

Abstract
Spinal cord injury (SCI) results not only in motor deficits, but produces, in many patients, excruciating chronic pain (SCI pain). We have previously shown, in a rodent model, that SCI causes suppression of activity in the GABAergic nucleus, the zona incerta (ZI), and concomitant increased activity in one of its main targets, the posterior nucleus of the thalamus (PO); the increased PO activity is correlated with the maintenance and expression of hyperalgesia after SCI. Here, we test the hypothesis that SCI causes a similar pathological increase in other thalamic nuclei regulated by the ZI, specifically the mediodorsal thalamus (MD), which is involved in the emotional-affective aspects of pain. We recorded single and multiunit activity from MD of either anesthetized or awake rats, and compared data from rats with SCI with data from sham-operated controls (anesthetized experiments) or with data from the same animals prelesion (awake experiments). Consistent with our hypothesis, MD neurons from rats with SCI show significant increases in spontaneous firing rates and in the magnitude and duration of responses to noxious stimuli. In a subset of anesthetized animals, similar changes in activity of MD neurons were produced by pharmacologically inactivating ZI in naive rats, suggesting that the changes in the MD after SCI are related to suppressed inhibition from the ZI. These data support our hypothesis that SCI pain results, at least in part, from a loss of inhibition to thalamic nuclei associated with both the sensory-discriminative and emotional-affective components of pain.
Introduction
Every year there are some 12,000 incidents of spinal cord injury (SCI) in the United States, and up to 80% of patients with SCI develop chronic pain (Siddall et al., 2003). Chronic pain after spinal cord injury (SCI pain) often presents as wide-spread allodynia, hyperalgesia, and spontaneous pain (Bowsher, 1996; Garcia-Larrea et al., 2002; Finnerup et al., 2003) that develop weeks or even months after the initial injury (Tasker et al., 1992; Falci et al., 2002). SCI pain has no cure and, in most patients, is resistant to conventional pharmacological treatment (Baastrup and Finnerup, 2008). The delayed expression of SCI pain, the diffuse localization of painful symptoms, and the presence of pain below the denervated spinal segment strongly suggest the occurrence of maladaptive plasticity in not only the spinal cord, but also in supraspinal structures (Masri and Keller, 2011). Though the cause of SCI pain remains unknown, it has long been hypothesized that chronic pain results from abnormally suppressed inhibition in the thalamus (Head and Holmes, 1911; Boivie, 2005).
To this end, we recently demonstrated in an animal model of SCI that pain results in a significant decrease of neuronal activity in the zona incerta (ZI) (Masri et al., 2009), which provides potent inhibition to select thalamic nuclei and that the loss of ZI activity correlates with a pathological increase of neuronal activity in the posterior thalamus (PO), a somatosensory thalamic nucleus critical for processing nociceptive information (Masri et al., 2009). Further, we demonstrated that these maladaptive changes are causally related to the development of chronic pain after SCI (Masri et al., 2009; Davoody et al., 2011; Lucas et al., 2011).
Pain is not only a sensory-discriminative experience, but also an unpleasant emotional experience (Merskey and Bogduk, 1994). We therefore predicted that after SCI, maladaptive changes in the ZI would correlate with pathological activity in both somatosensory and associative thalamic nuclei, which are important for nociceptive processing. These thalamic nuclei include the mediodorsal thalamus (MD). The MD, like the PO, receives dense GABAergic inputs from the ZI (Barthó et al., 2002; Erickson et al., 2004) and is heavily connected to cortical areas involved in processing the affective aspects of pain (Price and Slotnick, 1983; Cornwall and Phillipson, 1988; Groenewegen, 1988). In humans, chronic pain is associated with abnormal activity in the MD (Rinaldi et al., 1991). Similarly, lesions or inactivation of the MD reduces both thermal and mechanical hyperalgesia in a rat model of peripheral neuropathic pain (Saadé et al., 2007). These findings led to our hypothesis that pain after SCI is associated with abnormal increases in spontaneous and evoked activity of MD neurons.
Materials and Methods
All procedures were conducted according to Animal Welfare Act regulations and Public Health Service guidelines. Strict aseptic surgical procedures were used, according to the guidelines of the International Association for the Study of Pain, and approved by the University of Maryland School of Medicine Animal Care and Use Committee. A total of 31 adult male Sprague Dawley rats weighing 250–300 g were used in this study.
Behavioral confirmation of hyperalgesia.
All animals were tested on 3 consecutive days before the spinal lesion surgery, at day three after surgery, at day seven after surgery, and at weekly intervals thereafter. To minimize the animals' anxiety, they were habituated for 2 weeks before behavioral testing and were trained to stand upright with their forepaws on the experimenter's hand as described by Ren (1999). Calibrated von Frey filaments (Stoelting) were applied in ascending order to the hind paw. We applied the filaments to the dorsal surface of the paws based on studies demonstrating that the dorsal approach more reliably and consistently detects threshold changes (Ren, 1999). Mechanical withdrawal threshold was defined as the force at which the animal withdrew the paw to 3–5 stimuli delivered.
Spinal lesions.
Sterile surgery was performed as previously described (Masri et al., 2009; Lucas et al., 2011). Briefly, a laminectomy to expose the spinal cord immediately rostral to C7 was performed and a quartz-insulated platinum electrode (5 μm tip) was targeted to the spinothalamic tract (STT). Electrolytic lesions were made by passing DC (10 μA for 10 s, repeated 4×) in two locations, 0.4 mm apart. Sham surgery was performed without laminectomy. After completion of surgery, the incision sites were approximated and sutured in layers.
In vivo recordings.
At least 14 d after spinal lesion surgery, rats were anesthetized with urethane and prepared for extracellular recordings as previously described (Trageser et al., 2006; Masri et al., 2009). We selected urethane because it has no, or negligible, effects on glutamatergic and GABAergic transmission and therefore produces only minimal disruption of signal transmission (Sceniak and Maciver, 2006). For anesthetized experiments in sham-operated and spinal-lesioned animals (n = 15 male animals), rats were initially anesthetized with ketamine/xylazine (100/8 mg/kg, i.p.). We used intravenous infusion of a 6% urethane solution, delivered through a jugular catheter, to control the level of anesthetic, which was kept at level III-2 described by Friedberg et al. (1999). Anesthetic levels were monitored by continually recording electrocorticographs (ECoGs) obtained from a pair of screws implanted in the skull.
Extracellular recordings were obtained from the MD contralateral to the spinal lesion, through quartz-insulated tungsten electrodes (2–4 MΩ). We recorded from well isolated units, digitized (40 kHz) the waveforms through a Plexon data-acquisition system, and sorted units off-line with Plexon's off-line sorter using dual thresholds and principal component analyses. We generated autocorrelograms with Neuroexplorer software (Plexon) to confirm that we obtained recordings from single units. We exported time stamps of well isolated units and stimulus triggers to Matlab (MathWorks) for analyses using custom-written algorithms. Because we found wind-up-like responses in MD neurons (see Figs. 1C, 2A), we analyzed responses to each stimulus separately using modified peristimulus time histograms (PSTHs, 100 ms bins) and defined significant stimulus-evoked responses as PSTH bins that significantly exceeded (99% confidence interval) spontaneous activity levels.
Figure 1.
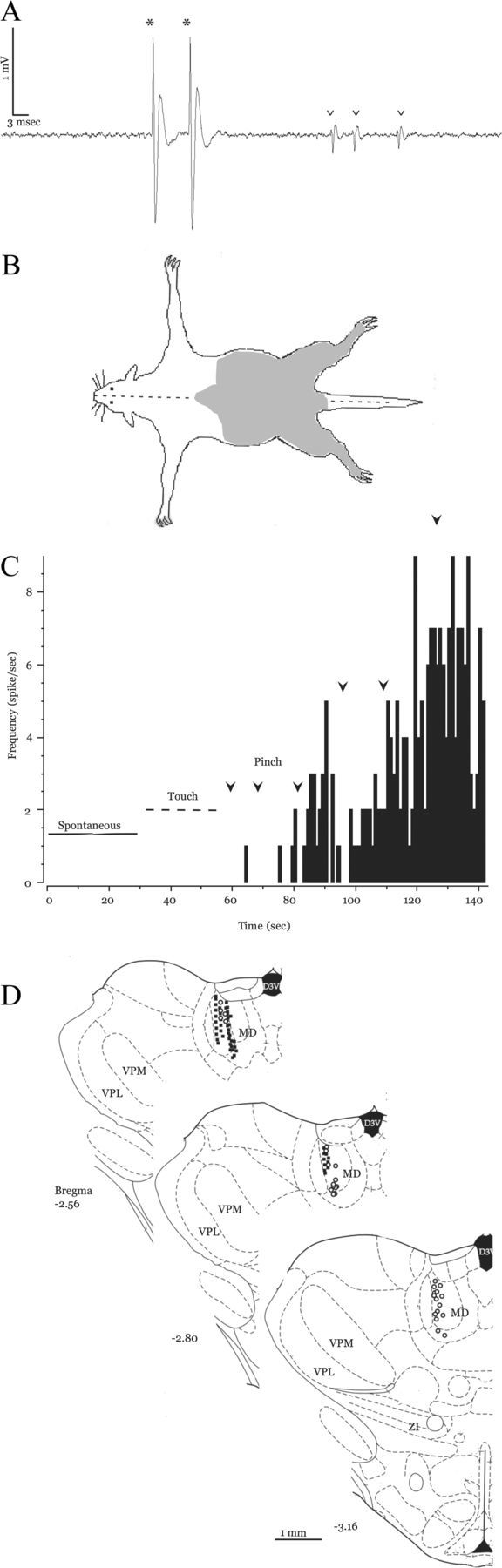
Neuronal activity in the MD of control animals. A, Representative extracellular recording from MD showing two single units: a typical MD neuron (asterisks; large biphasic wave, two positive peaks, >2 ms duration) and an atypical MD neuron (arrowheads). B, Receptive field map of an MD neuron from a sham-operated animal. Shaded area represents the regions from which responses were evoked. C, Rate histogram computed from the activity of an MD neuron from a sham-operated animal (1 s bins). The neuron has no spontaneous activity (solid bar), nor does it respond to innocuous touch (dashed bar). It displays robust and augmenting responses to repeated pinches (1 s pinch, onset designated by arrowheads). D, Recording locations of MD neurons: 52 from sham-operated (solid squares) and 58 from SCI animals (open circles, adapted from Paxinos and Watson, 1998).
