

Abstract
Dysfunctional tau accumulation is a major contributing factor in tauopathies, and the heat-shock protein 70 (Hsp70) seems to play an important role in this accumulation. Several reports suggest that Hsp70 proteins can cause tau degradation to be accelerated or slowed, but how these opposing activities are controlled is unclear. Here we demonstrate that highly homologous variants in the Hsp70 family can have opposing effects on tau clearance kinetics. When overexpressed in a tetracycline (Tet)-based protein chase model, constitutive heat shock cognate 70 (Hsc70) and inducible Hsp72 slowed or accelerated tau clearance, respectively. Tau synergized with Hsc70, but not Hsp72, to promote microtubule assembly at nearly twice the rate of either Hsp70 homologue in reconstituted, ATP-regenerating Xenopus extracts supplemented with rhodamine-labeled tubulin and human recombinant Hsp72 and Hsc70. Nuclear magnetic resonance spectroscopy with human recombinant protein revealed that Hsp72 had greater affinity for tau than Hsc70 (I/I0 ratio difference of 0.3), but Hsc70 was 30 times more abundant than Hsp72 in human and mouse brain tissue. This indicates that the predominant Hsp70 variant in the brain is Hsc70, suggesting that the brain environment primarily supports slower tau clearance. Despite its capacity to clear tau, Hsp72 was not induced in the Alzheimer's disease brain, suggesting a mechanism for age-associated onset of the disease. Through the use of chimeras that blended the domains of Hsp72 and Hsc70, we determined that the reason for these differences between Hsc70 and Hsp72 with regard to tau clearance kinetics lies within their C-terminal domains, which are essential for their interactions with substrates and cochaperones. Hsp72 but not Hsc70 in the presence of tau was able to recruit the cochaperone ubiquitin ligase CHIP, which is known to facilitate the ubiquitination of tau, describing a possible mechanism of how the C-termini of these homologous Hsp70 variants can differentially regulate tau triage. Thus, efforts to promote Hsp72 expression and inhibit Hsc70 could be therapeutically relevant for tauopathies.—Jinwal, U. K., Akoury, E., Abisambra, J. F., O'Leary, J. C., III, Thompson, A. D., Blair, L. J., Jin, Y., Bacon, J., Nordhues, B. A., Cockman, M., Zhang, J., Li, P., Zhang, B., Borysov, S., Uversky, V. N., Biernat, J., Mandelkow, E., Gestwicki, J. E., Zweckstetter, M., Dickey, C. A. Imbalance of Hsp70 family variants fosters tau accumulation.
Keywords: chaperones, CHIP, Hsc70, Hsp72, Alzheimer's
The microtubule-associated protein tau, when disengaged from microtubules, becomes deleterious in tauopathies (1–4). Part of this pathogenesis involves structural changes that promote its aggregation. Chaperone machinery, and in particular members of the heat-shock protein 70 (Hsp70) family, are engaged with tau during these pathogenic events, and these proteins dramatically affect tau stability and function at this critical juncture (5–7). In mammalian systems, there are ∼13 homologous Hsp70 genes, including the primary cytosolic variants heat-shock cognate 70 (Hsc70; HSPA8) and Hsp72 (HSPA1A). Though these two variants share nearly 80% sequence identity, some functional differences have been reported (8, 9). Hsc70 is constitutively expressed (10), whereas Hsp72 is expressed in response to stress (11). Moreover, the C termini of these chaperones, known to interact with key cochaperone effectors containing a tetratricopeptide (TPR) repeat motif, is where their sequences differ the most (12). Despite these observations, the term Hsp70 is often used to generically refer to both Hsc70 and Hsp72.
We and others have shown that the collective Hsp70 chaperone network, and in particular Hsp72 and Hsc70, influences tau conformation, degradation, and aggregation kinetics. But how these fates are chosen for tau has been challenging to define. For example, some studies strongly suggest that overexpression of an unspecified Hsp70 variant can facilitate tau clearance (5), whereas others suggest that both Hsp72 and Hsc70 restore tau's binding to microtubules (1, 7). Though differences in the cellular environment, such as stress (12) and client abundance (13) may contribute to this discrepancy, it has yet to be definitively shown how these different fates for tau are chosen by the Hsp70 machinery. We show here that tau fate is critically linked to both Hsc70 and Hsp72.
MATERIALS AND METHODS
Materials
Hsc70 and Hsp72 antibodies were purchased from Stressgen (Famingdale, NY, USA), and specificity was validated as described previously (8). Total tau H150 antibody was purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA). Actin and GAPDH antibodies were purchased from Sigma (St. Louis, MO, USA) and Biodesign International (Memphis, TN, USA), respectively. Myc and Flag antibodies were obtained from Roche (Indianapolis, IN, USA) and Sigma, respectively. Carboxyl terminus of Hsc70-interacting protein (CHIP) antibody was provided by Cell Signaling (Danvers, MA, USA). Phosphatase inhibitor cocktails (1 and 3), albendazole, and celastrol were purchased from Sigma. Cell lysis buffer M-PER was purchased from Fisher Scientific (Pittsburgh, PA, USA). Secondary antibodies horseradish peroxidase linked were purchased from Southern Biotech (Birmingham, AL, USA). Lipofectamine 2000 and secondary antibodies conjugated to flourophore were purchased from Invitrogen (Grand Island, NY, USA). Plasmids for wild-type human Hsc70, Hsp72, and chimera were generated in the pCMV6 vector (Origene, Rockville, MD, USA), which was then engineered to contain an N-terminal Flag tag. Mutant constructs were generated using the Site directed mutagenesis kit from Stratagene (Santa Clara, CA, USA). All other clones were in pCDNA3.1 plasmid. Human brain tissue was provided from the University of California at Irvine Alzheimer's Disease Research Center (UCI-ADRC) Institute for Memory Impairments and Neurological Disorders (MIND; Irvine, CA, USA).
