

The X-ray crystal structure of ribosome hibernation promoting factor from V. cholerae has been determined at 2.0 Å resolution. The crystal was phased by two-wavelength MAD using cocrystallized cobalt.
Keywords: Vibrio cholerae, ribosome hibernation, hibernation promotion factor, ribosome-binding proteins, stationary phase, cold shock, MAD, cobalt, metal-binding site
Abstract
The X-ray crystal structure of ribosome hibernation promoting factor (HPF) from Vibrio cholerae is presented at 2.0 Å resolution. The crystal was phased by two-wavelength MAD using cocrystallized cobalt. The asymmetric unit contained two molecules of HPF linked by four Co atoms. The metal-binding sites observed in the crystal are probably not related to biological function. The structure of HPF has a typical β–α–β–β–β–α fold consistent with previous structures of YfiA and HPF from Escherichia coli. Comparison of the new structure with that of HPF from E. coli bound to the Thermus thermophilus ribosome [Polikanov et al. (2012 ▶), Science, 336, 915–918] shows that no significant structural changes are induced in HPF by binding.
1. Introduction
When bacteria encounter adverse conditions such as a lack of nutrients or low temperature, a number of metabolic changes occur that prepare the cell for survival until favorable conditions return. One of these is that protein synthesis, a major energy-consuming process in active cells, is strongly down-regulated. Table 1 ▶ summarizes the properties and synonyms of three proteins involved in ribosome inhibition under these conditions. Other transcriptional changes that take place during entry to the stationary phase have been described by Lange & Hengge-Aronis (1991 ▶) and Chang et al. (2002 ▶).
Table 1. Key to ribosome hibernation factors.
| Name used here (explanation) | YfiA† (name of gene) | RMF (ribosome modulation factor) | HPF† (hibernation promoting factor) |
|---|---|---|---|
| Alternate names | pY, RaiA | YhbH | |
| Associated with | Ribosome monomer | Dimer | Dimer |
The sequences of the genes encoding both of these proteins have often been annotated as ‘sigma modulation factor’.
Wada et al. (1990 ▶) noticed that this decrease in translation activity was associated with dimerization of ribosomes to the 100S state and with the presence of a protein, which they called ribosome modulation factor (RMF), bound to the ribosome. In 1999, Agafonov and coworkers discovered protein Y (pY), the product of the yfiA gene, in association with 70S ribosomes (Agafonov et al., 1999 ▶). In 2001, the same group showed that pY was absent from cells growing at normal temperature but was induced upon downshift to 277 K, and that pY inhibited protein translation at the stage of aminoacyl-tRNA binding (Agafonov et al., 2001 ▶). They renamed pY RaiA (ribosome-associated inhibitor A; Agafonov et al., 2001 ▶), although in recent literature it is mainly called YfiA. It has been proposed that this protein inhibits translation indirectly by enforcing a more stringent ribosomal proofreading mechanism (Agafonov & Spirin, 2004 ▶), although the details have not been resolved.
Meanwhile, Maki et al. (2000 ▶) showed that YfiA and another protein, the product of the yhbH gene, were associated with ribosomes in stationary phase but not with active ribosomes. YfiA was associated with monomeric (70S) ribosomes, while YhbH was associated with dimeric (100S) ribosomes. It was proposed that both proteins are involved in the stabilization and preservation of ribosomes in the stationary phase. In 2008, it was confirmed genetically that YfiA stabilizes monomers while YhbH stabilizes 100S dimers (Ueta et al., 2008 ▶). In the double knockout, RMF was still expressed and 90S dimers were formed. Ueta and coworkers coined the name hibernation promoting factor (HPF) for the yhbH gene product.
In 2001, the solution structure of the Haemophilus influenzae homolog of YfiA was reported (Parsons et al., 2001 ▶). It proved to be a new fold, with a four-stranded β-sheet and two α-helices which occur between the first and second and after the last strand, both resting diagonally against one surface of the sheet and against each other in an antiparallel orientation. This fold, which they named the double-stranded RNA-binding domain (DRBD), is distinct from that of a family of cold-shock proteins (CSPA–E; Graumann & Marahiel, 1996 ▶; Horn et al., 2007 ▶), which consist of a five-stranded β-barrel and are believed to function by binding mRNA. Within a year, two groups had independently determined solution structures of Escherichia coli YfiA (Rak et al., 2002 ▶; Ye et al., 2002 ▶). As expected, the structure was very similar to that of the H. influenzae homolog. In 2003, Vila-Sanjurjo and coworkers rigid-body refined the structure of E. coli YfiA against the 11 Å resolution diffraction data from a crystal of YfiA bound to the E. coli 70S ribosome to obtain a model of its interaction with the ribosome (Vila-Sanjurjo et al., 2004 ▶).
Structural studies of HPF lagged behind, with the solution structure of E. coli HPF (PDB entry 2rql) being determined in 2009 (Sato et al., 2009 ▶) and a crystal structure of the Coxiella burnetii homolog becoming available in 2011 (PDB entry 3tqm; M. Rudolph, J. Cheung, M. Franklin, M. Cassidy, E. Gary, F. Burshteyn & J. Love, unpublished work). In 2012, Steitz and coworkers provided a 3.1 Å resolution structure of the Thermus thermophilus ribosome with HPF bound (PDB entries 3v26, 3v27, 3v28 and 3v29; Polikanov et al., 2012 ▶). The HPF proteins share the same DRBD fold as the YfiA proteins, but the C-terminus is shorter, giving sequences of 95–102 amino acids compared with 110–116. Steitz and coworkers concluded that the longer tail of YfiA blocks the binding of RMF and thus prevents the dimerization of ribosomes.
