

The crystal structure of inosine 5′-monophosphate dehydrogenase from P. aeruginosa has been determined to 2.25 Å resolution.
Keywords: inosine 5′-monophosphate dehydrogenase, Pseudomonas aeruginosa, antimicrobial drug targets
Abstract
Inosine 5′-monophosphate dehydrogenase (IMPDH) represents a potential antimicrobial drug target. The crystal structure of recombinant Pseudomonas aeruginosa IMPDH has been determined to a resolution of 2.25 Å. The structure is a homotetramer of subunits dominated by a (β/α)8-barrel fold, consistent with other known structures of IMPDH. Also in common with previous work, the cystathionine β-synthase domains, residues 92–204, are not present in the model owing to disorder. However, unlike the majority of available structures, clearly defined electron density exists for a loop that creates part of the active site. This loop, composed of residues 297–315, links α8 and β9 and carries the catalytic Cys304. P. aeruginosa IMPDH shares a high level of sequence identity with bacterial and protozoan homologues, with residues involved in binding substrate and the NAD+ cofactor being conserved. Specific differences that have been proven to contribute to selectivity against the human enzyme in a study of Cryptosporidium parvum IMPDH are also conserved, highlighting the potential value of IMPDH as a drug target.
1. Introduction
Inosine 5′-monophosphate dehydrogenase (IMPDH; EC 1.1.1.205) catalyzes the first committed reaction in de novo guanine-nucleotide biosynthesis. This enzyme converts inosine 5′-monophosphate (IMP) to xanthosine 5′-monophosphate (XMP), which can then be converted to guanosine 5′-monophosphate (GMP) by GMP synthetase. Guanine nucleotides are essential for signal transduction and DNA/RNA synthesis and IMPDH therefore plays a pivotal role in the growth and proliferation of both prokaryotic and eukaryotic cells (Hedstrom, 2009 ▶). The enzyme has been the subject of thorough kinetic and crystallographic studies. Structures of IMPDH from a variety of organisms are known, including pathogenic bacteria such as Borrelia burgdorferi (McMillan et al., 2000 ▶; PDB entry 1eep), Bacillus anthracis (Center for Structural Genomics of Infectious Diseases, unpublished work; PDB entry 3tsb) and Streptococcus pyogenes (Zhang et al., 1999 ▶), the protozoan parasites Cryptosporidium parvum (MacPherson et al., 2010 ▶; PDB entry 3ffs) and Tritrichomonas foetus (Whitby et al., 1997 ▶; PDB entry 1ak5), and human type II (Colby et al., 1999 ▶; PDB entry 1b30).
The enzyme mechanism is thought to start with a catalytic cysteine attacking the C2 position of IMP; an NAD+-dependent dehydrogenation then generates NADH and the covalent product intermediate E-XMP*. The expulsion of NADH promotes the insertion of a mobile flap to occupy the vacated cofactor-binding site. An arginine then acts as a general base catalyst and supports hydrolysis to convert E-XMP* into XMP and to recharge the enzyme (Hedstrom, 2009 ▶). IMPDH has been identified as a potential drug target for antimicrobial infections (Hedstrom et al., 2011 ▶). Progress has been made in developing highly potent inhibitors of the enzyme from C. parvum, which also display antiparasitic efficacy against Toxoplasma gondii (Gorla et al., 2012 ▶). Critically, these researchers have also addressed the issue of selectivity over the human homologue and a selectivity of greater than 1000-fold has been attained for the microbial enzymes.
Our interest is in the opportunistic Gram-negative pathogen Pseudomonas aeruginosa, which is widely recognized as a significant cause of hospital-acquired infections that often affect patients with compromised immune systems (Gaynes & Edwards, 2005 ▶). As part of a wide-ranging project to advance early-stage drug discovery against P. aeruginosa (Eadsforth et al., 2012 ▶; Moynie et al., 2013 ▶), we have adopted a structure-based approach (Hunter, 2009 ▶) to drive target assessment. Genetic evidence exists suggesting that the gene encoding IMPDH is essential in several bacteria, including P. aeruginosa (Liberati et al., 2006 ▶). We therefore sought to generate a recombinant source of the enzyme and to investigate its structure. Here, we report success in this endeavour and present the crystal structure of apo IMPDH from P. aeruginosa at 2.25 Å resolution. Comparisons with the structures of the enzyme from C. parvum (CpIMPDH) and Homo sapiens suggests that it may be possible to selectively inhibit the bacterial IMPDH over that of the host.