Cell culture, immunoblotting, dot blot, immunostaining, and immunoprecipitation
Human neuroblastoma IMR-32, human embryonic kidney (HEK), and HeLa cells were maintained in OptiMEM medium supplemented with 10% heat-inactivated fetal bovine serum (FBS) and 1% penicillin/streptomycin antibiotic solution. All the transfections, immunoblotting, and immunoprecipitation experiments were performed as described previously (14). Dot blots were performed using the Whatman Minifold I apparatus on nitrocellulose (Whatman, Piscataway, NJ, USA). All values were normalized to Ponceau S. Recombinant Hsp72 and Hsc70 were applied at indicated molar concentrations in PBS. Lysates from human Alzheimer's disease (AD) brain tissue and control brain tissue were spotted at 20 μg total protein. Quantification of Western blot and dot blot data was performed using replicate values from ScionImage (Scion Corp., Frederick, MD, USA) or ImageJ (U.S. National Insitutes of Health, Bethesda, MD, USA) densitometric software suites. Dot blot calculations were performed using a semilog linear regression analysis. Immunostaining of mouse brain tissue was performed as described previously (15, 16).
Tetracycline (Tet)-based protein chase HEK cell model
Inducible HEK cell line was generated by the insertion of human wild-type tau 4R0N DNA into a pCDNA 4/TO plasmid vector carrying Zeocin selection (Invitrogen). The tau/TO plasmid construct was transfected into the Tetracycline-Regulated Expression (T-Rex; Invitrogen) HEK cell lines stably expressing Tet repressor under the selection of blasticidin. After 24 h of DNA transfection, Zeocin (400 μg/ml) and blasticidin (5 μg/ml) were added to the medum (DMEM containing 10% FBS, 2 mM L-glutamine, and 1% Pen-Strep). Every 3-4 d medium was replaced with fresh medium containing Zeocin and blasticidin. After 3 wk, double-stable cell colonies were picked up and transferred into 6-well plates. These colonies were induced by Tet (1 μg/ml) and tested for tau expression by Western blot. Tau-expressing double-stable HEK cells were maintained under the selection of Zeocin and blasticidin and used for the experimentation. Following transfection with Hsp72 or Hsc70 clones, Tet-containing medium was removed and replaced with normal medium. Tau levels were reduced over time to near basal levels.
Live-cell imaging
HEK-293 cells were plated onto a 6-well plate and maintained in DMEM plus 10% FBS and 1% of 200 mM l-glutamine. Plasmid transfections were done utilizing Lipofectamine 2000 (Invitrogen). HEK 293 cells were transfected with 2 μg of Hsc70 DNA, 1 μg of tubulin-green fluorescent protein (GFP) DNA, and 1 μg of tau-red fluorescent protein (RFP) DNA. The cells were incubated overnight with the Lipofectamine/plasmid mixture in Opti-MEM medium, and replaced with fresh complete medium for an additional 36 h. An Olympus FV1000 MPE multiphoton laser scanning microscope (Olympus, Tokyo, Japan) was used to capture images. Image analysis was completed with the Olympus FV10-ASW software, and significance was assessed by Pearson's correlation coefficient. Cells were then lysed and analyzed by Western blot to confirm overexpression of Hsp72 and Hsc70.
Recombinant protein production
The coding regions for WT 4R0N tau, Hsp72, and Hsc70 were separately PCR amplified and subcloned into pET28A vectors with N-terminal His6 tags. Resulting plasmids were transformed into E. coli OneShot BL21 Star (DE3) cells (Invitrogen), and 10 ml starter cultures were grown overnight in Luria-Bertani (LB) medium with 30 μg/ml kanamycin. Cells were then inoculated into 1.0 L LB medium, grown at 37°C to A600 ∼ 0.7–0.8, and induced with 1 mM isopropyl β-d-1-thiogalactopyranoside. After 3 h of continued growth at 37°C, cells were harvested by centrifugation and resuspended in lysis buffer [500 mM NaCl, 20 mM Tris-HCl, 5 mM imidazole, 1× protease inhibitor cocktail III (Calbiochem, Billerica, MA, USA), and 1 mM PMSF, pH 8.0]. The cells were then lysed by sonication and centrifuged (18,200 g, 30 min, 4°C), and supernatant was loaded onto 5 ml of charged Ni-NTA agarose beads (Qiagen, Valencia, CA, USA). Protein was eluted in lysis buffer containing 250 mM imidazole. Eluent was concentrated to 2 ml and loaded onto a HiLoad 16/60 Superdex 200 pg size-exclusion column. Purified fractions were dialyzed in 2 steps into 10 mM phosphate buffer (pH 7.5), and concentrations were determined by BCA.
In vitro microtubule assembly assay
These assays were performed as described previously (1, 14). Briefly, S-phase Xenopus egg extracts were prepared according to a standard protocol (17) and supplemented with 5% DMSO to stimulate microtubule polymerization, as described previously (18). Purified recombinant wild-type tau, Hsp72, and Hsc70 proteins were added at the final concentrations of 40, 80, and 80 ng/μl. The extracts were incubated 30 min at room temperature, and obtained microtubule structures were visualized by fluorescence of incorporated rhodamine-labeled tubulin.
Nuclear magnetic resonance (NMR)
15N-labeled human tau protein was expressed and purified as described previously (19). NMR experiments were recorded at 5°C on a Bruker Avance 800 MHz spectrometer (Bruker Corp., Billerica, MA, USA) equipped with a cryogenic probe. NMR samples contained 50 μM 15N single-labeled tau protein in 50 mM phosphate buffer and 10 mM NaCl (pH 6.8), 1 mM DTT, and 10% (v/v) D2O. Unlabeled Hsp72 and Hsc70 samples were prepared in the same buffer. Two-dimensional 1H–15N heteronuclear single-quantum coherence (HSQC) spectra were acquired using 600 complex points and 128 scans/increment with spectral widths of 8012 and 1944 Hz in the 1H and 15N dimensions, respectively. Spectra were processed with NMRPipe (20) and analyzed using CcpNMR Analysis (21). NMR intensity ratio plots were reported with a 3-residues averaging window.
Immunostaining
Mouse brains were processed for immunohistochemical analysis as described previously (16, 22). Briefly, mouse brain sections were immersed in 3% H2O2 solution for 15 min at room temperature. After washing with PBS, sections were permeabilized by immersing in the 4% donkey serum blocking buffer (supplemented with 1.83% lysine and 2% Triton X-100) for 30 min at room temperature. Sections were treated with anti-HSC70 (1:20000) and anti-Hsp72 (1:300) primary antibodies overnight at 4°C. After washing with PBS, sections were incubated with biotinylated anti-rat (1:1000) and anti-mouse (1:3000) secondary antibodies (Southern Biotech) for 2 h and then with streptavidin-peroxidase. The peroxidase reactions consisted of 1.4 mM diaminobenzidine with 0.03% hydrogen peroxide in PBS for 5 min. Finally, stained sections were mounted on glass slides, dehydrated, cleaned using histoclean, and coverslipped. Imaging was performed with the Zeiss Mirax slide-scanning microscope (Carl Zeiss, Oberkochen, Germany).