γ-Proteobacteria such as E. coli and Vibrio cholerae have a short (95-residue) HPF and/or YfiA which is homologous but has a short C-terminal extension of about 15 residues (Ueta et al., 2008 ▶). On the other hand, α-proteobacteria, cyanobacteria and plant plastids have what is called long HPF, which consists of an ∼95-residue domain homologous to short HPF fused to a C-terminal domain of similar size and unknown function with no similarity to the short extension present in YfiA. These organisms do not have YfiA or RMF (Ueta et al., 2008 ▶).
Here, we report the crystal structure of HPF from the pathogenic enteric bacterium V. cholerae. The asymmetric unit contains two molecules cross-linked by a pair of cobalt ions from the crystallization mother liquor. This is the highest resolution structure available to date for a protein of this family.
2. Methods
HPF protein purified from a nonvirulent strain of V. cholerae (strain 0395-N1dToxT; Häse & Mekalanos, 1998 ▶; Hemp et al., 2005 ▶) was provided by Li-Shar Huang of our group as an unknown protein for practice in crystallization. (The protein had been saved as a byproduct of another purification project, and details of its purification and cell growth were not recorded.) Crystallization trays and precipitant screens were from Hampton Research.
2.1. Crystallization of HPF
In order to crystallize HPF, the protein was dissolved in 20 mM Tris pH 7.5, 0.5 mM EDTA and mixed with an equal volume of precipitant No. 25 from the Sigma Extension Kit for Proteins [1.8 M ammonium sulfate, 0.01 M cobalt(II) chloride, 0.1 M MES potassium salt pH 6.4] in a hanging drop. The addition of 5% 2 N sodium thiocyanate significantly increased the size and improved the appearance of the crystals. These conditions were used in a sitting-drop format to produce the crystal used in this study. The best resolution was obtained from a crystal cooled directly from the mother liquor; however, strong ice rings obliterated part of the data. Cryoprotection with 3 M ammonium sulfate and/or partial dehydration reduced the ice rings but the diffraction suffered, so the crystal cooled directly from the mother liquor was used for preliminary phasing and structure solution. Subsequent unsuccessful attempts to repeat the crystallization consumed the rest of the protein, of which no more was available. Because the protein turned out to be one of significant biological interest, we decided to refine and deposit the structure using the data we had.
2.2. Diffraction data collection, phasing and refinement
All diffraction data were collected on beamline F2 at Cornell High Energy Synchrotron Source (CHESS). Initially, 66 consecutive 1° oscillations were collected at a wavelength of 0.9790 Å and a crystal-to-detector distance of 200 mm with 100 s exposures (run 1, high resolution). Subsequently, 86 1° oscillations were collected at the same wavelength but with a crystal-to-detector distance of 400 mm and 20 s exposures to avoid saturating the strong low-resolution spots (run 2, low resolution). Following this, the crystal was dismounted and stored in liquid nitrogen. After the wavelength had been adjusted to a wavelength of 1.604 Å near the Co absorption edge, a third pass was made, collecting 97 images with a crystal-to-detector distance of 170 mm and an exposure time of 120 s to record the anomalous signal (run 3).
Diffraction spots were autoindexed and processed using DENZO from the HKL package (Otwinowski & Minor, 1997 ▶), which identified a tetragonal lattice. SCALEPACK from HKL scaled the data in point group P422. Systematic absences indicated that the space group was either P41212 or P43212. The high- and low-resolution runs 1 and 2 were scaled together (0.9790 Å data set) and the anomalous run 3 was scaled separately (1.604 Å data set). The data were output as unmerged intensities with the original indexing to allow local scaling by the phasing program. R merge was 0.044 (0.480 in the last shell) when Bijvoet mates were merged separately, or 0.051 (0.472 in the last shell) when they were merged together. The χ2 value also increased (from 1.436 to 1.754) when the Bijvoet mates were considered equivalent, indicating this is at least partly owing to anomalous differences even at the remote wavelength (f′′ ≃ 2 electrons).
For the final refinement, a thin shell of reflections in the resolution range 1.86–1.95 Å was deleted from the data set owing to a strong ice ring at 1.91 Å. Ice rings at lower resolutions did not seem to interfere as much, perhaps because the diffraction spots were stronger.
The cobalt present in the crystal was used to phase the diffraction data directly by two-wavelength MAD. Automated phasing was carried out with phenix.autosol (Adams et al., 2010 ▶), using the two SCALEPACK output files as ‘peak’ (1.604 Å) and ‘high-energy remote’ (0.9790 Å) wavelengths. No sequence was given at this time. Assuming space group P43212 gave an uninterpretable map in which only four residues were built. Assuming space group P41212, four heavy-atom sites were located and 122 residues of protein were built in 12 segments making up the four α-helices and eight β-strands of the final model, with no connecting linkers. Taking this as input to the ARP/wARP program (v.6.1.1; Cohen et al., 2008 ▶) with task ‘improve existing model’, 17 further residues were added, including four of the linkers, resulting in 139 residues in eight segments. Manual building into the density with O (Jones et al., 1991 ▶) using the now-recognized twofold noncrystallographic symmetry (NCS) gave a complete backbone for one of the monomers. This was submitted to the DALI server (Holm & Rosenström, 2010 ▶), which identified the fold as related to that of E. coli RaiA.
The sequence of E. coli RaiA was used in a BLAST (Altschul et al., 1990 ▶) search against the V. cholerae genome (Perez Chaparro et al., 2011 ▶), identifying two protein sequences initially both annotated as ‘sigma-54 modulation protein, putative’ and corresponding to E. coli YfiA (RaiA) and HPF. The sequence of YfiA could not be fitted into the density by phenix.autosol or ARP/wARP, but that of HPF could: phenix.autosol gave 126 residues with sequence assigned to 59. Starting with this model, ARP/wARP built 159 residues in four chains with sequence assigned to all. The two gaps were at different places in the two monomers, allowing each to be completed using NCS. Cobalt ions were placed at the four sites identified by phenix.autosol. Bulbous lobes of density connected to the metal sites and not accounted for by protein residues were initially modeled as Cl− ions. The density peaks were 4.7 Å from the metal but were strongly connected by a linear strand of density. These were later identified as thiocyanate ions, with electron-dense sulfur in the peaks and N ligating the metal in a linear end-on fashion.