Figure 2.

Neural activity in MD is enhanced in animals with SCI. A, Box-and-whisker plots show that MD neurons from spinal-lesioned animals (n = 58) have significantly higher spontaneous firing rates than those from sham-operated animals (n = 52; p = 0.011, MWU). B, Rate histogram computed from activity of an MD neuron in a spinal-lesioned animal (1 s bins). The neuron fires action potentials spontaneously (solid bar) but does not significantly increase its firing rate in response to innocuous stimulation (dashed bar, compare with Fig. 1C). Repeated pinches evoke augmenting responses (1 s pinch, onset designated by arrowheads). C, Response magnitude and duration to 1 s pinch were significantly larger in neurons from spinal-lesioned animals (n = 58, 226 pinches) than in those of sham-operated animals (n = 52, 204 pinches). D, Box plots show the magnitude of response of MD neurons in sham-operated (n = 52) and spinal-lesioned (n = 58) animals to each of a succession of 1 s pinches. Spinal-lesioned animals show significantly larger response magnitudes to pinches 2–4 compared with sham-operated animals.
We defined bursts of action potentials as clusters of at least three spikes with interspike intervals of ≤5 ms in which the first spike in the burst has a preceding interspike interval of ≥100 ms based on previous reports of bursting in thalamic nuclei (Rinaldi et al., 1991; Guido et al., 1995; Sherman, 1996). Extracellularly recorded bursts that meet these criteria are thought to correspond to calcium-dependent spike bursts characterized in thalamic neurons through intracellular recordings (Jahnsen and Llinas, 1984).
Sensory stimuli.
We recorded responses to noxious pinches (1 s application, 200–225 g) using electronic calibrated forceps (IITC Life Science). Stimuli were applied to both hind paws (≥2 stimuli on each paw) because MD neurons have large, bilateral receptive fields (Dostrovsky and Guilbaud, 1990; Hsu et al., 2000; Wang et al., 2003) and because hyperalgesia occurs in both hind paws in this animal model of SCI pain (Masri et al., 2009; Lucas et al., 2011). To minimize peripheral sensitization and avoid tissue inflammation, stimuli were applied to left and right hind paws in an alternating pattern, with at least 20 s between applications on each paw.
ZI inactivation.
In a different group of naive rats (n = 9), a microdialysis probe (CMA Microdialysis) was implanted into the ventral portion of zona incerta (ZIv; stereotaxic coordinates: anterior, −3.9 mm; lateral, 2.8 mm; deep, 7.1 mm (Paxinos and Watson, 1998). The microdialysis probe was used to administer 0.1 ml of muscimol or saline (2.5 μl/min) to ZIv while we recorded single-unit activity in the MD of urethane-anesthetized animals, as described above. To determine the effect of drug administration on neurons in MD, we compared mean firing rate during 30 s of baseline activity to mean firing rate during 30 s of activity after muscimol application.
In an additional group of naive rats (n = 6), we implanted a microdialysis probe in either the right ZIv (n = 4) or the right substantia nigra (n = 2). Several days after the rats completely recovered from surgery, we tested the effects of reversible inactivation on their responses to mechanical stimuli. To this end, we applied either muscimol (200 μm) or saline through these probes. Agents (50 μl volume) were applied with a micropump at a rate of 2.5 μl/min for 20 min. We then tested the effects of drug infusion on mechanical withdrawal thresholds, as described earlier.
Recordings from behaving animals.
In seven animals, a longitudinal incision was made along the midline of the skull to expose bregma and lambda. Bone overlaying the MD was removed and custom-made five-channel moveable (1.4–4 MΩ, 40–80 μm, insulated except at tip) recording electrodes were slowly (50 μm/5 min) lowered into the MD. Electrode locations were based on stereotaxic coordinates (A, −2.8 mm; L, 0.9 mm; D, 4.5–5.5 mm (Paxinos and Watson, 1998), based on data from our anesthetized recordings (see Fig. 1D), and based on previously published anatomical and electrophysiological studies (Dostrovsky and Guilbaud, 1990; Ray and Price, 1992) of areas in the MD that respond to noxious peripheral stimulation and project to limbic cortical areas. Electrodes were secured in place using six bone screws and acrylic resin and were protected by permanent custom-made shields. Before electrophysiological recording experiments began, animals were treated with oral antibiotics for 2 weeks after implant to reduce risk of infection.
During that time, animals were handled daily to become comfortable with experimenters and were trained to rest quietly in experimenters' hands to minimize stress and ease headstage connection. To minimize anxiety and stress at the start of recording experiments, animals were acclimated to the recording room for at least 30 min before behavioral testing and recording. Recordings were obtained during ≥3 d before spinal lesions, and weekly after the lesions. Rats were fitted with a wireless headstage (Triangle BioSystems International) and allowed to move freely in their home cage for 2–3 min, during which spontaneous activity of MD neurons was recorded. Next, the animals were placed on an elevated platform [0.25 m (width) × 0.6 m (length) × 0.5 m (height)] with a mesh floor, where they freely moved for 2 min before testing began to determine pain thresholds. During this unrestricted behavior, spontaneous activity of MD neurons was recorded.
We recorded wideband activity from MD with the wireless headstage (Triangle BioSystems International), and digitized the waveforms as described above. The wideband waveforms were filtered with a low-cut Butterworth filter (4-pole, 250 Hz) to obtain multiunit activity (MUA). The MUAs were classified offline with Plexon's off-line sorter, using thresholds set at least 6 SDs higher than mean background activity.
To test for mechanical withdrawal thresholds, confirm development of hyperalgesia after spinal lesion, and produce stimulus-evoked neuronal responses, von Frey filaments were applied to the plantar surface of the rats' hind paw through holes in the mesh floor of the elevated platform. Each filament (range, 26–180 g; intensities that in normal rats span from innocuous to noxious forces) was applied 5× when the animal was calm and still. Manual triggers were used to store a digital timestamp of the stimuli. We recorded frequency of hind paw withdrawal to ascending forces and threshold was defined as the force at which an animal withdrew to at least three of five stimuli. After a complete series of von Frey filaments was applied to the hind paw, a noxious thermal stimulus (42–45°C, 2 mm2 surface area, <5 s application) was presented 5× (10 s intervals) to evoke a robust behavioral response from the animals and to confirm that the neurons respond to noxious stimuli.
Because we and others have reported that neurons in MD have large, bilateral receptive fields (see Fig. 5C) (Dostrovsky and Guilbaud, 1990; Wang et al., 2003), because our model of SCI pain presents with significant bilateral hyperalgesia below the site of the spinal lesion (Masri et al., 2009), and to help prevent peripheral sensitization, we applied stimuli to alternating hind paws. We computed mean firing rate during spontaneous activity in both the home cage and on the elevated platform and in response to each level of stimulation delivered.
Figure 5.

ZI inactivation in naive rats produces hyperalgesia. A, Bars represent means (and SEM) of hind limb withdrawal thresholds in rats before and after drug infusion through microdialysis cannulas implanted over the right ZI or into the substantia nigra (pars reticulata). Infusion of muscimol resulted in a large and significant decrease in withdrawal of both hind limbs. In contrast, infusion of saline had no discernible effect. Similarly, muscimol injection into substantia nigra had no effect on withdrawal thresholds.
Histology.
All animals were deeply anesthetized and transcardially perfused with buffered saline followed by 4% buffered paraformaldehyde. We obtained coronal brain and spinal cord sections (80 μm thick) and Nissl-stained them. The sections were examined with a transmission microscope to identify stimulation sites, lesions sites, and microdialysis probe and recording implant locations. For electrophysiological experiments, all recordings sites were reconstructed and only neurons with confirmed placement within the desired thalamic nucleus were included in analysis.
Data analysis.
In all experiments performed, we determined the appropriate sample size by performing a power analysis using α = 0.05 and power = 0.85. In all experiments, p < 0.05 was considered significant. We performed statistical analyses in Stata (StataCorp), and assessed, in recordings of individual units and MUAs, changes occurring in sensory-evoked activity using the Mann–Whitney U (MWU) test. Differences in neuronal activity between groups were analyzed using the MWU test. Comparisons of proportions of bursting neurons were made using the Pearson's χ2 test. p < 0.05 was considered significant.
For each group, extreme outliers were identified using the fourth-spread method and removed (Hoaglin et al., 1986).
Results
Classification of MD neurons
MD neurons were particularly sensitive to level of anesthesia, such that when animals were deeper than level III-2 described by Friedberg et al. (1999), we recorded very few spontaneously active neurons in MDs and virtually no neurons with peripherally evoked responses. To address this issue and to be sure we were making appropriate comparisons between sham-operated and spinal-lesioned animals, we carefully monitored level of anesthesia in all experiments using several physiological metrics, including corneal reflex, breathing rate, pinch withdrawal as in (Friedberg et al., 1999), and ECoG activity (described in Materials and Methods).
We identified neurons in MDs using two main criteria: their location, based on stereotaxic coordinates (Paxinos and Watson, 1998) and confirmed histologically post hoc, and their responses to noxious peripheral stimulation. We focused our recordings on neurons near the border between the central and lateral subdivisions of MDs (Fig. 1D) because these regions have a high density of thalamocortical neurons projecting to the anterior cingulate and insular cortices (Krettek and Price, 1977; Groenewegen, 1988; Jones, 2007), regions that are important for nociceptive processing and pain perception (Treede et al., 1999, 2000; Price, 2000; Johansen et al., 2001; Wang et al., 2003, 2004; Jasmin et al., 2004; Shyu and Vogt, 2009; Zhang et al., 2011). In primates, this region of the MD also receives dense inputs from several other regions in the frontal cortex (Erickson and Lewis, 2004). In addition to its relationship with the frontal cortex, the primate lateral MD receives inputs from periaquedactal gray and from the ZI (Erickson et al., 2004). Thus, the lateral MD in both primates and rodents has extensive connections with pain-related brain regions.