2. Methods
2.1. Protein expression and purification
The full-length gene (PA3770) for P. aeruginosa IMPDH (PaIMPDH; amino acids 1–489; UniProt ID Q9HXM5) was synthesized and codon-optimized for expression in Escherichia coli (GenScript). It was cloned into a modified pET15b (Novagen) cloning vector which encodes an N-terminal His tag followed by a tobacco etch virus (TEV) protease cleavage site and the protein of interest (Rao et al., 2011 ▶). The integrity of the construct was verified by DNA sequencing (DNA Sequencing Unit, University of Dundee). Recombinant PaIMPDH was produced in E. coli BL21 (DE3) pLysS cells (Stratagene) at 310 K in LB medium supplemented with 100 µg ml−1 ampicillin. The cells were grown to an OD600 of ∼0.7 before induction with isopropyl β-d-1-thiogalactopyranoside at a final concentration of 1 mM and growth at 293 K for a further 24 h. The cells were harvested by centrifugation (3500g at 277 K for 40 min). The cell pellet was resuspended in buffer (25 mM Tris–HCl pH 8.5, 500 mM NaCl, 20 mM imidazole) supplemented with an EDTA-free protease-inhibitor cocktail tablet (Calbiochem. The cells were lysed by passage through a continuous-flow cell disruptor (Constant Systems) at 207 MPa and the cell debris was removed following centrifugation (40 000g at 277 K for 20 min). PaIMPDH was purified using affinity chromatography on a 5 ml HisTrap HP column (GE Healthcare) pre-charged with Ni2+ and equilibrated in buffer A (25 mM Tris–HCl pH 8.5, 500 mM NaCl, 20 mM imidazole). A linear concentration gradient of imidazole was applied using buffer B (25 mM Tris–HCl pH 8.5, 500 mM NaCl, 500 mM imidazole) to elute the protein, which was then dialyzed against buffer C (25 mM Tris–HCl pH 7.5, 250 mM NaCl) at 277 K. Fractions were analyzed using SDS–PAGE and those containing PaIMPDH were pooled. The protein was further purified, with the His tag still present, by size-exclusion chromatography using a Superdex 75 26/60 column (GE Healthcare) equilibrated with buffer C on an ÄKTApurifier (GE Healthcare). This column had previously been calibrated with molecular-weight standards: blue dextran (>2000 kDa), thyroglobulin (669 kDa), ferritin (440 kDa), aldolase (158 kDa), conalbumin (75 kDa), ovalbumin (43 kDa), carbonic anhydrase (29.5 kDa), ribonuclease A (13.7 kDa) and aprotinin (6.5 kDa) (GE Healthcare; data not shown). The protein eluted as a single species of approximate mass 200 kDa. Fractions containing the protein were pooled and concentrated to 22 mg ml−1 using Amicon Ultra devices (Millipore) for subsequent use. A final yield of 25 mg per litre of bacterial culture was obtained. The purity of the protein was confirmed by SDS–PAGE and mass spectrometry (Fingerprint Proteomics Facility, University of Dundee). A theoretical extinction coefficient of 22 450 M −1 cm−1 at 280 nm was used to estimate the protein concentration (ProtParam; Gasteiger et al., 2005 ▶); the theoretical mass of one subunit was estimated as 54 kDa, with a calculated isoelectric point of 6.8. The purified protein was stored at 277 K in buffer C for subsequent use.