Further rebuilding in O and refinement with CNS v.1.1 (Brünger et al., 1998 ▶) was carried out using twofold NCS, with numerous residues exempted because they showed obvious violation of NCS. The side chain of Arg47 in chain A showed clear density in two different positions and thus was modeled with two conformations. In conformation A the guanidino group lies between and interacts with Asp66 and Glu44, while in conformation B it is between Asp66 and Glu68.
Up until this point, the structure was refined against all of the data to 1.6 Å resolution, with the zone 1.86–1.95 Å excluded owing to ice problems as described above. The resulting structure, and the 1.6 Å resolution data, were deposited with the Protein Data Bank (Berman et al., 2012 ▶) as entry 4hei. According to the conventions of the PDB, the resolution of an X-ray structure is determined to be the highest resolution data used in refinement; hence, the resolution of 4hei is 1.6 Å. Data-reduction and refinement statistics at this stage, corresponding to the deposited structure, are presented in the second column of Table 1 ▶. However, exclusion of the ice ring together with the fact that 1.6 Å reflections were only present in the corners of the detector make the data set rather incomplete (81%). Owing to this incompleteness, and errors introduced by ice rings at lower resolution, the electron density does not resemble that of a high-resolution structure and the structure would not support evaluation as such.
In order to evaluate the structure solution in a resolution range that was more complete, we restricted the data to 1.96 Å resolution and continued refinement. Table 2 ▶ lists the data-reduction and refinement statistics for this reduced resolution range in the first column, and Supplementary Fig. S11 shows a density map calculated at 1.96 Å resolution using phases from the final model. The model did not change significantly with extensive refinement at the lower resolution, with the all-atom r.m.s.d. between the structures before and after limiting the resolution being 0.065 Å and the maximum deviation being 0.59 Å for a water molecule. The R factor and R free using all of the data were 0.262 and 0.281, respectively. Recalculating the R factors at limited resolution gave slightly better values (0.255 and 0.276, respectively), but upon refinement at the lower resolution R and R free diverged somewhat, presumably owing to the inferior data-to-parameter ratio, so that the final R free value was the same (0.281) with all of the data or with the restricted data range. This relatively high R free value even for a 2 Å resolution structure is probably caused by the failure of background and outlier rejection to completely eliminate errors in the data arising from the ice rings.
Table 2. Data-collection and refinement statistics for PDB entry 4hei .
| Remote | Peak | ||
|---|---|---|---|
| Data collection | |||
| Space group | P41212 | ||
| Unit-cell parameters (, ) | a = b = 46.35, c = 174.49, = = = 90 | ||
| Wavelength () | 0.9790 | 0.9790 | 1.604 |
| Resolution () | 43.61.96 (1.991.96) | 43.61.60 (1.631.60) | 992.70 (2.752.70) |
| R merge † | 0.048 (0.114) | 0.043 (0.408) | 0.053 (0.092) |
| I/(I) | 20.8 (10.1) | 15.4 (1.26) | 16.5 (9.5) |
| Completeness (%) | 98.0 (98.4) | 80.6 (18.5) | 98.8 (92.4) |
| Anomalous completeness‡ (%) | 95.8 (92.3) | 73.4 (12.9) | 85.5 (54.4) |
| Average multiplicity | 5.2 (3.2) | 4.1 (1.4) | 5.3 (2.9) |
| Wilson B factor (2) | 26.0 | 24.1 | 54.5 |
| Refinement | |||
| Resolution () | 43.61.96 (2.061.96) | 43.61.60 (1.681.60) | |
| No. of reflections | 14228 (1931) | 19305 (1032) | |
| R work | 0.252 (0.277) | 0.262 (0.432) | |
| R free | 0.281 (0.284) | 0.281 (0.394) | |
| No. of atoms | |||
| Protein | 1492 | 1492 | |
| Ligand/ion | 29 | 29 | |
| Water | 138 | 138 | |
| Solvent model | |||
| Density (e3) | 0.363 | 0.366 | |
| B factor (2) | 68.3 | 68.2 | |
| Average B factors (2) | |||
| Protein | 29.6 | 30.6 | |
| Ligand/ion | 24.8 | 25.7 | |
| Water | 28.8 | 31.4 | |
| R.m.s. deviations§ | |||
| Bond lengths () | 0.005 | 0.005 | |
| Bond angles () | 1.10 | 1.10 | |
| E.s.d. estimates | |||
| Cross-validated Luzzati plot¶ () | 0.33 | 0.33 | |
| Ramachandran plot (MolProbity) | |||
| Favorable | 172/180 [95.6%] | 172/180 [95.6%] | |
| Allowed | 180/180 [100%] | 180/180 [100%] | |
| Disallowed | 0 [0%] | 0 [0%] | |
| Rotamer outliers | 0 | 0 | |
| C deviations > 0.25 | 0 | ||
| All-atom clashscore†† | 18.71 | ||
R
merge = 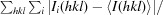
 for all hkl with two or more observations, merging data from individual diffraction images.
for all hkl with two or more observations, merging data from individual diffraction images.
Anomalous completeness is the completeness considering F + and F as separate non-equivalent reflections.
From the ideal stereochemistry values of Engh Huber (1991 ▶).
Steric overlaps of >0.4 per 1000 atoms.
Figures were produced with O and PyMOL (Schrödinger). Electrostatic maps were generated in UCSF Chimera (Pettersen et al., 2004 ▶) using the Coulombic Surface Coloring Tool, and overall charge was calculated using PDB2PQR (Dolinsky et al., 2004 ▶) and APBS (Baker et al., 2001 ▶). PISA (Protein Interfaces, Surfaces and Assemblies; Krissinel & Henrick, 2007 ▶) was used to identify residues and ligands involved in the dimer interface within the asymmetric unit.
