

Abstract
With age, the collagen content of the heart increases, leading to interstitial fibrosis. We have shown that CD44pos fibroblasts derived from aged murine hearts display reduced responsiveness to TGF-β but, paradoxically, have increased collagen expression in vivo and in vitro. We postulated that this phenomenon was due to the defect in mesenchymal stem cell (MSC) differentiation in a setting of elevated circulating insulin levels and production that we observed in aging mice. We discovered that cultured fibroblasts derived from aged but not young cardiac MSCs of nonhematopoietic lineage displayed increased basal and insulin-induced (1 nM) collagen expression (2-fold), accompanied by increased farnesyltransferase (FTase) and Erk activities. In a quest for a possible mechanism, we found that a chronic pathophysiologic insulin concentration (1 nM) caused abnormal fibroblast differentiation of MSCs isolated from young hearts. Fibroblasts derived from these MSCs responded to insulin by elevating collagen expression as seen in untreated aged fibroblast cultures, suggesting a causal link between increased insulin levels and defective MSC responses. Here we report an insulin-dependent pathway that specifically targets collagen type I transcriptional activation leading to a unique mechanism of fibrosis that is TGF-β and inflammation-independent in the aged heart.—Cieslik, K. A., Trial, J., Carlson, S., Taffet, G. E., Entman, M. L. Aberrant differentiation of fibroblast progenitors contributes to fibrosis in the aged murine heart: role of elevated circulating insulin levels.
Keywords: mesenchymal stem cell, collagen
Aging, even in the absence of disease, is associated with diastolic dysfunction (1). The old heart relaxes slowly and additionally has increased passive stiffness. With age, the collagen content of the heart increases in a pattern of diffuse, interstitial fibrosis (2). The pathophysiology of this diffuse fibrosis and the source of the fibroblasts remain uncertain; however, dysregulation of multipotent progenitor cells has been increasingly implicated as one of the common themes of aging (3).
Recently, we reported that endogenous mesenchymal stem cells (MSCs) derived from aged mouse hearts are characterized by reduced expression of Nanog, a transcription factor involved in silencing gene expression and preserving the primitive state, and by the highly up-regulated early preadipocytic marker delta-like 1 homologue (Dlk-1; ref. 4). These cells in vitro could differentiate into various lineages with two major distinctions from young cells: age favored adipocytic differentiation, and the fibroblast maturation into myofibroblasts was defective because aged fibroblasts had a decreased expression of transforming growth factor-β (TGF-β) receptors (4). Since TGF-β induces α-smooth muscle actin (α-SMA) in myofibroblasts, these fibroblasts exhibit a decreased expression of α-SMA, indicative of poor maturation into myofibroblasts. TGF-β is a canonical signal for collagen production (5), and inhibition of TGF-β1 activity in young animals attenuates cardiac (6), renal (7), lung (8), and hepatic fibrosis (9). The present studies arose from observations suggesting that cardiac fibroblasts derived from MSCs in aged animals actually demonstrated increased collagen type I (ColI) expression despite decreased TGF-β responsiveness. In this report, we propose and characterize an TGF-β-independent pathway that leads to fibrosis in the aged heart.
MSCs from the aging heart display preadipocytic lineage choice abnormalities (4) similar to those associated with increased insulin stimulation. We postulated therefore, that there is a functional link between the elevated circulating plasma insulin levels seen in aging (10–12), the increase of cardiac MSC differentiation, and the paradoxical appearance of TGF-β-independent increases in collagen synthesis. In this report, we present evidence that in aging animals insulin production and circulating insulin levels are increased, and this may be linked to a greater number of cardiac CD44posCD45neg ColIpos fibroblasts that arise from endogenous MSC precursors. We hypothesize that differentiation of fibroblast precursors (MSCs) into fibroblasts may be forced by insulin since cultured MSCs derived from the young heart when subjected in vitro to chronic pathophysiologic insulin levels (1 nM) demonstrated changes similar to observed in cardiac MSCs derived from untreated aged mice: their expression of Nanog was reduced, and fibroblasts derived from these MSCs resembled aged cardiac fibroblasts by responding to insulin by up-regulating procollagen α 1 chain type I (proCol1a1) expression. Furthermore, our studies demonstrated that insulin in CD44posCD45neg fibroblasts derived from aged but not young MSCs specifically targets transcriptional activation of both ColI chains (mature collagen type I fibrils comprised of collagen type 1a1 and 1a2 chains in 2:1 ratio) in contrast to other profibrotic genes such as connective tissue growth factor (Ctgf) and collagen α 1 chain type III (Col3a1). Moreover collagen deposition was also elevated in fibroblasts derived from aged MSCs in in vitro studies in basal and insulin-stimulated conditions. The insulin-targeted pathway leading to up-regulated collagen was identified as farnesyltransferase (FTase)/extracellular signal-regulated kinase (Erk)-dependent.
MATERIALS AND METHODS
Reagents
FTI-277 was obtained from EMD Millipore (Billerica, MA, USA) and from Sigma (St. Louis, MO, USA), and insulin and atorvastatin were obtained from Sigma. PD0325901 was purchased from Cayman Chemical Co. (Ann Arbor, MI, USA), and Akt inhibitor II, LY 294002, and GGTI-2147 were purchased from EMD Millipore.
Animals
Male C57BL/6 mice (3 mo old) were obtained from the Center for Comparative Medicine, Baylor College of Medicine. We purchased 13-, 20-, 24-, and 30-mo-old male C57BL/6 mice were purchased from the National Institute of Aging. For insulin and glucose measurements animals were fasted for 6 h. All animals were used in agreement with the Baylor College of Medicine Animal Care and Research Advisory Committee guidelines.
Serum insulin levels
Serum insulin concentration was determined by a commercially available ELISA kit (Alpco, Salem, NH, USA).
Glucose measurements
Mice were deprived of food for 6 h beginning at 8 AM. Animals were awake and restrained during the time when 2 μl of blood from the tail vein was drawn. Fasting blood glucose level was measured on an Accu-chek glucometer, Aviva system (Roche Diagnostics, Basel, Switzerland).
Homeostasis model assessment of insulin resistance (HOMA-IR)
HOMA-IR was performed using HOMA2 Calculator software obtained from the Diabetes Trial Unit (University of Oxford, Oxford, UK; http//www.dtu.ox.ac.uk/homa).
Histology
Tissue was fixed as described previously (13). Sections were incubated with the following antibodies overnight: anti-CD44-PE (731959; Beckman Coulter, Brea, CA, USA), anti-CD44-biotin (731958; Beckman Coulter), anti-CD45-PE (553081; BD Pharmingen, San Diego, CA, USA), anti-procollagen type I (sc-25973; Santa Cruz Biotechnology, Santa Cruz, CA, USA), anti-αSMA-FITC (F3777; Sigma), anti-pErk1/2 (Thr202/Tyr204) (4377; Cell Signaling, Danvers, MA, USA), anti-pAkt (Thr308) (2965; Cell Signaling), and anti-farnesyl epitope (ab12432; Abcam, Cambridge, MA, USA). A secondary reagent was applied if applicable, such as streptavidin-FITC (Beckman Coulter), anti-goat IgG DyLight-488, or anti-rabbit IgG DyLight-594 conjugated (Jackson ImmunoResearch, West Grove, PA, USA).