Most MD neurons had distinctive action potential waveforms. Figure 1A shows an extracellular recording from a representative MD neuron (asterisks) that has a large biphasic action potential with two positive peaks and a >2 ms duration. These waveform indices were characteristic of most (>85%) of the neurons recorded in MD. This likely reflects the rather homogeneous neuronal population of this nucleus, which contains stellate and fusiform excitatory neurons and, like some other thalamic nuclei in rodents, contains virtually no GABAergic interneurons (Ottersen and Storm-Mathisen, 1984; Kuroda et al., 1992). However, we occasionally recorded neurons with action potentials that did not have two positive peaks and that had a <1.5 ms duration (Fig. 1A, carets). Because these were rare (<15%) and because these cells were generally not responsive to peripheral stimulation, we excluded them from further analyses.
As reported by others, we found that neurons in MD are responsive to peripheral stimulation and that these responses occur at relatively long latencies [sham-operated: n = 52; interquartile range (IQR), 0.2–1.7 s; SCI: n = 58; IQR, 0.2–1.4 s; calculated using 100-ms-long bins], possibly reflecting their role in the affective—as opposed to sensory-discriminatory—component of pain perception (Dostrovsky and Guilbaud, 1990; Wang et al., 2003; Zhang et al., 2011). Note also that the latencies reported here (and in previous studies) are affected by the relatively large temporal bins used in the analyses (100 ms in the present study). The receptive fields of these neurons were always bilateral and often included limbs and parts of the torso (Fig. 1B). In some cases, stimulation to the entire trunk and all four limbs evoked responses from these cells. Though we did not systematically study areas above the neck, some MD neurons responded to parts of the head, both forepaws, and the upper torso. We focused our recordings on neurons that responded to stimulation of the hind paws because in this model of SCI pain bilateral thermal and mechanical hyperalgesia develops in dermatomes below the spinal lesion (Masri et al., 2009) and because in both anesthetized and awake experiments it was more feasible to control placement, duration, and intensity of stimulation to the hind paws than to areas of the torso or the forepaws.
Consistent with previous reports (Dostrovsky and Guilbaud, 1990; Wang et al., 2003; Zhang et al., 2011), we found that MD neurons responded almost exclusively to noxious peripheral stimulation: in preliminary experiments in naive rats, <13% (4 of 31 cells) responded weakly to innocuous stimulation. We therefore did not quantify responses to innocuous touch in subsequent experiments. Figure 1C shows a rate histogram of typical spontaneous and evoked firing of MD neurons in control (sham-operated) animals. This neuron had no spontaneous activity and was not activated by touch with a wooden probe (innocuous peripheral stimulation) on the hind paws or lower trunk. Noxious pinch to the hind paws elicited responses, and repeated pinches caused an increase in its firing rate (Fig. 1C, compare firing rate after arrowheads). As a group, MD neurons in sham-operated animals had similar response properties to those described above, and only frank noxious stimulation evoked increased firing rates. Individual neurons showed both increased firing rate and increased duration of response to later pinches compared with initial pinches, though the peripheral stimulation was always identical (1 s pinch, 200–225 g; Fig. 1C, compare firing rate and duration after arrowheads; quantified below).
Enhanced neuronal activity in MD
To test whether SCI affected neurons in MD, we compared spontaneous and evoked activity of MD neurons from sham-operated and SCI rats. Figure 2A shows a rate histogram computed from activity of an MD neuron in a spinal-lesioned animal with confirmed hyperalgesia. Note the presence of spontaneous firing rate in this neuron, compared with that in sham-operated animals (compare Fig. 1C “spontaneous” with Fig. 2A “spontaneous”). MD neurons from animals with SCI (58 neurons from 6 animals) had, on average, a 2.5-fold increase in spontaneous firing rates, compared with those from sham-operated animals (52 neurons from 3 animals) (sham-operated: mean, 1.9 ± 0.26 Hz; median, 1.3 Hz; SCI: mean, 4.8 ± 0.75 Hz; median, 2.3 Hz; p = 0.011, MWU; Fig. 2B).
Similar to MD neurons in sham-operated animals, MD neurons in spinal-lesioned rats did not respond to innocuous peripheral stimulation, but they did respond robustly to noxious pinch of the hind paws. As described above for neurons from sham-operated animals, neurons in animals with SCI responded to repeated noxious stimulation with increasing firing rate and increasing duration of response (Fig. 2B, compare firing rate and duration after arrowheads; quantified below).
To test whether animals with SCI had increased responses to peripheral stimulation, we compared magnitude and duration of neuronal activation in response to 1 s pinches between sham-operated and spinal-lesioned animals. Total magnitude of response (combined responses to pinches 1–4) was nearly sevenfold higher in SCI animals than in sham-operated animals (sham-operated: mean, 4.3 ± 1.2 spikes/stimulus; median, 1.7 spikes/stimulus; SCI: mean, 29.0 ± 8.0 spikes/stimulus; median, 3.5 spikes/stimulus; p < 0.0001, MWU; Fig. 2C). Not only did MD neurons from SCI animals have significantly higher firing rates in response to noxious stimulation, but they also had elevated firing rates for significantly longer periods of time. Figure 2D shows total duration of elevated firing rates after noxious stimulation (pinches 1–4 grouped: sham-operated: mean, 0.72 ± 0.15 s, median, 0.30 s; SCI: mean, 2.6 ± 0.76 s; median, 0.50 s; p = 0.0018, MWU).
Because we found augmenting responses in both sham-operated and spinal-lesioned animals, such that later pinches produced significantly elevated firing rates for prolonged periods, we thought it insufficient to simply compare magnitude and duration of responses for all pinches grouped together. To address whether these wind-up-like responses were enhanced in animals with SCI, we separately analyzed responses evoked by individual pinches. Response magnitude evoked by the first pinch was larger in spinal-lesioned animals when compared with that of sham-operated animals, and this difference approached significance (sham-operated: mean, 2.4 ± 0.35 spikes/stimulus; median, 1.5 spikes/stimulus; SCI: mean, 33.3 ± 21.3 spikes/stimulus, median, 2.29 spikes/stimulus; p = 0.068, MWU; Fig. 2E). However, spinal-lesioned animals had significantly larger evoked responses to each of pinches 2–4 (Pinch 2: sham-operated: mean, 2.52 ± 0.36 spikes/stimulus; median, 1.84 spikes/stimulus; SCI: mean, 18.3 ± 5.88 spikes/stimulus; median, 3.62 spikes/stimulus; p = 0.018, MWU. Pinch 3: sham-operated: mean, 3.01 ± 0.51 spikes/stimulus; median, 1.64 spikes/stimulus; SCI: mean, 48.6 ± 22.1 spikes/stimulus; median, 4.14 spikes/stimulus; p = 0.009, MWU. Pinch 4: sham-operated: mean, 9.48 ± 4.65 spikes/stimulus; median, 2.07 spikes/stimulus; SCI: mean, 14.4 ± 4.00 spikes/stimulus; median, 4.17 spikes/stimulus; p = 0.031, MWU; Fig. 2D). A similar augmenting pattern was seen with response duration (data not shown).
Burst properties of MD neurons
The function and prevalence of bursts in thalamic cells during conditions of pain have been debated. Some have suggested that chronic pain conditions are associated with increased incidence of bursting activity in somatosensory thalamic nuclei in both humans and rats (Lenz et al., 1989; Vierck et al., 1990; Weng et al., 2003; Hains et al., 2005, 2006; Lee et al., 2005; Iwata et al., 2011). However, this hypothesis is not universally accepted (Canavero and Bonicalzi, 2007; Dostrovsky, 2007). Bursting activity has been reported in MD of humans with chronic pain (Rinaldi et al., 1991), though these authors acknowledge that without similar recordings from healthy individuals, it is impossible to determine the significance of this finding. To our knowledge, nothing has been reported about bursting activity in MD of healthy individuals, and there are no published reports about bursts in MD of normal rats.
We therefore compared the prevalence of bursting cells between sham-operated and spinal-lesioned animals. Because the prevalence of bursts varies as animals transition through stages of wakefulness (McCarley et al., 1983; Ramcharan et al., 2000; Swadlow and Gusev, 2001; Massaux and Edeline, 2003), we carefully controlled and monitored the level of anesthetic for each animal, as described above.
We based our burst criteria on previously published analyses of bursts in somatosensory thalamic neurons (Guido et al., 1995; Lu et al., 1995; Sherman, 1996; Masri et al., 2009) and on burst properties reported from MD neurons in humans with chronic deafferentation pain (Rinaldi et al., 1991). We defined bursts as clusters of at least three action potentials with interspike intervals of ≤5 ms in which the first spike in the burst has a preceding interspike interval of ≥100 ms during spontaneous activity.
In sham-operated animals, 28% (15 of 52) of neurons exhibited burst firing during spontaneous activity. This was not different from the proportion of cells that exhibited burst firing in spinal-lesioned animals (33%, 19 of 58; p = 0.576, Pearson's x2). Figure 3A shows a burst of action potentials from an MD neuron. Burst firing was typically present during spontaneous activity, but the frequency of bursts was reduced when the cell was activated by pinch. In sham-operated animals, frequency of bursts was reduced by half during pinch-evoked responses (spontaneous: mean, 0.209 ± 0.075 Hz; median, 0.033 Hz; evoked: mean, 0.147 ± 0.07 Hz; median, 0.013 Hz; p = 0.02, MWU; Fig. 3A) compared with spontaneous activity. Similarly, in spinal-lesioned animals frequency of bursts was significantly lower during pinch-evoked responses than during spontaneous activity (spontaneous: mean, 0.26 ± 0.056 Hz; median, 0.15 Hz; evoked: mean, 0.14 ± 0.044 Hz; median, 0.073 Hz; p = 0.02, MWU; Fig. 3B). There was no difference in frequency of bursts between sham-operated and spinal-lesioned animals during either spontaneous or evoked activity (spontaneous: p = 0.12, MWU; evoked: p = 0.3, MWU). Together, these findings are not consistent with the hypothesis that SCI pain is associated with increased bursting in MD neurons.