3. Results
Fig. 1 ▶ shows crystals of HPF from V. cholerae. They were often in the shape of thick rods which were square or rectangular in cross section. Fig. 1 ▶(b) with the polarizers removed shows the true color, which is purple, presumably owing to the presence of cobalt ions.
Figure 1.

HPF crystals obtained in sitting-drop format. The color in (a) is false color from the polarizer, while the purplish color in (b) is the true color arising from bound cobalt.
3.1. Data statistics and refinement
Table 2 ▶ summarizes the refinement statistics. The second column reflects the refinement using all available data to 1.6 Å resolution, corresponding to the deposited structure. The overall R merge was 0.043, and there were dips in completeness but no spikes in R merge at the resolution of water rings, indicating that the integrating and scaling software was successfully rejecting spots in ice rings. Despite the excellent overall R merge, the crystallographic R factor was relatively high and spiked to around 0.6 in a narrow zone around the ice ring at 1.91 Å. This may have been owing to bad reflections that were measured only once, and therefore were not rejected as outliers and did not contribute to R merge. This prompted us to exclude the zone 1.86–1.95 Å from refinement and map calculation. Omitting this zone actually had little effect on the overall R factors, which were R = 0.262 and R free = 0.281. The high value of R free may still be owing to ice problems, as indicated by peaks in R cryst (but not R merge) in shells containing water spacings of 2.26 and 2.08 Å. The overall completeness of the data was low (80.6%) because the outer resolution shells were present only in the corners of the detector and a zone was excluded owing to an ice ring. However, in all shells below 2.27 Å resolution the completeness was greater than 95%. The total number of reflections used (19 305) would constitute a complete data set to 1.777 Å resolution; however, the effective resolution seemed to be lower than this, probably owing to errors introduced by ice rings. Column 1 of Table 2 ▶ evaluates the structure using only data to 1.96 Å resolution as described in §2.
The automatically determined (CNS v.1.1) solvent parameters were a density of 0.364 e Å−3 and a B factor of 68.3 Å2. Fixing the solvent density at 0.41 gave a slight improvement in R free, but this was not performed for the deposited structure. The stereochemical deviations from the values of Engh & Huber (1991 ▶) are 0.005 Å for bond lengths and 1.10° for bond angles.
In any case, the maps were unambiguous for most side chains. The ORF for V. cholerae HPF codes for 95 residues. Electron density was available to build residues 1–92 of chain A and residues 1–90 of chain B, with the last five residues being somewhat disordered in both chains. There was extra density on the N atom of Met1 in chain A which might represent a formyl group; however, we have not determined chemically whether the N-formyl group is actually retained. Apparently, the E. coli protein sequenced by Maki et al. (2000 ▶) was not blocked, implying that the formyl group is removed, so we left this density unmodeled.
The structure has 90.9% of the residues in the most favored region of the PROCHECK (Laskowski et al., 1993 ▶) Ramachandran plot and 95.6% in the favoured region of the MolProbity (Chen et al., 2010 ▶) Ramachandran plot (Supplementary Fig. S3). Lys45 in chain A is in the disallowed region of the PROCHECK plot. In the MolProbity plot no residues are disallowed, but residues 35 and 45 in both chains, residue 57 in chain A and residues 12, 56 and 90 in chain B are outside the favored region. Lys45 (in the loop between strands β2 and β3) and residue 90 (at the disordered C-terminus) are poorly ordered. The others are well defined by the density and are unlikely to be wrong. The Cα atoms of residues 1–90 in the two chains could be superimposed with an r.m.s.d. of 0.384 Å. Refining without NCS, this only increased to 0.410 Å. Since this is not much greater than the e.s.d. value of 0.33 Å estimated from the cross-validated (Kleywegt et al., 1994 ▶) Luzzati plot (Luzzati, 1952 ▶), this indicates a relatively rigid structure unperturbed by crystal packing. The overall APBS-derived charge on HPF of V. cholerae was −4.0 e, indicating the overall acidity of this protein (theoretical pI of 6.17).
There were three clashes closer than 2.2 Å, all in the same crystal contact, involving atoms in the vicinity of a twofold crystallographic axis. They are illustrated in Supplementary Fig. S2 and further described in the legend to this figure.
3.2. X-ray crystal structure of HPF
The overall structure in our crystal is shown in Fig. 2 ▶. The asymmetric unit contained two monomers of HPF, each having the DRBD fold β–α–β–β–β–α with both helices on the same side of the sheet. The sheets of the two monomers face towards each other and are linked by metal ions, as discussed in the next section. The helices which bind to the ribosome in the complex structures (Vila-Sanjurjo et al., 2004 ▶; Polikanov et al., 2012 ▶) are thus on the exterior surfaces of the dimer described here.
Figure 2.

(a) Cartoon structure of the HPF homodimer. Four cobalts (salmon spheres) coalesce the two monomers and the protein shows an overall β–α–β–β–β–α fold common to several ribosome-associated inhibitor A proteins. α-Helices and β-strands are labeled. (b) shows a close-up of the dimer interface; the spheres are cobalts, which are coordinated by histidines and glutamic acids as indicated. Co atoms are labeled with their PDB designation. (c) A surface representation of a monomer viewed on the dimer interface, with β-strands running from top right to bottom left. The prominent cavity is formed by residues 51-EAT in β3 and the side chains of the flanking residues Gln38 and Ile40 in β2 and His62 and His64 in β4. Slate and gray residues are those from monomers A and B, respectively, in (a) and (b). Figures were generated in PyMOL.