Fluorescent polarization (FP) assay
FP assays were carried out essentially as described previously (23). The peptide FITC-HLA (RENLRIARLY) was synthesized on Wang resin using microwave-assisted DIC/HOBt solid-phase peptide synthesis. It was capped with 2 β-alanine residues and labeled on resin via the N terminus with fluorescein-5-isothiocyanate (AnaSpec, Fremont, CA, USA). The final product was obtained in >90% purity. FP assays were carried out in 384-well plates (20 μl final volume) with 30 min incubation. Conditions were Hsp72 (2 μM) or Hsc70 (4 μM), 25 nM FITC-HLA probe, 100 mM Tris buffer, 20 mM KCl, 6 mM MgCl2, pH 7.4.
Sequence alignment
Global sequence alignment of H. sapiens Hsp72 (HspA1A/ A29160) and Hsc70 (HspA8/ A27077) was performed using Praline (http://www.ibi.vu.nl/programs/pralinewww/) with manual adjustment. Figures are drawn to scale, and the degree of residue conservancy is illustrated by color.
RESULTS
Hsp70 proteins have the capacity to both reduce and preserve tau in cells in addition to regulating its association with microtubules (1, 5, 7). The reasons for these functional differences are unclear. We speculated that one possible reason could be the unique effects of distinct Hsp70 variants. Since tau is a cytosolic protein, we investigated the relationship of tau with the two major cytosolic Hsp70 variants: Hsp72, which is induced by stress (11), and Hsc70, which is constitutively expressed (10). The effects of Hsp72 and Hsc70 overexpression on tau stability were initially tested using a Tet-based protein chase model in HEK cells (TauTO/6TR). As shown in Fig. 1A, tau levels are undetectable prior to Tet treatment and maximally induced 72 h after Tet treatment. Once the Tet is removed, tau levels are depleted by natural turnover processes over the course of 72 h (Fig. 1B). Using this model, we demonstrated that Hsc70 slowed while Hsp72 accelerated tau clearance (Fig. 1C, D). This indicated that these two highly similar proteins were distinctly affecting tau turnover. We then confirmed that endogenous tau could interact with endogenous Hsc70 and Hsp72 using coimmunoprecipitation assays in neuronal IMR-32 cell lysates analyzed by immunoblot. The neuronal cells were first treated for 24 h with celastrol, a compound that is known to enhance the expression of heat-shock proteins, including Hsp72 (24), and destabilize microtubules (25). This was done to determine whether inducing Hsp72 expression might facilitate the interaction of Hsp72 with tau. Then, 30 min prior to harvest, these cells were also treated with the microtubule destabilizer albendazole to release tau from microtubules, a process that was previously found to enhance the tau/Hsc70 interaction (1). As previously reported, Hsc70 bound more tau when microtubules were destabilized, but celastrol treatment had no impact on Hsc70 binding to tau (Fig. 1E). Conversely, for endogenous Hsp72 to bind tau in a similar manner, Hsp72 levels had to be induced by celastrol treatment for it to bind tau in a manner similar to Hsc70 (Fig. 1E). These data suggested that relative levels of these 2 proteins could play a critical part in deciding whether tau was to be preserved or degraded. Our data suggested that Hsp72 was accelerating tau clearance while Hsc70 was slowing it. Therefore, we speculated that increasing the levels of Hsp72 could overcome any preservative effects of Hsc70 on tau. Cells overexpressing tau were transfected with equivalent amounts of Hsc70 along with increasing amounts of Hsp72. Analysis of these lysates by immunoblot revealed that the enhanced steady-state levels of tau by Hsc70 could indeed be negated by Hsp72 overexpression (Fig. 1F, G).
Figure 1.

Hsp72 and Hsc70 have opposing effects on tau. A) Western blot of lysates from HEK cells transiently transfected with tau driven by the minimal CMV promoter controlled by the Tet response element (TauTO) or empty vector, and transiently transfected with the Tet repressor protein (6TR). Cells were then treated with Tet and harvested at indicated time points. B) HEK cells stably transfected with TauTO and the 6TR were treated with indicated concentration of Tet for 24 h. After 24 h, Tet-containing medium was replaced with medium either containing Tet (on) or without Tet (on/off), and these cells were incubated for an additional 72 h (96 h total). Lysates were analyzed by Western blot. C) TauTO/6TR stably transfected cells were treated with Tet for 24 h. Cells were then transfected with Hsc70 or Hsp72 for 24 h, and tetracycline was removed. Cells were harvested at indicated times following Tet removal. D) Quantification of tau levels in C shown as a percentage of empty vector transfected cells (Ctrl) ± sd (sd) after GAPDH normalization (n=3). Open bars, dark gray bars, and light gray bars indicate vector, Hsc70-, and Hsp72-transfected cells, respectively. E) Coimmunoprecipitation of tau from IMR-32 cell lysates treated with DMSO alone and albendazole with DMSO, or celastrol alone and albendazole with celastrol. Then 3 μM Celastrol was applied for 24 h, followed by 20 μM Albendazole treatment for 30 min. Levels of Hsp72 and Hsc70 bound to tau were assessed. Inputs indicate whole lysates. F) Western blot of tau in lysates of HEK cells cotransfected with Hsc70 and increasing amounts of Hsp72. G) Quantification of tau levels in F, shown as mean ± sd percentage of vector-transfected cells after GAPDH normalization.