Figure 3.

Burst activity in MD neurons is not altered in animals with SCI. A, Recording of a burst of action potentials elicited from an MD neuron. B, Box plots show that the frequency of bursts is significantly higher during spontaneous activity compared with evoked responses for both sham-operated and spinal-lesioned animals (sham-operated, n = 15; SCI, n = 20; p values computed using MWU). Frequency of bursts was not different between surgical groups (in either spontaneous or evoked epochs).
Reversible ZI inactivation: electrophysiology
We have previously shown that both spontaneous and evoked activity in ZI are significantly reduced after spinal lesion (Masri et al., 2009) and others have shown that ZI projects to thalamocortical neurons in MD. In monkey, Erickson et al. (2004) report that up to 10% of all neurons that project to MD originate from the ZI; this likely represents a major inhibitory input to the MD. In their original manuscript, Barthó et al. (2002) report that in the rat there are “scattered” ZI terminals in MD. In a personal communication, the senior author of that manuscript suggests that they likely underestimated the density of these inputs due to their restricted injection sites in the ZI. Their more recent unpublished data in mice demonstrate dense projections from more rostral ZI sectors to the MD (L. Acsády, personal communication).
Therefore, we hypothesized that the significant increase of neuronal activity in MD after spinal lesion was a result of decreased inhibition from the ZI. However, to the best of our knowledge, functional inhibition of the MD by ZI activity has never been demonstrated. To test whether the ZI exerts significant tonic inhibition on neurons in the MD, in naive animals we transiently inactivated ZI neurons with the GABAA receptor agonist muscimol, while recording spontaneous activity from MD neurons (described in detail in Materials and Methods). Figure 4A shows a rate histogram (5 s bins) of a representative MD neuron before and during ZI inactivation. The infusion of 0.1 ml of muscimol (arrow) into ZI caused a significant increase in MD firing rate. Infusion of the same volume of saline (arrowhead) did not significantly alter firing rate. On average, inactivation of the ZI caused a 2.5-fold increase of spontaneous neuronal activity in MD neurons when compared with baseline activity (baseline: mean, 1.61 ± 2.62 spikes/s; median, 0.85 spikes/s; after muscimol: mean, 4.37 ± 5.04 spikes/s; median, 1.95 spikes/s; n = 36; p = 0.0002, MWU; Fig. 4B). Infusion of an equivalent volume of saline (before or after the muscimol injections) had no significant effect on the activity of MD neurons (baseline: mean, 1.03 ± 1.14 spikes/s; median, 0.667 spikes/s; after saline: mean, 1.09 ± 0.758 spikes/s; median, 0.9 spikes/s; n = 11; p = 0.532, MWU). The significant increase in MD spontaneous activity during ZI inactivation indicates that under normal conditions the ZI tonically inhibits the MD, and is consistent with our hypothesis that decreased activity of the ZI is causally related to the increase of MD activity in conditions of chronic pain. We note, however, that we cannot exclude the possibility that muscimol inadvertently inactivated pathways other than the one from the ZI to the MD. We describe below the results of a control experiment that addresses this possibility.
Figure 4.
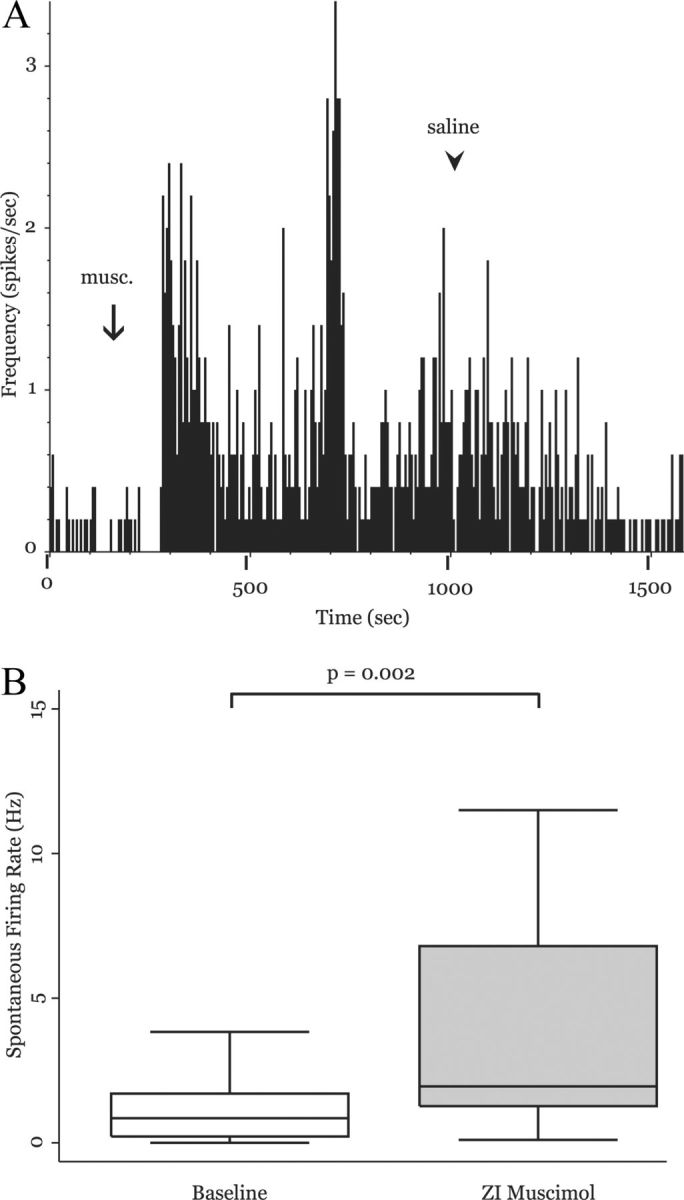
Neuronal activity in the MD is enhanced during transient ZI inactivation. A, Rate histogram computed from a representative MD neuron in a naive rat (5 s bins). After infusion of 0.1 ml of muscimol into the ZI (arrow), firing rate of this neuron significantly increased. Saline injection resulted in a gradual washout (see Results). B, Group data from 36 MD neurons shows that transiently inactivating ZI with muscimol significantly increases MD firing rate (p = 0.002, MWU).
Reversible ZI inactivation: behavior
To further test the causal relationship between ZI and MD activities, we asked whether reversible, pharmacological inactivation of the ZI affects behavioral hyperalgesia. In rats implanted with microdialysis probes over the right, ventral ZI, we compared responses to mechanical stimuli before and after infusion of muscimol (as described above). Muscimol infusion in the ZI resulted in a large and significant (p < 0.01, t test) reduction in withdrawal threshold of both the left and right hind limbs (Fig. 5). The bilateral effects of ipsilateral ZI inactivation may be related to the bilateral projections from the ZI to thalamic nuclei in both hemispheres (Power and Mitrofanis, 2001). This is consistent also with our findings that unilateral spinal lesions result in bilateral hyperalgesia (Masri et al., 2009) and that unilateral stimulation of motor cortex results in bilateral pain relief (Lucas et al., 2011).
Injections of saline had no effect on withdrawal thresholds of either limb
We observed also, in three of four rats, that immediately after muscimol injections they displayed typical nociceptive behaviors, including vigorous shaking of a hind limb and repetitive facial grooming. These behaviors, which were not quantified, ended within 30 min after drug application. They were never observed in animals that received saline injections.
As a negative control, we infused muscimol into the substantia nigra (pars reticulata) of two rats. We chose this nucleus because it neighbors the ZI, allowing us to test the possible diffusion of drugs distal to the intended injection region (ZI). Further, the substantia nigra contains inhibitory neurons that may affect MD activity indirectly through their projections to the superior colliculus (Edwards et al., 1979), which provides significant inputs to the MD (Groenewegen, 1988). In contrast to the effects of injections into the ZI, muscimol injection into substantia nigra had no discernible effects on paw withdrawal thresholds, nor on the rats' behavior (Fig. 5).
These findings are consistent with the conclusion that decreased activity of the ZI is causally related to the increase of MD activity in conditions of chronic pain.
Increased spontaneous neuronal activity in the MD of awake animals
Pain is a complex sensation, the individual components of which can be modified and modulated by behavioral states, such as arousal and attention (Miron et al., 1989; Buffington et al., 2005). For this reason, we next tested whether SCI affected neuronal activity in MD of awake, behaving animals. We obtained extracellular multiunit recordings from MD of seven awake animals both before and after spinal lesion. All animals included in this study developed clear and statistically significant mechanical hyperalgesia after the spinal lesion. We identified MUA in MD using two main criteria: recording location, based on stereotaxic coordinates (Paxinos and Watson, 1998) and confirmed histologically post hoc, and responses to mechanical and thermal peripheral stimulation. After histological reconstruction of recording sites, MUA recordings outside of MD were excluded from further analysis. Figure 6A depicts a rate histogram (100 ms bins) from a representative MUA recording taken from an awake animal before spinal lesion. Both spontaneous activity and activity evoked in response to a series of von Frey filaments are shown. All traces were taken from the same continuous recording and depict an increased firing rate in response to increased von Frey force. The experimental procedure (see Materials and Methods) did not allow us to record from the same site before and after SCI. However, to our knowledge, ours is the first demonstration of spontaneous and evoked activity—to innocuous and noxious stimuli—in the MD of a normal, awake animal.
Figure 6.