2.2. Crystallization, data collection and structure determination
A variety of commercial screens were employed to identify appropriate starting conditions for crystal growth. Diffraction-quality crystals of PaIMPDH were obtained at room temperature by sitting-drop vapour diffusion with a reservoir solution consisting of 0.2 M sodium acetate pH 4.5, 40% 2-methyl-2,4-pentanediol (MPD). Prismatic crystals appeared after a month from a mixture of equal volumes of protein solution (9 mg ml−1 in 25 mM Tris–HCl pH 7.5, 250 mM NaCl from the stock protein solution) and reservoir solution. The concentration of protein in the drop is therefore about 4.5 mg ml−1. The crystals grew to a maximum dimension of approximately 0.15 mm. The high concentration of MPD in the reservoir solution proved to be a suitable cryoprotectant and data were collected at 100 K using a Rigaku MicroMax-007 rotating-anode X-ray generator (Cu Kα, λ = 1.5418 Å) coupled to an R-AXIS IV++ image-plate detector. The data were indexed with XDS (Kabsch, 2010 ▶) and scaled using SCALA (Evans, 2006 ▶) from the CCP4 program suite (Winn et al., 2011 ▶). The structure was solved by molecular replacement with Phaser (McCoy et al., 2007 ▶) using the coordinates of a monomer (chain A) of IMPDH from B. anthracis (PDB entry 3tsb; Center for Structural Genomics of Infectious Diseases, unpublished work) as a search model. The search model shares 54% sequence identity with PaIMPDH. Model building and manipulation of the model were carried out using Coot (Emsley et al., 2010 ▶). Refinement calculations were performed using REFMAC5 (Murshudov et al., 2011 ▶) and translation/libration/screw analysis (TLS) was applied (Painter & Merritt, 2006 ▶). The refinement proceeded with the incorporation of water molecules following conservative criteria (Leonard & Hunter, 1993 ▶), a chloride and a number of side chains with dual conformers. The refinement was terminated when there were no significant changes in the R work and R free values and when inspection of the difference map suggested that no further corrections or additions were required. The stereochemistry and quality of the model were validated using MolProbity (Chen et al., 2010 ▶). Data-collection and structure-refinement statistics are shown in Table 1 ▶.
Table 1. Crystallographic statistics for PaIMPDH.
Values in parentheses are for the highest resolution shell.
| Space group | I4 |
| Unit-cell parameters (Å) | a = b = 115.5, c = 56.4 |
| Resolution (Å) | 19.1–2.25 (2.37–2.25) |
| No. of reflections recorded | 86989 (12454) |
| Unique reflections | 16612 (2476) |
| Completeness (%) | 94.6 (97.0) |
| Multiplicity | 5.2 (5.0) |
| 〈I/σ(I)〉 | 16.3 (4.3) |
| Wilson B (Å2) | 29.3 |
| No. of residues | 293 |
| No. of waters | 117 |
| R merge † (%) | 9.1 (48.9) |
| R work ‡ (%) | 14.8 |
| R free § (%) | 19.0 |
| Average B factor (Å2) | |
| Protein | 33.7 |
| Waters | 38.5 |
| Chloride | 72.1 |
| Cruickshank DPI¶ (Å) | 0.2 |
| Ramachandran plot | |
| Most favoured (%) | 97.3 |
| Additional allowed (%) | 2.0 |
| Outliers (%) | 0.7 |
| R.m.s.d. from ideal values†† | |
| Bond lengths (Å) | 0.01 |
| Bond angles (°) | 1.5 |
R
merge = 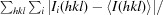
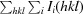 , where Ii(hkl) is the intensity of the ith measurement of reflection hkl and 〈I(hkl)〉 is the mean value of Ii(hkl) for all i measurements.
, where Ii(hkl) is the intensity of the ith measurement of reflection hkl and 〈I(hkl)〉 is the mean value of Ii(hkl) for all i measurements.
R
work = 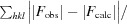
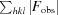 , where F
obs is the observed structure factor and F
calc is the calculated structure factor.
, where F
obs is the observed structure factor and F
calc is the calculated structure factor.