Cell isolation
The hearts were cut into 1-mm3 pieces, digested with Liberase TH (Roche Diagnostics) and incubated in a 37°C shaking water bath with regular trituration by pipette to obtain a single cell suspension (all nonmyocytes). Afterward, cells were centrifuged at 250 g for 5 min. The cell pellet was washed and then suspended in cell growth medium (13).
Tissue culture
For fibroblast culture, cells were cultured in DMEM/F12 (1:1; Life Technologies, Grand Island, NY, USA) medium supplemented with 10% FBS (Thermo Scientific, Rockford, IL, USA) and 1% antibiotic-antimycotic (Life Technologies).
MSCs were cultured in flasks coated with 0.1% poly-d-lysine (Trevigen, Gaithersburg, MD, USA) in low-glucose DMEM and F-12 nutrient mixed in a 3:1 ratio, supplemented with 20 ng/ml EGF, 10 ng/ml bFGF, 1× B27 and antibiotic/antimycotic (all reagents from Life Technologies), and 10 ng/ml leukemia inhibitory factor (LIF; Sigma) (14). MSC enrichment was performed as described previously (13).
Flow cytometry
Cells were incubated with antibodies to CD45-PE (553081; BD Pharmingen) and CD44-biotin (731958; Beckman Coulter) or the appropriate isotype controls followed by streptavidin-PE-Cy5 (Beckman Coulter). We added calceinAM (50 nM; Life Technologies) to samples to determine cell viability, and we proceeded with analysis of external antigens. Calcein was not added to samples that were analyzed for internal antigens, but instead these samples were fixed in 2% paraformaldehyde with 0.1% saponin for 20 min at 4°C, washed, and then incubated with antibodies to collagen type I (Rockland Immunochemicals, Gilbertsville, PA, USA) or an IgG control. Then the secondary antibody conjugated to AlexaFluor 488 (Invitrogen) was applied, and cells were analyzed on a Cell Lab Quanta SC flow cytometer (Beckman Coulter).
Whole-cell lysate
Cells were harvested with PBS containing a phosphatase and protease inhibitor cocktail (Halt Protease & Phosphatase Single Use Inhibitor Cocktail; Thermo Scientific). Next, cells were centrifuged at 250 g for 5 min. The cell pellet was resuspended in 50 μl of RIPA buffer (Thermo Scientific) or FTase lysis buffer (150 mM NaCl, 5 mM MgCl2, 1 mM NaPO4, 1% Triton X-100, 0.05% SDS, and 50 mM HEPES, pH 7.5; ref. 15) supplemented with inhibitor cocktail and then subsequently frozen, thawed, and centrifuged at 10,000 g for 5 min.
Whole-heart lysate
Hearts were snap-frozen in liquid nitrogen and then ground under liquid nitrogen to a fine powder. The tissue powder was resuspended in FTase lysis buffer supplemented with inhibitor cocktail. Lysates were then frozen, thawed, and centrifuged at 10,000 g for 5 min. Supernatant was portioned into aliquots and stored at −80°C.
Western blot analysis
Western blot analysis was conducted as described previously (4). The following antibodies were used: anti-procollagen type I (SP1.D8, developed by Dr. Heinz Furthmayr, acquired from the Developmental Studies Hybridoma Bank, Iowa City, IA, USA), anti-FTase subunit α (FNTA; ab109738; Abcam), anti-FTase subunit β (FNTB; ab74206; Abcam), anti-pErk1/2 (Thr202/Tyr204) (4377; Cell Signaling), anti-Erk1/2 (4695; Cell Signaling), anti-pAkt (Thr308) (2965, Cell Signaling), and anti-Akt (9272; Cell Signaling).
FTase activity assay
Cells were serum starved for 48 h; then the medium was supplemented with 1 nM insulin, and cells were incubated for 1 h. Subsequently, cells were lysed in FTase lysis buffer. We incubated 30 μg of protein in the reaction mixture containing 50 mM Tris-HCl (pH 7.5), 5 mM MgCl2, 5 mM DTT, 0.2% octyl-β-d-glucopyranoside, 10 μM dansyl-GCVLS (EMD Millipore), and 10 μM farnesylpyrophosphate (Enzo, New York, NY, USA) for 30 min. The activity of FTase was determined by measuring the value of fluorescence (excitation at 340 nm and emission at 505 nm; ref. 15) using a BioTek plate reader and Gen 5 Data Analysis Software (BioTek, Winooski, VT, USA).
Geranylgeranyltransferase (GGTase) activity assay
GGTase activity was determined as above except for the composition of reaction buffer, which was as follows: 50 mM Tris-HCl (pH 7.5), 5 mM MgCl2, 50 μM ZnCl2, 20 mM KCl, 1 mM DTT, 0.2% octyl-β-d-glucopyranoside, 10 μM dansyl-GCVLL (Biosynthesis, Lewisville, TX, USA), and 10 μM geranylgeranylpyrophosphate (Enzo). GGTase activity was measured using excitation at 360 nm and emission at 460 nm (15).
Picrosirius red staining
Cells at equal density were seeded in slide chambers. Quiescent cells were cultured in medium with 2% FBS supplemented with insulin or inhibitors as indicated and cultured for 4 d. Then cells were fixed in 2% paraformaldehyde and incubated in 0.1% picrosirius red for 1 h. The staining solution was eluted, and cells were washed with acidified water. Cells were photographed, and picrosirius red was removed by 0.1 N NaOH (200 μl/chamber). Optical density was measured at 540 nm. These reading were normalized to cell density, which was evaluated by 0.1% crystal violet staining. Optical density of eluted crystal violet was measured at 585 nm.
Quantitative PCR (qPCR)
Total RNA was isolated from whole hearts with TRIzol reagent (Life Technologies), purified by RNeasy kit (Qiagen, Valencia, CA, USA) and transcribed to cDNA by Verso cDNA Synthesis kit (Thermo Scientific). qPCR was performed on CFX96 Real Time PCR Detection System (Bio-Rad, Hercules, CA, USA) using Sso Advanced SYBR Green (Bio-Rad) and specific primers. Gene expression was measured by the ΔΔCq method to calculate the amount of target mRNA normalized to an endogenous reference (Hprt). Primers were designed using Beacon Designer software (version 7.0; Premier Biosoft, Palo Alto, CA, USA) or web-based Primer 3 (version 0.4.0) developed by Steve Rozen and Helen Skaletsky. All primers were evaluated according to MIQE guidelines (minimum information for publication of quantitative real-time PCR experiments) (16–19). PCR efficiency (E) and correlation efficiency (r2) for standard curve for each primer pair are reported below.