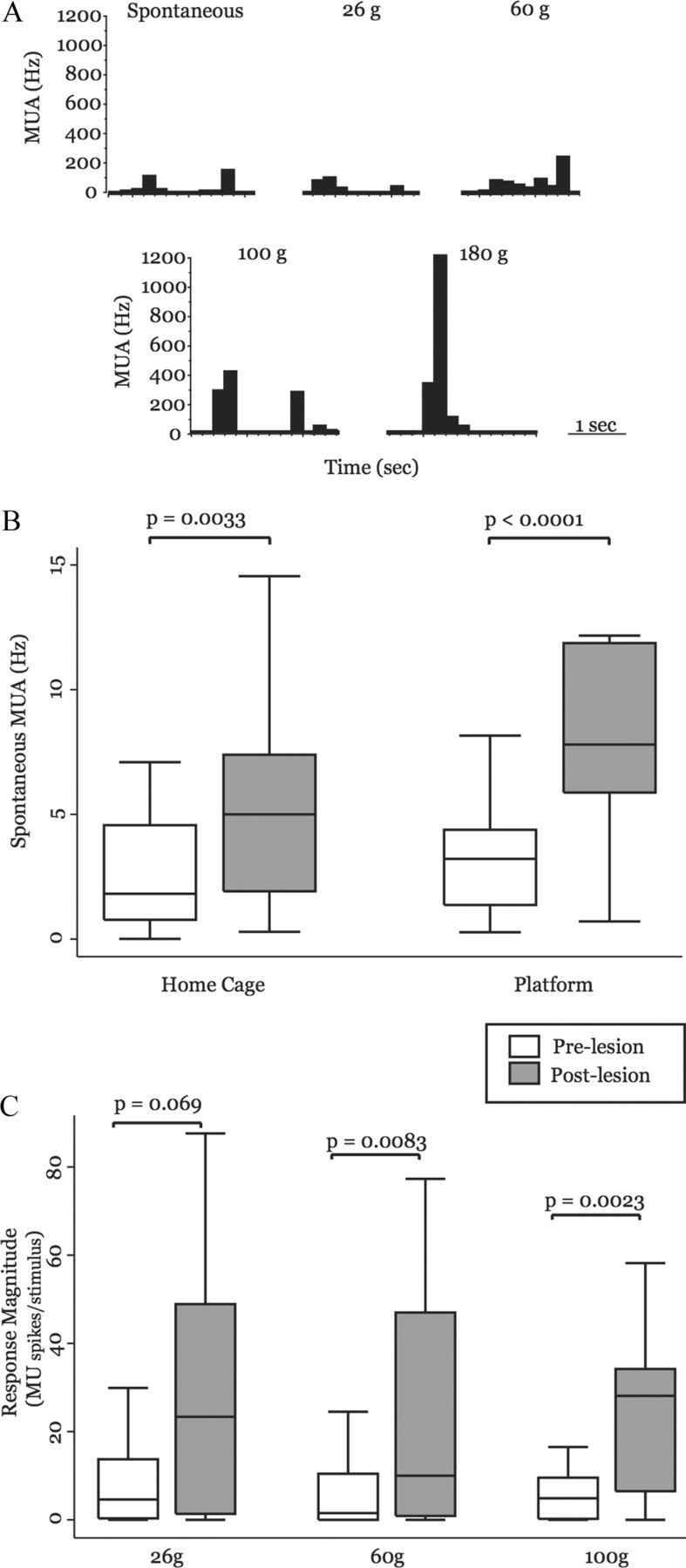
Neuronal activity in the MD is enhanced in behaving animals with SCI. A, Rate histogram computed from representative MUA in a naive rat (100 ms bins) showing spontaneous activity and activity evoked in response to a series of von Frey filaments. B, Box-and-whisker plots show that MD MUAs from animals before spinal lesion (n = 31) have significantly higher spontaneous firing rates in two environments: the animals' home cage and the elevated testing platform, compared with the same animals before spinal lesion (n = 29). C, Box plots show the magnitude of MUA-evoked responses in animals before spinal lesion and in the same animals after spinal lesion. After spinal lesion, animals have greater responses to 26 g of von Frey stimulation (before lesion, n = 16; after lesion, n = 22) and have significantly larger responses to 60 g (before lesion, n = 24; after lesion, n = 31) and 100 g (before lesion, n = 24; after lesion, n = 23) of von Frey stimulation. MU, multiunit response magnitude (MU spikes/stimulus).
We emphasize that MUAs reflect the summed activity of multiple neurons. Therefore, changes in the firing rate of MUAs may reflect changes in the firing rates of individual neurons in the recorded cluster, as well as changes in the number of neurons recorded in that cluster.
To test whether neurons in MD of behaving animals had increased spontaneous activity after spinal lesions, we compared spontaneous firing rates of MUAs in MD during two different behavioral conditions. First, we recorded 2 min of spontaneous activity while animals were in their home cages and then while animals were freely moving on the testing platform (described in detail in Materials and Methods). After spinal lesion, there was a significant increase in MUA spontaneous firing rate in MD when the animals were in the home cage (before lesion: mean, 2.69 ± 0.44 spikes/s; median, 1.82 spikes/s; n = 29 MUAs; after lesion: mean, 5.30 ± 0.65 spikes/s; median, 5.0 spikes/s; n = 31 MUAs; p = 0.0033, MWU; Fig. 6B). There was also a threefold increase in spontaneous firing rate recorded while animals were freely moving on the elevated testing platform (before lesion: mean, 3.45 ± 0.50 spikes/s; median, 3.21 spikes/s; n = 29 MUAs; after lesion: mean, 10.78 ± 1.71 spikes/s; median, 7.80 spikes/s; n = 31 MUAs; p = 0.0001, MWU; Fig. 6B).
These data show that spontaneous neuronal activity is increased after SCI both in environments associated with noxious peripheral stimulation and in less-anxiety-producing environments, such as the animals' home cage. To our knowledge, this is the first report of enhanced spontaneous neuronal activity in MD of awake behaving animals with hyperalgesia after SCI.
Increased responses to peripheral stimulation in MDs of behaving animals
We tested for changes in pain thresholds while the animals were on an elevated platform with a mesh floor (described in Materials and Methods). Briefly, we applied von Frey filaments in ascending order (range, 26–180 g) five times each to the plantar surface of the rats' hind paws to evoke neuronal activity in MD neurons and to obtain pain thresholds for each animal, both before and after spinal lesion.
For each stimulus intensity, we compared response magnitude of MUAs that had significant stimulus-evoked increases in firing rate (exceeding 99% confidence interval relative to spontaneous firing rates) for animals before lesion to those after spinal lesion. In response to 26 g of von Frey filament stimulation, there was a large increase in mean firing rate after lesion and the difference between the two groups approached statistical significance (before lesion: mean, 10.2 ± 3.62 spikes/stimulus; median, 4.61 spikes/stimulus; after lesion: mean, 26.7 ± 5.59 spikes/stimulus; median, 23.4 spikes/stimulus; p = 0.069, MWU; Fig. 6C).
When 60 g of force was applied to the hind paw of animals with SCI, there was a sixfold increase in MUA response magnitude compared with responses from the same animals before spinal lesion (before lesion: mean, 6.60 ± 1.95 spikes/stimulus; median, 1.52 spikes/stimulus, n = 24 MUAs; after lesion: mean, 44.7 ± 13.5 spikes/stimulus; median, 10.0 spikes/stimulus; n = 31 MUAs; p = 0.0083, MWU; Fig. 6C). Similarly, there was, on average, a 3.5-fold increase in response magnitude when 100 g of force was applied to the hind paws of animals with SCI, compared with before lesion responses (before lesion: mean, 7.62 ± 2.54 spikes/stimulus; median, 4.89 spikes/stimulus; n = 24 MUAs; after lesion: mean, 26.5 ± 5.22 spikes/stimulus; median, 28.1 spikes/stimulus; n = 23 MUAs; p = 0.0023, MWU; Fig. 6C).
After obtaining mechanical thresholds with von Frey filaments, we applied a clearly noxious heat stimulus (42–45°C, 10 s intervals), using a heat probe (see Materials and Methods), five times to alternating hind paws of the animals. In all animals, both before and after spinal surgery, the heat probe elicited a clear nocifensive response, including hind paw withdrawal, licking, biting, and shaking. There was no significant difference in the magnitude of neuronal responses elicited by the heat probe between animals before and after lesion (before lesion: mean, 14.5 ± 4.49 spikes/stimulus; median, 0.82 spikes/stimulus; n = 24 MUAs; after lesion: mean, 9.55 ± 2.04 spikes/stimulus; median, 5.95 spikes/stimulus; n = 31 MUAs; p = 0.64, MWU; data not shown). Fifty-four percent (13 of 24) of MUAs in MD of before-lesion animals and 70% (21 of 30) of MUAs in MD of after-lesion animals had significant increases in firing rate in response to the heat probe; this difference was not significant (p = 0.231, Pearson's x2). A possible explanation for the lack of effect of SCI on MD responses to noxious thermal stimuli may be that in both groups of rats these stimuli saturated the responses of the peripheral heat-sensitive receptors; that is, that these results are confounded by a “ceiling effect.”
Discussion
We have previously shown that hyperalgesia after SCI is associated with a reduction in neuronal activity in the GABAergic ZI and that this correlates with an increase in neuronal activity in somatosensory thalamic nuclei, such as the PO (Masri et al., 2009) (Fig. 5). Because pain is both a sensory-discriminative experience and an unpleasant emotional experience (Merskey and Bogduk, 1994), we tested the hypothesis that chronic pain produces an increase of activity also in associative thalamic nuclei involved in processing affective components of pain. We focused on the MD, which has been proposed to be involved in integrating somatosensory and affective components of pain (Price, 2002). Consistent with this hypothesis, we found that neurons in MD are strongly driven by noxious peripheral stimulation in both anesthetized animals and awake behaving animals. Consistent with this hypothesis, we observed that SCI causes a robust increase in both spontaneous and evoked MD activity in both anesthetized and awake behaving animals.