3.3. Metal-binding sites
The four anomalous scatterers identified by phenix.autosol are sandwiched between the β-sheets of the two monomers present in the asymmetric unit (see Fig. 2 ▶ a). They are provisionally assigned as Co atoms, since Co was the only element known to be present that has a strong anomalous signal at the wavelengths used. Their location in a crystal contact could explain why omitting cobalt chloride from the precipitant yielded no crystals. The presence of cobalt presumably imparts the purple color (Fig. 1 ▶ b) to the HPF crystals.
The metal ions make up the dimer interface in our asymmetric unit. We applied our structure to PISA in order to estimate the probability that the dimerization was of biological significance. Four hydrogen bonds and nine salt bridges were found between monomers A and B within 3.5 Å, which involved only the histidines and glutamic acids involved in metal binding. The complex-formation significance score (CSS) was 0.051. Details of the dimer interface with metal atoms are depicted in Fig. 2 ▶(b).
Co atom 1 (A101 in the deposited structure) is ligated by Glu60 in one monomer and His62 and His64 in the other. In addition there is one nonprotein ligand, which we have modeled as a thiocyanate ion (Fig. 3 ▶). Co atom 2 (A102) is ligated by His55 of the first monomer, Glu51 of the second monomer and two SCN− ions. His55 and Glu51 are located within β3, while Glu60, His62 and His64 are located within β4. The SCN− ions, oriented with N toward the cobalt and the electron-dense S in the bulbous distal expansion, gave an excellent fit to the density for one of the ligands on each cobalt (Fig. 3 ▶). This orientation is consistent with the known tendency of thiocyanate to ligate class A metals such as Co2+ with its N atom. The second nonprotein ligand on cobalt A102 and B102 was also modeled as thiocyanate, but the fit was less satisfactory. This position may have a mixture of thiocyanate ions and water. Co atoms 3 and 4 (B101 and B102) are related to atoms 1 and 2 by NCS, i.e. they occupy similar positions and density with the protein ligands symmetrically swapped. The NCS here at the interface between monomers is not very accurate, however. In particular, Glu60 interacts with the Co atoms differently, with Glu60 of chain A binding cobalt 101 with both carboxylate O atoms, while the O∊2 atom of Glu60 in chain B is closer to cobalt 102 (Fig. 2 ▶).
Figure 3.

Stereoviews of electron density around metal-binding sites. (a) Cobalt 1 (large gray sphere, B101 in the PDB file) and surroundings. Thiocyanate is modeled as a ball-and-stick representation, with its N atom binding to the cobalt ion. (b) Cobalt 2 (A102) with two thiocyanate ions. Selected residues of the protein are shown with blue-gray (chain A) or pink (chain B) C atoms. Co atoms 3 and 4 (A101 and B102) are symmetry-related to these two. The map is a 2F o − F c map is contoured at 0.58 e Å−3 (1.5σ). The figures were generated with the programs O (Jones et al., 1991 ▶), MolScript (Kraulis, 1991 ▶) and Raster3D (Merritt & Murphy, 1994 ▶).
4. Discussion
4.1. Metal-binding sites
What is the significance of the metal-binding sites in the crystal? In 2005, it was shown that cyclen, a small-molecule cobalt chelate, inhibited RNA translation (Delehanty et al., 2005 ▶). In this case, however, the inhibitor seems to interact with the mRNA, not with the ribosome. The Mg2+ concentration is critical for ribosome assembly and hibernation factor binding, with stable binding to ribosomes at 10 mM but ribosome dissociation and release of HPF and YfiA at 1 mM (Maki et al., 2000 ▶; Ueta et al., 2008 ▶). However, no metal was bound to our cobalt sites in the structure of E. coli HPF bound to ribosomes in the presence of ∼5 mM MgCl2 (Polikanov et al., 2012 ▶). In fact, the sites only really exist in the dimer, while only monomers were bound in the ribosome. Finally, the five residues involved in the binding sites (highlighted with an orange background in the alignment in Fig. 4 ▶) are not well conserved among γ-proteobacteria. E. coli has Asp in place of Glu51 and Ser in place of His64, and in the Coxiella protein none of these five residues are conserved. It seems likely that the dimer that we observe is purely a result of crystal packing and the metal-binding sites generated by dimerization have no biological significance.
Figure 4.

ClustalW alignment. Sequences are as follows: A, E. coli HPF; B, V. cholerae HPF; C, H. influenzae YfiA; D, E. coli YfiA; E, V. cholerae YfiA; F, C. burnetii HPF. The symbols below the sequences are as follows: *, identity; :, strongly similar; ., similar; a space indicates difference. β-Strands and α-helices are indicated as transparent green arrows and red bars, respectively. The numbering scheme follows the V. cholerae HPF sequence. Underlined residues are basic residues in the C-terminal portions of α1 and α2 of YfiA of E. coli involved in the proposed ribosome-recognition site, while gray boxes highlight highly conserved residues according to Ye et al. (2002 ▶). Alignments were made using ClustalW (Thompson et al., 1994 ▶).
4.2. Comparison with previous structures of ribosome hibernation factors
A number of structures of YfiA are available, but structures of HPF are more limited. As mentioned in §1, there is an NMR structure of E. coli HPF (Sato et al., 2009 ▶) and an X-ray structure of ‘ribosome subunit interface protein’ from C. burnetii at 2.45 Å resolution. While this paper was in preparation, the 3.1 Å resolution structure of the T. thermophilus ribosome with E. coli HPF bound became available (PDB entries 3v26, 3v27, 3v28 and 3v29; Polikanov et al., 2012 ▶). Fig. 4 ▶ shows the amino-acid sequence of V. cholerae HPF aligned with the sequences of C. burnetii and E. coli HPF and V. cholerae and E. coli YfiA using ClustalW (Thompson et al., 1994 ▶).