Previous work suggested that Hsp70 proteins could promote the association of tau with microtubules (7); however, based on our results, we speculated that Hsc70 would affect this more potently than Hsp72. Using colocalization analyses of GFP-tagged tubulin and RFP-tagged tau in live cells, we found a slight increase in tau-tubulin associations induced by Hsc70 (Fig. 2A–C). However, Hsp72 overexpression significantly decreased the association of tau with tubulin, further suggesting that Hsp72 promoted tau degradation (Fig. 2A–C). Western blot confirmed overexpression of both Hsp72 and Hsc70 (Fig. 2D). While these studies supported opposing roles for these two Hsp70 variants on tau function, it still was not clear whether Hsp72 and Hsc70 had direct effects on the ability of tau to regulate microtubule formation. To better evaluate this, we utilized microscopy of in vitro cell-free Xenopus egg extracts supplemented with fluorescently labeled tubulin and combinations of either recombinant 4R0N tau, Hsp72, or Hsc70 alone, or 4R0N tau combined with Hsp72 or Hsc70. Hsc70, Hsp72, and tau were all able to promote microtubule assembly alone relative to control, but Hsc70 and tau together appeared to dramatically synergize this process, producing densely packed microtubule filaments (Fig. 3A, B). Conversely, Hsp72 did not appear to synergize with tau: In fact, there was a slight reduction in microtubule network formation when Hsp72 alone was added with tau.
Figure 2.

Hsc70, but not Hsp72, enhances the colocalization of tau with tubulin in cells. A) Representative 2-photon fluorescent images of live HEK cells cotransfected with tubulin-GFP and tau-RFP fusion constructs. Cells transfected with vector (Ctrl) or Hsc70 showed significant colocalization between tau and tubulin as compared to cells transfected with Hsp72. B) Higher-magnification imaging of areas boxed in red in A. C) Graph demonstrating the Pearson's colocalization coefficient derived from 12 different images. Bars represent means ± sd. *P < 0.05. D) Western blot of cell lysates confirming overexpression of Flag-tagged Hsp72 and Hsc70.
Figure 3.
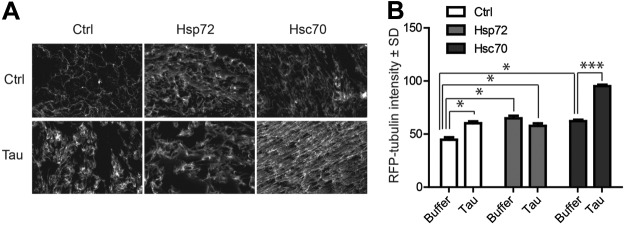
Hsc70, but not Hsp72, synergizes with tau to promote microtubule assembly. A) Representative fluorescent images of Xenopus extracts supplemented with rhodamine-labeled tubulin and indicated combinations of recombinant tau, Hsp72, and Hsc70 (n=10). B) Graph demonstrating that tau, Hsp72, and Hsc70 alone could promote microtubule assembly, but only Hsc70 combined with tau to dramatically influence microtubule formation. *P < 0.05, ***P < 0.001.
These findings suggested that despite their sequence similarity, Hsp72 and Hsc70 caused distinct and even opposing effects on tau's function and stability. To determine whether this was caused by differing binding locations on tau, NMR spectroscopy was used to identify tau residues essential for Hsp72 and Hsc70 interactions. Changes in NMR signal position and intensity in 2-dimensional 1H–15N HSQC spectra of the 441-residue full length tau protein were observed for both Hsp70 variants in a concentration-dependent manner (Fig. 4A). The observed changes were caused by an exchange of tau between the free and the Hsp72/Hsc70-bound state. Strong signal broadening was particularly evident in the second and third repeats of tau comprising the 2 hexapeptides 275VQIINK280 and 306VQIVYK311, essential for aggregation of tau into paired helical filaments (Fig. 4B, C). These data were consistent with previous studies belying the importance of these motifs in chaperone-mediated autophagy (CMA; ref. 26) and interaction with Hsc70 proteins (27). Strikingly, much more pronounced changes in repeats 2 and 3 were observed for Hsp72 than Hsc70 (Fig. 4B), indicating differences in the interaction between Hsp72 and Hsc70 with tau (Fig. 4C). In addition, weaker interactions were observed with the residue stretches 346FKDRVQSK353 and 375KLTFRE380.
Figure 4.

Hsp72 binds tau with higher affinity than Hsc70. A) Superposition of 2D 1H–15N HSQC spectra of tau protein (50 μM) in the absence (black) and presence of an equimolar (green) and 2-fold excess ratios of Hsp72 (orange). Inset highlights the resonances of the 2 hexapeptides and their attenuation in a concentrationdependent manner. B, C) NMR intensity ratios I/I0 (I, intensity of tau resonances in presence of Hsp72 or Hsc70; I0, peak intensities of free tau) from HSQC spectra. Signal broadening (I/I0<1.0) is due to exchange of tau between the free conformations and those in the presence of Hsp72 or Hsc70. NMR signal broadening of tau at increasing concentrations of Hsp72 (B) and Hsc70 (C) (1:1 green, 2:1 orange). Domain organization of tau is shown above (I, insert, P, proline-rich region, and R, pseudorepeat). Asterisk corresponds to the residues stretch 346FKDRVQSK353.
We sought to better understand the mechanism responsible for these distinct direct effects of Hsp72 and Hsc70 on tau, and why in particular Hsp72 could outcompete Hsc70 for tau binding. We performed fluorescence polarization assays in which recombinant Hsp72 and Hsc70 were incubated with both a labeled peptide known to interact with their substrate-binding domains (SBDs) and increasing concentrations of recombinant tau. Tau was able to compete with the fluorescent mock-substrate peptide in a dose-dependent manner, suggesting that tau was interacting with the SBDs of Hsp72 and Hsc70 (Fig. 5A). While these data showed where tau interacted with Hsp72 and Hsc70, it did not explain why these two proteins were causing differential effects on tau biology in cells. Using the Praline algorithms, we found that while these two proteins shared > 80% similarity in their complete protein sequence, their C-terminal regions had the highest sequence variation (30%; Fig. 5B, C). Therefore, chimeras were constructed to blend the nucleotide-binding domains (NBDs), SBDs, and the most variable C-terminal domains (CTDs) from Hsp72 and Hsc70 (Fig. 5D) to determine which region was most essential for the distinct tau effects. Overexpression of these chimeras in cells revealed that constructs possessing the CTD of Hsc70 preserved tau, while those containing the CTD of Hsp72 facilitated tau clearance (Fig. 5E, F). The NBD and SBD of both proteins appeared to be interchangeable.
Figure 5.