The subdivisions and cortical projections of MD are largely conserved between species, though the anatomical nomenclature can vary. Considering thalamocortical projections and cellular architecture, Ray and Price (1993) identified the primate MD pars caudodorsalis as analogous to the rat medial and dorsolateral MD studied here. Although there is evidence that the MD plays a critical role in processing affective components of pain, there have been few studies on the extracellular electrophysiological properties of MD neurons (Mogenson et al., 1987; Lavin and Grace, 1998; Vaculín et al., 2000), and even fewer focused on responses to noxious peripheral stimuli (Dostrovsky and Guilbaud, 1990; Wang et al., 2003; Zhang et al., 2011). To the best of our knowledge, no studies have been conducted on the activity of MD neurons in animal models of SCI pain.
In addition to its role in processing noxious stimuli, the MD has been implicated in diverse functions, including learning and memory and other cognitive functions (Jones, 2007). We therefore cannot exclude the possibility that the electrophysiological changes we report in MD might be related also to other consequences of SCI.
Enhanced spontaneous activity in the MD
In humans, SCI pain presents not only as exaggerated responses to peripheral stimuli, but also as chronic spontaneous pain (Bowsher, 1996; Garcia-Larrea et al., 2002; Finnerup et al., 2003). We have shown that in this model of SCI pain, rats develop mechanical and thermal hyperalgesia as well as a tonic aversive state, analogous to the human condition of spontaneous pain (Davoody et al., 2011). Here, we report that spinal lesions result in significant increases in spontaneous firing of MD neurons. Because the MD is densely interconnected with several areas of the limbic circuit critical for nociceptive processing (Treede et al., 1999, 2000; Price, 2000; Johansen et al., 2001; Wang et al., 2003, 2004; Jasmin et al., 2004; Shyu and Vogt, 2009; Zhang et al., 2011), it is possible that this contributes to the ongoing spontaneous pain in this condition.
We hypothesized that the increase in spontaneous activity is causally related to the dramatic loss of spontaneous activity we have recorded from ZI in animals with SCI (Masri et al., 2009). Consistent with this, here we show that, in naive animals, the ZI exerts significant tonic inhibition over the MD. Even so, we considered the possibility that, in SCI pain, there is a reduction in other inhibitory inputs to the MD. There are no GABAergic interneurons within the MD (Kuroda et al., 1992) and, therefore, all GABAergic inhibition is mediated by extrinsic afferents. An important source of GABAergic afferents is the reticular nucleus of the thalamus (TRN). Unlike the ZI, the TRN does not receive spinothalamic inputs, and its major source of excitatory input is the somatosensory cortex (Liu and Jones, 1999). Moreover, we found that responses evoked by innocuous stimuli in the ventrobasal somatosensory nuclei that receive inhibition exclusively from the TRN are unaffected by spinal lesions (Masri et al., 2009). These findings suggest that the enhanced activity in the thalamus after SCI is related primarily to thalamic nuclei that normally receive strong inhibition from the ZI.
However, other thalamic nuclei may also have a causal role in SCI pain. We have previously reported that after SCI spontaneous activity increases modestly in the ventrobasal thalamus (4-fold increase, compared with 30-fold increase in the PO thalamus), and that SCI is associated with an increase in the incidence of bursting cells in the ventrobasal thalamus (Masri et al., 2009). These findings are consistent with reports of similar changes in the ventrobasal thalamus from the Waxman group (Hains et al., 2005, 2006).
Moreover, TRN neurons can be inhibited by noxious stimuli (Yen and Shaw, 2003), and the inhibitory drive from the TRN upon ventrobasal neurons regulates bursting in ventrobasal neurons (Steriade, 2000; Halassa et al., 2011). Therefore, SCI-associated alterations in inhibitory inputs from the TRN may also affect the activity of ventrobasal (“first order”) thalamic nuclei and contribute to chronic pain.
Evoked activity in the MD
An early study by Dostrovsky and Guilbaud (1990) examined electrophysiological activity in several midline thalamic nuclei, including the MD, in anesthetized normal and arthritic rats. In agreement with our findings, they found MD neurons respond exclusively to nociceptive stimulation over large, bilateral receptive fields. Similar to the long-duration, augmenting responses we describe (Figs. 1B, 2B, 6C), these authors also report that MD neurons have long after-discharges. Though the authors' description of normal electrophysiological activity in the MD agrees with what we have presented here, they found no change in spontaneous firing rate or magnitude of response in arthritic animals.
There are two critical differences between the Dostrovsky study and ours. First, they used an arthritic rat model in which inflammation-related hyperalgesia lasts several weeks (Colpaert, 1987). This hyperalgesia is largely driven by peripheral inflammation and can be relieved by administration of NSAIDs (Hirose and Jyoyama, 1971; Winter et al., 1979; Capetola et al., 1980), indicating that peripheral tissue damage is a driving force for maintenance of pain. In contrast, we use an animal model of SCI pain to produce long-lasting pain characterized by a tonic aversive state, and that has significant central component (Masri et al., 2009). Second, the authors presented continuous noxious stimulation for 15 s, which may have maximized the responses, producing a “ceiling effect.” Because we found that MD neurons quickly changed their responses to repeated stimulation, we limited the duration of peripheral stimulation to <1 s of stimulation.
Burst activity in the MD
We found no difference between spinal-lesioned and sham-operated groups in the proportion of MD neurons that had spontaneous burst activity. Neither was there a difference in the number or frequency of bursts during spontaneous activity or of any burst property that we tested (number of action potentials per burst, interspike interval during burst, etc.). This is consistent with our previous report on posterior thalamus, where we found no change in bursting activity in animals with SCI (Masri et al., 2009), though we and others have reported a moderate increase of bursting in first-order somatosensory nuclei (ventrobasal thalamus) in this model of SCI pain (Wang and Thompson, 2008; Masri et al., 2009). This selective change in burst activity following lesion of the STT may be a result of different anatomical connections or physiological properties between first-order and higher-order thalamic nuclei (Sherman and Guillery, 2005).
MD neurons from both sham-operated and spinal-lesioned animals had significantly lower frequency of burst firing in response to noxious peripheral stimulation, compared with spontaneous bursts. Because thalamic burst firing requires deinactivation of T-type calcium channels, neurons only fire in burst mode when they are relatively hyperpolarized (Sherman and Guillery, 2005). It is possible that after noxious pinch ascending excitation from the STT prevents thalamocortical neurons in the MD from becoming sufficiently depolarized, thereby preventing neurons from firing in burst mode in response to peripheral simulation.
The role of the ZI
As discussed above, the ZI potently regulates the activity of both the PO and MD thalamus. We have previously shown that SCI is associated with significant decreases in the activity of ZI neurons (Masri et al., 2009). Here, we demonstrate that reversible inactivation of the ZI results in immediate and profound hyperalgesia, as well as in increased activity of MD neurons. Further, we previously demonstrated that stimulation of the ZI in rats with SCI results in immediate reversal of behavioral signs of hyperalgesia (Lucas et al., 2011). Thus, the present demonstration of abnormal neuronal activity in the MD, together with findings from our previous reports, support the conclusion that the pathophysiology of SCI pain involves abnormal reduction in GABAergic inhibition of thalamic nuclei regulated by the ZI (Masri and Keller, 2011).
We are actively investigating the mechanisms through which SCI results in suppressed ZI activity. Preliminary findings (Keller, 2011) suggest that SCI is associated with a reduction of ∼30% in the expression of GAD (the rate-limiting enzyme in GABA synthesis) in ZI neurons, and a subsequent, gradual apoptosis of GABAergic neurons in the ZI. Our preliminary findings also suggest that the changes result in a significant decrease in the frequency of miniature IPSPs recorded from PO and MD neurons, supporting the conclusion that SCI results in suppression of inhibitory control of these thalamic nuclei by the ZI.
We cannot exclude the possibility that the enhanced activity of thalamic neurons reflects increased nociceptive inputs from upstream structures, such as sensitized dorsal horn neurons. Preliminary findings (Keller, 2011) suggest that this may not be the case, because acute suppression of spinal activity with injections of tetrodotoxin did not reverse the enhanced spontaneous activity of thalamic neurons in SCI rats.
It is also possible that the sensitization of thalamic neurons reflects more than just disinhibitory mechanisms. For example, Waxman and collaborators reported that sensitization of ventroposterolateral neurons after SCI is related to upregulation of chemokines, microglia, and unique sodium channels (Zhao et al., 2007; Dib-Hajj et al., 2010). Thus, chronic pain after SCI likely represents a multifaceted pathophysiology involving converging processes in several thalamic nuclei.
Footnotes
This work was supported by National Institute of Neurological Disorders and Stroke Fellowship F31NS-070458 to J.L.W.; Research Grants R01-051799 to A.K. and R01-NS069568 to R.M.; Department of Defense Grant SC090126 to R.M.; and the Christopher and Dana Reeve Foundation.
The authors declare no competing financial interests.