R free is the same as R cryst except that it was calculated with a subset (5%) of data that were excluded from the refinement calculations.
Diffraction precision index (Cruickshank, 1999 ▶).
Engh & Huber (1991 ▶).
3. Results and discussion
3.1. Structure determination
A highly efficient recombinant protein-expression system has been prepared and protocols for purification and crystallization have been established. The crystal structure of PaIMPDH was determined at 2.25 Å resolution. The asymmetric unit consists of a single polypeptide chain comprising residues 1–91, 205–370 and 426–467, with an estimated solvent content of 60% and a V M (Matthews, 1968 ▶) of 2.94 Å3 Da−1. The initial model consisted of 261 residues, with a correlation coefficient of 0.75 and R work and R free values of 37.2% and 41.8%, respectively. Subsequent model building and refinement extended this to 293 residues, with a correlation coefficient of 0.96 and improved R work and R free values of 14.8% and 19.0%, respectively. Analysis of the Ramachandran plot revealed that 99.3% of the residues were in the allowed region, with only two outliers: Gly226 and Gln458. These two residues are located on flexible loops linking α8 to β9 and α13 to β12, respectively, and the associated electron density is relatively poor. A greater degree of disorder was evident at several positions, where it was not even possible to interpret the electron density. Consequently, 196 residues are absent from the final model. A further comment on this will be made below.
3.2. Overall structure
The crystal structures of IMPDHs from several species have previously been determined and an excellent review has been published (Hedstrom, 2009 ▶). In brief, IMPDH forms homotetramers. The subunit is constructed from two segments: a catalytic domain which displays the (β/α)8-barrel fold and a subdomain containing two cystathionine β-synthase (CBS)/Bateman domains (Bateman, 1997 ▶; Hedstrom, 2009 ▶). The function of the CBS domains in this enzyme system is a source of debate (Hedstrom et al., 2011 ▶). It has been determined that the deletion of these subdomains has no effect on the enzymatic activity (Nimmesgern et al., 1999 ▶). This subdomain protrudes out from the catalytic domain and is usually disordered in IMPDH structures (Hedstrom, 2009 ▶).
PaIMPDH displays the canonical (β/α)8-barrel fold of the catalytic domain, with approximate dimensions of 40 × 40 × 50 Å (Fig. 1 ▶ a). One subunit comprises the asymmetric unit and the tetramer is formed by the crystallographic fourfold axis with symmetry operators −y, x, z; −x + 1/2, −y + 1/2, z − 1/2 and −y + 1/2, x − 1/2, z − 1/2 (Fig. 1 ▶ b). The tetramer has a square-planar shape with a disordered region of 114 residues located at the corners of the tetramer (Fig. 1 ▶ b). A comparison with the crystal structure of S. pyogenes IMPDH (PDB entry 1zfj; Zhang et al., 1999 ▶) confirms this missing region as corresponding to the disordered CBS domain (Fig. 2 ▶ a). The structure of PaIMPDH that we report is therefore consistent with other IMPDH structures (Hedstrom, 2009 ▶).
Figure 1.

(a) The crystal structure of PaIMPDH displays a (β/α)8-barrel fold. β-Strands and α-helices are coloured blue and red, respectively. (b) View parallel to the crystallographic fourfold axis showing the PaIMPDH tetramer.
Figure 2.

(a) Superposition of PaIMPDH (grey main-chain trace with red helices and blue strands) and S. pyogenes IMPDH (all yellow; PDB entry 1zfj) matches 293 residues with an r.m.s.d. of 1.1 Å based on a least-squares fit of Cα positions and highlights the missing residues in PaIMPDH as corresponding to the CBS subdomain, a mobile flap and C-terminal regions. (b) Close-up of the active site. The active-site loop of PaIMPDH (grey) is fully ordered and adopts an ‘open’ conformation compared with the ‘closed’ conformation observed when a ligand is bound to the S. pyogenes enzyme (yellow). The distances from the catalytic cysteine Sγ atoms to IMP C2 are approximately 6 and 3 Å in PaIMPDH and SpIMPDH, respectively.