C-terminal domains from Hsc70 and Hsp72 are essential for tau triage. A) FP assays with recombinant Hsp72 or Hsc70 in the presence of 1 mM ADP, the FITC-HLA (RENLRIARLY) peptide, and increasing amounts of recombinant tau were carried out in 384-well plates (20 μl final volume) with 30 min incubation. Results are the average of experiments performed in triplicate; error bars represent sem. B, C) Sequence alignment showing 29% sequence variability in the C termini of Hsp72 and Hsc70. D) Schematic of Hsc70/Hsp72 chimera design. E) Western blot of lysates from HeLa cells stably expressing tau and transfected with Flag-tagged Hsc70/Hsp72 chimeras. F) Quantification of tau levels in D shown as mean ± sd percentage of vector-transfected cells (Ctrl) after GAPDH normalization (n=3).
This C-terminal interaction suggested an important role for cofactors/cochaperones of Hsp72 and Hsc70 that bind to this same region in regulating tau stability. To test this, cells overexpressing tau and either Hsp72 or Hsc70 were transfected with siRNA targeting CHIP, a ubiquitin ligase cochaperone known to regulate tau turnover (5, 6, 28). Western blot of these lysates showed that knocking down CHIP preserved tau despite overexpression of Hsp72 or Hsc70 (Fig. 6A, B). This suggested that CHIP expression was important for the effects of Hsp72 and Hsc70 on tau in cells. To assess whether CHIP binding to Hsp72 and Hsc70 was different, we performed coimmunoprecipitation using anti-Flag antibody in cells overexpressing myc-tagged CHIP, tau, and either Flag-tagged Hsc70 or Flag-tagged Hsp72. In cells overexpressing tau, the number of CHIP/Hsp72 complexes was significantly greater than the number of CHIP/Hsc70 complexes (Fig. 6C), but surprisingly, in cells not overexpressing tau, the binding of CHIP to Hsc70 and Hsp72 was equivalent (Fig. 6D). In this way, Hsp72, but not Hsc70, in the presence of tau and other cofactors in the cell would favor CHIP binding to tau.
Figure 6.
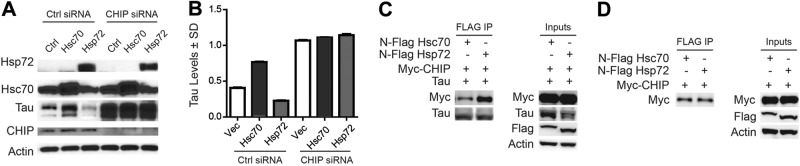
Tau influences the composition of Hsp70/CHIP complexes, affecting its own triage. A) Western blots of HeLa cells that were transfected with Hsp72, Hsc70, and siRNA for CHIP. B) Quantification of tau levels in A, shown as mean ± sd ratio to actin derived from 3 repeated experiments. C, D) Western blot of samples coimmunoprecipitated with anti-Flag antibody with (C) or without (D) tau overexpression shows that tau selectively increases the association of Hsp72 with CHIP.
To better understand the physiological relevance of these physical differences, we investigated the protein levels of Hsc70 and Hsp72 in brain homogenates from normal and AD brains. Using a dot blot, we generated a standard curve for antibodies targeting Hsc70 and Hsp72 to account for differences in antibody affinity (Fig. 7A–C). We then spotted 20 μg of total protein from AD and control brains onto dot blot membranes, probed with Hsp72 or Hsc70 antibodies, and plotted the resulting intensities along the standard curves generated with recombinant proteins (Fig. 7A–C). Relative quantitation of these proteins revealed that levels of Hsc70 were 30 times greater than Hsp72, with no difference between normal and AD brains (Fig. 7D). Staining of mouse brain tissue produced similar results, revealing an excess of Hsc70 relative to Hsp72 (Fig. 7E).
Figure 7.

Hsc70 levels are much higher than Hsp72 in human brain. A, B) Recombinant Hsc70 (A) and Hsp72 (B) were spotted onto nitrocellulose at indicated concentrations alongside 20 μg of total protein from normal and AD brain lysates. Ponceau S was performed, followed by blocking and immunoblot with anti-Hsc70 or anti-Hsp72 antibodies. C) Semilog linear regression analysis was performed using the values of the recombinant protein to normalize for difference in antibody affinity. D) Abundance of Hsc70 (dark gray bars) and Hsp72 (light gray bars) immunoreactivity in lysates from normal and AD human brain (n=3/group) normalized to the standard curve composed of recombinant Hsc70 and Hsp72, respectively, in A and B. E) Representative images of immunostaining of mouse brain (n=4) with Hsc70 and Hsp72 antibodies.
DISCUSSION
We have found that Hsc70 and Hsp72 have opposing effects on tau that are driven by their relative expression levels, affinities for tau, and their C-terminal interaction domains. The data presented here suggest that the C-terminal dynamics of Hsc70 and Hsp72 are essential for whether tau degradation is accelerated or slowed. Because the CTDs of these proteins form the lid of the substrate binding pocket and host interactions with cochaperones that contain the TPR motif (29, 30), we predicted that levels of cochaperones known to interact with the C-terminal portions of Hsp72 and Hsc70 could also have a major influence on tau triage decisions. One such TPR-containing protein, CHIP, has been extensively linked by our laboratory and others to both Hsp70 proteins and tau (5, 6, 31, 32). This protein has ubiquitin ligase activity and has been shown to facilitate tau clearance. Here we discovered that CHIP is able to bind to Hsc70 and Hsp72 differently when tau is present. This suggests that there are unique structural changes to both Hsp70 variants brought on by tau and other cochaperones that can dictate CHIP association. Future studies will determine what these structures are and what components are needed; however, these cellular findings point to this phenomenon as an important component for tau triage. Perhaps this represents a mechanism for tau to maintain its own proteostasis; as tau levels become overabundant and exceed the capacity of the microtubule network, tau interacts more with Hsp72 and recruits a distinct subset of cochaperones including CHIP to promote its self-destruction. This would prevent its accumulation inside neurons. However, this same recruitment of CHIP did not occur when tau interacted with Hsc70, suggesting that restoring or boosting the levels of Hsp72 may supplant Hsc70 and promote tau clearance.
Interestingly, we also found that Hsp72 levels were not induced in AD, despite the chronic stress conditions that must occur during the disease course. It is unclear why tau accumulation failed to promote more robust expression of Hsp72 in contrast to what has been reported for other neurodegenerative diseases brought on by protein misfolding and accumulation (11, 33). It is possible that Hsp72 levels are elevated early in the disease process, but then attenuate over time back to baseline levels due to the chronic proteotoxic stress. We would predict that even a small increase in Hsp72 could counterbalance tau burden by subverting the preservative effects of Hsc70 due to the enhanced affinity of Hsp72 for Hsc70, but not even slight increases were observed in human AD or tau transgenic mouse brain tissue. This could be indicative of the failing heat shock response that occurs in general with age (34, 35). Thus, selective enhancement of Hsp72 levels with small molecules or gene therapies could favor an environment that removes excess tau.