References
- Baastrup C, Finnerup NB. Pharmacological management of neuropathic pain following spinal cord injury. CNS Drugs. 2008;22:455–475. doi: 10.2165/00023210-200822060-00002. [DOI] [PubMed] [Google Scholar]
- Barthó P, Freund TF, Acsády L. Selective GABAergic innervation of thalamic nuclei from zona incerta. Eur J Neurosci. 2002;16:999–1014. doi: 10.1046/j.1460-9568.2002.02157.x. [DOI] [PubMed] [Google Scholar]
- Boivie J. Central pain. In: McMahon S, Koltzenburg M, editors. Wall and Melzack's textbook of pain. Oxford: Churchill Livingstone; 2005. pp. 1057–1074. [Google Scholar]
- Bowsher D. Central pain: clinical and physiological characteristics. J Neurol Neurosurg Psychiatry. 1996;61:62–69. doi: 10.1136/jnnp.61.1.62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buffington AL, Hanlon CA, McKeown MJ. Acute and persistent pain modulation of attention-related anterior cingulate fMRI activations. Pain. 2005;113:172–184. doi: 10.1016/j.pain.2004.10.006. [DOI] [PubMed] [Google Scholar]
- Canavero S, Bonicalzi V. Central pain syndrome: pathophysiology, diagnosis and management. New York: Cambridge UP; 2007. [Google Scholar]
- Capetola RJ, Shriver DA, Rosenthale ME. Suprofen, a new peripheral analgesic. J Pharmacol Exp Ther. 1980;214:16–23. [PubMed] [Google Scholar]
- Colpaert FC. Evidence that adjuvant arthritis in the rat is associated with chronic pain. Pain. 1987;28:201–222. doi: 10.1016/0304-3959(87)90117-5. [DOI] [PubMed] [Google Scholar]
- Cornwall J, Phillipson OT. Afferent projections to the dorsal thalamus of the rat as shown by retrograde lectin transport—I. The mediodorsal nucleus. Neuroscience. 1988;24:1035–1049. doi: 10.1016/0306-4522(88)90085-1. [DOI] [PubMed] [Google Scholar]
- Davoody L, Quiton RL, Lucas JM, Ji Y, Keller A, Masri R. Conditioned place preference reveals tonic pain in an animal model of central pain. J Pain. 2011;12:868–874. doi: 10.1016/j.jpain.2011.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dib-Hajj SD, Cummins TR, Black JA, Waxman SG. Sodium channels in normal and pathological pain. Annu Rev Neurosci. 2010;33:325–347. doi: 10.1146/annurev-neuro-060909-153234. [DOI] [PubMed] [Google Scholar]
- Dostrovsky JO. The thalamus and human pain. In: Henry JL, Panju A, Yashpal K, editors. Central neuropathic pain: focus on Poststroke pain. Seattle: IASP; 2007. pp. 101–112. [Google Scholar]
- Dostrovsky JO, Guilbaud G. Nociceptive responses in medial thalamus of the normal and arthritic rat. Pain. 1990;40:93–104. doi: 10.1016/0304-3959(90)91056-O. [DOI] [PubMed] [Google Scholar]
- Edwards SB, Ginsburgh CL, Henkel CK, Stein BE. Sources of subcortical projections to the superior colliculus in the cat. J Comp Neurol. 1979;184:309–329. doi: 10.1002/cne.901840207. [DOI] [PubMed] [Google Scholar]
- Erickson SL, Lewis DA. Cortical connections of the lateral mediodorsal thalamus in cynomolgus monkeys. J Comp Neurol. 2004;473:107–127. doi: 10.1002/cne.20084. [DOI] [PubMed] [Google Scholar]
- Erickson SL, Melchitzky DS, Lewis DA. Subcortical afferents to the lateral mediodorsal thalamus in cynomolgus monkeys. Neuroscience. 2004;129:675–690. doi: 10.1016/j.neuroscience.2004.08.016. [DOI] [PubMed] [Google Scholar]
- Falci S, Best L, Bayles R, Lammertse D, Starnes C. Dorsal root entry zone microcoagulation for spinal cord injury-related central pain: operative intramedullary electrophysiological guidance and clinical outcome. J Neurosurg. 2002;97:193–200. doi: 10.3171/spi.2002.97.2.0193. [DOI] [PubMed] [Google Scholar]
- Finnerup NB, Johannesen IL, Fuglsang-Frederiksen A, Bach FW, Jensen TS. Sensory function in spinal cord injury patients with and without central pain. Brain. 2003;126:57–70. doi: 10.1093/brain/awg007. [DOI] [PubMed] [Google Scholar]
- Friedberg MH, Lee SM, Ebner FF. Modulation of receptive field properties of thalamic somatosensory neurons by the depth of anesthesia. J Neurophysiol. 1999;81:2243–2252. doi: 10.1152/jn.1999.81.5.2243. [DOI] [PubMed] [Google Scholar]
- Garcia-Larrea L, Convers P, Magnin M, André-Obadia N, Peyron R, Laurent B, Mauguière F. Laser-evoked potential abnormalities in central pain patients: the influence of spontaneous and provoked pain. Brain. 2002;125:2766–2781. doi: 10.1093/brain/awf275. [DOI] [PubMed] [Google Scholar]
- Groenewegen HJ. Organization of the afferent connections of the mediodorsal thalamic nucleus in the rat, related to the mediodorsal-prefrontal topography. Neuroscience. 1988;24:379–431. doi: 10.1016/0306-4522(88)90339-9. [DOI] [PubMed] [Google Scholar]
- Guido W, Lu SM, Vaughan JW, Godwin DW, Sherman SM. Receiver operating characteristic (ROC) analysis of neurons in the cat's lateral geniculate nucleus during tonic and burst response mode. Vis Neurosci. 1995;12:723–741. doi: 10.1017/s0952523800008993. [DOI] [PubMed] [Google Scholar]
- Hains BC, Saab CY, Waxman SG. Changes in electrophysiological properties and sodium channel Nav1.3 expression in thalamic neurons after spinal cord injury. Brain. 2005;128:2359–2371. doi: 10.1093/brain/awh623. [DOI] [PubMed] [Google Scholar]
- Hains BC, Saab CY, Waxman SG. Alterations in burst firing of thalamic VPL neurons and reversal by Na(v)1.3 antisense after spinal cord injury. J Neurophysiol. 2006;95:3343–3352. doi: 10.1152/jn.01009.2005. [DOI] [PubMed] [Google Scholar]
- Halassa MM, Siegle JH, Ritt JT, Ting JT, Feng G, Moore CI. Selective optical drive of thalamic reticular nucleus generates thalamic bursts and cortical spindles. Nat Neurosci. 2011;14:1118–1120. doi: 10.1038/nn.2880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Head H, Holmes G. Sensory disturbances from cerebral lesions. Brain. 1911;34:102–254. [Google Scholar]
- Hirose K, Jyoyama H. Measurement of arthritic pain and effects of analgesics in the adjuvant-treated rat. Jpn J Pharmacol. 1971;21:717–720. doi: 10.1254/jjp.21.717. [DOI] [PubMed] [Google Scholar]
- Hoaglin DC, Iglewicz B, Tukey JW. Performance of some resistant rules for outlier labeling. J Am Stat Assoc. 1986;81:991–999. [Google Scholar]
- Hsu MM, Kung JC, Shyu BC. Evoked responses of the anterior cingulate cortex to stimulation of the medial thalamus. Chin J Physiol. 2000;43:81–89. [PubMed] [Google Scholar]
- Iwata M, LeBlanc BW, Kadasi LM, Zerah ML, Cosgrove RG, Saab CY. High-frequency stimulation in the ventral posterolateral thalamus reverses electrophysiologic changes and hyperalgesia in a rat model of peripheral neuropathic pain. Pain. 2011;152:2505–2513. doi: 10.1016/j.pain.2011.07.011. [DOI] [PubMed] [Google Scholar]
- Jahnsen H, Llinás R. Electrophysiological properties of guinea-pig thalamic neurones: an in vitro study. J Physiol. 1984;349:205–226. doi: 10.1113/jphysiol.1984.sp015153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jasmin L, Burkey AR, Granato A, Ohara PT. Rostral agranular insular cortex and pain areas of the central nervous system: a tract-tracing study in the rat. J Comp Neurol. 2004;468:425–440. doi: 10.1002/cne.10978. [DOI] [PubMed] [Google Scholar]
- Johansen JP, Fields HL, Manning BH. The affective component of pain in rodents: direct evidence for a contribution of the anterior cingulate cortex. Proc Natl Acad Sci U S A. 2001;98:8077–8082. doi: 10.1073/pnas.141218998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones EG. The thalamus. Cambridge: Cambridge UP; 2007. [Google Scholar]
- Keller A. Thalamic and cortical plasticity in pain models. Neuroscience Meeting Planner. 2011. [Accessed on January 29, 2013]. Program 524.05. Available online at http://goo.gl/p87Oq.