Hypoxanthine phosphoribosyltransferase (Hprt), NM_013556.2, E = 93%, r2 = 0.987, forward primer: 5′-GCCCCAAAATGGTTAAGGTT-3′, reverse primer: 5′-TTGCGCTCATCTTAGGCTTT-3′; Col1a1, NM_007742, E = 97%, r2 = 0.993,forward primer: 5′-GTATGCTTGATCTGTATCTG-3′, reverse primer: 5′-CGACTCCTACATCTTCTG-3′; Nanog, NM_028016.2, E = 111%, r2 = 0.984, forward primer: 5′-ATTTGGAGGTGAATTTGG-3′, reverse primer: 5′-TGTTGCGTAAGTCTCATA-3′; MMP8 NM_008611.4, E = 107%, r2 = 0.94, forward primer: 5′-AAACGGAGTGAGAGGTGTGG-3′, reverse primer: 5′-TCTGCCTGGGAACTTATTGG-3′; MMP9, NM_013556.2, E = 99%, r2 = 0.978, forward primer: 5′-TGAATCAGCTGGCTTTTGTG-3′, reverse primer: 5′-GTGGATAGCTCGGTGGTGTT-3′; Col3a1, NM_ NM_009930.2, E = 106%, r2 = 0.977, forward primer: 5′-GATGAGGAGCCACTAGACTG-3′, reverse primer: 5′-GCCATCAGGAAGCACAGG-3′; Ctgf, NM_ 010217.2, E = 90%, r2 = 0.997, forward primer: 5′-ATCCCACCAAAGTGAGAACG-3′, reverse primer: 5′-TAATTTCCCTCCCCGGTTAC-3′.
PCR array
Genes involved in fibrosis and remodeling were examined by Mouse Fibrosis PCRArray (Qiagen). RNA was isolated from cardiac cultured fibroblasts following the manufacturer's protocol. cDNA was prepared from 180 μg total RNA by using a RT2 PCR array first strand kit (Qiagen). Data were analyzed by web-based software (Qiagen) using the comparative cycle threshold method normalized to expression of housekeeping genes. All samples passed the required quality control.
Insulin binding and immunofluorescent staining
Fibroblasts were cultured in serum-free medium for 24 h and then were treated with 100 nM FITC-labeled insulin (Sigma) or nonspecific FITC-labeled IgG (BD Pharmingen) for 30 min. Cells next were washed and fixed in methanol. Then cells were incubated with anti-insulin receptor subunit α antibody (IRα; ab7842; Abcam) for 2 h. Cells were incubated with an anti-rabbit DyLight-594 antibody (Jackson Immunoresearch) for 45 min.
Statistical analysis
Results are presented as means ± se. A comparison between 2 groups was analyzed by an unpaired Student's t test or Mann-Whitney test, and comparisons among 3 or 4 groups were done using 1-way ANOVA. Differences were considered statistically significant at values of P < 0.05.
Densitometry
Densitometry analysis was performed using ImageJ 1.46x software (U.S. National Institutes of Health, Bethesda, MD, USA).
RESULTS
Increased number of CD44posCD45negcells in the aged myocardium
We demonstrated previously in our myocardial infarction model that injury to the young heart mobilizes endogenous MSCs and causes an increase in number of CD44posCD45neg cells (progenitor cells not of hematopoietic origin). These cells differentiate into fibroblasts (ColIpos) and mature into myofibroblasts (ColIposα-SMApos) (13). Overall, the process enables the repair mechanisms necessary to mend the injured heart by promoting scar formation (by collagen-depositing cells) and scar contraction (via α-SMA expression). In the young uninjured heart the total number of CD44posCD45neg cells is quite low (< 2%; Fig. 1A, right panel), and these cells are rarely identified in heart sections (Fig. 1B, left panel). Interestingly, CD44posCD45neg cells were much more easily identified in the aged uninjured heart (Fig. 1B, right panel), and their number was increased to ∼6% of total viable nonmyocyte cells (Fig. 1A, right panel), suggesting augmented progenitor differentiation. The percentage of total CD44posCD45neg cells differentiating into ColI expressing fibroblasts was also increased with age, from 26% (3-mo-old animals) to 93% (30-mo-old animals) (Fig. 2A, B). These CD44posCD45neg proCol1a1pos cells did not express α-SMA (Fig. 2C), suggesting that in the aged heart TGF-β-signaling response of CD44posCD45neg cell differentiation was defective.
Figure 1.

Increased number of CD44posCD45neg cells in the aged myocardium. A) Cells isolated from hearts were analyzed by flow cytometry. Left panel: representative histogram of viable CD44posCD45neg nonmyocytes. Right panel: analysis of viable CD44posCD45neg nonmyocytes isolated from mice of various ages, n = 5, 4, 5, 5. B) Immunofluorescence staining of heart paraffin sections from young (3-mo-old) and aged (30-mo-old) mice revealed an increased number of CD44posCD45neg cells in the aged heart. Arrows point to CD44posCD45neg nonmyocytes, n = 3, 3. Scale bars = 20 μm; n = number of animals used per experiment in appropriate age (starting from the youngest age). *P < 0.05; $P < 0.001.
Figure 2.

Increased differentiation of CD44posCD45neg nonmyocytes into proCol1a1pos-expressing fibroblasts. A) Flow cytometry analysis of viable CD44posCD45neg nonmyocytes isolated from mice of various ages, n = 5, 4, 5, 5. B) Immunofluorescence staining of heart paraffin sections from young (3-mo-old) and aged (30-mo-old) mice revealed increased numbers of CD44posproCol1a1pos cells in the aged heart. Arrows indicate double-stained cells, n = 3, 3. C) Fibroblasts in the aged hearts do not mature into myofibroblasts because they are negative for the myofibroblast marker α-SMA, n = 3, 3. Scale bars = 20 μm; n = number of animals used per experiment in appropriate age (starting from the youngest age). $P < 0.001.
Evidence of elevated circulating insulin levels in the aged mouse
Insulin is a critical factor effecting differentiation of precursor cells (20, 21). Elevated circulating insulin levels in aging have been reported in humans and rodents, but not in 30-mo-old C57BL/6 mice, so we tested young and old mice (3 and 30 mo old, respectively). We found that the fasting circulating insulin level (Supplemental Fig. S1A, left panel) as well as HOMA-IR (Supplemental Fig. S1A, right panel) were elevated up to 3-fold in the aged mice, whereas the fasting blood glucose levels were not significantly different than in young controls (Supplemental Fig. S1B, middle panel). As we analyzed the pancreas by H&E staining, we observed an increased size of pancreatic islets in 30-mo-old animals (Supplemental Fig. S1B). The islet area compared to total pancreatic area was increased by ∼3-fold in the aged animals, as shown in Supplemental Fig. S1B, right panel. The expression of insulin and glucagon was evaluated to assess the islets' function. In islets from 30-mo-old animals there was a clear increase of insulin production from β-cells when compared to islets from young animals (Supplemental Fig. S1C); however, the function of α-cells (producing glucagon) seemed to be unaltered by aging (Supplemental Fig. S1C). Although the elevated HOMA-IR index suggests insulin resistance, cardiac fibroblasts derived from young and aged MSCs were fully responsive to insulin as evaluated by FITC-labeled insulin binding to its receptor and/or insulin uptake. We found no significant difference in insulin binding and expression of IRα between cells derived from young and aged MSCs (Supplemental Fig. S2), suggesting that aging does not promote insulin resistance in cardiac CD44posCD45neg fibroblasts.