The structure and sequence of the Coxiella protein are clearly related to ribosome hibernation proteins and the closest sequence match in E. coli or V. cholerae is the HPF protein of these organisms. However, the sequence homology is rather low, with the Coxiella protein being less similar to HPF from V. cholerae than are YfiA from V. cholerae or E. coli. The overall fold is similar to HPF from V. cholerae or E. coli, but there are significant differences. As shown in Fig. 5 ▶, the last one-and-a-half turns of α1 are melted into random coil in the Coxiella protein and there is a single turn of helix just before the start of β2 that does not exist in V. cholerae HPF. These differences are not likely to be crystal-packing artefacts, as the same features are present in all four monomers in the Coxiella asymmetric unit, and the Vibrio features are present in both monomers of our crystal and are essentially the same as the E. coli HPF structures in PDB entries 2rql and 3v26.
Figure 5.

Stereoviews of the Cα-trace alignment of chain A of V. cholerae HPF (green) with chain A of C. burnetii HPF (yellow; PDB entry 3tqm). Notice the differences in secondary structure from the end of α1 to the start of β2. The figure was generated with O, MolScript and Raster3D. Residue numbering for the V. cholerae protein is indicated.
In fact, the E. coli HPF is quite similar to that of V. cholerae. Residues 2–90 of chain A of our structure superimpose on the first model of the NMR structure 2rlq with an r.m.s.d. of 1.22 Å. The structural alignment is shown in Fig. 6 ▶(a). No major secondary-structure differences were found, although β2 is fanned out with respect to 2rql.
Figure 6.

(a, c) HPF from E. coli (PDB entry 2rql, sky blue) and (b, d) YfiA from E. coli (PDB entry 1n3g, magenta) superimposed on one monomer of HPF from V. cholerae (our structure, green). β1 is shown in black in (b) and the C-terminal extension of 1n3g projects towards the bottom of the page. (c) and (d) show differences in contacts between the α1–β2 loop and α1 in the structures shown in (a) and (b), respectively. In (d), Leu85 is represented as sticks in each structure for ease of viewing; other key residues are represented as thick sticks. The structures were overlaid using the align command in PyMOL.
In Fig. 6 ▶(b), our structure of HPF from V. cholerae is aligned with that of YfiA from E. coli (PDB entry 1n3g; Rak et al., 2002 ▶). No major secondary-structure differences are present, although there are important differences in the way that the residues within the α1–β2 loop interact with α2. Strand β1 of 1n3g is assigned to only two residues in the deposited file and is therefore depicted this way in Fig. 6 ▶(b), but the Cα-atom positions do not deviate greatly from those of HPF (from E. coli or V. cholerae); this is thus probably a consequence of small inaccuracies in the orientation of the backbone atoms and the actual secondary structure is the same for the three proteins.
Next, we generated an electrostatic surface map of our structure in PyMOL (Fig. 7 ▶). Residues that are proposed to recognize rRNA are labeled in this figure, as well as the acidic Asp79 which separates the two basic patches generated by Lys80, Arg83 and Lys87 and by Lys23, Lys26 and Arg29.
Figure 7.
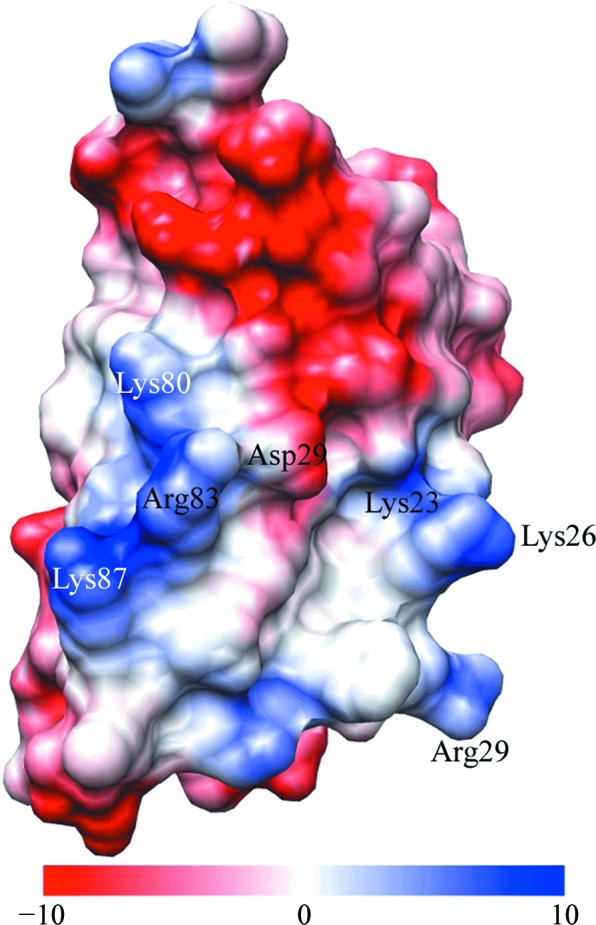
Electrostatic surface map of our structure, viewing the residues of the basic patch on the surface that faces towards the ribosome. Residues in the basic patch (blue) and Asp79 (red, acidic) are indicated; the electrostatic potential is given in kcal mol−1 e−1. This figure was generated using UCSF Chimera.
Additional biochemical studies would be useful in order to discern whether the presence of a C-terminal extension on its own is necessary and sufficient to generate the functional difference between YfiA and HPF. For example, does truncation of YfiA to the length of HPF make it perform as an HPF and stabilize dimers? Does fusion of the C-terminal extension of YfiA onto HPF make it function as YfiA? If not, then site-directed mutagenesis of the residues discussed above might be needed in order to determine the specificity-conferring factors.