Using NMR spectroscopy, we identified the binding motifs for a chaperone on tau. As expected, both Hsc70 and Hsp72 shared binding sites within the microtubule-binding domains of tau. In particular, consistent with previous studies using traditional mutagenesis, the 275VQIINK280 and 306VQIVYK311 hexapeptide motifs were found to be critical for binding these chaperones to tau (26, 27). These motifs are particularly important for tau in that they are necessary for tau aggregation (36, 37) and triage toward an inefficient CMA pathway (26). These proamyloid motifs reside in most proteins (38) and may serve as beacons for chaperones (39, 40). Thus, as tau disengages from microtubules, these motifs are exposed, recruiting available chaperones. It is at this point that the balance between Hsp72 and Hsc70 may be most critical. Hsc70 is necessary for CMA in that it facilitates the triage of certain proteins toward Lamp2a, a lysosomal membrane protein for CMA substrates (41); however, Hsp72 does not have a similar function. While facilitating CMA would at first seem beneficial in promoting tau clearance, tau actually is anchored to the lysosomal membrane following its triage to Lamp2a, whereby it is then proteolytically cleaved and primed for aggregation (26).
Using NMR, we were also able to identify some additional consensus sites 346FKDRVQSK353 and 375KLTFRE380 that were previously unidentified. Most intriguingly, the lysine at 353 is part of the known KXGS consensus sequence for the microtubule affinity regulating kinase 2 (MARK2). We previously demonstrated that CHIP is less prone to interact with tau when it is phosphorylated at MARK2 consensus sites (42–44). MARK2 promotes the release of tau from microtubules (45) while also altering tau sorting within neurons (46). Since tau phosphorylated at these sites is protected from CHIP interaction, it suggests that this is a strategy adopted by the neuron to preserve tau during plasticity to conserve energy. We speculate that to promote plasticity, tau must detach from microtubules, but without a post-translational modification of some sort, a CHIP/Hsp72 complex would quickly bind and promote tau clearance, a step that would require new tau protein to be synthesized after every axonal plastic event. By promoting phosphorylation of tau near the CHIP/Hsp72 binding sites, the neuron can preserve tau and conserve energy by allowing tau recycling: But this comes with a price: Phosphorylation of tau destabilizes the axonal diffusion barrier that prevents tau localization to the soma and dendrites (46), the subcellular location where tau pathology is typically found in AD and other tauopathies. Therefore, while preventing CHIP/Hsp72 binding through MARK2 phosphorylation may be energetically favorable for the neuron, it allows tau to move into the somatodendritic compartment where it is known to accumulate and become toxic. A delicate balance is required to meet energy demands while also preventing proteotoxicity. Expression levels of Hsp70 variants, tau phosphorylation, microtubule integrity, and neuronal trafficking all must be managed to prevent tau from accumulating to the point of being neurotoxic. If any of these processes are altered, the toxic sequelae of tau can be accelerated. Thus, interventions aimed at any of these pathways could be effective therapeutics for managing tauopathies.
Acknowledgments
C.A.D. was supported by grants from the U.S. National Institutes of Health (NIH)/National Institute on Aging (NIA; R00AG031291), NIH/National Institute of Neurological Disorders and Stroke (NINDS; R01NS073899), the Alzheimer's Association, and CurePSP. U.K.J. was supported by the Alzheimer's Association. J.E.G. was supported by NIH/NINDS (NS059690) and the Alzheimer's Association. Funding for the University of California at Irvine Alzheimer's Disease Research Center (UCI-ADRC; Irvine, CA, USA) was provided by NIH/NIA grant P50 AG16573.
The authors declare no conflicts of interest.
Footnotes
- AD
- Alzheimer's disease
- CHIP
- carboxyl terminus of Hsc70-interacting protein
- CMA
- chaperone-mediated autophagy
- CTD
- C-terminal domains
- FBS
- fetal bovine serum
- FP
- fluorescent polarization
- GFP
- green fluorescent protein
- HEK
- human embryonic kidney
- Hsc
- heat-shock cognate
- Hsp70
- heat-shock protein 70
- HSQC
- heteronuclear single-quantum coherence
- LB
- Luria-Bertani
- MARK2
- microtubule affinity regulating kinase 2
- NBD
- nucleotide-binding domain
- NMR
- nuclear magnetic resonance
- RFP
- red fluorescent protein
- SBD
- substrate-binding domain
- Tet
- tetracycline
- TPR
- tetratricopeptide
REFERENCES
- 1. Jinwal U. K., O'Leary J. C., 3rd, Borysov S. I., Jones J. R., Li Q., Koren J., 3rd, Abisambra J. F., Vestal G. D., Lawson L. Y., Johnson A. G., Blair L. J., Jin Y., Miyata Y., Gestwicki J. E., Dickey C. A. (2010) Hsc70 rapidly engages tau after microtubule destabilization. J. Biol. Chem. 285, 16798–16805 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Ebneth A., Godemann R., Stamer K., Illenberger S., Trinczek B., Mandelkow E. (1998) Overexpression of tau protein inhibits kinesin-dependent trafficking of vesicles, mitochondria, and endoplasmic reticulum: implications for Alzheimer's disease. J. Cell Biol. 143, 777–794 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Mattson M. P. (1992) Effects of microtubule stabilization and destabilization on tau immunoreactivity in cultured hippocampal neurons. Brain Res. 582, 107–118 [DOI] [PubMed] [Google Scholar]
- 4. Brion J. P., Octave J. N., Couck A. M. (1994) Distribution of the phosphorylated microtubule-associated protein tau in developing cortical neurons. Neuroscience 63, 895–909 [DOI] [PubMed] [Google Scholar]