Role of insulin in aberrant differentiation of CD44posCD45neg MSCs derived from the aged heart
Augmented differentiation of CD44posCD45neg cells in the aged heart (Fig. 1A) coincides with elevated circulating insulin levels as presented in Supplemental Fig. S1A. To study the potential mechanism by which insulin affects this cell population, MSCs were isolated from both young and aged hearts. In in vitro conditions, in the presence of serum, MSCs give rise to CD44posCD45neg fibroblasts. The basal proCol1a1 expression was up-regulated in fibroblasts isolated from 30-mo-old hearts (compared with fibroblasts derived from 3-mo-old hearts), and 24 h treatment with pathophysiological insulin concentration (1 nM) increased that expression even further. Fibroblasts isolated from young animals did not respond to insulin stimulation by increasing the expression of proCol1a1 (Fig. 3A). However, when cardiac MSCs derived from young hearts were subjected to chronic pathophysiologic insulin levels for 4 wk and after that differentiated into fibroblasts, only then did fibroblasts derived from young MSCs respond to insulin by up-regulating proCol1a1 expression (Fig. 3B).
Figure 3.

Insulin-dependent differentiation of cardiac MSCs. A) Quiescent cells were stimulated with 1 nM insulin for 24 h in serum-free medium, n = 3, 3. B) Fibroblasts derived from young MSCs subjected to chronic insulin exposure (1 nM) resemble fibroblasts derived from aged hearts. After 4 wk of chronic insulin treatment MSCs were differentiated into fibroblasts. C) Nanog expression level was reduced by chronic insulin stimulation. MSCs derived from young mice were cultured for 4 wk in medium supplemented with 1 nM insulin, then mRNA was isolated and qPCR was performed with 30 ng of reverse transcribed cDNA. Data were normalized to Hprt transcript level, n = 4. Right panel: Ponceau staining. C, control; INS, insulin (1 nM). *P < 0.05.
Our working hypothesis presumed that excessive insulin signaling forces aberrant MSC differentiation and allows escape from the control of genes assuring a primitive state. We tested that hypothesis by evaluating Nanog expression (a multipotency marker) in MSCs derived from young hearts cultured in medium supplemented with pathophysiologic insulin concentration (as above). We found that Nanog expression was indeed reduced by ∼70% (Fig. 3C) in cells subjected to chronic pathophysiologic insulin concentration. We have previously reported a similar degree of Nanog reduction in MSCs derived from aged MSCs when compared to young controls (4).
Insulin and age-dependent farnesylation
Mitogenic but nonmetabolic insulin action is associated with Ras-Erk pathway that is controlled by FTase activity (22). FTase and GGTase transfer the prenyl chain formed during cholesterol biosynthesis onto a variety of proteins (23). Both enzymes share the same regulatory subunit α (FNTA; ref. 23), but their substrate specificity is different and depends on the catalytic subunit β (FNTB). It has been demonstrated that insulin stimulates the activity of FTase (24).
The activity of GGTase was unaffected by aging or insulin either in fibroblasts or in the whole-heart lysate (Fig. 4A). However, cardiac fibroblasts derived from MSCs isolated from 30-mo-old mice had ∼2-fold greater basal activity of FTase when compared to fibroblasts isolated from young controls (Fig. 4B, left panel). FTase activity was even further increased (∼50%) when fibroblasts isolated from aged MSCs were stimulated with a pathophysiologic concentration of insulin (1 nM). Fibroblasts derived from young controls did not exhibit significant up-regulation of FTase activity in response to insulin. The pattern of FTase activity in fibroblasts derived from young and aged MSCs mirrored the changes of proCol1a1 expression (Fig. 3A). Interestingly, FTase activity in the whole heart lysate was not affected by age (Fig. 4B, right panel). These results suggest that insulin in the heart may activate FTase mostly in fibroblasts and their precursors.
Figure 4.

Prenyl transferases activity in fibroblast and whole-heart lysates. A) GGTase activity was unaffected by age or insulin stimulation, n = 3, 3. B) FTase activity in fibroblasts derived from aged hearts (30 mo old) was elevated by insulin stimulation (1 nM). Insulin was introduced to quiescent cells 1 h prior to the assay (left panel, n=5, 5). FTase activity in whole heart lysates was unaffected by age (right panel, n=5, 3, 3). C) Expression of FNTA is up-regulated in quiescent fibroblasts derived only from 30-mo-old animals (top panel, n=3, 3), and expression of FNTB is elevated by 24 h insulin treatment (1 nM), n = 3, 3. These are representative immunoblots. Ponceau staining (right panel) demonstrates equal protein loading. D) qPCR analysis of quiescent fibroblasts derived from young and aged hearts stimulated with 1 nM insulin for 24 h. Left panel: FNTA mRNA level (n=3, 4). Right panel: FNTB transcript level (n=3, 4). n = number of animals used per experiment in appropriate age (starting from the youngest age). *P < 0.05.
Basal expression of FNTA was up-regulated in cultured fibroblasts isolated from aged MSCs (Fig. 4C, top Western blot). In addition, insulin (1 nM) treatment increased the expression of FTase catalytic subunit FNTB in fibroblasts derived from aged MSCs (Fig. 4C, bottom Western blot). As we previously reported, fibroblasts isolated from aged hearts are characterized by altered expression of housekeeping genes, and therefore Ponceau staining was included to assure equal protein loading (Fig. 4C, right panel). qPCR analysis demonstrated that neither age nor 1 nM insulin treatment changed the FNTA mRNA level in cultured fibroblasts (Fig. 4D, left panel) whereas expression of the FNTB transcript was up-regulated by insulin in fibroblasts derived from aged animals (Fig. 4D, right panel). These results indicate that the level of the FNTA protein may be regulated by post-translational modifications.
FTase inhibition attenuates proCol1a1 expression in fibroblasts derived from aged hearts
As expected, insulin-dependent stimulation of proCol1a1 expression was largely reduced by FTase inhibition (Fig. 5A) in fibroblasts derived from aged MSCs. Drugs, such as statins, that reduce cholesterol biosynthesis inhibit prenylation reactions by inhibiting synthesis of farnesyl and geranylgeranyl pyrophosphates, the isoprenyl donors. It has been demonstrated that statin treatment reduces fibrosis in several animal models (25, 26). Interestingly, when quiescent cells were cultured in medium supplemented with the FTase inhibitor FTI-277 or with atorvastatin and incubated for 24 h, basal proCol1a1 expression was reduced by ∼50% (Fig. 5B), which suggests that both basal and insulin-dependent augmentation in proCol1a1 expression as observed in aged cells is FTase and farnesylation dependent. Therefore, heart sections were examined for presence of N-acetyl-S-farnesyl-L-cysteine. We found that the farnesyl moiety is expressed in young and aged hearts, but in the aged heart farnesyl expression localizes with the CD44 marker (Fig. 5C) suggesting an active prenyl chain synthesis pathway in CD44pos cells.