4.3. Comparison of our crystalline and ribosome-bound HPF
Given the structural similarities between the structures of YfiA and HPF, it has been assumed that HPF binds a similar site in 30S ribosomes, and this has been confirmed recently by the abovementioned structure of the T. thermophilus ribosome with HPF or YfiA bound (Polikanov et al., 2012 ▶). This binding mode explains the fact that HPF does not inhibit translation in the presence of translation-initiation factors, which bind the A and P sites (Ueta et al., 2008 ▶). YfiA and YhbH show similar copy numbers when either is deleted (Ueta et al., 2005 ▶). The most significant difference between HPF and YfiA is the C-terminal extension of the latter, and this is believed to prevent RMF from binding in the presence of YfiA (Polikanov et al., 2012 ▶). However, these authors found that HPF, but not YfiA, induced a conformational change in the 30S subunit similar to that observed with RMF. Whether the C-terminal extension alone is responsible for the functional differences apparently has not been tested by expressing truncated YfiA or expressing HPF with the C-terminal extension of YfiA fused on, so the possibility remains that amino-acid differences contribute to the conformation that stabilizes dimers in one case and monomers in the other.
Although the HPF model in the structure of ribosome-bound HPF (PDB entries 3v26, 3v27, 3v28 and 3v29; Polikanov et al. 2012 ▶) was started with the solution structure 2rql, it was refined against 3.1 Å resolution data from the T. thermophilus ribosome cocrystallized with E. coli HPF and therefore represents the structure of the ribosome-bound HPF. Thus, comparing our crystal structure and/or the solution structure 2rql with the ribosome-bound structure can delineate any structural changes induced by binding. In fact there is very little change in the overall structure, with the Cα atoms of residues 2–90 of chain A superimposing on the ribosome-bound structure with an r.m.s.d. of 1.112 Å. The major deviations are in the loops between α1 and β2, between β2 and β3 and between β3 and β4 and at the end of α2. Excluding these and superimposing residues 2–7, 17–42, 48–56 and 61–90 gives an r.m.s.d. of 0.64 Å. Thus, the overall impression is that HPF binds as a rigid body, with no contortion required on the part of HPF, and inhibits translation by directly blocking the A and P sites.
Many side chains were truncated at Cβ in the ribosome-bound structure, presumably because of poor density owing to disorder or multiple conformations. Those that were modeled are generally in much the same conformation as in our structure, even when the residue type was not conserved. Exceptions include Met1, which extends in opposite directions in the two structures for no obvious reason, and Lys26 in the basic patch, which makes a salt bridge with an RNA phosphate in chain A of the ribosome. Asp32 in the α1–β2 loop is pointing in the opposite direction, possibly under the influence of an RNA phosphate 5 Å away. His62, which ring-stacks with a base in the ribosome, shows a different conformer in our structure. The deviation in the β2–β3 loop appears to arise from pressure from nucleic acids that would otherwise clash. The C-terminal five residues which are disordered in our structure are ordered in the ribosome structure, apparently owing to interactions between Lys93 and perhaps His95 and the C-terminal carboxylate with RNA in chains A and G of the ribosome.
5. Conclusions
The crystal structure in comparison with solution and ribosome-bound structures confirms that HPF is a rather rigid protein with little conformational change upon packing into the crystal or binding to the ribosome. Apparently, it is a rigid key fitting into its specific pocket at the interface of the ribosome subunits and stabilizing them, and does not interfere with the binding of RMF the way the similar hibernation factor YfiA does. This structure from the pathogenic V. cholerae is the first ribosome hibernation protein solved from any Vibrio species. Ribosome hibernation is likely to be important in the long-term survival of these typically marine disease-causing bacteria.
Supplementary Material
PDB reference: ribosome hibernation promoting factor, 4hei
Supporting information file. DOI: 10.1107/S1744309113000961/kw5057sup1.pdf
Acknowledgments
During the time of this work EAB was supported by new faculty start-up funds from the State University of New York (SUNY). HDB was supported by a student line from SUNY. We acknowledge the help of the beamline staff at CHESS for advice and help in beamline setup for the MAD experiment and Li-Shar Huang for supplying the protein, supervision of the crystallization experiments and help in data collection.
Footnotes
Supplementary material has been deposited in the IUCr electronic archive (Reference: KW5057).
References
- Adams, P. D. et al. (2010). Acta Cryst. D66, 213–221.
- Agafonov, D. E., Kolb, V. A., Nazimov, I. V. & Spirin, A. S. (1999). Proc. Natl Acad. Sci. USA, 96, 12345–12349. [DOI] [PMC free article] [PubMed]
- Agafonov, D. E., Kolb, V. A. & Spirin, A. S. (2001). EMBO Rep. 2, 399–402. [DOI] [PMC free article] [PubMed]
- Agafonov, D. E. & Spirin, A. S. (2004). Biochem. Biophys. Res. Commun. 320, 354–358. [DOI] [PubMed]
- Altschul, S. F., Gish, W., Miller, W., Myers, E. W. & Lipman, D. J. (1990). J. Mol. Biol. 215, 403–410. [DOI] [PubMed]
- Baker, N. A., Sept, D., Joseph, S., Holst, M. J. & McCammon, J. A. (2001). Proc. Natl Acad. Sci. USA, 98, 10037–10041. [DOI] [PMC free article] [PubMed]
- Berman, H. M., Kleywegt, G. J., Nakamura, H. & Markley, J. L. (2012). Biopolymers, 99, 218–222. [DOI] [PMC free article] [PubMed]
- Brünger, A. T., Adams, P. D., Clore, G. M., DeLano, W. L., Gros, P., Grosse-Kunstleve, R. W., Jiang, J.-S., Kuszewski, J., Nilges, M., Pannu, N. S., Read, R. J., Rice, L. M., Simonson, T. & Warren, G. L. (1998). Acta Cryst. D54, 905–921. [DOI] [PubMed]
- Chang, D. E., Smalley, D. J. & Conway, T. (2002). Mol. Microbiol. 45, 289–306. [DOI] [PubMed]
- Chen, V. B., Arendall, W. B., Headd, J. J., Keedy, D. A., Immormino, R. M., Kapral, G. J., Murray, L. W., Richardson, J. S. & Richardson, D. C. (2010). Acta Cryst. D66, 12–21. [DOI] [PMC free article] [PubMed]
- Cohen, S. X., Ben Jelloul, M., Long, F., Vagin, A., Knipscheer, P., Lebbink, J., Sixma, T. K., Lamzin, V. S., Murshudov, G. N. & Perrakis, A. (2008). Acta Cryst. D64, 49–60. [DOI] [PMC free article] [PubMed]
- Delehanty, J. B., Stuart, T. C., Knight, D. A., Goldman, E. R., Thach, D. C., Bongard, J. E. & Chang, E. L. (2005). RNA, 11, 831–836. [DOI] [PMC free article] [PubMed]
- Dolinsky, T. J., Nielsen, J. E., McCammon, J. A. & Baker, N. A. (2004). Nucleic Acids Res. 32, W665–W667. [DOI] [PMC free article] [PubMed]
- Engh, R. A. & Huber, R. (1991). Acta Cryst. A47, 392–400.