- 5. Petrucelli L., Dickson D., Kehoe K., Taylor J., Snyder H., Grover A., De Lucia M., McGowan E., Lewis J., Prihar G., Kim J., Dillmann W. H., Browne S. E., Hall A., Voellmy R., Tsuboi Y., Dawson T. M., Wolozin B., Hardy J., Hutton M. (2004) CHIP and Hsp70 regulate tau ubiquitination, degradation and aggregation. Hum. Mol. Genet. 13, 703–714 [DOI] [PubMed] [Google Scholar]
- 6. Dickey C. A., Yue M., Lin W. L., Dickson D. W., Dunmore J. H., Lee W. C., Zehr C., West G., Cao S., Clark A. M., Caldwell G. A., Caldwell K. A., Eckman C., Patterson C., Hutton M., Petrucelli L. (2006) Deletion of the ubiquitin ligase CHIP leads to the accumulation, but not the aggregation, of both endogenous phospho- and caspase-3-cleaved tau species. J. Neurosci. 26, 6985–6996 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Dou F., Netzer W. J., Tanemura K., Li F., Hartl F. U., Takashima A., Gouras G. K., Greengard P., Xu H. (2003) Chaperones increase association of tau protein with microtubules. Proc. Natl. Acad. Sci. U. S. A. 100, 721–726 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Koren J., 3rd, Miyata Y., Kiray J., O'Leary J. C., 3rd, Nguyen L., Guo J., Blair L. J., Li X., Jinwal U. K., Cheng J. Q., Gestwicki J. E., Dickey C. A. (2012) Rhodacyanine derivative selectively targets cancer cells and overcomes tamoxifen resistance. PloS ONE 7, e35566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Goldfarb S. B., Kashlan O. B., Watkins J. N., Suaud L., Yan W., Kleyman T. R., Rubenstein R. C. (2006) Differential effects of Hsc70 and Hsp70 on the intracellular trafficking and functional expression of epithelial sodium channels. Proc. Natl. Acad. Sci. U. S. A. 103, 5817–5822 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Black M. M., Chestnut M. H., Pleasure I. T., Keen J. H. (1991) Stable clathrin: uncoating protein (Hsc70) complexes in intact neurons and their axonal transport. J. Neurosci. 11, 1163–1172 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Williams G. T., Morimoto R. I. (1990) Maximal stress-induced transcription from the human HSP70 promoter requires interactions with the basal promoter elements independent of rotational alignment. Mol. Cell. Biol. 10, 3125–3136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Tavaria M., Gabriele T., Kola I., Anderson R. L. (1996) A hitchhiker's guide to the human Hsp70 family. Cell Stress Chaperones 1, 23–28 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Qian S. B., McDonough H., Boellmann F., Cyr D. M., Patterson C. (2006) CHIP-mediated stress recovery by sequential ubiquitination of substrates and Hsp70. Nature 440, 551–555 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Jinwal U. K., Koren J., 3rd, Borysov S. I., Schmid A. B., Abisambra J. F., Blair L. J., Johnson A. G., Jones J. R., Shults C. L., O'Leary J. C., 3rd, Jin Y., Buchner J., Cox M. B., Dickey C. A. (2010) The Hsp90 cochaperone, FKBP51, increases Tau stability and polymerizes microtubules. J. Neurosci. 30, 591–599 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. O'Leary J. C., 3rd, Li Q., Marinec P., Blair L. J., Congdon E. E., Johnson A. G., Jinwal U. K., Koren J., 3rd, Jones J. R., Kraft C., Peters M., Abisambra J. F., Duff K. E., Weeber E. J., Gestwicki J. E., Dickey C. A. (2010) Phenothiazine-mediated rescue of cognition in tau transgenic mice requires neuroprotection and reduced soluble tau burden. Mol. Neurodegener. 5, 45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Dickey C., Kraft C., Jinwal U., Koren J., Johnson A., Anderson L., Lebson L., Lee D., Dickson D., de Silva R., Binder L. I., Morgan D., Lewis J. (2009) Aging analysis reveals slowed tau turnover and enhanced stress response in a mouse model of tauopathy. Am. J. Pathol. 174, 228–238 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Desai A., Verma S., Mitchison T. J., Walczak C. E. (1999) Kin I kinesins are microtubule-destabilizing enzymes. Cell 96, 69–78 [DOI] [PubMed] [Google Scholar]
- 18. Budde P. P., Desai A., Heald R. (2006) Analysis of microtubule polymerization in vitro and during the cell cycle in Xenopus egg extracts. Methods 38, 29–34 [DOI] [PubMed] [Google Scholar]
- 19. Mukrasch M. D., Biernat J., von Bergen M., Griesinger C., Mandelkow E., Zweckstetter M. (2005) Sites of tau important for aggregation populate β-structure and bind to microtubules and polyanions. J. Biol. Chem. 280, 24978–24986 [DOI] [PubMed] [Google Scholar]
- 20. Delaglio F., Grzesiek S., Vuister G. W., Zhu G., Pfeifer J., Bax A. (1995) NMRPipe: a multidimensional spectral processing system based on UNIX pipes. J. Biomol. NMR 6, 277–293 [DOI] [PubMed] [Google Scholar]
- 21. Vranken W. F., Boucher W., Stevens T. J., Fogh R. H., Pajon A., Llinas M., Ulrich E. L., Markley J. L., Ionides J., Laue E. D. (2005) The CCPN data model for NMR spectroscopy: development of a software pipeline. Proteins 59, 687–696 [DOI] [PubMed] [Google Scholar]
- 22. Abisambra J. F., Blair L. J., Hill S. E., Jones J. R., Kraft C., Rogers J., Koren J., 3rd, Jinwal U. K., Lawson L., Johnson A. G., Wilcock D., O'Leary J. C., Jansen-West K., Muschol M., Golde T. E., Weeber E. J., Banko J., Dickey C. A. (2010) Phosphorylation dynamics regulate Hsp27-mediated rescue of neuronal plasticity deficits in tau transgenic mice. J. Neurosci. 30, 15374–15382 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Ricci L., Williams K. P. (2008) Development of fluorescence polarization assays for the molecular chaperone Hsp70 family members: Hsp72 and DnaK. Curr. Chem. Genomics 2, 90–95 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Westerheide S. D., Bosman J. D., Mbadugha B. N., Kawahara T. L., Matsumoto G., Kim S., Gu W., Devlin J. P., Silverman R. B., Morimoto R. I. (2004) Celastrols as inducers of the heat shock response and cytoprotection. J. Biol. Chem. 279, 56053–56060 [DOI] [PubMed] [Google Scholar]