Figure 5.

Effect of aging and insulin on proCol1a1 expression in cardiac fibroblasts derived from 3- and 30-mo-old mice. A) FTase inhibition (FTI) reduces insulin-dependent proCol1a1 expression. Cells derived from 30-mo-old hearts were pretreated with FTI 45 min prior to insulin treatment, n = 3. B) Augmented basal expression of proCol1a1 in aged cardiac fibroblasts was reduced by FTase inhibition via FTI or statin but not by GGTase inhibition, n = 3. GGTI, GGTI-2147 (1 μM, GGTase inhibitor); FTI, FTI-277 (100 nM); statin, atorvastatin (10 μM); C, control; INS, insulin (1 nM). *P < 0.05. C) Heart sections from young (3-mo-old) and aged (30-mo-old) mice were stained for CD44 and N-acetyl-S-farnesyl-l-cysteine (farnesyl epitope). Arrows indicate double-positive cells. Scale bars = 20 μm, n = 3, 3. n = number of animals used per independent experiment in appropriate age (starting from the youngest age).
Insulin-dependent pathway targeting Col1a1 transcription relies on Erk phosphorylation
It has been shown that the insulin-dependent mitogenic effect is mediated by the FTase-Ras-Erk pathway, while its metabolic effect is mediated by the PI3K-Akt signaling (22). Therefore, we examined the expression of phosphorylated Erk (at Thr202 and Tyr204 residues) in young and aged heart lysates (Fig. 6A) and heart sections (Fig. 6B). We found no significant difference in the expression level of activated Erk between young and aged hearts (Fig. 6A), but the phosphorylated Erkpos cells also stained for CD44 marker in the aged heart sections (Fig. 6B), suggesting a possible involvement of this pathway in CD44pos cell-dependent fibrosis. Further testing revealed that insulin stimulation resulted in Erk1/2 phosphorylation and thereby its activation (Fig. 6C) and fibroblasts derived from aged MSCs pretreated with Erk inhibitor PD0325901 showed a decrease in the insulin-dependent Col1a1 mRNA expression (Fig. 6D).
Figure 6.

Insulin-dependent Erk phosphorylation leads to Col1a1 expression in fibroblasts derived from aged MSCs. A) Phosphorylated Erk (Thr202/Tyr204) and Erk expression in heart lysates from 3-, 14-, and 30-mo-old mice. B) Immunofluorescence staining of heart sections colocalized pErk and proCol1a1 markers in the aged (30) heart section. Arrows point at double-positive cells. n = 5, 4. C) Representative Western blot analysis showing insulin-dependent Erk phosphorylation in fibroblasts derived from aged MSCs. C, control, INS, insulin, PD, pretreatment with 1 μM PD0325901. D) Erk inhibition reduces insulin-dependent Col1a1 expression in fibroblasts derived from aged MSCs. Results were normalized to Hprt level, n = 3. n = number of animals used per experiment in appropriate age (starting from the youngest age). Scale bars = 20 μm.
Because the PI3K-Akt pathway may contribute to the activation of the FTase-Ras-Erk pathway (27), we applied the same approach as described above: phosphorylated Akt (pAkt) was not significantly up-regulated in heart lysates from middle aged (14-mo-old) or aged (30-mo-old) mice, but there was a trend toward increased Akt phosphorylation in the middle aged and aged hearts (Supplemental Fig. S3A). Staining of the heart sections uncovered an increased number of phosphorylated Akt in the aged heart (compared to the young heart). A few of these pAktpos cells in the aged heart were also positive for proCol1a1 marker (Supplemental Fig. S3B). Despite this, however, analysis of Col1a1 expression in fibroblasts (derived from 30-mo-old hearts) incubated in the presence of PI3K inhibitor (LY 294002) or Akt inhibitor (Akt inhibitor II) was not statistically significant (Supplemental Fig. S3C), suggesting that this pathway was not rate-limiting.
Insulin stimulation specifically targets ColI genes in fibroblasts derived from aged MSCs
We used PCR array and qPCR to further characterize MSC-derived fibroblasts from aged hearts. These cells in quiescent condition expressed up-regulated levels of several profibrotic markers when compared to untreated fibroblasts derived from MSCs isolated from the young heart. Specifically expression levels of Col1a1 and Col1a2 were up-regulated (Fig. 7A). Interestingly insulin treatment further increased expression of both collagen chains in fibroblasts derived from aged MSCs.
Figure 7.
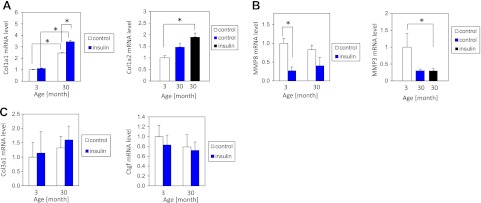
Insulin-activated pathway targets specifically ColI in fibroblasts derived from aged MSCs. A) qPCR analysis of Col1a1 level (left panel), n = 4, 5, and PCR array analysis of Col1a2 transcript (right panel) in quiescent fibroblasts derived from young and aged MSCs stimulated with 1 nM insulin for 24 h, n = 3, 4, 5. B) qPCR analysis of MMP8 (left panel) and PCR array analysis of MMP3 (right panel) n = 4, 5. C) qPCR analysis of Col3a1 (left panel) and Ctgf (right panel), n = 4, 5. n = number of animals used per treatment of the appropriate age group (starting from the youngest age).
We observed an intriguing expression pattern of enzymes involved in collagen degradation, such as metalloproteinases (MMPs). In fibroblasts derived from aged MSCs, the expression level of MMP8 and MMP3 (Fig. 7B) were down-regulated whereas expression of MMP9 and MMP13 (Supplemental Fig. S4A, B) were elevated by ∼50% in aged fibroblasts, but the changes were not statistically significant. Expression of other genes associated with TGF-β-dependent fibrosis such as Col3a1 and Ctgf was not affected by age or insulin treatment (Fig. 7C).
The presented results prompted the conclusion that aging-related changes specifically target genes encoding ColI chains and promote collagen production. Because of the complex pattern of MMP expression, we evaluated collagen deposition by picrosirius red staining in cells cultured for 4 d (Fig. 8). The experiment was started by seeding an equal amount of cells in 4-well slide chambers and the final readings were normalized to cell density at the end of the experiment. We found that cardiac fibroblasts derived from aged MSCs deposited a significantly higher amount of collagens than fibroblasts derived from young MSCs and that insulin treatment increased collagen deposition in wells seeded with aged fibroblasts. FTase and Erk inhibition reduced the amount of insulin-dependent collagen deposition (Fig. 8), providing additional proof that FTase and Erk participate in insulin-activated collagen production.
Figure 8.