- Graumann, P. & Marahiel, M. A. (1996). Arch. Microbiol. 166, 293–300. [DOI] [PubMed]
- Häse, C. C. & Mekalanos, J. J. (1998). Proc. Natl Acad. Sci. USA, 95, 730–734. [DOI] [PMC free article] [PubMed]
- Hemp, J., Christian, C., Barquera, B., Gennis, R. B. & Martínez, T. J. (2005). Biochemistry, 44, 10766–10775. [DOI] [PubMed]
- Holm, L. & Rosenström, P. (2010). Nucleic Acids Res. 38, W545–W549. [DOI] [PMC free article] [PubMed]
- Horn, G., Hofweber, R., Kremer, W. & Kalbitzer, H. R. (2007). Cell. Mol. Life Sci. 64, 1457–1470. [DOI] [PMC free article] [PubMed]
- Jones, T. A., Zou, J.-Y., Cowan, S. W. & Kjeldgaard, M. (1991). Acta Cryst. A47, 110–119. [DOI] [PubMed]
- Kleywegt, G. J., Bergfors, T., Senn, H., Le Motte, P., Gsell, B., Shudo, K. & Jones, T. A. (1994). Structure, 2, 1241–1258. [DOI] [PubMed]
- Kraulis, P. J. (1991). J. Appl. Cryst. 24, 946–950.
- Krissinel, E. & Henrick, K. (2007). J. Mol. Biol. 372, 774–797. [DOI] [PubMed]
- Lange, R. & Hengge-Aronis, R. (1991). Mol. Microbiol. 5, 49–59. [DOI] [PubMed]
- Laskowski, R. A., MacArthur, M. W., Moss, D. S. & Thornton, J. M. (1993). J. Appl. Cryst. 26, 283–291.
- Luzzati, V. (1952). Acta Cryst. 5, 802–810.
- Maki, Y., Yoshida, H. & Wada, A. (2000). Genes Cells, 5, 965–974. [DOI] [PubMed]
- Merritt, E. A. & Murphy, M. E. P. (1994). Acta Cryst. D50, 869–873. [DOI] [PubMed]
- Otwinowski, Z. & Minor, W. (1997). Methods Enzymol. 276, 307–326. [DOI] [PubMed]
- Parsons, L., Eisenstein, E. & Orban, J. (2001). Biochemistry, 40, 10979–10986. [DOI] [PubMed]
- Pérez Chaparro, P. J., McCulloch, J. A., Cerdeira, L. T., Al-Dilaimi, A., Canto de Sá, L. L., de Oliveira, R., Tauch, A., de Carvalho Azevedo, V. A., Cruz Schneider, M. P. & da Silva, A. L. (2011). J. Microbiol. Methods, 87, 208–212. [DOI] [PubMed]
- Pettersen, E. F., Goddard, T. D., Huang, C. C., Couch, G. S., Greenblatt, D. M., Meng, E. C. & Ferrin, T. E. (2004). J. Comput. Chem. 25, 1605–1612. [DOI] [PubMed]
- Polikanov, Y. S., Blaha, G. M. & Steitz, T. A. (2012). Science, 336, 915–918. [DOI] [PMC free article] [PubMed]
- Rak, A., Kalinin, A., Shcherbakov, D. & Bayer, P. (2002). Biochem. Biophys. Res. Commun. 299, 710–714. [DOI] [PubMed]
- Sato, A., Watanabe, T., Maki, Y., Ueta, M., Yoshida, H., Ito, Y., Wada, A. & Mishima, M. (2009). Biochem. Biophys. Res. Commun. 389, 580–585. [DOI] [PubMed]
- Thompson, J. D., Higgins, D. G. & Gibson, T. J. (1994). Nucleic Acids Res. 22, 4673–4680. [DOI] [PMC free article] [PubMed]
- Ueta, M., Ohniwa, R. L., Yoshida, H., Maki, Y., Wada, C. & Wada, A. (2008). J. Biochem. 143, 425–433. [DOI] [PubMed]
- Ueta, M., Yoshida, H., Wada, C., Baba, T., Mori, H. & Wada, A. (2005). Genes Cells, 10, 1103–1112. [DOI] [PubMed]
- Vila-Sanjurjo, A., Schuwirth, B.-S., Hau, C. W. & Cate, J. H. D. (2004). Nature Struct. Mol. Biol. 11, 1054–1059. [DOI] [PubMed]
- Wada, A., Yamazaki, Y., Fujita, N. & Ishihama, A. (1990). Proc. Natl Acad. Sci. USA, 87, 2657–2661. [DOI] [PMC free article] [PubMed]
- Ye, K., Serganov, A., Hu, W., Garber, M. & Patel, D. J. (2002). Eur. J. Biochem. 269, 5182–5191. [DOI] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
PDB reference: ribosome hibernation promoting factor, 4hei
Supporting information file. DOI: 10.1107/S1744309113000961/kw5057sup1.pdf