- 25. Jo H., Loison F., Hattori H., Silberstein L. E., Yu H., Luo H. R. (2010) Natural product Celastrol destabilizes tubulin heterodimer and facilitates mitotic cell death triggered by microtubule-targeting anti-cancer drugs. PloS ONE 5, e10318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Wang Y., Martinez-Vicente M., Kruger U., Kaushik S., Wong E., Mandelkow E. M., Cuervo A. M., Mandelkow E. (2009) Tau fragmentation, aggregation and clearance: the dual role of lysosomal processing. Hum. Mol. Genet. 18, 4153–4170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Sarkar M., Kuret J., Lee G. (2008) Two motifs within the tau microtubule-binding domain mediate its association with the hsc70 molecular chaperone. J. Neurosci. Res. 86, 2763–2773 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Dolan P. J., Johnson G. V. (2010) A caspase cleaved form of tau is preferentially degraded through the autophagy pathway. J. Biol. Chem. 285, 21978–21987 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Rudiger S., Buchberger A., Bukau B. (1997) Interaction of Hsp70 chaperones with substrates. Nat. Struc. Biol. 4, 342–349 [DOI] [PubMed] [Google Scholar]
- 30. Zhu X., Zhao X., Burkholder W. F., Gragerov A., Ogata C. M., Gottesman M. E., Hendrickson W. A. (1996) Structural analysis of substrate binding by the molecular chaperone DnaK. Science 272, 1606–1614 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Ballinger C. A., Connell P., Wu Y., Hu Z., Thompson L. J., Yin L. Y., Patterson C. (1999) Identification of CHIP, a novel tetratricopeptide repeat-containing protein that interacts with heat shock proteins and negatively regulates chaperone functions. Mol. Cell. Biol. 19, 4535–4545 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Sahara N., Murayama M., Mizoroki T., Urushitani M., Imai Y., Takahashi R., Murata S., Tanaka K., Takashima A. (2005) In vivo evidence of CHIP up-regulation attenuating tau aggregation. J. Neurochem. 94, 1254–1263 [DOI] [PubMed] [Google Scholar]
- 33. Hammond G. L., Lai Y. K., Markert C. L. (1982) Diverse forms of stress lead to new patterns of gene expression through a common and essential metabolic pathway. Proc. Natl. Acad. Sci. U. S. A. 79, 3485–3488 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Fawcett T. W., Sylvester S. L., Sarge K. D., Morimoto R. I., Holbrook N. J. (1994) Effects of neurohormonal stress and aging on the activation of mammalian heat shock factor 1. J. Biol. Chem. 269, 32272–32278 [PubMed] [Google Scholar]
- 35. Heydari A. R., Wu B., Takahashi R., Strong R., Richardson A. (1993) Expression of heat shock protein 70 is altered by age and diet at the level of transcription. Mol. Cell. Biol. 13, 2909–2918 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Von Bergen M., Friedhoff P., Biernat J., Heberle J., Mandelkow E. M., Mandelkow E. (2000) Assembly of tau protein into Alzheimer paired helical filaments depends on a local sequence motif ((306)VQIVYK(311)) forming beta structure. Proc. Natl. Acad. Sci. U. S. A. 97, 5129–5134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Von Bergen M., Barghorn S., Li L., Marx A., Biernat J., Mandelkow E. M., Mandelkow E. (2001) Mutations of tau protein in frontotemporal dementia promote aggregation of paired helical filaments by enhancing local beta-structure. J. Biol. Chem. 276, 48165–48174 [DOI] [PubMed] [Google Scholar]
- 38. Goldschmidt L., Teng P. K., Riek R., Eisenberg D. (2010) Identifying the amylome, proteins capable of forming amyloid-like fibrils. Proc. Natl. Acad. Sci. U. S. A. 107, 3487–3492 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Song Y., Wu Y. X., Jung G., Tutar Y., Eisenberg E., Greene L. E., Masison D. C. (2005) Role for Hsp70 chaperone in Saccharomyces cerevisiae prion seed replication. Eukaryot. Cell 4, 289–297 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Eisenberg D., Nelson R., Sawaya M. R., Balbirnie M., Sambashivan S., Ivanova M. I., Madsen A. O., Riekel C. (2006) The structural biology of protein aggregation diseases: fundamental questions and some answers. Acc. Chem. Res. 39, 568–575 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Cuervo A. M., Dice J. F. (1996) A receptor for the selective uptake and degradation of proteins by lysosomes. Science 273, 501–503 [DOI] [PubMed] [Google Scholar]
- 42. Dickey C. A., Dunmore J., Lu B., Wang J. W., Lee W. C., Kamal A., Burrows F., Eckman C., Hutton M., Petrucelli L. (2006) HSP induction mediates selective clearance of tau phosphorylated at proline-directed Ser/Thr sites but not KXGS (MARK) sites. FASEB J. 20, 753–755 [DOI] [PubMed] [Google Scholar]
- 43. Dickey C. A., Kamal A., Lundgren K., Klosak N., Bailey R. M., Dunmore J., Ash P., Shoraka S., Zlatkovic J., Eckman C. B., Patterson C., Dickson D. W., Nahman N. S., Jr., Hutton M., Burrows F., Petrucelli L. (2007) The high-affinity HSP90-CHIP complex recognizes and selectively degrades phosphorylated tau client proteins. J. Clin. Invest. 117, 648–658 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Jinwal U. K., Miyata Y., Koren J., 3rd, Jones J. R., Trotter J. H., Chang L., O'Leary J., Morgan D., Lee D. C., Shults C. L., Rousaki A., Weeber E. J., Zuiderweg E. R., Gestwicki J. E., Dickey C. A. (2009) Chemical manipulation of hsp70 ATPase activity regulates tau stability. J. Neurosci. 29, 12079–12088 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Drewes G., Ebneth A., Preuss U., Mandelkow E. M., Mandelkow E. (1997) MARK, a novel family of protein kinases that phosphorylate microtubule-associated proteins and trigger microtubule disruption. Cell 89, 297–308 [DOI] [PubMed] [Google Scholar]
- 46. Li X., Kumar Y., Zempel H., Mandelkow E. M., Biernat J., Mandelkow E. (2011) Novel diffusion barrier for axonal retention of Tau in neurons and its failure in neurodegeneration. EMBO J. 30, 4825–4837 [DOI] [PMC free article] [PubMed] [Google Scholar]