Insulin stimulation increases collagen deposition. A) Representative image of picrosirius red staining of cells stimulated by 1 nM insulin for 4 d. Cells were pretreated with inhibitors as indicated before insulin (INS) was applied. B) Picrosirus red stain was eluted with 0.1 N NaOH, and absolute optical density (measured at 540 nm) was measured and normalized to cell density (evaluated by crystal violet staining). PD, Erk inhibitor (1 μM PD0325901); FTI, FTase inhibitor (100 nM FTI-277).
DISCUSSION
Progressive fibrosis has been described in various organs as a feature of aging (28–32), but the mechanism responsible for increased collagen content in the aging heart is unclear. We recently proposed that immune-inflammatory dysregulation contributes to development of interstitial cardiac fibrosis by promoting infiltration of CD45pos (hematopoietic cells from the bone marrow) into the heart tissue that then differentiate into CD45pos ColIpos fibroblasts (33). We found that by 30 mo of age the amount of CD45posColIpos cells in murine hearts doubles. More recently, we found another cell population, CD44posCD45neg cardiac fibroblasts (arising from resident MSCs), which is characterized by excessive basal (pro)Col1a1 synthesis in vivo and in vitro in aging. The number of these cells increases by 3-fold in 30-mo-old animals compared to young animals. This suggests that both CD44posCD45neg and CD45pos (33) cell populations can contribute to cardiac fibrosis.
In a recent paper (4), we have shown that MSCs from aging hearts responded poorly to TGF-β1 and fibroblasts arising from them made very little α-SMA. Others have noted that α-SMA deficiency is associated with enhanced renal (34) and pulmonary (35) fibrosis. While the mechanism leading to increased ColI expression and organ fibrosis concomitant with reduced α-SMA expression is not known, it suggested the presence of a common TGF-β-independent pathway. We have demonstrated that MSCs isolated from the aged heart have an altered lineage choice; in their quiescent condition they are in a preadipocytic state, and even a low concentration of insulin drives them in vitro toward the adipocytic lineage (4) similar to age-dependent changes seen by others (36). Concomitantly, aged animals have increased circulating insulin levels (Supplemental Fig. S1A) that may contribute to this altered differentiation of cardiac MSCs. We decided to evaluate the role of insulin in age-related fibrosis because of that altered differentiation pattern and the following reported facts: insulin up-regulates collagen production (37, 38); circulating plasma insulin levels are increased in aging (10, 11); insulin can drive stem cell differentiation (21); hyperinsulinemia has been associated with cardiac fibrosis (39); and hyperinsulinemia increases FTase activity (40) in vivo.
Abnormal stem cell differentiation with aging has been reported in satellite cells, murine hematopoietic stem cells and bone marrow MSCs (3, 36). Our results indicate that insulin affects not only fibroblasts but also cardiac MSCs. After treatment with chronic, pathophysiologic insulin levels, MSCs derived from young animals displayed reduced expression of Nanog, a transcription factor that silences genes involved in differentiation (41 and Fig. 3C), similar to what we observed in MSCs derived from aged hearts (4), suggesting that insulin may contribute to aberrant MSC differentiation. Interestingly, it has been demonstrated that Erk signaling negatively controls Nanog expression (42), suggesting that in a scenario of reduced TGF-β signaling (TGF-β promotes Nanog transcription (43) and in the presence of signals that increase Erk activation (such as insulin), Nanog expression is restrained and MSCs are drifting toward differentiation.
Defects from impaired MSCs are passed on into progeny cells; CD44posCD45neg cardiac fibroblasts derived from young MSCs responded to insulin by elevated proCol1a1 expression (Fig. 3B), which resemble fibroblasts derived from aged hearts (Fig. 3A).
In a search for an additional mechanism behind CD44posCD45neg-dependent fibrosis, we found that expression of enzymes that control collagen turnover and degradation (such as MMPs) were variably regulated by age or insulin (Fig. 7B and Supplemental Fig. S4). Although not all MMPs are equally important for collagen degradation, it has been demonstrated that MMP8 and MMP13 are “true” collagenases, while MMP3 plays a role in activating other MMPs (44), and MMP9 (which is a gelatinase) has been implicated in age-dependent fibrosis (45). In our study, the expression of MMP3, MMP8 and MMP13 was down-regulated either by age or insulin treatment and only the MMP9 level was not reduced by insulin. It has been demonstrated that MMP9 is involved in increasing collagen content in the left ventricle via post-translational regulation of Col1a1 expression by TGF-β-induced Ctgf and periostin expressions (45). In our study, which involves a unique CD44posCD45neg cell population (as opposed to the entire left ventricle), Ctgf levels were unchanged by age or insulin (Fig. 7C). The mRNA expression of lysyl oxidase (Lox), an enzyme involved in collagen cross-linking, was unchanged by age or insulin (data not shown), suggesting that studied CD44posCD45neg-dependent fibrosis is not due to increased cross-linking.
The role of mechanical stimulation in proliferation and differentiation of fibroblasts has been also implicated. To perceive mechanical signals from the extracellular matrix, fibroblasts utilize integrins that transmit signals through focal adhesion kinase (FAK) (46). As we previously reported, we found in fibroblasts derived from aged MSCs that FAK signaling was defective and their responses to stress in collagen lattices were impaired (4). However, others have shown that FAK phosphorylation can be bypassed, leading to direct activation of Erk pathway (47). This may well contribute to the mechanism of fibrosis.
To our knowledge this is the first report linking aging-related fibrosis with pathological MSC differentiation and insulin-dependent FTase-Erk signaling in fibroblasts. The described signaling pathway in CD44posCD45neg fibroblasts is very exclusive (Figs. 3A, 5, 6, and 7A, B); an additional examination of matrix and adhesion proteins demonstrated that only 2 genes (Col1a1a and Col1a2) were up-regulated by insulin treatment. This suggests a highly focused common transcriptionally responsive element that is preferentially targeted by the insulin-FTase-Erk signaling.
Acknowledgments
This work was supported by U.S. National Institutes of Health grants R01HL089792 (M.L.E.) and T32HL07816 (S.C.), a Medallion Foundation grant (J.T.), and the Hankamer Foundation.
The authors are grateful to Dr. Heinrich Taegtmeyer for critical reading of this manuscript. The authors also thank Dorellyn B. Lee and Hao Zhang for excellent technical assistance. The authors declare no conflicts of interest.
This article includes supplemental data. Please visit http://www.fasebj.org to obtain this information.