3.3. The active site
The active site of PaIMPDH is located at the C-terminal end of the β-barrel, as is common for this (β/α)8-barrel fold (Hall et al., 2002 ▶), and at the edge there is a flexible loop (residues 297–315; Fig. 2 ▶ b). This loop adopts different conformations at different stages of the catalytic cycle and is often disordered in crystal structures (Hedstrom, 2009 ▶). Recent studies have shown that the active-site loop controls the transition between the dehydrogenase and hydrolase activities (Josephine et al., 2010 ▶). This loop is fully ordered in PaIMPDH, with an average B factor of 33.6 Å2, which is comparable to the average for the whole protein (33.7 Å2), and adopts an ‘open’ conformation in contrast to the ‘closed’ conformation observed in other IMPDH structures (Fig. 2 ▶ b).
3.4. Potential for selective inhibition of PaIMPDH
A sequence alignment of PaIMPDH with selected homologues shows that the key residues involved in catalysis are conserved across species (Fig. 3 ▶). However, variation in the residues interacting with the adenosine and pyrophosphate portions of the NAD+ cofactor have been observed (Fig. 3 ▶) and are believed to be good targets for achieving selective IMPDH inhibition (Hedstrom et al., 2011 ▶). Furthermore, PaIMPDH possesses a structural motif that indicates that this enzyme belongs to a group of IMPDHs that are susceptible to inhibitors developed to target CpIMPDH (MacPherson et al., 2010 ▶; Hedstrom et al., 2011 ▶). The compound N-(4-bromophenyl)-2-[2-(1,3-thiazol-2-yl)-1H-benzimidazol-1-yl]acetamide, labelled C64, is particularly relevant. This compound is a potent inhibitor of CpIMPDH, with an IC50 of about 30 nM, but not of the human type II enzyme. Several residues have been identified that contribute to this observation (MacPherson et al., 2010 ▶). Of particular importance are two residues that interact with the bromoaniline moiety of C64: Ala165 and Tyr358 (Gollapalli et al., 2010 ▶; Fig. 4 ▶). The alanine and tyrosine residues create a hydrophobic surface that forms van der Waals interactions with the bromoaniline group. These residues are conserved in bacterial homologues, including PaIMPDH (Ala249 and Tyr446; Figs. 3 ▶ and 4 ▶), but differ in the human enzyme (Ser276 and Asp470; Fig. 3 ▶). A further seven residues in CpIMPDH are implicated in assisting C64 binding and these are Ser22, Pro26, Thr221, Ser354, Gly357, Met308 and Glu329 (data not shown). In PaIMPDH these residues correspond to Gly19, Pro25, Thr306, Ala442, Gly445, Met393 and Glu417 (Fig. 3 ▶). This high degree of conservation extends to other bacterial homologues but not to the human enzyme (Fig. 3 ▶). This reinforces the idea that selectivity for the bacterial enzymes over human IMPDH is possible. With an efficient recombinant expression system for PaIMPDH and established crystallization conditions, the scene is set for a structure-based approach, noting that the reagents and data generated in the study of protozoan homologues might now be exploited to fast-track the discovery of new IMPDH inhibitors relevant to antibacterial drug discovery.
Figure 3.

Sequence alignment of PaIMPDH with homologues from T. foetus (GenBank ABI11203.1), H. sapiens (NCBI Reference Sequence NP_000875.2), B. anthracis (UniProt Q81W29) and S. pyogenes (UniProt P0C0H6). Residues involved in interactions with IMP, including the catalytic cysteine, are coloured green. Conserved residues interacting with NAD+ are coloured blue and nonconserved residues interacting with the adenosine and pyrophosphate portions are coloured magenta and orange, respectively. Two conserved residues on the mobile flap are coloured red. Residues involved in selectively binding the inhibitor C64 are highlighted in yellow. Residues missing in the PaIMPDH structure have a dashed line beneath them. The sequence alignment was generated and annotated using ClustalW (Larkin et al., 2007 ▶) and ALINE (Bond & Schüttelkopf, 2009 ▶), respectively.
