Presta M., Rusnati M. (2005) Integrin alphavbeta3 as a target for blocking HIV-1 Tat-induced endothelial cell activation in vitro and angiogenesis in vivo. Arterioscler. Thromb. Vasc. Biol. 25, 2315–2320 [DOI] [PubMed] [Google Scholar]
- 62. Milner R., Campbell I. L. (2002) Cytokines regulate microglial adhesion to laminin and astrocyte extracellular matrix via protein kinase C-dependent activation of the alpha6beta1 integrin. J. Neurosci. 22, 1562–1572 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Milner R., Campbell I. L. (2003) The extracellular matrix and cytokines regulate microglial integrin expression and activation. J. Immunol. 170, 3850–3858 [DOI] [PubMed] [Google Scholar]
- 64. Birukov K. G., Csortos C., Marzilli L., Dudek S., Ma S. F., Bresnick A. R., Verin A. D., Cotter R. J., Garcia J. G. (2001) Differential regulation of alternatively spliced endothelial cell myosin light chain kinase isoforms by p60(Src). J. Biol. Chem. 276, 8567–8573 [DOI] [PubMed] [Google Scholar]