- α-SMA
- α-smooth muscle actin
- Col1a1
- collagen α 1 chain type I
- Col1a2
- collagen α 2 chain type II
- Col3a1
- collagen α 1 chain type III
- ColI
- collagen type I
- Ctgf
- connective tissue growth factor
- Erk1/2
- extracellular signal-regulated kinase 1/2
- FNTA
- farnesyltransferase subunit α
- FNTB
- farnesyltransferase subunit β
- FTase
- farnesyltransferase
- FTI
- FTase inhibitor
- GGTase
- geranylgeranyltransferase
- HOMA-IR
- homeostasis model assessment of insulin resistance
- Hprt
- hypoxanthine phosphoribosyltransferase
- IRα
- insulin receptor subunit α
- Lox
- lysyl oxidase
- MMP
- metalloproteinase
- MSC
- mesenchymal stem cell
- TGF-β
- transforming growth factor-β
REFERENCES
- 1. Lakatta E. G., Levy D. (2003) Arterial and cardiac aging: major shareholders in cardiovascular disease enterprises: part II: the aging heart in health: links to heart disease. Circulation 107, 346–354 [DOI] [PubMed] [Google Scholar]
- 2. Song Y., Yao Q., Zhu J., Luo B., Liang S. (1999) Age-related variation in the interstitial tissues of the cardiac conduction system: an autopsy study of 230 Han Chinese. Forensic Sci. Int. 104, 133–142 [DOI] [PubMed] [Google Scholar]
- 3. Jones D. L., Rando T. A. (2011) Emerging models and paradigms for stem cell ageing. Nat. Cell Biol. 13, 506–512 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Cieslik K. A., Trial J., Entman M. L. (2011) Defective myofibroblast formation from mesenchymal stem cells in the aging murine heart rescue by activation of the AMPK pathway. Am. J. Pathol. 179, 1792–1806 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Chen S. J., Yuan W., Mori Y., Levenson A., Trojanowska M., Varga J. (1999) Stimulation of type I collagen transcription in human skin fibroblasts by TGF-beta: involvement of Smad 3. J. Invest. Dermatol. 112, 49–57 [DOI] [PubMed] [Google Scholar]
- 6. Teekakirikul P., Eminaga S., Toka O., Alcalai R., Wang L., Wakimoto H., Nayor M., Konno T., Gorham J. M., Wolf C. M., Kim J. B., Schmitt J. P., Molkentin J. D., Norris R. A., Tager A. M., Hoffman S. R., Markwald R. R., Seidman C. E., Seidman J. G. (2010) Cardiac fibrosis in mice with hypertrophic cardiomyopathy is mediated by non-myocyte proliferation and requires Tgf-beta. J. Clin. Invest. 120, 3520–3529 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Fukasawa H., Yamamoto T., Suzuki H., Togawa A., Ohashi N., Fujigaki Y., Uchida C., Aoki M., Hosono M., Kitagawa M., Hishida A. (2004) Treatment with anti-TGF-beta antibody ameliorates chronic progressive nephritis by inhibiting Smad/TGF-beta signaling. Kidney Int. 65, 63–74 [DOI] [PubMed] [Google Scholar]
- 8. Kang H. R., Cho S. J., Lee C. G., Homer R. J., Elias J. A. (2007) Transforming growth factor (TGF)-beta1 stimulates pulmonary fibrosis and inflammation via a Bax-dependent, bid-activated pathway that involves matrix metalloproteinase-12. J. Biol. Chem. 282, 7723–7732 [DOI] [PubMed] [Google Scholar]
- 9. Nakamura T., Sakata R., Ueno T., Sata M., Ueno H. (2000) Inhibition of transforming growth factor beta prevents progression of liver fibrosis and enhances hepatocyte regeneration in dimethylnitrosamine-treated rats. Hepatology 32, 247–255 [DOI] [PubMed] [Google Scholar]
- 10. Barzilai N., Banerjee S., Hawkins M., Chen W., Rossetti L. (1998) Caloric restriction reverses hepatic insulin resistance in aging rats by decreasing visceral fat. J. Clin. Invest. 101, 1353–1361 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Fink R. I., Kolterman O. G., Griffin J., Olefsky J. M. (1983) Mechanisms of insulin resistance in aging. J. Clin. Invest. 71, 1523–1535 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Fraze E., Chiou Y. A., Chen Y. D., Reaven G. M. (1987) Age-related changes in postprandial plasma glucose, insulin, and free fatty acid concentrations in nondiabetic individuals. J. Am. Geriatr. Soc. 35, 224–228 [DOI] [PubMed] [Google Scholar]
- 13. Carlson S., Trial J., Soeller C., Entman M. L. (2011) Cardiac mesenchymal stem cells contribute to scar formation after myocardial infarction. Cardiovasc. Res. 91, 99–107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Shih D. T., Lee D. C., Chen S. C., Tsai R. Y., Huang C. T., Tsai C. C., Shen E. Y., Chiu W. T. (2005) Isolation and characterization of neurogenic mesenchymal stem cells in human scalp tissue. Stem Cells 23, 1012–1020 [DOI] [PubMed] [Google Scholar]
- 15. Zhou X. P., Wu K. Y., Liang B., Fu X. Q., Luo Z. G. (2008) TrkB-mediated activation of geranylgeranyltransferase I promotes dendritic morphogenesis. Proc. Natl. Acad. Sci. U. S. A. 105, 17181–17186 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Zuker M. (2003) Mfold web server for nucleic acid folding and hybridization prediction. Nucleic Acids Res. 31, 3406–3415 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. SantaLucia J., Jr. (1998) A unified view of polymer, dumbbell, and oligonucleotide DNA nearest-neighbor thermodynamics. Proc. Natl. Acad. Sci. U. S. A. 95, 1460–1465 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Peyret N. (2000) Melt curve analysis for each primer pair resulted in a single peak indicating a single PCR product. Ph.D. dissertation, Wayne State University [Google Scholar]
- 19. Bustin S. A., Benes V., Garson J. A., Hellemans J., Huggett J., Kubista M., Mueller R., Nolan T., Pfaffl M. W., Shipley G. L., Vandesompele J., Wittwer C. T. (2009) The MIQE guidelines: minimum information for publication of quantitative real-time PCR experiments. Clin. Chem. 55, 611–622 [DOI] [PubMed] [Google Scholar]
- 20. Fulzele K., Riddle R. C., DiGirolamo D. J., Cao X., Wan C., Chen D., Faugere M. C., Aja S., Hussain M. A., Bruning J. C., Clemens T. L. (2010) Insulin receptor signaling in osteoblasts regulates postnatal bone acquisition and body composition. Cell 142, 309–319 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Klemm D. J., Leitner J. W., Watson P., Nesterova A., Reusch J. E., Goalstone M. L., Draznin B. (2001) Insulin-induced adipocyte differentiation. Activation of CREB rescues adipogenesis from the arrest caused by inhibition of prenylation. J. Biol. Chem. 276, 28430–28435 [DOI] [PubMed] [Google Scholar]
- 22. Goalstone M. L., Draznin B. (2004) Insulin and the ras-dependent signaling pathway. In Diabetes Mellitus: A Fundamental and Clinical Text (LeRoith D., Taylor S. I., Olefsky J. M., eds) pp. 422–428, Lippincott Williams & Wilkins, Philadelphia [Google Scholar]