Figure 4.

PaIMPDH (green) superimposed onto the structure of CpIMPDH (PDB entry 3khj; cyan) with the substrate IMP and the inhibitor C64 bound. Two residues involved in the selectivity of C64 towards the parasite enzyme IMPDH are shown and are also present in PaIMPDH. Residues marked with a prime are from the adjacent subunit that forms the catalytic tetramer.
Supplementary Material
PDB reference: inosine 5′-monophosphate dehydrogenase, 3zfh
Acknowledgments
This work was primarily funded by the European Commission Seventh Framework Programme (FP7/2007–2013) with contributions from The Wellcome Trust (grant Nos. 082596, 083481 and 094090). We thank Dr Paul Fyfe for excellent support.
References
- Bateman, A. (1997). Trends Biochem. Sci. 22, 12–13. [DOI] [PubMed]
- Bond, C. S. & Schüttelkopf, A. W. (2009). Acta Cryst. D65, 510–512. [DOI] [PubMed]
- Chen, V. B., Arendall, W. B., Headd, J. J., Keedy, D. A., Immormino, R. M., Kapral, G. J., Murray, L. W., Richardson, J. S. & Richardson, D. C. (2010). Acta Cryst. D66, 12–21. [DOI] [PMC free article] [PubMed]
- Colby, T. D., Vanderveen, K., Strickler, M. D., Markham, G. D. & Goldstein, B. M. (1999). Proc. Natl Acad. Sci. USA, 96, 3531–3536. [DOI] [PMC free article] [PubMed]
- Cruickshank, D. W. J. (1999). Acta Cryst. D55, 583–601. [DOI] [PubMed]
- Eadsforth, T. C., Gardiner, M., Maluf, F. V., McElroy, S., James, D., Frearson, J., Gray, D. & Hunter, W. N. (2012). PLoS One, 7, e35973. [DOI] [PMC free article] [PubMed]
- Emsley, P., Lohkamp, B., Scott, W. G. & Cowtan, K. (2010). Acta Cryst. D66, 486–501. [DOI] [PMC free article] [PubMed]
- Engh, R. A. & Huber, R. (1991). Acta Cryst. A47, 392–400.
- Evans, P. (2006). Acta Cryst. D62, 72–82. [DOI] [PubMed]
- Gasteiger, E., Hoogland, C., Gattiker, A., Duvaud, S., Wilkins, M. R., Appel, R. D. & Bairoch, A. (2005). The Proteomics Protocols Handbook, edited by J. M. Walker, pp. 571–607. Totowa: Humana Press.
- Gaynes, R. & Edwards, J. R. (2005). Clin. Infect. Dis. 41, 848–854. [DOI] [PubMed]
- Gollapalli, D. R., MacPherson, I. S., Liechti, G., Gorla, S. K., Goldberg, J. B. & Hedstrom, L. (2010). Chem. Biol. 17, 1084–1091. [DOI] [PMC free article] [PubMed]
- Gorla, S. K., Kavitha, M., Zhang, M., Liu, X., Sharling, L., Gollapalli, D. R., Striepen, B., Hedstrom, L. & Cuny, G. D. (2012). J. Med. Chem. 55, 7759–7771. [DOI] [PMC free article] [PubMed]
- Hall, D. R., Bond, C. S., Leonard, G. A., Watt, C. I., Berry, A. & Hunter, W. N. (2002). J. Biol. Chem. 277, 22018–22024. [DOI] [PubMed]
- Hedstrom, L. (2009). Chem. Rev. 109, 2903–2928. [DOI] [PMC free article] [PubMed]
- Hedstrom, L., Liechti, G., Goldberg, J. B. & Gollapalli, D. R. (2011). Curr. Med. Chem. 18, 1909–1918. [DOI] [PMC free article] [PubMed]