- Krettek JE, Price JL. The cortical projections of the mediodorsal nucleus and adjacent thalamic nuclei in the rat. J Comp Neurol. 1977;171:157–191. doi: 10.1002/cne.901710204. [DOI] [PubMed] [Google Scholar]
- Kuroda M, López-Mascaraque L, Price JL. Neuronal and synaptic composition of the mediodorsal thalamic nucleus in the rat: a light and electron microscopic Golgi study. J Comp Neurol. 1992;326:61–81. doi: 10.1002/cne.903260106. [DOI] [PubMed] [Google Scholar]
- Lavin A, Grace AA. Response of the ventral pallidal/mediodorsal thalamic system to antipsychotic drug administration: involvement of the prefrontal cortex. Neuropsychopharmacology. 1998;18:352–363. doi: 10.1016/S0893-133X(97)00165-6. [DOI] [PubMed] [Google Scholar]
- Lee JI, Ohara S, Dougherty PM, Lenz FA. Pain and temperature encoding in the human thalamic somatic sensory nucleus (ventral caudal): inhibition-related bursting evoked by somatic stimuli. J Neurophysiol. 2005;94:1676–1687. doi: 10.1152/jn.00343.2005. [DOI] [PubMed] [Google Scholar]
- Lenz FA, Kwan HC, Dostrovsky JO, Tasker RR. Characteristics of the bursting pattern of action potentials that occurs in the thalamus of patients with central pain. Brain Res. 1989;496:357–360. doi: 10.1016/0006-8993(89)91088-3. [DOI] [PubMed] [Google Scholar]
- Liu XB, Jones EG. Predominance of corticothalamic synaptic inputs to thalamic reticular nucleus neurons in the rat. J Comp Neurol. 1999;414:67–79. [PubMed] [Google Scholar]
- Lu SM, Guido W, Vaughan JW, Sherman SM. Latency variability of responses to visual stimuli in cells of the cat's lateral geniculate nucleus. Exp Brain Res. 1995;105:7–17. doi: 10.1007/BF00242177. [DOI] [PubMed] [Google Scholar]
- Lucas JM, Ji Y, Masri R. Motor cortex stimulation reduces hyperalgesia in an animal model of central pain. Pain. 2011;152:1398–1407. doi: 10.1016/j.pain.2011.02.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masri R, Quiton RL, Lucas JM, Murray PD, Thompson SM, Keller A. Zona incerta: a role in central pain. J Neurophysiol. 2009;102:181–191. doi: 10.1152/jn.00152.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masri R, Keller A. Chronic pain following spinal cord injury. In: Jandial R, editor. Frontiers in spinal cord and spine repair. Austin, TX: Landers Bioscience; 2011. [Google Scholar]
- Massaux A, Edeline JM. Bursts in the medial geniculate body: a comparison between anesthetized and unanesthetized states in guinea pig. Exp Brain Res. 2003;153:573–578. doi: 10.1007/s00221-003-1516-3. [DOI] [PubMed] [Google Scholar]
- McCarley RW, Benoit O, Barrionuevo G. Lateral geniculate nucleus unitary discharge in sleep and waking: state- and rate-specific aspects. J Neurophysiol. 1983;50:798–818. doi: 10.1152/jn.1983.50.4.798. [DOI] [PubMed] [Google Scholar]
- Merskey H, Bogduk N. Classification of chronic pain: descriptions of chronic pain syndromes and definitions of pain terms. Seattle: IASP; 1994. [Google Scholar]
- Miron D, Duncan GH, Bushnell MC. Effects of attention on the intensity and unpleasantness of thermal pain. Pain. 1989;39:345–352. doi: 10.1016/0304-3959(89)90048-1. [DOI] [PubMed] [Google Scholar]
- Mogenson GJ, Ciriello J, Garland J, Wu M. Ventral pallidum projections to mediodorsal nucleus of the thalamus: an anatomical and electrophysiological investigation in the rat. Brain Res. 1987;404:221–230. doi: 10.1016/0006-8993(87)91373-4. [DOI] [PubMed] [Google Scholar]
- Ottersen OP, Storm-Mathisen J. Glutamate and GABA-containing neurons in the mouse and rat brain, as demonstrated with a new immunocytochemical technique. J Comp Neurol. 1984;229:374–392. doi: 10.1002/cne.902290308. [DOI] [PubMed] [Google Scholar]
- Paxinos G, Watson C. The rat brain in stereotaxic coordinates. San Diego: Academic; 1998. [DOI] [PubMed] [Google Scholar]
- Power BD, Mitrofanis J. Zona incerta: substrate for contralateral interconnectivity in the thalamus of rats. J Comp Neurol. 2001;436:52–63. [PubMed] [Google Scholar]
- Price DD. Psychological and neural mechanisms of the affective dimension of pain. Science. 2000;288:1769–1772. doi: 10.1126/science.288.5472.1769. [DOI] [PubMed] [Google Scholar]
- Price DD. Central neural mechanisms that interrelate sensory and affective dimensions of pain. Mol Interv. 2002;2:392–403. 339. doi: 10.1124/mi.2.6.392. [DOI] [PubMed] [Google Scholar]
- Price JL, Slotnick BM. Dual olfactory representation in the rat thalamus: an anatomical and electrophysiological study. J Comp Neurol. 1983;215:63–77. doi: 10.1002/cne.902150106. [DOI] [PubMed] [Google Scholar]
- Ramcharan EJ, Gnadt JW, Sherman SM. Burst and tonic firing in thalamic cells of unanesthetized, behaving monkeys. Vis Neurosci. 2000;17:55–62. doi: 10.1017/s0952523800171056. [DOI] [PubMed] [Google Scholar]
- Ray JP, Price JL. The organization of the thalamocortical connections of the mediodorsal thalamic nucleus in the rat, related to the ventral forebrain-prefrontal cortex topography. J Comp Neurol. 1992;323:167–197. doi: 10.1002/cne.903230204. [DOI] [PubMed] [Google Scholar]
- Ray JP, Price JL. The organization of projections from the mediodorsal nucleus of the thalamus to orbital and medial prefrontal cortex in macaque monkeys. J Comp Neurol. 1993;337:1–31. doi: 10.1002/cne.903370102. [DOI] [PubMed] [Google Scholar]
- Ren K. An improved method for assessing mechanical allodynia in the rat. Physiol Behav. 1999;67:711–716. doi: 10.1016/s0031-9384(99)00136-5. [DOI] [PubMed] [Google Scholar]
- Rinaldi PC, Young RF, Albe-Fessard D, Chodakiewitz J. Spontaneous neuronal hyperactivity in the medial and intralaminar thalamic nuclei of patients with deafferentation pain. J Neurosurg. 1991;74:415–421. doi: 10.3171/jns.1991.74.3.0415. [DOI] [PubMed] [Google Scholar]
- Saadé NE, Al Amin H, Abdel Baki S, Chalouhi S, Jabbur SJ, Atweh SF. Reversible attenuation of neuropathic-like manifestations in rats by lesions or local blocks of the intralaminar or the medial thalamic nuclei. Exp Neurol. 2007;204:205–219. doi: 10.1016/j.expneurol.2006.10.009. [DOI] [PubMed] [Google Scholar]
- Sceniak MP, Maciver MB. Cellular actions of urethane on rat visual cortical neurons in vitro. J Neurophysiol. 2006;95:3865–3874. doi: 10.1152/jn.01196.2005. [DOI] [PubMed] [Google Scholar]
- Sherman SM. Dual response modes in lateral geniculate neurons: mechanisms and functions. Vis Neurosci. 1996;13:205–213. doi: 10.1017/s0952523800007446. [DOI] [PubMed] [Google Scholar]
- Sherman SM, Guillery RW. Exploring the thalamus and its role in cortical function. Cambridge, MA: MIT; 2005. [Google Scholar]
- Shyu BC, Vogt BA. Short-term synaptic plasticity in the nociceptive thalamic-anterior cingulate pathway. Mol Pain. 2009;5:51. doi: 10.1186/1744-8069-5-51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siddall PJ, McClelland JM, Rutkowski SB, Cousins MJ. A longitudinal study of the prevalence and characteristics of pain in the first 5 years following spinal cord injury. Pain. 2003;103:249–257. doi: 10.1016/S0304-3959(02)00452-9. [DOI] [PubMed] [Google Scholar]
- Steriade M. Corticothalamic resonance, states of vigilance and mentation. Neuroscience. 2000;101:243–276. doi: 10.1016/s0306-4522(00)00353-5. [DOI] [PubMed] [Google Scholar]
- Swadlow HA, Gusev AG. The impact of ‘bursting’ thalamic impulses at a neocortical synapse. Nat Neurosci. 2001;4:402–408. doi: 10.1038/86054. [DOI] [PubMed] [Google Scholar]
- Tasker RR, DeCarvalho GT, Dolan EJ. Intractable pain of spinal cord origin: clinical features and implications for surgery. J Neurosurg. 1992;77:373–378. doi: 10.3171/jns.1992.77.3.0373. [DOI] [PubMed] [Google Scholar]
- Trageser JC, Burke KA, Masri R, Li Y, Sellers L, Keller A. State-dependent gating of sensory inputs by zona incerta. J Neurophysiol. 2006;96:1456–1463. doi: 10.1152/jn.00423.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Treede RD, Kenshalo DR, Gracely RH, Jones AK. The cortical representation of pain. Pain. 1999;79:105–111. doi: 10.1016/s0304-3959(98)00184-5. [DOI] [PubMed] [Google Scholar]
- Treede RD, Apkarian AV, Bromm B, Greenspan JD, Lenz FA. Cortical representation of pain: functional characterization of nociceptive areas near the lateral sulcus. Pain. 2000;87:113–119. doi: 10.1016/S0304-3959(00)00350-X. [DOI] [PubMed] [Google Scholar]
- Vaculín S, Franek M, Rokyta R. Dorsal rhizotomy changes the spontaneous neuronal activity of nuclei in the medial thalamus. Physiol Res. 2000;49:279–283. [PubMed] [Google Scholar]
- Vierck CJ, Jr, Greenspan JD, Ritz LA. Long-term changes in purposive and reflexive responses to nociceptive stimulation following anterolateral chordotomy. J Neurosci. 1990;10:2077–2095. doi: 10.1523/JNEUROSCI.10-07-02077.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang G, Thompson SM. Maladaptive homeostatic plasticity in a rodent model of central pain syndrome: thalamic hyperexcitability after spinothalamic tract lesions. J Neurosci. 2008;28:11959–11969. doi: 10.1523/JNEUROSCI.3296-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang JY, Luo F, Chang JY, Woodward DJ, Han JS. Parallel pain processing in freely moving rats revealed by distributed neuron recording. Brain Res. 2003;992:263–271. doi: 10.1016/j.brainres.2003.08.059. [DOI] [PubMed] [Google Scholar]
- Wang JY, Zhang HT, Han JS, Chang JY, Woodward DJ, Luo F. Differential modulation of nociceptive neural responses in medial and lateral pain pathways by peripheral electrical stimulation: a multichannel recording study. Brain Res. 2004;1014:197–208. doi: 10.1016/j.brainres.2004.04.029. [DOI] [PubMed] [Google Scholar]
- Weng HR, Lenz FA, Vierck C, Dougherty PM. Physiological changes in primate somatosensory thalamus induced by deafferentation are dependent on the spinal funiculi that are sectioned and time following injury. Neuroscience. 2003;116:1149–1160. doi: 10.1016/s0306-4522(02)00796-0. [DOI] [PubMed] [Google Scholar]
- Winter CA, Kling PJ, Tocco DJ, Tanabe K. Analgesic activity of diflunisal [MK-647; 5-(2,4-difluorophenyl)salicylic acid] in rats with hyperalgesia induced by Freund's adjuvant. J Pharmacol Exp Ther. 1979;211:678–685. [PubMed] [Google Scholar]
- Yen CT, Shaw FZ. Reticular thalamic responses to nociceptive inputs in anesthetized rats. Brain Res. 2003;968:179–191. doi: 10.1016/s0006-8993(03)02235-2. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Wang N, Wang JY, Chang JY, Woodward DJ, Luo F. Ensemble encoding of nociceptive stimulus intensity in the rat medial and lateral pain systems. Mol Pain. 2011;7:64. doi: 10.1186/1744-8069-7-64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao P, Waxman SG, Hains BC. Modulation of thalamic nociceptive processing after spinal cord injury through remote activation of thalamic microglia by cysteine cysteine chemokine ligand 21. J Neurosci. 2007;27:8893–8902. doi: 10.1523/JNEUROSCI.2209-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]