- 23. Seabra M. C., Reiss Y., Casey P. J., Brown M. S., Goldstein J. L. (1991) Protein farnesyltransferase and geranylgeranyltransferase share a common alpha subunit. Cell 65, 429–434 [DOI] [PubMed] [Google Scholar]
- 24. Goalstone M., Carel K., Leitner J. W., Draznin B. (1997) Insulin stimulates the phosphorylation and activity of farnesyltransferase via the Ras-mitogen-activated protein kinase pathway. Endocrinology 138, 5119–5124 [DOI] [PubMed] [Google Scholar]
- 25. Tang X. L., Sanganalmath S. K., Sato H., Bi Q., Hunt G., Vincent R. J., Peng Y., Shirk G., Dawn B., Bolli R. (2011) Atorvastatin therapy during the peri-infarct period attenuates left ventricular dysfunction and remodeling after myocardial infarction. PLoS ONE 6, e25320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Watts K. L., Sampson E. M., Schultz G. S., Spiteri M. A. (2005) Simvastatin inhibits growth factor expression and modulates profibrogenic markers in lung fibroblasts. Am. J. Respir. Cell Mol. Biol. 32, 290–300 [DOI] [PubMed] [Google Scholar]
- 27. Grammer T. C., Blenis J. (1997) Evidence for MEK-independent pathways regulating the prolonged activation of the ERK-MAP kinases. Oncogene 14, 1635–1642 [DOI] [PubMed] [Google Scholar]
- 28. Gagliano N., Arosio B., Santambrogio D., Balestrieri M. R., Padoani G., Tagliabue J., Masson S., Vergani C., Annoni G. (2000) Age-dependent expression of fibrosis-related genes and collagen deposition in rat kidney cortex. J. Gerontol. A Biol. Sci. Med. Sci. 55, B365–372 [DOI] [PubMed] [Google Scholar]
- 29. Gagliano N., Grizzi F., Annoni G. (2007) Mechanisms of aging and liver functions. Dig. Dis. 25, 118–123 [DOI] [PubMed] [Google Scholar]
- 30. Hinton D. E., Williams W. L. (1968) Hepatic fibrosis associated with aging in four stocks of mice. J. Gerontol. 23, 205–211 [DOI] [PubMed] [Google Scholar]
- 31. Glaser J., Stienecker K. (2000) Pancreas and aging: a study using ultrasonography. Gerontology 46, 93–96 [DOI] [PubMed] [Google Scholar]
- 32. Gazoti Debessa C. R., Mesiano Maifrino L. B., Rodrigues de Souza R. (2001) Age related changes of the collagen network of the human heart. Mech. Ageing Dev. 122, 1049–1058 [DOI] [PubMed] [Google Scholar]
- 33. Cieslik K. A., Taffet G. E., Carlson S., Hermosillo J., Trial J., Entman M. L. (2011) Immune-inflammatory dysregulation modulates the incidence of progressive fibrosis and diastolic stiffness in the aging heart. J. Mol. Cell. Cardiol. 50, 248–256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Takeji M., Moriyama T., Oseto S., Kawada N., Hori M., Imai E., Miwa T. (2006) Smooth muscle alpha-actin deficiency in myofibroblasts leads to enhanced renal tissue fibrosis. J. Biol. Chem. 281, 40193–40200 [DOI] [PubMed] [Google Scholar]
- 35. Liu T., Warburton R. R., Guevara O. E., Hill N. S., Fanburg B. L., Gaestel M., Kayyali U. S. (2007) Lack of MK2 inhibits myofibroblast formation and exacerbates pulmonary fibrosis. Am. J. Respir. Cell Mol. Biol. 37, 507–517 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Moerman E. J., Teng K., Lipschitz D. A., Lecka-Czernik B. (2004) Aging activates adipogenic and suppresses osteogenic programs in mesenchymal marrow stroma/stem cells: the role of PPAR-gamma2 transcription factor and TGF-beta/BMP signaling pathways. Aging Cell 3, 379–389 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Kjellstrom T., Malmquist J. (1984) Insulin effects on collagen and protein production in cultured human skin fibroblasts from diabetic and non-diabetic subjects. Horm. Metab. Res. 16, 168–171 [DOI] [PubMed] [Google Scholar]
- 38. Krupsky M., Fine A., Kuang P. P., Berk J. L., Goldstein R. H. (1996) Regulation of type I collagen production by insulin and transforming growth factor-beta in human lung fibroblasts. Connect. Tissue Res. 34, 53–62 [DOI] [PubMed] [Google Scholar]
- 39. Mizushige K., Yao L., Noma T., Kiyomoto H., Yu Y., Hosomi N., Ohmori K., Matsuo H. (2000) Alteration in left ventricular diastolic filling and accumulation of myocardial collagen at insulin-resistant prediabetic stage of a type II diabetic rat model. Circulation 101, 899–907 [DOI] [PubMed] [Google Scholar]
- 40. Draznin B., Miles P., Kruszynska Y., Olefsky J., Friedman J., Golovchenko I., Stjernholm R., Wall K., Reitman M., Accili D., Cooksey R., McClain D., Goalstone M. (2000) Effects of insulin on prenylation as a mechanism of potentially detrimental influence of hyperinsulinemia. Endocrinology 141, 1310–1316 [DOI] [PubMed] [Google Scholar]
- 41. Liang J., Wan M., Zhang Y., Gu P., Xin H., Jung S. Y., Qin J., Wong J., Cooney A. J., Liu D., Songyang Z. (2008) Nanog and Oct4 associate with unique transcriptional repression complexes in embryonic stem cells. Nat. Cell Biol. 10, 731–739 [DOI] [PubMed] [Google Scholar]
- 42. Burdon T., Smith A., Savatier P. (2002) Signalling, cell cycle and pluripotency in embryonic stem cells. Trends Cell Biol. 12, 432–438 [DOI] [PubMed] [Google Scholar]
- 43. Xu R. H., Sampsell-Barron T. L., Gu F., Root S., Peck R. M., Pan G., Yu J., Antosiewicz-Bourget J., Tian S., Stewart R., Thomson J. A. (2008) NANOG is a direct target of TGFbeta/activin-mediated SMAD signaling in human ESCs. Cell Stem Cell 3, 196–206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Spinale F. G. (2007) Myocardial matrix remodeling and the matrix metalloproteinases: influence on cardiac form and function. Physiol. Rev. 87, 1285–1342 [DOI] [PubMed] [Google Scholar]
- 45. Chiao Y. A., Ramirez T. A., Zamilpa R., Okoronkwo S. M., Dai Q., Zhang J., Jin Y. F., Lindsey M. L. (2012) Matrix metalloproteinase-9 deletion attenuates myocardial fibrosis and diastolic dysfunction in ageing mice. Cardiovasc. Res. 96, 444–455 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Hinz B. (2007) Formation and function of the myofibroblast during tissue repair. J. Invest. Dermatol. 127, 526–537 [DOI] [PubMed] [Google Scholar]
- 47. Lin T. H., Aplin A. E., Shen Y., Chen Q., Schaller M., Romer L., Aukhil I., Juliano R. L. (1997) Integrin-mediated activation of MAP kinase is independent of FAK: evidence for dual integrin signaling pathways in fibroblasts. J. Cell Biol. 136, 1385–1395 [DOI] [PMC free article] [PubMed] [Google Scholar]