- Hunter, W. N. (2009). J. Biol. Chem. 284, 11749–11753. [DOI] [PMC free article] [PubMed]
- Josephine, H. R., Ravichandran, K. R. & Hedstrom, L. (2010). Biochemistry, 49, 10674–10681. [DOI] [PMC free article] [PubMed]
- Kabsch, W. (2010). Acta Cryst. D66, 125–132. [DOI] [PMC free article] [PubMed]
- Larkin, M. A., Blackshields, G., Brown, N. P., Chenna, R., McGettigan, P. A., McWilliam, H., Valentin, F., Wallace, I. M., Wilm, A., Lopez, R., Thompson, J. D., Gibson, T. J. & Higgins, D. G. (2007). Bioinformatics, 23, 2947–2948. [DOI] [PubMed]
- Leonard, G. A. & Hunter, W. N. (1993). J. Mol. Biol. 234, 198–208. [DOI] [PubMed]
- Liberati, N. T., Urbach, J. M., Miyata, S., Lee, D. G., Drenkard, E., Wu, G., Villanueva, J., Wei, T. & Ausubel, F. M. (2006). Proc. Natl Acad. Sci. USA, 103, 2833–2838. [DOI] [PMC free article] [PubMed]
- MacPherson, I. S., Kirubakaran, S., Gorla, S. K., Riera, T. V., D’Aquino, J. A., Zhang, M., Cuny, G. D. & Hedstrom, L. (2010). J. Am. Chem. Soc. 132, 1230–1231. [DOI] [PMC free article] [PubMed]
- Matthews, B. W. (1968). J. Mol. Biol. 33, 491–497. [DOI] [PubMed]
- McCoy, A. J., Grosse-Kunstleve, R. W., Adams, P. D., Winn, M. D., Storoni, L. C. & Read, R. J. (2007). J. Appl. Cryst. 40, 658–674. [DOI] [PMC free article] [PubMed]
- McMillan, F. M., Cahoon, M., White, A., Hedstrom, L., Petsko, G. A. & Ringe, D. (2000). Biochemistry, 39, 4533–4542. [DOI] [PubMed]
- Moynie, L., Schnell, R., McMahon, S. A., Sandalova, T., Boulkerou, W. A., Schmidberger, J. W., Alphey, M., Cukier, C., Duthie, F., Kopec, J., Liu, H., Jacewicz, A., Hunter, W. N., Naismith, J. H. & Schneider, G. (2013). Acta Cryst. F69, 25–34. [DOI] [PMC free article] [PubMed]
- Murshudov, G. N., Skubák, P., Lebedev, A. A., Pannu, N. S., Steiner, R. A., Nicholls, R. A., Winn, M. D., Long, F. & Vagin, A. A. (2011). Acta Cryst. D67, 355–367. [DOI] [PMC free article] [PubMed]
- Nimmesgern, E., Black, J., Futer, O., Fulghum, J. R., Chambers, S. P., Brummel, C. L., Raybuck, S. A. & Sintchak, M. D. (1999). Protein Exp. Purif. 17, 282–289. [DOI] [PubMed]
- Painter, J. & Merritt, E. A. (2006). J. Appl. Cryst. 39, 109–111.
- Rao, V. A., Shepherd, S. M., English, G., Coulthurst, S. J. & Hunter, W. N. (2011). Acta Cryst. D67, 1065–1072. [DOI] [PMC free article] [PubMed]
- Whitby, F. G., Luecke, H., Kuhn, P., Somoza, J. R., Huete-Perez, J. A., Phillips, J. D., Hill, C. P., Fletterick, R. J. & Wang, C. C. (1997). Biochemistry, 36, 10666–10674. [DOI] [PubMed]
- Winn, M. D. et al. (2011). Acta Cryst. D67, 235–242.
- Zhang, R., Evans, G., Rotella, F. J., Westbrook, E. M., Beno, D., Huberman, E., Joachimiak, A. & Collart, F. R. (1999). Biochemistry, 38, 4691–4700. [DOI] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
PDB reference: inosine 5′-monophosphate dehydrogenase, 3zfh